

Abstract
Chinese medicines are an important source of secondary metabolites with excellent antitumour activity. Evodia rutaecarpa, from the family Rutaceae, exhibits antitumour activity. Evodiamine (EVO), which was isolated from the fruit of E. rutaecarpa, exhibits robust antitumour activity. However, the antitumour mechanism of EVO remains unclear. In this study, we assessed the growth-inhibiting effect of EVO on two renal carcinoma cell lines. We found that EVO could change the morphology and decrease the viability and proliferation of cells in a time- and concentration-dependent manner in vitro. In addition, transcriptome analysis indicated that EVO can modulate the transcriptome of Caki-1 cells. In total, 7,243 differentially expressed genes were found, among which 3,347 downregulated genes and 3,896 upregulated genes were mainly involved in cell migration, apoptosis, cell cycle, and DNA replication. Furthermore, we demonstrated that EVO can cause apoptosis, arrest cells in the G2/M phase, and regulate the expression of apoptosis- and cell cycle-related genes in Caki-1 cells. Our study reveals the anticancer effects of EVO using cellular and molecular data, and indicates the potential uses of this compound as a resource to characterize the antitumour mechanisms of E. rutaecarpa.
Introduction
Renal cell carcinoma (RCC) is one of the most serious cancers in adults, accounting for ~90% of all renal malignancies1. The annual incidence of RCC is continually increasing2. Because of its highly invasive and metastatic characteristics, the initial diagnosis is often delayed. For example, nearly 30% of patients have distant metastases and 25% have locally advanced disease when they are initially diagnosed3–5. The survival rate of patients with metastatic RCC can be as low as 9.5%6. Because RCC is insensitive to radiotherapy and chemotherapy7, therapy with targeted agents (e.g., interleukin-2, temsirolimus, or pazopanib) has proven to be superior compared to other treatments8,9. However, not all of treatments can produce better therapeutic effects or achieve complete responses10–13. Therefore, an urgent need exists to develop novel therapeutics that overcome resistance to the currently administered anticancer drugs for RCC.
Evodiamine (EVO) is an indoloquinazoline alkaloid isolated from fruit of Evodia rutaecarpa 14. For millennia, it has been considered an effective Chinese medicine that can be used for the treatment of gastropathy, hypertension, and eczema. In the previous decade, increasing evidence has revealed that EVO exhibits various biological effects, including antibacterial, antiobesity, and antinociceptive activities15–17. Recently, it has been established that EVO exhibits antitumour activity against gastric adenocarcinoma, colorectal carcinoma, and breast cancer cells18–21. Recently, Wu and colleagues investigated the anticancer mechanism of EVO against renal carcinoma cells at the cellular and protein levels. Their results showed that EVO induces renal carcinoma cell apoptosis via disruption of the mitochondrial membrane potential, resulting from JNK- and PERK-mediated phosphorylation of the Bcl-2 protein22. However, the mechanism by which EVO exerts its anticancer effects on renal carcinoma cells requires further research.
In this study, cytological experiments and a transcriptome profiling study were performed to reveal the anticancer mechanism of EVO. First, a series of experiments investigating the viability and proliferation of human renal carcinoma 786-O cells and Caki-1 cells and the human renal epithelial cell line HK-2 was performed under EVO treatment. Then, a transcriptome profiling study was used to analyse EVO-regulated genes and signalling pathways responsible for growth and apoptosis using RNA sequencing. In addition, we verified the molecular mechanisms through which EVO inhibits growth and induces apoptosis using ultrastructural observations, flow cytometry analysis, and qPCR.
Materials and Methods
Cell culture and EVO treatments
Three cell lines (human renal carcinoma cell lines (786-O and Caki-1 cells) and the human renal epithelial cell line HK-2) were purchased from the Chinese Academy of Sciences Committee on Type Culture Collection Cell Bank (Shanghai, China). The 786-O and Caki-1 cells were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% foetal bovine serum (FBS; Gibco, Carlsbad, CA, USA) and the HK-2 cells were maintained in keratinocyte serum-free medium (Gibco BRL, Grand Island, NY, USA) supplemented with 50 μg/ml bovine pituitary extract (BPE) and 5 ng/ml recombinant epithelial growth factor. All cells were maintained in a humidified atmosphere with 95% air and 5% CO2 at 37 °C.
EVO with a purity of more than 99% was obtained from J&K Scientific (Beijing, China). Before use, 20 mg of EVO powder was dissolved in dimethyl sulphoxide (DMSO) to prepare a 30.36 mg/ml stock solution. For EVO treatment and antitumour analysis, cells were treated with serially diluted doses of EVO for 24, 48, or 72 h. All control cells were cultured in medium containing 0.2% DMSO. A separate evaluation of the effects of 0.2% DMSO in this study did not reveal any significant differences between DMSO-treated and untreated cells.
Cell viability analysis
The Cell Counting Kit-8 (CCK-8) colorimetric method was used to determine the sensitivity of cells to EVO. Briefly, the three cell lines were inoculated into 96-well plates at 3.0 × 104 cells per well. All treatments were performed in triplicate and in parallel. After 24 h, supernatants were replaced, and cells were exposed to different concentrations of EVO (0, 5, 10, 20, 40, 60, 80, and 100 μg/ml). After 24, 48, and 72 h, 10 μL of 5 g/L CCK-8 solution was added to each well and then the cells were incubated at 37 °C for 1.5 h in the dark. A microplate spectrophotometer reader (Multickan GO, Thermo Scientific, Waltham, MA, USA) was used to measure the absorbance of cells per well at 490 nm, which was used to calculate the percent inhibition of cell growth for each group.
