

Streptococcus suis (S. suis), a zoonotic bacterium found primarily in pigs and is now recognized as an emerging pathogen of humans.1 Human S. suis infections have been reported from more than 30 countries and/or regions, and the number of cases has increased considerably in the last decade, particularly in southeast Asian countries, such as China, Thailand, and Vietnam.1-3 While most sporadic S. suis infections in humans have been reported to be occupational, especially in Western countries, 2 outbreaks in China (Sichuan in 2005, and Jiangsu in 1998) and one in Thailand (Phayao in 2007) were reported. The outbreaks aroused considerable public health concern about S. suis emerging as a zoonotic agent.2
In a global estimate, the numbers of human infections of S. suis reported from China, Thailand and Vietnam account for 22-36% of all cases reported worldwide.2 Nearly 97% of all human cases of streptococcosis relate to serotype 2, but it has become increasingly important to genetically classify S. suis isolates according to their sequence type (ST) by multilocus sequence typing (MLST).1,4 To date, 33 and 10 STs have been recorded in Thailand and Vietnam, respectively (cf. MLST database; http://ssuis.mlst.net), showing considerable genetic diversity within S. suis from humans.4 However, only 3 sequence types, ST1, ST7 and ST377, have been found to be responsible for human infections in mainland China. Indeed, since the last outbreak in 2005, there has been no update on genetic diversity within S. suis and their geographic distribution in China.4-6 Therefore, it is timely and meaningful to explore the genetic composition of S. suis in China, where 2 outbreaks have occurred in the past 2 decades.
ST7 strains of S. suis associated with the 2 epidemics (1998 and 2005 in China) have only been found in China.4 Since the last outbreak in 2005, although sporadic human cases were reported,5-7 the nature and extent of sequence types and their relationships have not been explored.7 Here, the STs of the 76 S. suis isolates from patients during 2006-2016 and their geographic distribution/s were investigated and are shown in Figure 1A and Table S1. MLST was performed and STs were defined as described.8 Among the 76 S. suis isolates, the major STs characterized were ST1 (n = 32, 42.1%) and ST7 (n = 43, 56.6%). Interestingly, ST7 was the most dominant in Sichuan (ST1 = 4.0%, ST7 = 96.0%), where the 2005 epidemic occurred; ST1 was the most dominant in Guangdong (ST1 = 59.3%, ST7 = 40.7%) and Guangxi (ST1 = 57.1%, ST7 = 35.7%); ST1 was reported from humans in 8 provinces, and ST7 in 5 provinces of China (Fig. 1A). It is notable that sequence types ST1 and ST7 predominate in mainland China, and ST7, recognized to be a highly virulent and linked to the largest S. suis outbreak,9 did not appear to spread substantially since the outbreak of 2005.
Figure 1.
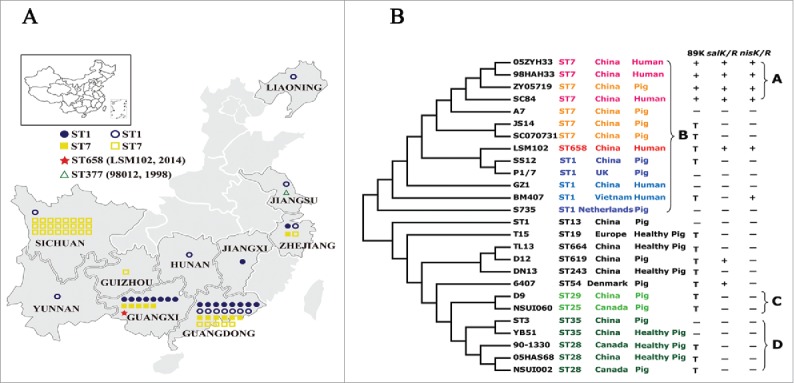
(A) Geographic distribution and sequence types of human S. suis isolates from 2006-2016 in this study. One symbol represents an isolate. Closed symbols denote isolates collected in this study, and open symbols denote isolates reported by literatures or MLST database. (B) The phylogeny of 26 completely sequenced S. suis strains. The Maximum likelihood tree was estimated by the SNPs of a core genome contained 885 genes. Bootstrapping was conducted using 500 replicates. The SC84 strain of S. suis was used as a reference. The label of each branch orderly showed the sequence type, the country and the host from which the S. suis strain was obtained. Then it marks whether or not the strain harbors the 89K PAI, salKR or nisKR (+ : the complete 89K PAI is included, salKR or nisKR; − : the complete 89K PAI is absent, salKR or nisKR; T : partial of 89K PAI associated region is detected).
