

Abstract
Purpose
Targeting the B-cell receptor (BCR) pathway with inhibitors of BTK and PI3K-delta is highly effective for the treatment of chronic lymphocytic leukemia (CLL). However, deep remissions are uncommon and drug resistance with single-agent therapy can occur. In vitro studies support the effectiveness of combing PI3K-delta and BTK inhibitors.
Experimental design
As CLL proliferation and survival depends on the microenvironment, we used murine models to assess the efficacy of the BTK inhibitor acalabrutinib combined with the PI3K-delta inhibitor ACP-319 in vivo. We compared single-agent with combination therapy in TCL1-192 cell-injected mice, a model of aggressive CLL.
Results
We found significantly larger reductions in tumor burden in the peripheral blood and spleen of combination-treated mice. While single-agent therapy improved survival compared with control mice by a few days, combination therapy extended survival by over two weeks compared to either single agent. The combination reduced tumor proliferation, NF-KB signaling and expression of BCL-xL and MCL-1 more potently than single-agent therapy.
Conclusion
The combination of acalabrutinib and ACP-319 was superior to single-agent treatment in a murine CLL model, warranting further investigation of this combination in clinical studies.
Keywords: CLL, BTK, PI3K
Introduction
Chronic Lymphocytic Leukemia (CLL) is the most common leukemia in western countries with more than 15,000 newly diagnosed cases each year in the US alone.(1) CLL is characterized by the accumulation of mature, clonal B cells in blood, bone marrow and lymphoid tissues. The proliferative compartment of CLL is mainly located in the lymphoid tissues, wherein CLL cells undergo B-cell receptor (BCR) and NF-κB activation, emphasizing the importance of microenvironmental interactions in lymphoid tissues for disease progression.(2, 3)
While chemo-immunotherapy is an effective first-line treatment for most patients with CLL, patients with TP53 aberrations or relapsed disease have benefited significantly from targeted therapies.(4, 5) The Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib and the phosphoinositide 3-kinase δ (PI3Kδ) inhibitor idelalisib are approved for clinical use and have demonstrated impressive results.(6–8) However, indefinite treatment seems required with single-agent therapy and drug resistance develops in a subset of patients.(9, 10)
In vitro studies with ibrutinib and idelalisib demonstrated synergy of combined PI3Kδ and BTK inhibition.(11, 12) However, in vivo data on the efficacy of combined PI3Kδ and BTK inhibition are missing. Acalabrutinib is a novel, potent and highly selective BTK inhibitor that, like ibrutinib, binds covalently to Cys481 in the ATP-binding pocket and has shown similar efficacy in murine models of CLL.(13) In a phase I/II clinical trial, acalabrutinib had an objective response rate of 95% at median follow-up of 14.3 months and 90% progression-free survival at 18 months.(14) ACP-319 (AMG319), a novel PI3Kδ inhibitor, was tolerable and showed promising activity in a phase I trial with 13/15 CLL patients remaining on treatment after a median follow up of 30 weeks.(15)
Because of the critical importance of the tumor-microenvironment in the pathogenesis of CLL, in vivo models that recapitulate tumor-microenvironment interactions are valuable tools to investigate the effects of targeted agents.(2, 13, 16–19) Therefore, we investigated the combination of acalabrutinib and ACP-319 in a murine CLL model derived from the well-established transgenic Eμ-TCL1 (TCL1) mouse model.(20) Through serial passaging of Eμ-TCL1 splenocytes in immunodeficient mice, a BCR-dependent cell line, TCL1-192, was derived. Adoptive transfer of TCL1-192 cells into immunodeficient mice leads to the development of an aggressive CLL-like disease resulting in death within five to six weeks.(18)
Herein, we utilized a murine model of CLL to investigate the efficacy and on-target effects of dual PI3Kδ and BTK inhibition in vivo by ACP-319 and acalabrutinib. Our data demonstrates a significant benefit of combination treatment, which is likely due to more potent inhibition of survival mechanisms that result in increased tumor cell death.
Materials and Methods
Murine allotransplant of CLL-like TCL1-192 cells
All animal handling and housing followed the guidelines set forth by the animal care and use committee of the NHLBI. Allografting of TCL1-192 cells (provided by N. Chiorazzi)(18) into 2-10 week old male NOD-SCID mice (Jax-1303; Jackson Laboratory) was carried out as previously described.(18) Briefly, 5×106 freshly thawed TCL1-192 splenic cells, in PBS, were injected retro-orbitally into 16–20 mice recipient mice for each experimental cohort, split equally between four treatment groups, vehicle, acalabrutinib, ACP-319 and combination. TCL1-192 cell proliferation (leukemia) in peripheral blood was proven by flow cytometry one or two weeks after cell injection.
Treatment of mice with kinase inhibitors or vehicle and handling of samples
Treatment for TCL1-192 allografted NOD-SCID mice was initiated 7–21 days after cell injection with drinking water (provided by Acerta Pharma, Redwood City, CA, USA) containing vehicle (2% w/v HPβCD) and 0.15 mg/mL acalabrutinib, vehicle and ACP-319 0.15 mg/mL, a combination of the two drugs or vehicle alone. This regimen results in an average daily dose of 25 mg/kg of each drug.(3, 19) TCL1-192 allografted mice were bled weekly or biweekly and followed for survival until death or predetermined morbidity endpoints, with the exception of two cohorts that were sacrificed four weeks post cell injection. Peripheral blood (PB) and spleens were harvested at sacrifice. Single-cell suspensions of splenocytes were obtained by manually homogenizing spleens and filtering through 70 nm nylon sieves (BD Falcon, Franklin Lakes, NJ, USA). Cells were analyzed immediately after erythrocyte lysis using ACK buffer (Quality Biological, Inc., Gaithersburg, MD, USA).
Flow cytometry
To determine tumor burden and the percentage of live cells, PBMCs and tissue-derived, single-cell suspensions were stained with LIVE/DEAD fixable violet solution (VIVID; Invitrogen, Grand Island, NY, USA) and Annexin-V (BD Biosciences, Franklin Lakes, NJ, USA) according to manufacturer’s instructions. Mouse anti- CD45R/B220 and CD5 were used to identify TCL1-192 cells. Determination of absolute cell counts was done using AccuCount blank particles (Spherotech, Lake Forest, IL, USA). Intracellular staining was done as previously described.(3) Briefly, cells were fixed in 4% paraformaldehyde, permeabilized in 90% methanol or 80% ethanol at −20°C and stained with surface antibodies and intracellular stains. Intracellular stains included anti- Ki67, phospho-PLCγ2(Y759), phospho-ERK1/2(T202/Y204) and phospho-p65(S529) (BD Biosciences). Cut-off for positive cells was determined by isotype controls. Cells were analyzed on a FACS Canto II flow cytometer using FACS-DIVA (BD Biosciences) and FlowJo software (version 9; TreeStar, Ashland, OR, USA). For some experiments, cells were barcoded with different concentrations of pacific orange and pacific blue (Invitrogen) to allow for analysis of multiple samples in one tube.(21)
BTK Occupancy Probe ELISA
This method was run as previously described.(14) Briefly, 96-well OptiPlates were coated overnight with a mouse anti-human BTK antibody (BD Biosciences). Cyropreserved splenocyte cell pellets were lysed with buffer containing digitonin and a Protease Inhibitor Cocktail. Lysates were incubated for one hour on ice in the presence or absence of a saturating concentration of acalabrutinib (10−6 M), followed by incubation with a biotinylated derivate (ACP-4016; 10−7 M) serving as a probe. After incubation of lysates in OptiPlates and a subsequent incubation with Streptavidin-HRP (Invitrogen; ELISA grade), SuperSignal ELISA Femto Substrate (Thermo Fisher Scientific) was added and chemiluminescence was measured on an EnVision® multilabel plate reader (PerkinElmer, Waltham, MA, USA). The percentage BTK occupancy for the drug-treated mice was calculated relative to the average signal from the vehicle control group. The sample without exogenous acalabrutinib represents 100% free BTK (or 0% occupied BTK), and the samples with exogenous acalabrutinib represents 0% free BTK (or 100% occupied BTK).