Colony formation assay
Cell proliferation in response to EVO was estimated using the colony formation assay. The concentration of human renal carcinoma cells (786-O and Caki-1 cells) was adjusted to 1 × 105 cells/ml per well. Cells were cultured in six-well plates and exposed to various concentrations of EVO for 48 h. The medium was replaced regularly and all cultures were incubated for 2 weeks. Colonies were fixed using 70% ethanol solution and then stained with Giemsa. The surviving fraction was calculated and compared to that of the control to estimate the clonogenic efficiency.
Flow cytometry analysis
Caki-1 cells exposed to 40 μg/ml EVO for 24, 48, and 72 h were collected at a concentration of 1 × 106 cells/ml. Cells were washed three times using phosphate-buffered saline (PBS). For cell cycle analysis, fixed cells were stained with propidium iodide (PI) solution (20 μg/mL) containing 0.1% Triton X-100 (Sigma–Aldrich, St. Louis, MO, USA) and 100 μg/mL RNase A (Fermentas International Inc., Burlington, ON, Canada). For Annexin V-FITC/PI analysis, cells were stained with FITC-Annexin V Apoptosis Detection Kit I (BD Pharmingen) following the manufacturer’s instructions. A FACScan flow cytometer was used to detect the stained cells (at least 10,000 cells), and CXP analysis software (BD Biosciences, San Jose, CA, USA) was used to analyse the flow cytometry data.
Fluorescence microscopy
The morphology of Caki-1 cells was observed using Hoechst 33258, acridine orange/ethidium bromide (AO/EB), and transferase-mediated deoxyuridine triphosphate-biotin nick end labelling (TUNEL) staining, as described previously23–26. Briefly, Caki-1 cells were first incubated for 24 h and then exposed to 40 μg/ml EVO for 24, 48, and 72 h for assessments via fluorescence microscopy. For AO/EB staining, cells were trypsinized and resuspended in 0.1 ml of serum-free RIPA 1640 medium. Then, the cells were stained with 4 μL of AO/EB solution (100 μg/ml; Sigma–Aldrich) at 37 °C for 15 min. The remaining Caki-1 cells were fixed with 70% ethanol for Hoechst 33258 and TUNEL staining. The magnitude of DNA damage was examined via TUNEL assay, using a commercial kit (Roche, Basel, Switzerland) following the manufacturer’s instructions. Subsequently, the cells were stained with 4 ng/ml 4′,6-diamidino-2-phenylindole (DAPI) or 1 μg/ml Hoechst 33258 solution (Sigma–Aldrich) at 4 °C for 5–10 min. Stained cells in each group were observed using a confocal laser scanning microscope (Leica Microsystems, Hessen Wetzlar, Germany).
Cellular ultrastructure examination
Cellular ultrastructure was investigated using transmission electron microscopy (TEM), as described previously21. Briefly, Caki-1 cells exposed to 40 μg/ml EVO for 24, 48, and 72 h were harvested as mentioned above. The cells were fixed with 2% glutaraldehyde (containing 0.1 M sucrose and 0.2 M sodium cacodylate) overnight at 4 °C, followed by 10 g/l osmium tetroxide. The samples were dehydrated and embedded in epoxy resin. Ultra-thin sections were observed using an H700 TEM (Hitachi, Tokyo, Japan).
RNA extraction
Total RNA of cells was extracted using a Cell RNA Kit (Omega Bio-Tek, Inc., Norcross, GA, USA) according to the manufacturer’s instructions. The integrity of the RNA was determined on 1% agarose gels. RNA quality and concentration were determined using a NanoPhotometer spectrophotometer (IMPLEN, Westlake Village, CA, USA).
Library construction for DGE sequencing
To explore the mechanism underlying the effects of EVO on human renal carcinoma cells, Caki-1 cells exposed to 40 μg/ml EVO for 48 h or without EVO treatment were collected for RNA extraction. For library construction, 5 μg RNA of each sample was used. Libraries were constructed using a NEBNext Ultra RNA Library Prep Kit for Illumina (NEB, Ipswich, MA, USA) according to the manufacturer’s instructions. When constructing the libraries, a unique barcode was added to each sample. First, mRNA was isolated from total RNA with poly(T)-oligo-conjugated magnetic beads. After purification, the mRNA was fragmented and used to synthesize the first strand using random hexamer primers and M-MuLV reverse transcriptase (RNase H). Subsequently, the second strand of cDNA was generated using DNA polymerase I and RNase H. Before adenylation of the 3ʹ ends of cDNA, exonuclease/polymerase was used to repair cohesive ends. Then, the specific adaptor for the Illumina library was added to each sample. cDNA fragments with a length of 150–200 bp were selected and purified using an AMPure XP system (Beckman Coulter, Beverly, MA, USA). Finally, these libraries were amplified and then subjected to purification. The quality and quantity of the final libraries were assessed on an Agilent Bioanalyzer 2100.
Analysis of differentially expressed genes
Differential expression analysis between the experimental and control groups was performed using the DESeq R package (1.18.0). Fragments per kilobase of transcript (FPKM) were used to measure gene expression levels. P-values were generated for each gene and the adjusted P-value was set at 0.05 for the subsequent analysis.
Functional annotation and GO/KEGG enrichment analysis
For functional annotation of differentially expressed genes, gene ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes analyses were implemented using the GOseq R package and KOBAS software, respectively27–30.