According to the MLST database and NCBI, in Thailand, Vietnam, France and the Netherlands, totals of 33, 10, 6 and 6 STs have been recorded from humans and 9, 51, 6 and 34 STs from pigs, respectively. By contrast, in China, 354 STs have been recorded from swine, but only 4 distinct STs from human, namely ST1, ST7 and ST377 and the newly designated ST658 identified by us in the present study. A recent review shows that most strains isolated from humans are phenotypically and genotypically similar to those recovered from pigs within the same geographical region.9 However, the present finding of a major difference in the number of sequence types of S. suis from humans (n = 4; ST1, ST7, ST377 and ST658) compared with the 354 sequence types from pigs in China was unexpected. Although the reason for this discrepancy is unclear, it is possible that sampling bias may play a role. However, it is also possible that the limited numbers of STs in humans might relate to a particular or relatively specific host affiliation in China. Moreover, although the ST1 and ST7 found in human infection, were also the most important S. suis serotype 2 sequence types isolated from clinical pig in mainland China.4 It is striking that ST377 and ST658 were found exclusively in humans (not pigs), which is interesting considering S. suis is a zoonotic pathogen primarily and usually found in pigs, but with no direct evidence to date showing human-to-human infection.9 Therefore, future work should focus on a broader sampling from humans (from different countries in Asia) and comparative analyses with S. suis from pigs.
In this study, S. suis LSM102, isolated from a patient who had handled and eaten pork before admission, was characterized and designated as ST658. The clinical symptoms of the patient included fever, nausea, and general malaise. ST658 has a unique thrA, constituting a single locus variant consistent with ST1. Sequence types ST1 and ST658 differ in thrA1 and thrA162 by a point mutation at position 212 (G212 > A212; Fig. S1). The eBURST analysis assigned ST658 to the ST1 complex, which also contains ST7.4,9 The ST1 complex represents principally human cases associated with septicaemia and meningitis in pigs and humans in different parts of the world.4,9
The genome of S. suis LSM102 was sequenced using PacBio RS II (250-fold coverage) and Illumina HiSeq 2500 (400-fold coverage).10 First, de novo assembly of the sequencing reads from PacBio RS II was carried out using SMRT Analysis software(https://github.com/PacificBiosciences/SMRT-Analysis/wiki/SMRT-Pipe-Reference-Guide-v2.3.0). Then, single nucleotide and short indel errors were corrected using the programs SOAPsnp and SOAPindel, respectively, using HiSeq short reads. Gaps and low coverage areas were filled/verified using all available reads. Finally, the complete genome of LSM102 was deposited in GenBank under accession number CP016175.
The genome of S. suis LSM102 (ST658) is a single, circular chromosome of 2,067,932 bp with a GC content of 41.23% (CP016175). Screening of the complete genome of S. suis LSM102 using the ARGs database revealed a single tetracycline resistance gene (tetM) (Table S2). Consistently, LSM102 was resistant to tetracycline (data not shown). tetM is recognized as a selective factor that provides considerable advantages for the clonal emergence and spread of S. suis ST7.5 Comparative analysis showed that the tetM also exists in S. suis strains from human (05ZYH33, 98HAH33 and SC84) linked to 2 outbreaks,1,11 but is absent from the human strain GZ1 relating to sporadic infections (Table S2).12
To predict the virulence of S. suis LSM102, its genome was screened for 15 virulence factors linked to immune evasion or systemic infection, including SalKR, NisKR, Epf, Fhb, IgA1, IdeSsuis, MRP, Sly, Nudp, SsnA, EndA, ScpA and SsadS.1 All of the ‘outbreak’ strains from humans harboured these virulence factors, whereas those from sporadic cases contained 10 (Table S2), indicating the virulence potential of S. suis LSM102. The growth curve of LSM102 is similar to P1/7 (data not shown), a highly virulent, international reference strain of S. suis.11 To evaluate the virulence of S. suis strains, 10 6-week-old BALB/c mice (per group) were inoculated intraperitoneally with 2×108 colony forming units (CFU) of S. suis P1/7 or LSM102, as described previously.13 All the animal experimental protocols were approved by the Laboratory Animal Monitoring Committee of Huazhong Agricultural University and performed accordingly. The mortality rates of LSM102 and P1/7 infection groups were 90% and 70%, respectively. The morbidity and mortality analysis showed that almost all mice in LSM102 or P1/7 group presented severe clinical signs of sepsis, such as depression, rough hair coat, swollen eyes, weakness and prostration during the first 3 d post-infection. In addition, bacterial cytotoxicity to Caco-2 cell line was evaluated with Cyto-Tox 96 Cytoxicity Kit (Promega) according to the manufacturer's instructions. Caco-2 was incubated with log-phase strains (multiplicity of infection = 100:1) and kept at 37 °C for 4 h.8 The cytotoxicity levels of LSM102 and P1/7 were 75.5 ± 12 .6% and 4.4 ± 4.6%, respectively. Results above indicate that LSM102 is highly pathogenic.