SDS-PAGE Western Blot analysis
Whole cell lysates were made from splenocytes in a RIPA lysis buffer with PhosSTOP phosphatase inhibitor cocktail and complete Protease Inhibitor Cocktail (Roche Applied Science, Indianapolis, IN, USA). Proteins were separated on Novex® NuPAGE® sodium dodecyl sulfate-polyacrylamide gel (Invitrogen), transferred to polyvinylidene fluoride membranes and immunoblotted using the indicated antibodies and horseradish peroxidase–labeled secondary antibodies (GE Healthcare Biosciences, Pittsburgh, PA, USA). Blots were developed by chemiluminescence (Thermo Fisher Scientific), recorded on an LAS-4000 imaging system (Fujifilm Global, Valhalla, NY, USA), and quantified using Multi Gauge software (Version 3.1; Fujifilm). Antibodies to γ-Tubulin (Sigma), IκBα, Bcl-xL, Mcl-1 and GapdH (all from Cell Signaling Technology, Inc, Danvers, MA, USA), were used for immunoblotting.
Statistical analysis
To compare treatment effect across mice, an unpaired t-test was applied taking into account the random batch effect (JMP, SAS, Cary, NC, USA). For survival analysis, estimates of overall survival were obtained using the Kaplan-Meier method, and the log-rank test (Mantel-Cox) was used to evaluate differences between curves (Prism 6).
Results
Superior anti-leukemic effects of combined inhibition of BTK and PI3Kδ in the aggressive TCL1-192 mouse model
We compared the anti-tumor activity of acalabrutinib, ACP-319 and their combination head-to-head in the aggressive TCL1-192 adoptive transfer model.(18) TCL1-192 cells were engrafted into NOD-SCID mice 1–2 weeks before treatment was initiated. Mice were followed until death or pre-determined morbidity endpoints. The proportion of TCL1-192 cells (CD5+B220hi) in the PB four weeks after cell injection was assessed by flow cytometry (Figure 1a). As summarized in Figure 1b, mice treated with single-agent acalabrutinib or ACP-319 had lower tumor burden than vehicle-treated mice (P<0.0001). No significant difference was seen between the mice treated with each agent individually (P=0.14). However, mice treated with the combination of acalabrutinib and ACP-319 had significantly fewer leukemic cells in the PB than mice treated with vehicle or single-agents (Figure 1b; P<0.0001). In concordance with the observed change in cell numbers, all treated mice demonstrated a significant decrease in the proportion of proliferating TCL1-192 cells compared with vehicle-treated mice (Figure S1a; P<0.0001). Similarly, the proportion of viable circulating TCL1-192 cells was significantly decreased in all treated mice compared with vehicle controls (Figure S1b; P≤.02). Of note, in combination-treated mice compared to mice treated with single agents, the absolute numbers of proliferating leukemic cells in the PB decreased more than 15-fold (Figure 1C; P<0.0001). In accordance with the change in proliferation, the absolute numbers of viable leukemic cells in the PB decreased by more than 25-fold (Figure 1d; P<0.0001). Thus, mice treated with the combination showed substantially reduced tumor burden and proliferation as well as increased cell death in the PB compared with both single-agent and vehicle-treated mice.
Figure 1. Effects of inhibiting BTK, PI3Kδ or both on tumor burden, cell death and proliferation in mice carrying TCL1-192 cells.
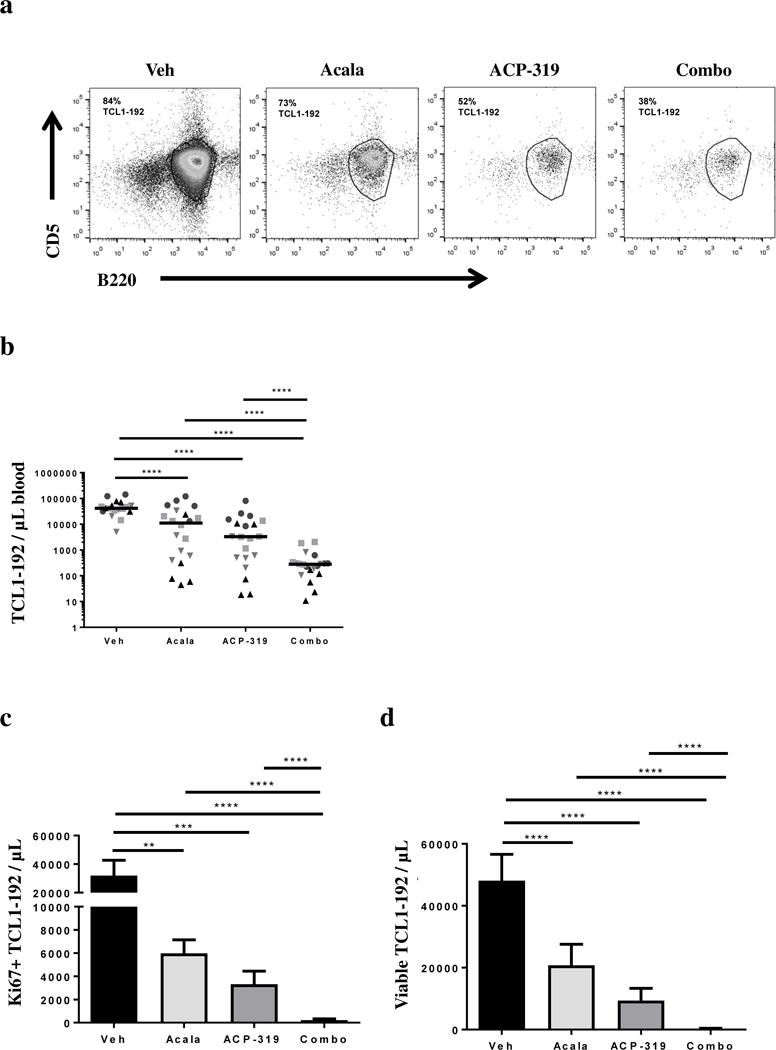
All data shown are at four weeks after TCL1-192 cell injection in NOD/SCID mice. Each experimental cohort consisted of 3-5 mice per treatment group. (a) Representative dot plots of CD5/B220 staining in mice receiving treatment as indicated. Percentage TCL1-192 cells (CD5+B220hi) are indicated for each treatment. (b) Absolute numbers of TCL1-192 cells/μL in the peripheral blood of mice receiving treatment as indicated, measured as in panel a. Each symbol represents one mouse; different symbols represent different experimental cohorts. Line represents median. (c) Mean ± SEM absolute number of proliferating (KI67+) TCL1-192 cells in each treatment group, n=40 split evenly across treatment groups in two experimental cohorts. (d) Mean ± SEM absolute number of viable TCL1-192 cells in the peripheral blood for each treatment group, measured as Annexin-V and VIVID double negative cells, n=80 split evenly across treatment groups in four experimental cohorts. Abbreviations: Veh, vehicle; Acala, acalabrutinib; Combo, combination treatment with acalabrutinib and ACP-319. All comparisons by an unpaired t-test using a linear model to take into account the random batch effect: **P<0.01, ***P<0.001 and ****P<0.0001.