Real-time quantitative PCR validation
Total RNA extraction was performed as described above. Briefly, 1 μg of total RNA was reverse-transcribed for validation of differentially expressed genes. In total, 16 genes were amplified using custom-designed primers. Two other pairs of primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin were used to amplify the internal reference genes, as shown in Supplementary File S5. The qPCR was performed using LightCycle 480. The reaction volume was 20 μl and included the following: 10 μl of SYBR mix, 0.6 μl of each primer, 2 μl of cDNA template, and 6.8 μl of RNA-free water. The thermocycling program was as follows: 95 °C for 5 min, followed by 45 cycles of 95 °C for 10 s, 57 °C for 10 s, and 72 °C for 20 s. Relative gene expression levels were calculated using the 2−ΔΔCt method31.
Statistical analysis
In our study, triplicate experiments were performed in parallel. For data analysis, the SPSS 21.0 software package (Chicago, IL, USA) was used to detect statistically significant differences between means. Compared with negative controls, differences were considered statistically significant if P < 0.05.
Results
EVO reduced cell viability and impaired renal carcinoma cell proliferation
First, we investigated the inhibitory activity of EVO against the proliferation of three cell lines (human renal carcinoma cell lines (786-O and Caki-1 cells), and the human renal epithelial cell line HK-2) using the CCK-8 assay. The three cell lines were exposed to increasing concentrations (5–100 μg/mL) of EVO for different incubation times (24, 48, and 72 h). We found that EVO decreased the viability of these three cell lines in a time- and concentration-dependent manner (Fig. 1a,b). However, the sensitivity to EVO was different among the three cells. The most remarkable effect was detected for Caki-1 cells, whereas a minimal growth-inhibiting effect was observed for HK-2 cells (Fig. 1c). The IC50 was 23.707 μg/mL at 48 h in Caki-1 cells. In addition, the colony-formation assay demonstrated that EVO significantly inhibited the proliferation of Caki-1 and 786-O cells compared with the control group (Fig. 1d,e), which was consistent with the results of the CCK-8 assay. Based on this finding, Caki-1 cells were used in subsequent experiments.
Figure 1.

Effect of EVO on indicated renal carcinoma cell lines. (a–c) Inhibitory effect of EVO on three cell lines (renal carcinoma cell lines Caki-1 and 786-O and the human renal epithelial cell HK-2). The survival rate of Caki-1 and 786-O cells after treatment with EVO (d,e) based on a colony formation assay (n = 5; *P < 0.05, **P < 0.01).
EVO-induced modulation of Caki-1 cells at the transcriptional level
To explore the mechanism underlying the antitumour effects of EVO, a transcriptional profiling analysis between EVO-treated Caki-1 cells and untreated cells was performed. In total, 27,366,108 reads in the EVO-treated group and 30,288,463 reads in the control group were generated via RNA-seq. These reads were mapped to the human genome, and the unique mapping rates reached 80%, which represented 14,705 and 15,703 expressed genes, respectively. Then, FPKM was used to quantify gene expression levels. The differences in gene expression levels between the treated and control groups are shown in Fig. 2a and Table S1. These results indicated that 480 and 1478 genes were uniquely expressed in the EVO-treated and control groups, respectively (Fig. 2b). According to the standard of P < 0.05 and fold change (FC) > 2, 7243 differentially expressed genes were found, among which 46.21% (3347) were downregulated and 53.79% (3896) were upregulated (Fig. 2c). Comparisons between the treated and control groups were performed using heatmaps. The standard normal distribution of expression signals for every gene was used for normalization (Fig. 2d).
Figure 2.
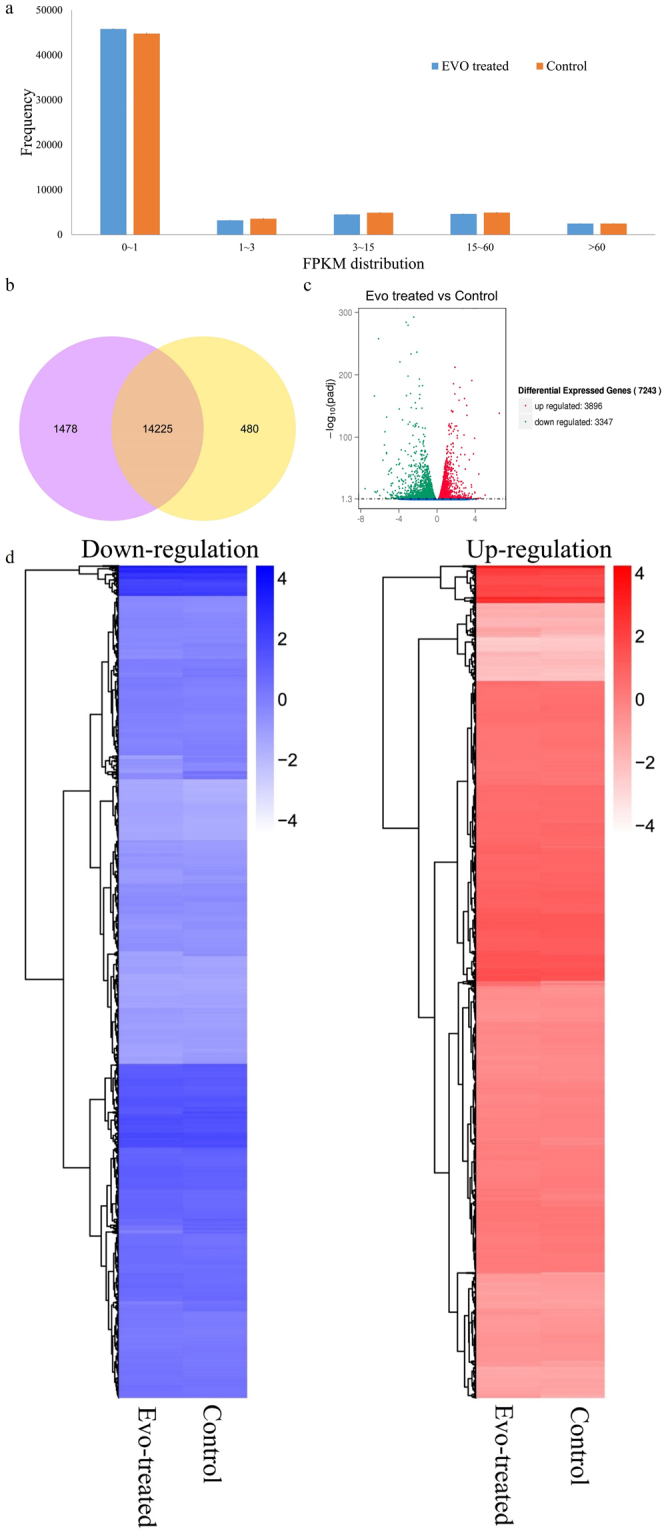