To investigate the phylogenetic relationships of S. suis LSM102 (ST658) and other strains, trees (by maximum composite likelihood model14) were constructed using 63,061 SNPs from the 885 core genes of the 26 complete genomes available (Fig. 1B). Consistent with observations that the 4 closely related strains (98HAH33, SC84, 05ZYH33, and ZY05719) were responsible for outbreaks in China1,11 and grouped together in Clade A. ST7, ST658 and ST1 represented Clade B, revealing an intrinsic relationship among the isolates, and suggesting that ST658 originates from the ST1 complex. In addition, human and pig isolates representing the ST1 complex grouped as Clade B, indicating their intrinsic relationship. Strain D9 (ST29) from China and NSUI060 (ST25) from Canada belonging to the ST25 complex grouped within Clade C, whereas strains ST3, 05HAS68, and YB51 from China and strains 90-1330 and NSUI002 from Canada belonging to the ST28 complex grouped together in Clade D. Taken together, these observations imply that the strains from China and Canada derive from a recent common ancestor or transfer between countries.
The global comparison of the 26 complete genomes of S. suis available showed that 89 kb pathogenicity island (89 K PAI)-associated region displayed a high degree of diversity (Fig. 2A). The 89 K PAI is specific to highly pathogenic S. suis linked to Chinese epidemics; 89 K PAI was found in strains 98HAH33, SC84, 05ZYH33; and, ZY05719 related to 2 outbreaks in China, but not sporadic infections (e.g., GZ1), while it is absent from isolates from North America and Europe.1 The 4 ST7 strains (98HAH33, SC84, 05ZYH33, and ZY05719) responsible for 2 outbreaks (in 1998 and 2005) carry full-length 89K PAIs. However, the highly pathogenic strain GZ1 (ST1), isolated in 2005 from a sporadic human case (causing septicaemia) in Guizhou Province of China did not harbour 89K PAI (Table S2).
Figure 2.
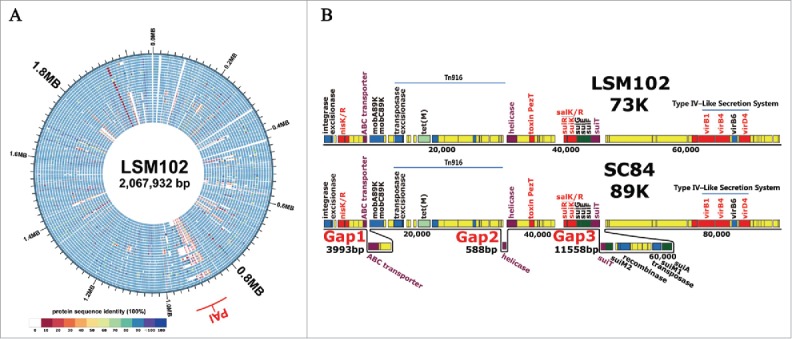
(A) Genomic comparison of 26 S. suis genomes. The circular diagrams showed the variations of LSM102 compared with the other completely sequenced S. suis strains. From outside to the inside, the order of the genomes was: LSM102, SS12, P1/7, ZY05719, SC84, 98HAH33, 05ZYH33, A7, SC070731, JS14, GZ1, BM407, S735, ST1, TL13, D12, DN13, T15, 6407, NSUI060, D9, NSUI002, 05HAS68, 90-1330, YB51, ST3. The different colors stand for the percentage of protein sequence identity. (B), Gene organization of 73 kb pathogenicity island (73K PAI) of LSM102. Virulence-related factors including SalKR, NisKR and Type IV–Like Secretion System (VirD4, VirB1, VirB4) that have been reported to be involved in full virulence of STSS-causing Chinese S. suis were included in 73K PAI of LSM102. Compared with 89K, 3 fragment losses/deletions occur in 73K PAI of LSM102, which were designated as Gap1, Gap2, and Gap3 respectively.