The combination of acalabrutinib and ACP-319 significantly improved tumor control in the tissue microenvironment as compared with single agents
We hypothesized that the superior anti-leukemic effect of the combination therapy resulted from more potent inhibition of tumor-microenvironment interactions. To address this, we analyzed TCL1-192 cells harvested from the spleens of mice four weeks after cell injection and two weeks after treatment start. We first measured BTK occupancy in mice treated with acalabrutinib or the combination of acalabrutinib and ACP-319. More than 90% of BTK was drug bound in the spleens of mice from both treatment groups, indicating that target occupancy was neither enhanced nor hindered by the addition of ACP-319 (Figure 2a). We next assessed the degree of tumor infiltration in the spleen. Overall, the absolute number of TCL1-192 cells was modestly reduced in single-agent acalabrutinib- or ACP-319-treated mice compared to vehicle-treated mice (Figure 2b; P<0.05). In contrast, mice treated with the combination of acalabrutinib and ACP-319 showed more than an 11-fold reduction in spleen-residing TCL1-192 cells on average compared with all other treatment groups (Figure 2b; P<0.0001). In agreement with the decrease in splenic tumor burden, tumor proliferation in the spleen was significantly reduced with combination treatment compared with vehicle or single-agent treatment (Figure 2c, P<0.0001). Similarly, in mice receiving combination therapy TCL1-192 cell viability was significantly lower as compared with vehicle or single-agent treated mice (Figure 2d; P≤0.0006). The change in spleen size was quite variable, and overall small, for single-agent treated mice (P≤0.03); however, the combination of acalabrutinib and ACP-319 resulted in a significant reduction in spleen size compared with both single-agent and vehicle-treated mice; in agreement with the observed reductions in proliferation and tumor cell viability (Figure 2e–f; P<0.0001). In fact, the median spleen weight decreased from 780 mg in vehicle-treated mice to 212 mg in combination-treated mice, approaching normal spleen size for NOD-SCID mice.
Figure 2. Impact of acalabrutinib, ACP-319 or their combination on tissue-resident TCL1-192 cells.
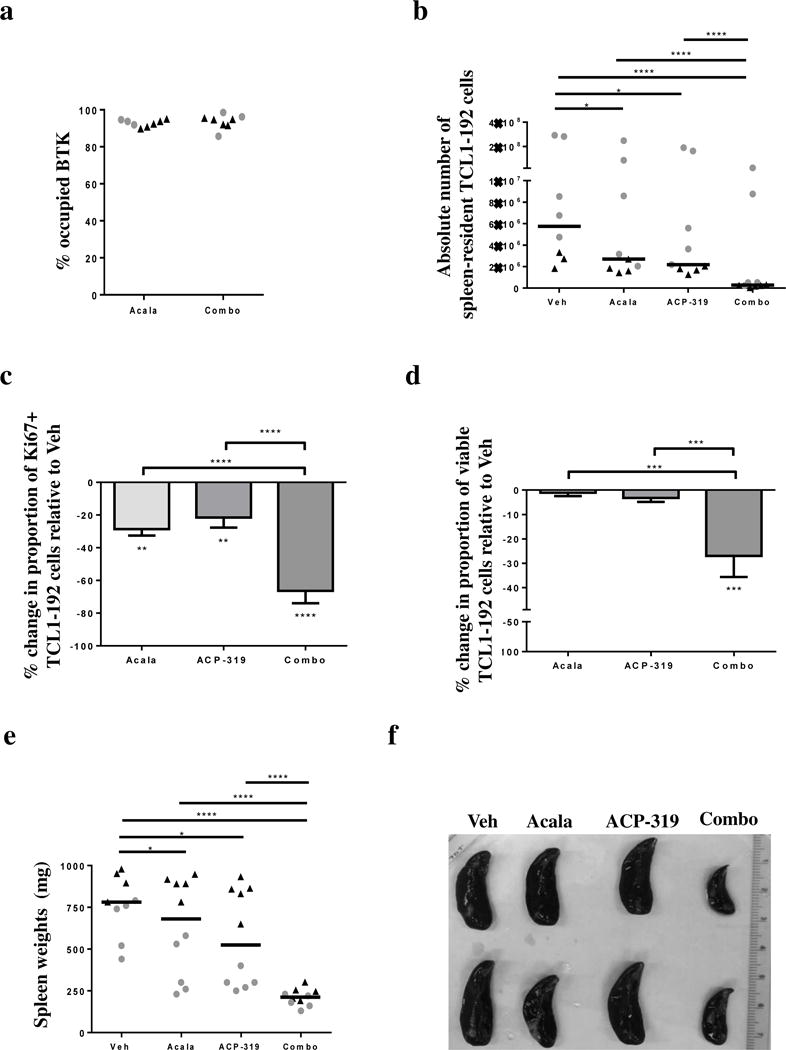
TCL1-192 cells were harvested from the spleen four weeks after cell injection. (a–b) Each symbol represents one mouse; different symbols represent different experimental cohorts. (a) Occupancy of BTK in mice treated with either acalabrutinib alone or the combination of acalabrutinib and ACP-319. (b) Absolute number of recovered TCL1-192 cells harvested from the spleen as measured by flow cytometry, based on counting beads and the volume used for preparation of single cell suspension from the spleens. Line represents median. (c–d) Measurements were normalized to vehicle control. (c) Mean ± SEM percentage change in the proportion of KI67+ TCL1-192 cells in each treatment group compared with vehicle treated mice (n=20 split evenly across treatment groups) are shown. (d) Mean ± SEM percentage change in the proportion of viable TCL1-192 cells in each treatment group compared with vehicle treated mice (n=40 split evenly across treatment groups in two experimental cohorts). Measured as Annexin-V and VIVID double negative cells. (e) Spleen weights at time of sacrifice by treatment group. Each symbol corresponds to one mouse; the type of symbol/color represents the experimental cohort. Line represents median. (f) Spleens from representative mice in the different treatment groups. Abbreviations: Veh, vehicle; Acala, Acalabrutinib; Combo, combination treatment with acalabrutinib and ACP-319. All comparisons by an unpaired t-test using a linear model to take into account the random batch effect. Statistics comparing treatment to vehicle control are shown either above the treatment groups (panels b and f) or below the treatment bars (panels c and d); statistics comparing treatments are shown with comparison brackets: *P<0.05, ***P<0.001 and ****P<0.0001.
The combination of acalabrutinib and ACP-319 significantly reduced tumor burden in a humanized CLL xenograft model without diminishing T cells
We validated the efficacy of BTK and PI3Kδ inhibition with acalabrutinib and ACP-319 on human CLL cells representing the clinical spectrum in a patient-derived xenograft model. This model based on the transfer of PBMCs includes autologous T cells. The number of CLL cells in the PB was significantly decreased in the treated mice as compared with vehicle (Figure S2a–b; P=0.03). In parallel, tumor burden in the spleen assessed as percentage CLL cells of total human CD45+ cells was also significantly decreased in the inhibitor-treated mice compared with vehicle-treated controls (Figure S2c, P=0.02). Concurrently, the proportion of human CD45+ cells to murine cells in blood and spleen was higher in inhibitor-treated mice compared to vehicle-treated mice (Figure S2d–e). To further elucidate the effect of combination treatment with acalabrutinib and ACP-319 on non-tumor cells we evaluated changes in the co-engrafted CLL T cells. There was a trend towards an increase in the absolute number of T cells in the PB in the treated-mice as compared with vehicle-treated controls (Figure S2f). Further, the percentage of T cells of total human CD45+ cells significantly increased in the inhibitor-treated mice compared with vehicle-treated controls (Figure S2g, P=0.02).