Overview of genes differentially expressed between EVO-treated Caki-1 cells and untreated cells. (a) Column diagram of gene expression values. (b) Venn diagram of the genes differentially expressed between EVO-treated Caki-1 cells and untreated cells. (Cc Volcano diagram of genes differentially expressed between EVO-treated Caki-1 cells and untreated cells. (d) Heatmaps of upregulated and downregulated genes in EVO-treated Caki-1 cells and untreated cells.
Functional analysis of differentially expressed genes
To understand the function of genes differentially expressed between the treated and control groups, GO category and KEGG pathway analyses for the differentially expressed genes were conducted. The enrichment of GO categories for these genes showed that the upregulated genes were mainly involved in protein polymerization, cell adhesion, negative regulation of the cell cycle, apoptosis, and the DNA damage response (Fig. 3a), and the downregulated genes were mainly related to cell migration, cell motility, cell and tube morphogenesis, cell cycle, and DNA replication (Fig. 3b). Analysis of the metabolic pathways that these differentially expressed genes participated in showed that the most enriched upregulated pathways were involved in mitogen-activated protein kinase (MAPK) signalling, endoplasmic reticulum, phosphatidylinositol signalling, apoptosis, and endocytosis (Fig. 3c), whereas the most enriched downregulated gene pathways included DNA replication, phosphatidylinositol-3 kinases/protein kinase B (PI3K/Akt) signalling, transforming growth factor-β (TGF-β) signalling protein digestion and absorption, and the cell cycle (Fig. 3d). The effects of EVO were most closely associated with the pathways of apoptosis (Figure S1), cell cycle (Figure S2), and DNA replication (Figure S3), all of which can decrease cell proliferation capacity and viability. These findings indicate that EVO might modulate Caki-1 cell biological processes by inhibiting the cell cycle and inducing apoptosis.
Figure 3.

GO and KEGG analysis of differentially expressed genes. (a,b) Upregulated and downregulated genes linked with GO enrichment terms. (c,d) Upregulated and downregulated genes in KEGG pathway analysis.
EVO induced G2/M-phase arrest in Caki-1 cells
To assess the effects of EVO on the cell cycle, Caki-1 cells were treated with 40 μg/ml EVO for 24, 48, and 72 h, respectively, and evaluated via flow cytometry. As shown in Fig. 4a,b, cell population in the G2 phase significantly increased upon treatment with EVO by 10.41%, 70.1%, 73.36%, and 80.31% compared with that in the control group. Additionally, EVO-treated cells exhibited a reduction in the population in S phase (30.08%, 19.8%, 19.25%, and 15.28%) and G1 phase (59.51%, 10.1%, 7.29%, and 4.41%). These findings indicate that EVO inhibits Caki-1 cell proliferation by inducing cell cycle arrest at the G2/M phase.
Figure 4.

Cell cycle profiling of Caki-1 cells exposed to EVO. (a) Cell cycle distribution of EVO-treated Caki-1 cells. (b) Cell cycle profile of EVO-treated Caki-1 cells.
EVO induces apoptosis
To further explore the role of apoptosis in EVO treatment of Caki-1 cells, morphological changes were observed via fluorescence microscopy and TEM. We found that Caki-1 cells exhibited time-dependent apoptosis-like morphological changes (Fig. 5) after exposure to 40 μg/ml EVO at times ranging from 24 to 72 h. The observed morphological features included bright staining (Fig. 5a), condensed chromatin (Fig. 5a), elevated permeability (Fig. 5b), microvillus disappearance (Fig. 5c), ultrastructural disorganization (Fig. 5c), vacuolation (Fig. 5c), and apoptotic body formation (Fig. 5c). Moreover, apoptosis was investigated via TUNEL assay and flow cytometry to quantitatively analyse apoptotic cells. TUNEL was used to evaluate fragmented DNA in apoptotic cells. After EVO treatment for 24, 48, and 72 h, cells were stained with TUNEL and analysed via fluorescence microscopy (Fig. 5d,f). The proportions of apoptotic cells were 30.9%, 37.12%, and 45.51% after treatment with EVO ranging from 24 to 72 h, respectively, compared with 3.36% for the control group (Fig. 5e,g).
Figure 5.