Distinct from GZ1, a mobile pathogenicity island of 72,679 bp (designated 73K PAI) likely derives from 89K PAI found in S. suis LSM102 (from a sporadic human case from Guangxi Province in China; see Fig. 2B). Compared with 89K PAI, the 73K PAI lacks 3 fragments (designated as Gap1, Gap2 and Gap3); however, elements including Integrase, Excisionase, MobA, MobC, Helicase, VirB4, VirB6 and VirD4 which are required for the 89K PAI excision and circularization, as well as virulence factors, including SalKR, NisKR and Type IV–Like Secretion System which are involved in ‘complete’ virulence in streptococcal toxic shock syndrome (STSS)-causing Chinese S. suis, are all encoded in 73K PAI of S. suis LSM102 (Fig. 2B).1,11,15 This observation suggests that horizontal transfer of 89K PAI occurred from ST7 to ST658, or from ST7 to ST1, which have evolved to become ST658. Furthermore, comparison of the 89K PAI-associated region from available genomes from different countries showed that PAI regions of strains are variegated and can be grouped into 4 types according to the features and structures of the PAI-associated region (Fig. S2). The comparative results provided evidence that strains can exhibit gene loss or gain in the 89K PAI-associated region, implying the 89K PAI associated region is disseminating globally and evolving in circulation.
In summary, this study demonstrates a predominance of ST1 and ST7 in a panel of S. suis isolates (n = 76) from Chinese patients collected since the last outbreak of human streptococcosis in China (2005). Comparison of the 89K PAI-associated region among 26 strains with 73K PAI in ST658 (a novel sequence type) provides some evidence that 89K PAI was transferred horizontally from ST7 to ST658 (or ST1) and has diversified during transmission. A better understanding of novel STs in human patients and the evolution of 89K PAI linked to 2 outbreaks in humans should provide insights into the emergence, virulence and evolution of this important pathogen. Furthermore, it may also provide some clues as to the pathogenesis of S. suis.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We acknowledge Sisi Xie and Shuqing Liang for genomic sequencing at Shenzhen Total Genomics Solution Institute. We would like to thank Dr. Stephan Willias for critical reading the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (31502080, 31402341), the Special Fund for Agro-scientific Research in the Public Interest (2014BBB016), the Open Project Program of Jiangsu Key Laboratory of Zoonosis (R1605), the Fundamental Research Funds for the Central Universities (2662016QD010) and the China Postdoctoral Science Foundation (2015T80819).
References
- [1].Feng Y, Zhang H, Wu Z, Wang S, Gao M, Hu D, Wang C. Streptococcus suis infection: an emerging/reemerging challenge of bacterial infectious diseases? Virulence 2014; 5:477-97; PMID:24667807; http://dx.doi.org/ 10.4161/viru.28595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Huong VT, Ha N, Huy NT, Horby P, Nghia HD, Thiem VD, Zhu X, Hoa NT, Hien TT, Zamora J, et al.. Epidemiology, Clinical Manifestations, and Outcomes of Streptococcus suis Infection in Humans. Emerg Infect Dis 2014; 20:1105-14; PMID:24959701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Praphasiri P, Owusu JT, Thammathitiwat S, Ditsungnoen D, Boonmongkon P, Sangwichian O, Prasert K, Srihapanya S, Sornwong K, Kerdsin A, et al.. Streptococcus suis infection in hospitalized patients, Nakhon Phanom Province, Thailand. Emerg Infect Dis 2014; 21:345-8; http://dx.doi.org/ 10.3201/eid2102.140961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Goyette-Desjardins G, Auger JP, Xu J, Segura M, Gottschalk M. Streptococcus suis, an important pig pathogen and emerging zoonotic agent-an update on the worldwide distribution based on serotyping and sequence typing. Emerg Microbes Infect 2014; 3:e45; PMID:26038745; http://dx.doi.org/ 10.1038/emi.2014.