Combined inhibition of BTK and PI3Kδ significantly improved survival of tumor bearing mice compared with single-agent treated mice
Mice were followed for survival or the development of terminal morbidity. Single-agent inhibition of BTK or PI3Kδ prolonged survival for mice bearing TCL1-192 cells from a median of 32 days (vehicle-treated mice) to a median 38 days, for ACP-319 or acalabrutinib (Figure 3; P<0.0001), in line with what has been previously observed with ibrutinib. Notably, the combination regimen of acalabrutinib and ACP-319 significantly improved survival over both single-agent treatments to a median 55.5 days (Figure 3; P<0.0001). If counted from the start of treatment, the median survival more than doubled from 17 days for vehicle-treated mice to 40.5 days for combination treated mice; in comparison, survival was approximately 21 days for single-agent treated mice.
Figure 3. The combination of acalabrutinib and ACP-319 improves survival of mice injected with TCL1-192 cells compared with single-agents.
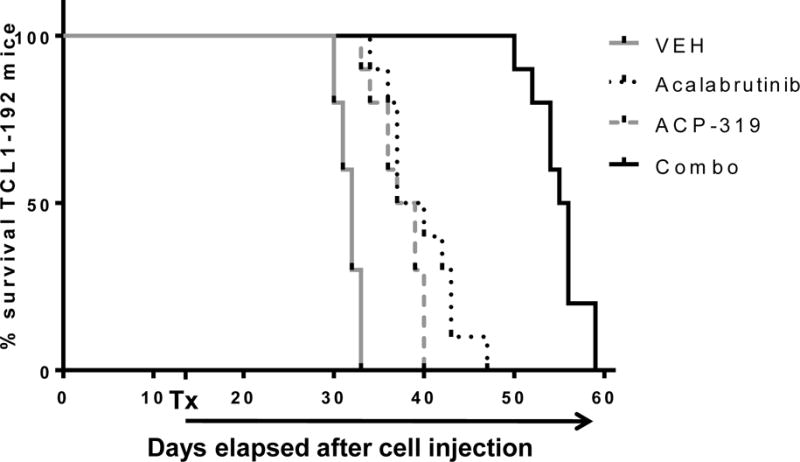
Kaplan-Meier survival curves for mice injected with TCL1-192 cells and treated with either vehicle, acalabrutinib, ACP-319, or the combination of the two (n=40 split evenly across treatment groups in two experimental cohorts). Tx indicates start of treatment (15 days from cell injection). Abbreviations: Veh; vehicle, Acala; Acalabrutinib; Combo, combination treatment with acalabrutinib and ACP-319. Comparisons by log-rank test (Mantel-Cox); P<0.0001 for each treatment group vs vehicle and for combination treated mice vs each single-agent treated group.
Inhibition of BTK and PI3Kδ reduces phosphorylation of downstream targets in the BCR pathway
To investigate mechanistic differences that could lead to the enhanced efficacy of combination therapy, we first examined the impact of BTK and/or PI3Kδ inhibition on phosphorylation of target molecules downstream of the BCR pathway. This was done by flow cytometry in TCL1-192 cells obtained two weeks after the start of therapy. All treated mice demonstrated a significant decrease in the proportion of TCL1-192 cells positive for phosphorylation of ERK in the PB compared with vehicle-treated mice (Figure S3a; P<0.0001). The mean absolute numbers of leukemic cells in the PB carrying pERK was more than 15-fold lower in mice treated with the combination of acalabrutinib and ACP-319 compared to each group of single-agent treated mice (Figure 4a; P<0.0001). For the more proliferative tumor population in the spleen, only mice treated with the combination of acalabrutinib and ACP-319 demonstrated a significant decrease in the percentage of TCL1-192 cells expressing pERK, compared with vehicle controls (Figure 4b; P=0.006). We next sought to confirm inhibition of BCR signaling in the context of functional immune cells in xenografted CLL cells harvested from the spleen microenvironment. We found that phosphorylation of both PLCγ2 and ERK were significantly inhibited in the combination treated-mice compared to vehicle-treated controls (Figure S3b–c; P<0.05 and P=0.04, respectively).
Figure 4. Effects of inhibiting BTK, PI3Kδ or both on ERK and NF-κB signaling in TCL1-192 cells.
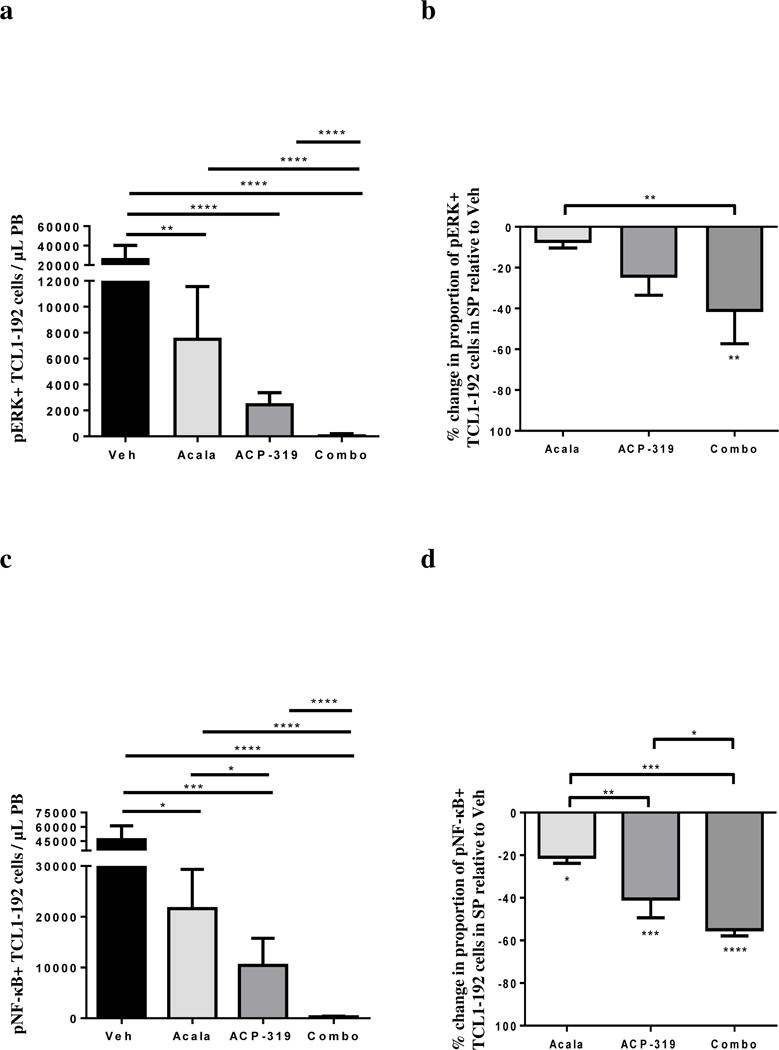
Treatment was started two weeks after TCL1-192 cell injection and mice were sacrificed four weeks after cell injection. (a) Mean ± SEM absolute numbers of pERK+ TCL1-192 cells in the peripheral blood, n=40 split evenly across treatment groups in two experimental cohorts. (b) Mean ± SEM percentage change in the proportion of pERK+ TCL1-192 cells in the spleen of mice in each treatment group compared with vehicle treated mice, n=20 split evenly across groups. (c) Mean ± SEM absolute numbers of phospho-NF-κB+ (p65) TCL1-192 cells in the PB (n=40 split evenly across treatment groups in two experimental cohorts). (d) Mean ± SEM percentage change in the proportion of phospho-NF-κB+ TCL1-192 cells in each treatment group compared with vehicle treated mice in the spleen (n=20 split evenly across treatment groups). Abbreviations: Acala, Acalabrutinib; Combo, combination treatment with acalabrutinib and ACP-319. All comparisons by an unpaired t-test using a linear model to take into account the random batch effect. Statistics comparing treatment to vehicle control are shown either above the treatment groups (panels a and c) or below the treatment bars (panels b and d); statistics comparing treatments are shown with comparison brackets: *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001.