EVO induced apoptosis in Caki-1 cells. (a) Apoptosis-related morphological changes were detected by staining cells with Hoechst 33258. Apoptotic cells were defined as those with blue-stained nuclei that exhibited a fragmented/condensed appearance (red arrow). Original magnification, × 200. (b) Elevated plasma membrane permeability was detected by staining cells in the AO/EB double-staining assay (white arrow). Original magnification, × 200. (c) Ultrastructural changes in Caki-1 cells were detected via TEM. a, apoptotic body; c, condensed chromatin; mv, microvillus; N, nucleus; v, vacuole. (d) The 3ʹ OH ends of fragmented DNA of apoptotic cells were labelled in the TUNEL assay and viewed under a fluorescence microscope. Original magnification, × 400. (e) Bar graph showing the quantification of TUNEL-positive Caki-1 cells. (f) Annexin V-FITC and PI reactivity was used to detect and quantify apoptosis induced by EVO. The percentage of cells undergoing apoptosis was determined via flow cytometry. Mean values are shown with the SD and significant differences between the EVO-treated and untreated groups are indicated as *(P < 0.05) or **(P < 0.01).
Validation of the expression of apoptosis- and cell cycle-associated genes
To validate the differentially expressed genes identified by RNA-Seq analysis, qRT-PCR was performed to determine the expression levels of 16 genes associated with apoptosis and the cell cycle. We measured the mRNA transcript levels of genes involved in apoptosis, including interleukin-1 (IL1), tumour necrosis factor receptor type 1-associated death domain protein (TRADD), interleukin-1 receptor-associated kinase (IRAK), cytochrome C (cyto-c), apoptosis-inducing factor (AIF), PI3K, endonuclease G (ENDOG), and protein kinase A (PKA), and the cell cycle, including tumour protein (p53), cell-division cycle protein (Cdc14), TGF-β, mothers against decapentaplegic homolog 2 (Smad2), growth arrest and DNA damage protein 45 (GADD45), checkpoint kinase 1 (Chk1), mini-chromosome maintenance 5 (Mcm5), and mini-chromosome maintenance 6 (Mcm6).
We observed a significant increase in the expression of genes involved in the induction of apoptosis (IL1, TRADD, IRAK, cyto-c, AIF, and ENDOG), whereas there was a reduction in the expression of genes involved in the inhibition of apoptosis (PI3K and PKA). Additionally, we detected a significant reduction of p53, Cdc14, TGF-β, Smad2, GADD45, Chk1, Mcm5, and Mcm6 transcript levels; these genes play important roles in cell cycle arrest (Fig. 6b). The relative expression levels of these genes were consistent with the RNA-Seq findings (Table S2).
Figure 6.
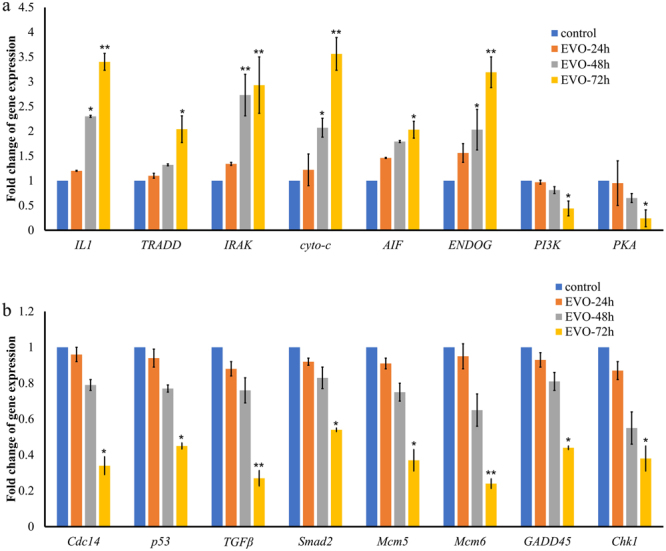
Validation of expression levels of genes related to apoptosis and the cell cycle. (a) Increased gene expression of apoptosis-related genes induced by EVO. (b) Decreased gene expression of cell cycle-related genes induced by EVO.
Discussion
Currently, the incidence and mortality of renal carcinoma are increasing globally, and existing treatments have not yet substantially prolonged survival32,33. The development of novel therapeutic agents for renal carcinoma is urgently needed32,34. The antibacterial, antiobesity, and antinociceptive effects of EVO have been investigated in previous studies14–21. However, the mechanism by which EVO exerts anticancer effects on renal carcinoma cells requires further research22. Herein, we provide evidence that EVO suppresses proliferation and induces apoptosis in renal carcinoma cells by affecting multiple cell signalling molecules based on cytology experiments and a transcriptome profiling study.
To characterize the antitumour mechanism of EVO, we first investigated cell viability by adding EVO to cultures of human renal carcinoma cell lines (Caki-1 and 786-O) and the human renal epithelial cell line HK-2. EVO decreased the viability of 786-O and Caki-1 cells, which is in line with the previous finding that EVO decreased the viability of various renal carcinoma cells22. The most obvious antitumour effects of EVO on Caki-1 cells were observed in the CCK-8 assay after comparison of cell viability. Additionally, the cell proliferation findings, based on a colony formation assay, were consistent with our cell viability findings. Preliminary tests showed that EVO could decrease the cell viability. To identify the mechanism that accounts for the EVO-induced antitumour effect, genes differentially expressed between EVO-treated and untreated groups were identified based on transcriptome analysis. In total, 7,243 differentially expressed genes were observed, and their functions were further analysed by GO and KEGG analysis. We found that EVO could affect Caki-1 cells by affecting the following biological processes: apoptosis, the cell cycle, translation, nuclear division, and cell division. Thus, EVO influenced the expression of genes related to apoptosis and cell cycle. The effects of EVO on Caki-1 cells were similar to those observed for polysaccharides on a non-small cell lung cancer cell line35.