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ye C, Bai X, Zhang J, Jing H, Zheng H, Du H, Cui Z, Zhang S, Jin D, Xu Y, et al.. Spread of Streptococcus suis sequence type 7, China. Emerg Infect Dis 2008; 14:787-91; PMID:18439362; http://dx.doi.org/ 10.3201/eid1405.070437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Li W, Ye C, Jing H, Cui Z, Bai X, Jin D, Zheng H, Zhao A, Xu Y, Gottschalk M, et al.. Streptococcus suis outbreak investigation using multiple-locus variable tandem repeat number analysis. Microbiol Immunol 2010; 54:380-8; PMID:20618684; http://dx.doi.org/ 10.1111/j.1348-0421.2009.00178.x [DOI] [PubMed] [Google Scholar]
- [7].Feng Y, Shi X, Zhang H, Zhang S, Ma Y, Zheng B, Han H, Tang J, Cheng J, Gao GF, et al.. Recurrence of Human Streptococcus suis Infections in 2007: Three Cases of Meningitis and Implications that Heterogeneous S. suis 2 Circulates in China. Zoonoses Public Health 2009; 56:506-14; PMID:19538458; http://dx.doi.org/ 10.1111/j.1863-2378.2008.01225.x [DOI] [PubMed] [Google Scholar]
- [8].Ye C, Zhu X, Jing H, Du H, Segura M, Zheng H, Kan B, Wang L, Bai X, Zhou Y, et al.. Streptococcus suis sequence type 7 outbreak, Sichuan, China. Emerg Infect Dis 2006; 12:1203-8; PMID:16965698; http://dx.doi.org/ 10.3201/eid1708.060232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gottschalk M, Xu J, Calzas C, Segura M. Streptococcus suis: a new emerging or an old neglected zoonotic pathogen? Future Microbiol 2010; 5:371-91; PMID:20210549; http://dx.doi.org/ 10.2217/fmb.10.2 [DOI] [PubMed] [Google Scholar]
- [10].Shim D, Park SG, Kim K, Bae W, Lee GW, Ha BS, Ro HS, Kim M, Ryoo R, Rhee SK, et al.. Whole genome de novo sequencing and genome annotation of the world popular cultivated edible mushroom, Lentinula edodes. J Biotechnol 2016; 223:24-5; PMID:26924240; http://dx.doi.org/ 10.1016/j.jbiotec.2016.02.032 [DOI] [PubMed] [Google Scholar]
- [11].Chen C, Tang J, Dong W, Wang C, Feng Y, Wang J, Zheng F, Pan X, Liu D, Li M, et al.. Glimpse of streptococcal toxic shock syndrome from comparative genomics of S. suis 2 Chinese isolates. PLoS One 2007; 2(3):e315; PMID:17375201; http://dx.doi.org/ 10.1371/journal.pone.0000315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ye C, Zheng H, Zhang J, Jing H, Wang L, Xiong Y, Wang W, Zhou Z, Sun Q, Luo X, et al.. Clinical, experimental, and genomic differences between intermediately pathogenic, and epidemic Streptococcus suis. J Infect Dis 2009; 199(1):97-107; http://dx.doi.org/ 10.1086/594370 [DOI] [PubMed] [Google Scholar]
- [13].Li J, Xia J, Tan C, Zhou Y, Wang Y, Zheng C, Chen H, Bei W. Evaluation of the immunogenicity and the protective efficacy of a novel identified immunogenic protein, SsPepO, of Streptococcus suis serotype 2. Vaccine 2011; 29:6514-9; PMID:21767591; http://dx.doi.org/ 10.1016/j.vaccine.2011.07.010 [DOI] [PubMed] [Google Scholar]
- [14].Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011; 28(10):2731-9; PMID:21546353; http://dx.doi.org/ 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li M, Shen X, Yan J, Han H, Zheng B, Liu D, Cheng H, Zhao Y, Rao X, Wang C, et al.. GI-type T4SS-mediated horizontal transfer of the 89K pathogenicity island in epidemic Streptococcus suis serotype 2. Mol Microbiol 2011; 79:1670-83; http://dx.doi.org/ 10.1111/j.1365-2958.2011.07553.x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.