In addition to readouts of BCR signaling we sought to evaluate changes in downstream activation of the NF-κB pathway. All treated mice demonstrated a significant reduction in the proportion of TCL1-192 cells expressing phosphorylated p65 (an NF-κB subunit) in the PB compared with vehicle-treated mice. However, combination treated mice demonstrated significantly more inhibition of NF-κB than mice treated with single agents (Figure S4a; P<0.007). In addition, the mean absolute numbers of leukemic cells in the PB expressing phospho-NF-κB was at least 9-fold lower in mice treated with the combination of acalabrutinib and ACP-319 compared to mice treated with either agent alone (Figure 4c; P<0.0001). Moreover, mice treated with the combination of acalabrutinib and ACP-319 had a significantly lower proportion of TCL1-192 cells in the spleen bearing phospho-NF-κB compared to all other treatment groups (Figure 4d; P≤0.04). In addition to changes in phosphorylation of p-65 we found that treated-mice demonstrated a significant reduction in nuclear p50 (Figure S4b; P<0.006). Lastly, to confirm the inhibition of the canonical NF-κB pathway, we assessed the expression of six known NF-κB target genes (CCND2, BCL2A, CCL3, CCL4, RGS1, and TNF) by quantitative RT-PCR and computed a gene signature score as the averaged expression of these six genes as previously described.(22) While we found a trend towards reduction in the NF-κB signature across all treatment groups compared to vehicle-treated controls, only the combination-treated mice showed a significant reduction (Figure S4c; P=0.01). In summary, inhibiting BTK, PI3Kδ or both resulted in reduction in phosphorylation of downstream targets in the BCR signaling pathway; however, combination treatment induced a greater reduction in downstream NF-κB signaling than single-agent treatment.
More potent inhibition of anti-apoptotic pathways downstream of NF-κB signaling with combined BTK and PI3Kδ inhibition
Unphosphorylated IκBα binds to NF-κB subunits, preventing them from translocating into the nucleus. Upon phosphorylation, IκBα is degraded and NF-κB is free to move into the nucleus. Consistent with the inhibition of BCR-dependent phosphorylation and degradation, we found that the combination of acalabrutinib and ACP-319, but not the single-agents, significantly increased the levels of IκBα in spleen-residing TCL1-192 cells (Figure 5a; P=0.006). Next, we evaluated expression of Bcl-xL, a pro-survival protein that is transcriptionally regulated by NF-κB. We found variable reductions in Bcl-xL in single-agent treated mice with a significant reduction only in combination-treated mice (Figure 5b; P=0.04). The expression of anti-apoptotic Mcl-1, whose expression is regulated by PI3K signaling, was also evaluated. We found a trend towards reduction in Mcl-1 in both single-agent treated-mice; however, only the combination-treated mice showed significant inhibition compared with control mice (Figure 5c; P=0.006). Collectively, these results suggest improved survival with the combination treatment is at least in part due to inhibition of NF-κB signaling and consequent downstream expression of anti-apoptotic proteins.
Figure 5. Inhibition of anti-apoptotic mechanisms in spleen-resident TCL1-192 cells.
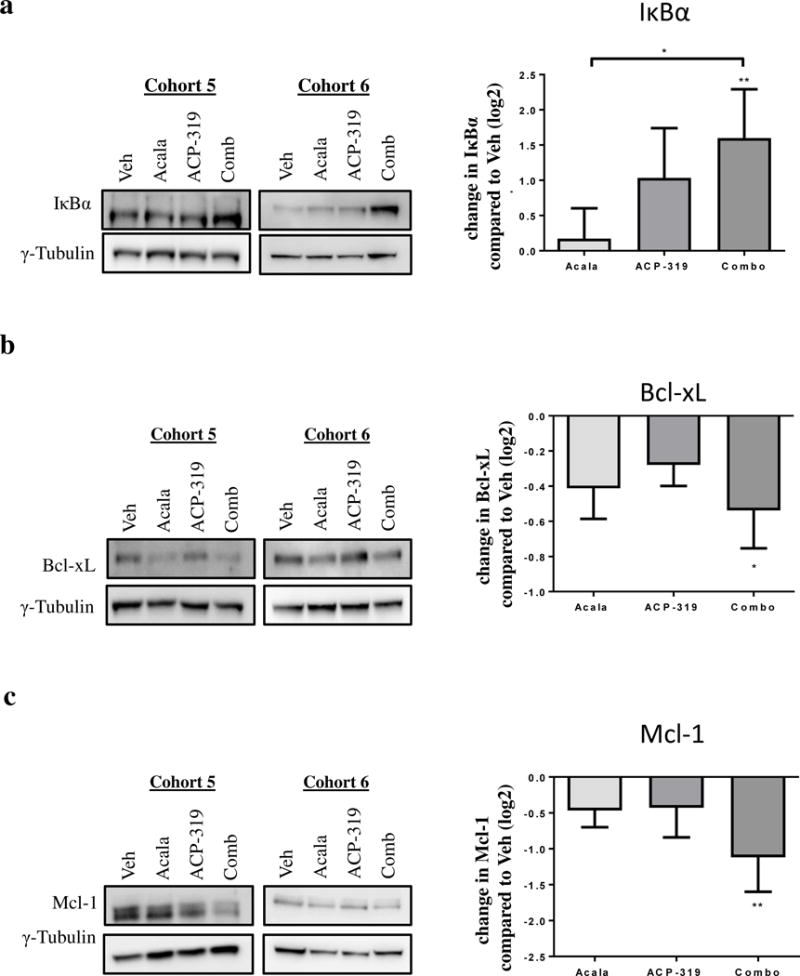
TCL1-192 cells were harvested from the spleen four weeks after cell injection and two weeks after treatment start. Left panels show representative immunoblots stained for each protein of interest and the loading control, γ-tubulin. Bands were quantitated by densitometry and normalized to loading control. Right panels show the mean ± SEM change in normalized expression as compared with vehicle-treated mice. (a) IκBα, n=24 split across treatment groups in two experimental cohorts, (b) Bcl-xL, n=24 split across treatment groups in two experimental cohorts and (C) Mcl-1, n=16 split across treatment groups in two experimental cohorts. Abbreviations: Veh, vehicle, Acala, Acalabrutinib and Combo, combination treatment with acalabrutinib and ACP-319. All comparisons by an unpaired t-test using a linear model to take into account the random batch effect. Statistics comparing treatment to vehicle control are shown above (panel a) or below (panels b and c) the treatment bars; statistics comparing treatments are shown with comparison brackets: *P<0.05 and **P<0.01.