Changes in the expression levels of cycle-related genes have often been reported to be related to DNA damage36. Our TUNEL assay results indicated that EVO could induce DNA damage over time; moreover, DNA damage has been found to be induced by EVO treatment of other renal carcinoma cells (i.e., 786-O cells and ACHN cells)22. The fidelity of replication is affected by DNA damage, and severe DNA damage could cause cells to undergo cell cycle arrest37,38. Additionally, our findings indicated that EVO could arrest the cell cycle of Caki-1 cells at the G2/M phase, which is consistent with previous research showing that EVO could induce G2/M arrest in human A498 RCC cells22. Moreover, our RNA-seq and qRT-PCR analysis showed that p53, Cdc14, TGF-β, Smad2, GADD45, Chk1, Mcm5, and Mcm6 were all downregulated in EVO-treated Caki-1 cells. Among the identified genes, p53 plays a vital role in regulation of the cell cycle by controlling the expression of GADD45 39 , one of the activators in the MAPK signalling pathway. TGF-β is a key regulator of cell fate and transmits its signals via Smad2 40. Minichromosome maintenance protein complex (MCM) is a eukaryotic DNA helicase complex that is essential for DNA replication and partially contributes to the chromatin association and phosphorylation of Chk141. p53 and CDC14 can arrest cells at the G2/M phase through PI3K and Chk1 42. The downregulation of these genes was closely related to the cell cycle, leading to decreased proliferation and viability of Caki-1 cells, which is in accordance with the results of the TUNEL and cell cycle assays. These cellular and transcriptional level data all support the hypothesis that EVO can arrest the cell cycle of renal carcinoma cells. It is well established that a cell may undergo apoptosis if it cannot progress through the G2/M checkpoint43. Therefore, the cell cycle arrest, combined with reduced viability and proliferative capacity, imply that EVO may have an apoptosis-inducing effect on Caki-1 cells in vitro.
Induction of cancer cell apoptosis is one of the main modes of cancer treatment44,45. Apoptotic cells present characteristics such as elevated plasma membrane permeability, phosphatidylserine (PS) externalization, and apoptotic body formation46–48. Therefore, plasma membrane permeability, PS orientation, DNA integrity, and the ultrastructure of EVO-treated Caki-1 cells were determined via AO/EB staining, flow cytometry analysis of Annexin-V/PI staining, PI staining, and TEM, respectively, to detect the effects of EVO on apoptosis. Our findings indicate that EVO could induce elevation of plasma membrane permeability in Caki-1 cells, which is similar to the effects previously observed for antitumour agents such as Schisandra chinensis polysaccharide and quercetin49,50. EVO could also induce PS externalization along with typical apoptotic-like ultrastructural changes, such as structural disorganization, vacuolation, and apoptotic body formation in Caki-1 cells. Moreover, we observed that the transcriptional levels of IL-1 mRNA transcripts increased, and the production of IL-1 can induce growth reduction and apoptosis by regulation of the downstream substrate IRAK 51. In addition, we observed that EVO could induce apoptosis by increasing the expression of genes involved in NF-κB-mediated apoptosis (i.e., TRADD) and mitochondrial apoptosis (i.e., AIF, ENDO-G, and cyto-c) pathways52 , 53. In contrast, genes that block the apoptotic death of cancer cells were all shown to be downregulated54. Overall, our data support the conclusion that EVO prominently regulated the expression of these genes and induced apoptosis, which underlies its antitumour effects.
The study findings reveal how EVO exerts its effects against renal carcinoma cells through cell viability and genome-wide differential gene expression analyses. As EVO is a new antitumour modality, the correlation between these findings in vitro and in vivo toxicology assessments should be further studied.
Electronic supplementary material
Acknowledgements
This work was supported by the Science Foundation for Young Scholars of Institute of Tobacco Research of CAAS (No. 2016A02), the Technology Project of China National Tobacco Corp. (No. 110201402007) and the Agricultural Science and Technology Innovation Program of CAAS (No. 20603020001002).