Discussion
Targeting the BCR pathway with single-agent BTK or PI3Kδ inhibitors has proven clinically effective in CLL.(6–8, 14) However, the majority of responses are partial and indefinite treatment is currently required. Further, resistance to single-agent therapy and toxicity with chronic therapy is a concern leading to approximately 20% of patients discontinuing single-agent treatment within two years.(10, 23, 24) In order to deepen responses and forestall the emergence of drug resistance, combination therapy appears necessary, especially for high risk CLL.(10, 25) We here present the first in vivo efficacy data on the combination of BTK and PI3Kδ inhibitors, specifically acalabrutinib and ACP-319, in murine models of CLL. Combination therapy resulted in significant reductions in tumor burden compared with vehicle and single-agent treatment; resulting in a nearly 2-fold increase in survival time for the combination compared with single-agent therapy. Molecular data from these mice suggests that the increased efficacy of the combination is at least in part due to more potent inhibition of pro-survival signals, including decreased NF-κB signaling and reduced expression of anti-apoptotic proteins.
Progression of CLL results from a combination of apoptosis resistance and uncontrolled proliferation supported by tumor-microenvironment interactions in lymphoid tissues.(2, 26) In order to investigate the anti-leukemic effects of combined BTK and PI3Kδ inhibition with acalabrutinib and ACP-319, we chose a murine model that recapitulate the contribution of the microenvironment to CLL progression.(27) The TCL1-192 adoptive transfer model corresponds to a rapidly progressive CLL with fatal outcome and is therefore well suited for the comparison of multiple treatments in a controlled manner, with overall survival as a final endpoint.(18) The TCL1-192 cells do not proliferate in vitro, thus demonstrating dependency on the microenvironment similarly to primary CLL cells.(2) However, the TCL1-192 adoptive transfer model lacks functional T cells. To assess the treatment effects in the presence of T cells, we utilized the patient-derived xenograft model which recapitulates the biology of CLL in the lymph node.(3) While the requirement for large numbers of human cells, the variability in engraftment, and the lack of survival as a treatment endpoint are limiting, the patient-derived xenograft model recreates a humanized microenvironment comprising patient-derived T cells, which may modulate the response to kinase inhibitors. In these experiments, the combination of acalabrutinib and ACP-319 reduced tumor burden in the presence of activated immune cells and demonstrated selectivity for tumor cells while T cells expanded on treatment. This is of particular importance as microenvironmental cells including T lymphocytes may modulate the response to kinase inhibitors. For example, PI3Kδ is expressed and mediates effects in some T cell subsets.(28–30) Likewise, BTK inhibitors impact a range of immune cells through on- and off-target mechanisms.(16, 31–36) Thus, testing the combination of acalabrutinib and ACP-319 in a model encompassing the microenvironment is an important step towards clinical use.
We here demonstrate for the first time that the combination of acalabrutinib and ACP-319 provided more potent tumor control in vivo, significantly extending the survival compared to single-agent therapy. This survival advantage with combination therapy is at least in part attributable to more potent inhibition of NF-κB signaling and down-regulation of anti-apoptotic regulators resulting in decreased tumor proliferation and increased cell death. Notably, in the spleen microenvironment combination treatment, but not single-agent therapy, significantly reduced the proportion of viable TCL1-192 cells. These data suggest that tumor cells are protected from the effects of single-agent therapy through signaling pathways, activated within the tissue microenvironment, for which BTK and PI3Kδ assume partially redundant functions.(2) This view is further supported by the more potent inhibition of downstream targets in tumor cells obtained from the spleen of mice receiving combination treatment compared to single-agents.
In vitro and ex vivo analyses have attributed the efficacy of PI3Kδ inhibition by idelalisib and BTK inhibition by ibrutinib to inhibition of tumor-microenvironment interactions and BCR signaling.(30, 37, 38) Interestingly, work done by Suzuki and colleagues demonstrated that mice with dual knockout of BTK and PI3K (PI3K−/−Btk−/−) had substantially reduced B cell numbers (particularly in the spleen compartment), cell proliferation, and intracellular NF-κB signaling, indicating partially redundant functions of these kinases.(39) Further supporting the more potent effect of dual targeting are the synergistic effects of combined BTK and PI3K inhibition on lymphoma cell survival in vitro,(11) and the limited efficacy of sequential BTK/PI3Kδ targeting after clinical progression on one agent.(40) Furthermore, our data are consistent with recent in vitro studies reporting that ibrutinib and idelalisib can synergistically target BCR-controlled adhesion.(12) Here we demonstrated improved therapeutic efficacy of the combination of acalabrutinib with ACP-319 in vivo that was especially evident against the aggressive disease within the tumor-microenvironment. Whether the effects of the combination result from inhibition of sequential steps within one pathway, for example BCR signaling, or from the concurrent targeting of parallel pathways that converge on shared effectors such as Bcl-xL, a target of both BTK and PI3K signaling in activated B cells, remains to be determined.(39)
Single-agent acalabrutinib has demonstrated significant clinical efficacy in relapsed CLL.(14) Similarly, ACP-319 has shown efficacy and tolerability in a phase I trial, with early data suggesting that colitis and transaminitis were manageable upon re-exposure to treatment.(15) Translational data from trials with single-agent idelalisib (a compound similar to ACP-319) indicate that suppression of regulatory T cells and consequent emergence of autoimmunity may be responsible for some adverse events; including hepatotoxicity, pneumonitis, colitis, and increased rates of infection.(28, 41, 42) In addition, the dose limiting toxicity of pneumonitis in the combination trial of idelalisib with the SYK inhibitor entospletinib has been linked to increased levels of IFNγ and interleukins- 6, 7 and 8, likely released from accessory cells.(43) We have previously shown that ibrutinib, inhibits generation of Th17 cells, reduces overall T cell activation and decreases inflammatory cytokines while possibly increasing regulatory T cells.(16) Conceivably, immune mediated adverse events associated with PI3Kδ inhibitors could be attenuated in the less inflammatory microenvironment we observed in ibrutinib treated patients; a hypothesis that is supported by the lack of changes in behavior or signs of morbidity in the combination treated TCL1-192 injected mice compared to vehicle treated mice. However, the safety of the combination should be tested in future studies. Further, the first safety data from a clinical phase I trial combining BTK and PI3Kδ inhibitors demonstrated that the combination of ibrutinib and TGR-1202 treatment was well tolerated with no dose limiting toxicities.(44) Increased expression of activation-induced cytidine deaminase (AID) in patients treated with a PI3Kδ inhibitor but not a BTK inhibitor could promote genomic instability (45). The impact of dual kinase inhibition remains to be investigated.
Despite the impressive clinical results of single-agent BTK and PI3Kδ inhibitors, the combination is of particular interest for patients with high-risk CLL. Complete remissions, without minimal residual disease, are rare with single-agent therapy, and occur only after prolonged treatment leading to an increased risk of progression for high-risk patients.(10, 46) This is further supported by data on subclonal development in CLL(47) and the development of pathway specific mutations upon BTK inhibition.(48–50) Our data support the rationale of combining BTK and PI3Kδ inhibitors to achieve more potent anti-leukemic effects. Conceivably, benefit of this approach may be best realized in patients with TP53 aberrations, del(11q) and/or complex karyotype,(10, 25) who are at higher risk of failing single-agent BTK inhibitors, while minimizing toxicity for patients who can be expected to have durable responses to single-agent therapy. The recently initiated phase I/II trials combining acalabrutinib and ACP-319 (Clinicaltrials.gov: NCT02157324 and NCT02328014) will provide further insights on how the benefit of the combination may be realized in the clinic.
Supplementary Material
Statement of significance.
Combined inhibition of BTK and PI3Kδ triples the survival advantage and reduces NF-κB signaling, compared with single agents in an aggressive murine CLL model, supporting the continuation of clinical trials with the combination of BTK and PI3Kδ inhibitors.
Statement of translational relevance.
Targeting the BCR pathway with single-agent BTK or PI3Kδ inhibitors has proven clinically effective in CLL. However, the majority of responses are partial and indefinite treatment is currently required while resistance to single-agent therapy and toxicity with chronic therapy is a concern. We herein demonstrate for the first time that the combination of the BTK inhibitor acalabrutinib and the PI3Kδ inhibitor ACP-319 provides more potent tumor control in vivo and significantly extends the survival compared to single-agent therapy. Thus, combining BTK and PI3Kδ inhibitors to achieve more potent anti-leukemic effects may benefit patients with TP53 aberrations, del(11q) and/or complex karyotype, who are at higher risk of failing single-agent BTK inhibitors. The pre-clinical in vivo data presented herein support the continuation of clinical trials with the combination of BTK and PI3Kδ inhibitors.
Acknowledgments
We thank our patients for participating and donating samples to make this research possible. We acknowledge Acerta Pharma for providing study drug and especially Tim Ingallinera for formulating the drinking water. We thank Allard Kaptein for sharing his expertise and assistance. This research was supported by the Intramural Research Program of the National, Heart, Lung and Blood Institute, the Danish Cancer Society and Acerta Pharma.
Financial Support:
AW received intramural National Institutes of Health funding. CN received funding from the Danish Cancer Society and the Novo Nordisk Foundation.
Footnotes
Authorship contributions
CN, HMJ, AW and SH planned the research, performed experiments, and analyzed data; FK, TC, RI, RU and BL took part in planning, performing and analyzing parts of experiments. SSC and NC provided TCL1-192 cells and assisted with experimental design. CN and HMJ wrote the first draft manuscript, AW and SH contributed to writing the final version. All authors approved the final version of the manuscript.
Conflicts of Interest
FK, TC, RI are employees and equity holders of Acerta Pharma. RU is an equity holder and member of the board of directors of Acerta Pharma. BL is a former employee of Acerta and entitled to milestone payments. AW received research funding from Pharmacylics and Acerta Pharma. NC has received research funding from Pharmacylics. CN received funding from the Danish Cancer Society and the Novo Nordisk Foundation, and has received grants/consultancy fees from Abbvie, Janssen, Gilead, Roche and Novartis outside of this study. HMJ, SSC, and SH declare no competing financial interests.
References
- 1.NCI. Surveillance Epidemiology and End Results. [website] 2012 [cited 2013 10–17]; Available from: [Google Scholar]
- 2.Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011;117:563–74. doi: 10.1182/blood-2010-05-284984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herman SE, Sun X, McAuley EM, Hsieh MM, Pittaluga S, Raffeld M, et al. Modeling tumor-host interactions of chronic lymphocytic leukemia in xenografted mice to study tumor biology and evaluate targeted therapy. Leukemia. 2013;27:1769–73. doi: 10.1038/leu.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wiestner A. The role of B-cell receptor inhibitors in the treatment of patients with chronic lymphocytic leukemia. Haematologica. 2015;100:1495–507. doi: 10.3324/haematol.2014.119123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown JR, Hallek MJ, Pagel JM. Chemoimmunotherapy Versus Targeted Treatment in Chronic Lymphocytic Leukemia: When, How Long, How Much, and in Which Combination? American Society of Clinical Oncology educational book/ASCO American Society of Clinical Oncology Meeting. 2016;35:e387–98. doi: 10.1200/EDBK_159018. [DOI] [PubMed] [Google Scholar]
- 6.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and Rituximab in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2014 doi: 10.1056/NEJMoa1315226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farooqui MZ, Valdez J, Martyr S, Aue G, Saba N, Niemann CU, et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol. 2015;16:169–76. doi: 10.1016/S1470-2045(14)71182-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maddocks KJ, Ruppert AS, Lozanski G, Heerema NA, Zhao W, Abruzzo L, et al. Etiology of Ibrutinib Therapy Discontinuation and Outcomes in Patients With Chronic Lymphocytic Leukemia. JAMA Oncol. 2015;1:80–7. doi: 10.1001/jamaoncol.2014.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Byrd JC, Furman RR, Coutre SE, Burger JA, Blum KA, Coleman M, et al. Three-year follow-up of treatment-naive and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125:2497–506. doi: 10.1182/blood-2014-10-606038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathews Griner LA, Guha R, Shinn P, Young RM, Keller JM, Liu D, et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:2349–54. doi: 10.1073/pnas.1311846111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Rooij MF, Kuil A, Kater AP, Kersten MJ, Pals ST, Spaargaren M. Ibrutinib and idelalisib synergistically target BCR-controlled adhesion in MCL and CLL: a rationale for combination therapy. Blood. 2015;125:2306–9. doi: 10.1182/blood-2014-12-619163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herman SEM, Montraveta A, Niemann CU, Mora-Jensen H, Gulrajani M, Krantz F, et al. The Bruton’s tyrosine kinase (BTK) inhibitor acalabrutinib demonstrates potent on-target effects and efficacy in two mouse models of chronic lymphocytic leukemia. Clinical Cancer Research. 2016 doi: 10.1158/1078-0432.CCR-16-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016;374:323–32. doi: 10.1056/NEJMoa1509981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lanasa MC, Glenn M, Mato AR, Allgood SD, Wong S, Amore B, et al. First-In-Human Study Of AMG 319, a Highly Selective, Small Molecule Inhibitor Of PI3Kδ, In Adult Patients With Relapsed Or Refractory Lymphoid Malignancies. Blood. 2013;122:678. [Google Scholar]
- 16.Niemann CU, Herman SE, Maric I, Gomez-Rodriguez J, Biancotto A, Chang BY, et al. Disruption of in vivo Chronic Lymphocytic Leukemia Tumor-Microenvironment Interactions by Ibrutinib - Findings from an Investigator-Initiated Phase II Study. Clin Cancer Res. 2016;22:1572–82. doi: 10.1158/1078-0432.CCR-15-1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bachireddy P, Wu CJ. Arresting the Inflammatory Drive of Chronic Lymphocytic Leukemia with Ibrutinib. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22:1547–9. doi: 10.1158/1078-0432.CCR-15-3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen SS, Batliwalla F, Holodick NE, Yan XJ, Yancopoulos S, Croce CM, et al. Autoantigen can promote progression to a more aggressive TCL1 leukemia by selecting variants with enhanced B-cell receptor signaling. Proc Natl Acad Sci U S A. 2013;110:E1500–7. doi: 10.1073/pnas.1300616110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119:1182–9. doi: 10.1182/blood-2011-10-386417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bichi R, Shinton SA, Martin ES, Koval A, Calin GA, Cesari R, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci U S A. 2002;99:6955–60. doi: 10.1073/pnas.102181599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krutzik PO, Nolan GP. Intracellular phospho-protein staining techniques for flow cytometry: monitoring single cell signaling events. Cytometry A. 2003;55:61–70. doi: 10.1002/cyto.a.10072. [DOI] [PubMed] [Google Scholar]