Author Contributions
Z.-F.Z. conceived and designed the project. X.-L.Y. performed the experiments. X.-M.L. and Y.-M.D. drafted and revised the manuscript. Z.Z., X.-D.H., S.C., X.-L.Y., X.-M.L., and Y.-M.D. analysed and interpreted the data. All authors have read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-12918-y.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ljungberg B, et al. The epidemiology of renal cell carcinoma. European urology. 2011;60:615–621. doi: 10.1016/j.eururo.2011.06.049. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 3.Gandaglia G, Ravi P, Abdollah F, Abd-El A-E-RM. Contemporary incidence and mortality rates of kidney cancer in the United States. Canadian Urological Association Journal. 2014;8:247. doi: 10.5489/cuaj.1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ljungberg B, et al. EAU guidelines on renal cell carcinoma: 2014 update. European urology. 2015;67:913–924. doi: 10.1016/j.eururo.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Sun M, et al. Age-adjusted incidence, mortality, and survival rates of stage-specific renal cell carcinoma in North America: a trend analysis. European urology. 2011;59:135–141. doi: 10.1016/j.eururo.2010.10.029. [DOI] [PubMed] [Google Scholar]
- 6.Russo P. Delayed systemic treatment in metastatic renal-cell carcinoma. Lancet Oncology. 2016;17:1187. doi: 10.1016/S1470-2045(16)30247-9. [DOI] [PubMed] [Google Scholar]
- 7.Yim N-H, Jung YP, Kim A, Kim T, Ma JY. Induction of apoptotic cell death by betulin in multidrug-resistant human renal carcinoma cells. Oncology reports. 2015;34:1058–1064. doi: 10.3892/or.2015.4045. [DOI] [PubMed] [Google Scholar]
- 8.Drake CG, Lipson EJ, Brahmer JR. Breathing new life into immunotherapy: review of melanoma, lung and kidney cancer. Nature reviews Clinical oncology. 2014;11:24–37. doi: 10.1038/nrclinonc.2013.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alwan LM, et al. Comparison of acute toxicity and mortality after two different dosing regimens of high-dose interleukin-2 for patients with metastatic melanoma. Targeted oncology. 2014;9:63–71. doi: 10.1007/s11523-013-0276-7. [DOI] [PubMed] [Google Scholar]
- 10.Motzer RJ, Hutson TE, McCann L, Deen K, Choueiri TK. Overall survival in renal-cell carcinoma with pazopanib versus sunitinib. New England Journal of Medicine. 2014;370:1769–1770. doi: 10.1056/NEJMc1400731. [DOI] [PubMed] [Google Scholar]
- 11.Ravaud A, et al. Adjuvant sunitinib in high-risk renal-cell carcinoma after nephrectomy. New England Journal of Medicine. 2016;375:2246–2254. doi: 10.1056/NEJMoa1611406. [DOI] [PubMed] [Google Scholar]
- 12.Motzer RJ, et al. Tivozanib versus sorafenib as initial targeted therapy for patients with metastatic renal cell carcinoma: results from a phase III trial. Journal of clinical oncology. 2013;31:3791–3799. doi: 10.1200/JCO.2012.47.4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidinger M, et al. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. Journal of Clinical Oncology. 2008;26:5204–5212. doi: 10.1200/JCO.2007.15.6331. [DOI] [PubMed] [Google Scholar]
- 14.Jiang J, Hu C. Evodiamine: a novel anti-cancer alkaloid from Evodia rutaecarpa. Molecules. 2009;14:1852–1859. doi: 10.3390/molecules14051852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu J-Y, et al. Topoisomerase I inhibitor evodiamine acts as an antibacterial agent against drug-resistant Klebsiella pneumoniae. Planta medica. 2013;79:27–29. doi: 10.1055/s-0032-1327925. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi Y, et al. Capsaicin-like anti-obese activities of evodiamine from fruits of Evodia rutaecarpa, a vanilloid receptor agonist. Planta medica. 2001;67:628–633. doi: 10.1055/s-2001-17353. [DOI] [PubMed] [Google Scholar]
- 17.Wei J, Li Z, Yuan F. Evodiamine might inhibit TGF‐beta1‐induced epithelial–mesenchymal transition in NRK52E cells via Smad and PPAR‐gamma pathway. Cell biology international. 2014;38:875–880. doi: 10.1002/cbin.10270. [DOI] [PubMed] [Google Scholar]
- 18.Hu CQ, et al. Evodiamine sensitizes BGC-823 gastric cancer cells to radiotherapy in vitro and in vivo. Molecular medicine reports. 2016;14:413–4191. doi: 10.3892/mmr.2016.5237. [DOI] [PubMed] [Google Scholar]
- 19.Mohan V, et al. A novel alkaloid, evodiamine causes nuclear localization of cytochrome-c and induces apoptosis independent of p53 in human lung cancer cells. Biochemical and biophysical research communications. 2016;477(4):1065–1071. doi: 10.1016/j.bbrc.2016.07.037. [DOI] [PubMed] [Google Scholar]
- 20.Chien C-C, et al. Activation of JNK contributes to evodiamine-induced apoptosis and G2/M arrest in human colorectal carcinoma cells: a structure-activity study of evodiamine. PloS one. 2014;9(6):e99729. doi: 10.1371/journal.pone.0099729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du J, et al. Evodiamine induces apoptosis and inhibits metastasis in MDA-MB-231 human breast cancer cells in vitro and in vivo. Oncology reports. 2013;30(2):685–694. doi: 10.3892/or.2013.2498. [DOI] [PubMed] [Google Scholar]
- 22.Wu WS, et al. Protein kinase RNA-like endoplasmic reticulum kinase-mediated Bcl-2 protein phosphorylation contributes to evodiamine-induced apoptosis of human renal cell carcinoma cells. PloS one. 2016;11(8):e0160484. doi: 10.1371/journal.pone.0160484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deng H, et al. Sodium fluoride induces apoptosis in cultured splenic lymphocytes from mice. Oncotarget. 2016;7:67880–67900. doi: 10.18632/oncotarget.12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zuo D, et al. Amphipathic silica nanoparticles induce cytotoxicity through oxidative stress mediated and p53 dependent apoptosis pathway in human liver cell line HL-7702 and rat liver cell line BRL-3A. Colloids and Surfaces B: Biointerfaces. 2016;145:232–240. doi: 10.1016/j.colsurfb.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, et al. Anti-proliferation of breast cancer cells with itraconazole: Hedgehog pathway inhibition induces apoptosis and autophagic cell death. Cancer Letters. 2017;385:128–136. doi: 10.1016/j.canlet.2016.10.034. [DOI] [PubMed] [Google Scholar]