- 22.Herman SE, Mustafa RZ, Gyamfi JA, Pittaluga S, Chang S, Chang B, et al. Ibrutinib inhibits BCR and NF-kappaB signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood. 2014;123:3286–95. doi: 10.1182/blood-2014-02-548610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winqvist M, Asklid A, Andersson PO, Karlsson K, Karlsson C, Lauri B, et al. Real–world results of ibrutinib in patients with relapsed or refractory chronic lymphocytic leukemia: Data from 95 consecutive patients treated in a compassionate use program. Haematologica. 2016 doi: 10.3324/haematol.2016.144576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Furman RR, Cheng S, Lu P, Setty M, Perez AR, Guo A, et al. Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med. 2014;370:2352–4. doi: 10.1056/NEJMc1402716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson PA, O’Brien SM, Wierda WG, Ferrajoli A, Stingo F, Smith SC, et al. Complex karyotype is a stronger predictor than del(17p) for an inferior outcome in relapsed or refractory chronic lymphocytic leukemia patients treated with ibrutinib-based regimens. Cancer. 2015;121:3612–21. doi: 10.1002/cncr.29566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Calissano C, Damle RN, Hayes G, Murphy EJ, Hellerstein MK, Moreno C, et al. In vivo intraclonal and interclonal kinetic heterogeneity in B-cell chronic lymphocytic leukemia. Blood. 2009;114:4832–42. doi: 10.1182/blood-2009-05-219634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herman SEM, Wiestner A. Preclinical modeling of novel therapeutics in chronic lymphocytic leukemia: the tools of the trade. Semin Oncol. 2016;43:222–32. doi: 10.1053/j.seminoncol.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lampson BL, Kasar SN, Matos TR, Morgan EA, Rassenti L, Davids MS, et al. Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-mediated hepatotoxicity. Blood. 2016;128:195–203. doi: 10.1182/blood-2016-03-707133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcon F, Okkenhaug K. PI3Kdelta promotes CD4(+) T-cell interactions with antigen-presenting cells by increasing LFA-1 binding to ICAM-1. Immunol Cell Biol. 2016;94:486–95. doi: 10.1038/icb.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116:2078–88. doi: 10.1182/blood-2010-02-271171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013;122:2539–49. doi: 10.1182/blood-2013-06-507947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sagiv-Barfi I, Kohrt HE, Burckhardt L, Czerwinski DK, Levy R. Ibrutinib enhances the antitumor immune response induced by intratumoral injection of a TLR9 ligand in mouse lymphoma. Blood. 2015;125:2079–86. doi: 10.1182/blood-2014-08-593137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kohrt HE, Sagiv-Barfi I, Rafiq S, Herman SE, Butchar JP, Cheney C, et al. Ibrutinib antagonizes rituximab-dependent NK cell-mediated cytotoxicity. Blood. 2014;123:1957–60. doi: 10.1182/blood-2014-01-547869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Da Roit F, Engelberts PJ, Taylor RP, Breij EC, Gritti G, Rambaldi A, et al. Ibrutinib interferes with the cell-mediated anti-tumor activities of therapeutic CD20 antibodies: implications for combination therapy. Haematologica. 2015;100:77–86. doi: 10.3324/haematol.2014.107011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borge M, Belen Almejun M, Podaza E, Colado A, Fernandez Grecco H, Cabrejo M, et al. Ibrutinib impairs the phagocytosis of rituximab-coated leukemic cells from chronic lymphocytic leukemia patients by human macrophages. Haematologica. 2015;100:e140–2. doi: 10.3324/haematol.2014.119669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skarzynski M, Niemann CU, Lee YS, Martyr S, Maric I, Salem D, et al. Interactions between Ibrutinib and Anti-CD20 Antibodies: Competing Effects on the Outcome of Combination Therapy. Clin Cancer Res. 2016;22:86–95. doi: 10.1158/1078-0432.CCR-15-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117:6287–96. doi: 10.1182/blood-2011-01-328484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herman SE, Mustafa RZ, Jones J, Wong DH, Farooqui M, Wiestner A. Treatment with Ibrutinib Inhibits BTK- and VLA-4-Dependent Adhesion of Chronic Lymphocytic Leukemia Cells In Vivo. Clin Cancer Res. 2015;21:4642–51. doi: 10.1158/1078-0432.CCR-15-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki H, Matsuda S, Terauchi Y, Fujiwara M, Ohteki T, Asano T, et al. PI3K and Btk differentially regulate B cell antigen receptor-mediated signal transduction. Nat Immunol. 2003;4:280–6. doi: 10.1038/ni890. [DOI] [PubMed] [Google Scholar]
- 40.Mato AR, Nabhan C, Barr PM, Ujjani CS, Hill BT, Lamanna N, et al. Outcomes of CLL patients treated with sequential kinase inhibitor therapy: a real world experience. Blood. 2016 doi: 10.1182/blood-2016-05-716977. [DOI] [PubMed] [Google Scholar]
- 41.Cheah CY, Nastoupil LJ, Neelapu SS, Forbes SG, Oki Y, Fowler NH. Lenalidomide, idelalisib, and rituximab are unacceptably toxic in patients with relapsed/refractory indolent lymphoma. Blood. 2015;125:3357–9. doi: 10.1182/blood-2015-03-633156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lampson BL, Matos T, Kim HT, Kasar S, Morgan EA, Hirakawa M, et al. Idelalisib Given Front-Line for the Treatment of Chronic Lymphocytic Leukemia Results in Frequent and Severe Immune-Mediated Toxicities. Blood. 2015;126:497. [Google Scholar]
- 43.Barr PM, Saylors GB, Spurgeon SE, Cheson BD, Greenwald DR, O’Brien SM, et al. Phase 2 study of idelalisib and entospletinib: pneumonitis limits combination therapy in relapsed refractory CLL and NHL. Blood. 2016;127:2411–5. doi: 10.1182/blood-2015-12-683516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davids MS, Kim HT, Nicotra A, Savell A, Francoeur K, Hellman J, et al. TGR-1202 in Combination with Ibrutinib in Patients with Relapsed or Refractory CLL or MCL: Preliminary Results of a Multicenter Phase I/Ib Study. Blood. 2016;128:641. [Google Scholar]
- 45.Compagno M, Wang Q, Pighi C, Cheong TC, Meng FL, Poggio T, et al. Phosphatidylinositol 3-kinase delta blockade increases genomic instability in B cells. Nature. 2017;542:489–93. doi: 10.1038/nature21406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farooqui M, Valdez J, Soto S, Stetler-Stevenson M, Yuan CM, Thomas F, et al. Single Agent Ibrutinib in CLL/SLL Patients with and without Deletion 17p. Blood. 2015;126:2937. [Google Scholar]
- 47.Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–30. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Albitar A, Ma W, De Dios I, Estrella J, Farooqui M, Wiestner A, et al. High Sensitivity Testing Shows Multiclonal Mutations in Patients with CLL Treated with BTK Inhibitor and Lack of Mutations in Ibrutinib-Naive Patients. Blood. 2015;126:716. [Google Scholar]
- 49.Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 2014;370:2286–94. doi: 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Komarova NL, Burger JA, Wodarz D. Evolution of ibrutinib resistance in chronic lymphocytic leukemia (CLL) Proc Natl Acad Sci U S A. 2014;111:13906–11. doi: 10.1073/pnas.1409362111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.