- 26.Zhang M, et al. Cytotoxicity and apoptosis induced by nanobacteria in human breast cancer cells. Int J Nanomedicine. 2014;9:265–271. doi: 10.2147/IJN.S54906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Young MD, et al. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome biology. 2010;11(2):R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanehisa M, et al. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Research. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanehisa M, et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Research. 2016;44:D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Alt AL, et al. Survival after complete surgical resection of multiple metastases from renal cell carcinoma. Cancer. 2011;117:2873–2882. doi: 10.1002/cncr.25836. [DOI] [PubMed] [Google Scholar]
- 33.Escudier B, et al. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of oncology. 2012;23:vii65–vii71. doi: 10.1093/annonc/mdr034. [DOI] [PubMed] [Google Scholar]
- 34.Patard, J. J., Pouessel, D., Bensalah, K. & Culine, S. Targeted therapy in renal cell carcinoma. World Journal of Urology26, 135–140 (2008). [DOI] [PubMed]
- 35.Kang Y, et al. Transcriptome Profiling Reveals the Antitumor Mechanism of Polysaccharide from Marine Algae Gracilariopsis lemaneiformis. PloS one. 2016;11(6):e0158279. doi: 10.1371/journal.pone.0158279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shkreta L, Toutant J, Durand M, Manley JL, Chabot B. SRSF10 connects DNA damage to the alternative splicing of transcripts encoding apoptosis, cell-cycle control, and DNA repair factors. Cell Reports. 2016;17:1990–2003. doi: 10.1016/j.celrep.2016.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu B, et al. Mechanisms of mutagenesis: DNA replication in the presence of DNA damage. Mutation Research/Reviews in Mutation Research. 2016;768:53–67. doi: 10.1016/j.mrrev.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Puente BN, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salvador, J. M., Brown-Clay, J. D. & Fornace Jr, A. J. in Gadd45 Stress Sensor Genes 1–19 (Springer, 2013).
- 40.Chen H-S, Bai M-H, Zhang T, Li G-D, Liu M. Ellagic acid induces cell cycle arrest and apoptosis through TGF-β/Smad3 signaling pathway in human breast cancer MCF-7 cells. International journal of oncology. 2015;46:1730–1738. doi: 10.3892/ijo.2015.2870. [DOI] [PubMed] [Google Scholar]
- 41.Han X, Aslanian A, Fu K, Tsuji T, Zhang Y. The interaction between checkpoint kinase 1 (Chk1) and the minichromosome maintenance (MCM) complex is required for DNA damage-induced Chk1 phosphorylation. Journal of Biological Chemistry. 2014;289:24716–24723. doi: 10.1074/jbc.M114.575035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Syam S, et al. β-Mangostin induces p53-dependent G2/M cell cycle arrest and apoptosis through ROS mediated mitochondrial pathway and NfkB suppression in MCF-7 cells. Journal of Functional Foods. 2014;6:290–304. doi: 10.1016/j.jff.2013.10.018. [DOI] [Google Scholar]
- 43.Nicolini F, et al. Induction of G2/M phase arrest and apoptosis by the flavonoid tamarixetin on human leukemia cells. Molecular carcinogenesis. 2014;53:939–950. doi: 10.1002/mc.22055. [DOI] [PubMed] [Google Scholar]
- 44.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nature reviews Molecular cell biology. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 45.Green DR. Cancer and Apoptosis: Who Is Built to Last? Cancer Cell. 2017;31:2–4. doi: 10.1016/j.ccell.2016.12.007. [DOI] [PubMed] [Google Scholar]
- 46.Shan M, Fan T-J. Cytotoxicity of carteolol to human corneal epithelial cells by inducing apoptosis via triggering the Bcl-2 family protein-mediated mitochondrial pro-apoptotic pathway. Toxicology in Vitro. 2016;35:36–42. doi: 10.1016/j.tiv.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 47.Robles-Escajeda E, et al. A novel curcumin-like dienone induces apoptosis in triple-negative breast cancer cells. Cellular Oncology. 2016;39:265–277. doi: 10.1007/s13402-016-0272-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wani ZA, et al. A novel quinazolinone chalcone derivative induces mitochondrial dependent apoptosis and inhibits PI3K/Akt/mTOR signaling pathway in human colon cancer HCT-116 cells. Food and Chemical Toxicology. 2016;87:1–11. doi: 10.1016/j.fct.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 49.Liu SJ, Qu H-M, Ren YP. SCP, a polysaccharide from Schisandra chinensis, induces apoptosis in human renal cell carcinoma Caki-1 cells through mitochondrial-dependent pathway via inhibition of ERK activation. Tumor Biology. 2014;35(6):5369. doi: 10.1007/s13277-014-1699-1. [DOI] [PubMed] [Google Scholar]
- 50.Baghel SS, et al. A review of quercetin: antioxidant and anticancer properties. World J Pharm Pharmaceutical Sci. 2012;1(1):146–60. [Google Scholar]
- 51.Rhyasen GW, Starczynowski DT. IRAK signalling in cancer. British journal of cancer. 2015;112(2):232–237. doi: 10.1038/bjc.2014.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cabal-Hierro L, et al. TRAF-mediated modulation of NF-kB AND JNK activation by TNFR2. Cellular signalling. 2014;26(12):2658–2666. doi: 10.1016/j.cellsig.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 53.Lopez J, Tait SWG. Mitochondrial apoptosis: killing cancer using the enemy within. British journal of cancer. 2015;112(6):957–962. doi: 10.1038/bjc.2015.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mayer IA, Arteaga CL. The PI3K/AKT pathway as a target for cancer treatment. Annual review of medicine. 2016;67:11–28. doi: 10.1146/annurev-med-062913-051343. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.