

ABSTRACT
Electron bifurcation is the coupling of exergonic and endergonic redox reactions to simultaneously generate (or utilize) low- and high-potential electrons. It is the third recognized form of energy conservation in biology and was recently described for select electron-transferring flavoproteins (Etfs). Etfs are flavin-containing heterodimers best known for donating electrons derived from fatty acid and amino acid oxidation to an electron transfer respiratory chain via Etf-quinone oxidoreductase. Canonical examples contain a flavin adenine dinucleotide (FAD) that is involved in electron transfer, as well as a non-redox-active AMP. However, Etfs demonstrated to bifurcate electrons contain a second FAD in place of the AMP. To expand our understanding of the functional variety and metabolic significance of Etfs and to identify amino acid sequence motifs that potentially enable electron bifurcation, we compiled 1,314 Etf protein sequences from genome sequence databases and subjected them to informatic and structural analyses. Etfs were identified in diverse archaea and bacteria, and they clustered into five distinct well-supported groups, based on their amino acid sequences. Gene neighborhood analyses indicated that these Etf group designations largely correspond to putative differences in functionality. Etfs with the demonstrated ability to bifurcate were found to form one group, suggesting that distinct conserved amino acid sequence motifs enable this capability. Indeed, structural modeling and sequence alignments revealed that identifying residues occur in the NADH- and FAD-binding regions of bifurcating Etfs. Collectively, a new classification scheme for Etf proteins that delineates putative bifurcating versus nonbifurcating members is presented and suggests that Etf-mediated bifurcation is associated with surprisingly diverse enzymes.
IMPORTANCE Electron bifurcation has recently been recognized as an electron transfer mechanism used by microorganisms to maximize energy conservation. Bifurcating enzymes couple thermodynamically unfavorable reactions with thermodynamically favorable reactions in an overall spontaneous process. Here we show that the electron-transferring flavoprotein (Etf) enzyme family exhibits far greater diversity than previously recognized, and we provide a phylogenetic analysis that clearly delineates bifurcating versus nonbifurcating members of this family. Structural modeling of proteins within these groups reveals key differences between the bifurcating and nonbifurcating Etfs.
KEYWORDS: electron-transferring flavoprotein, flavin, nitrogen fixation, nitrogenase, electron bifurcation
INTRODUCTION
Electron bifurcation, or the coupling of exergonic and endergonic redox reactions to simultaneously generate (or utilize) low- and high-potential electrons, has been proposed as the third fundamental form of energy conservation (1, 2). Bifurcating (Bf) enzymes play central roles in the energy metabolism of anaerobic bacteria and archaea, allowing them to reduce the low-potential [4Fe-4S] clusters of ferredoxin (Fd) using higher-potential electron donors. To date, a total of nine Bf enzymes have been identified. The nine currently demonstrated bifurcating enzymes employ diverse substrates and feature in a wide variety of pathways but share the common features of employing NAD(P)H and Fd as redox substrates/products and possessing at least one flavin, proposed to be the site of bifurcation (1, 3).
The first bifurcating enzyme to be characterized in detail was electron-transferring flavoprotein (Etf)-butyryl coenzyme A (butyryl-CoA) dehydrogenase (Bcd) from Clostridium kluyveri and Acidaminococcus fermentans (4–6). Etf-Bcd was shown to couple the oxidation of NADH (Em = −320 mV) to the simultaneous endergonic reduction of Fd (Em = −500 mV) and exergonic reduction of crotonyl-CoA (Em = −10 mV) during acetate or ethanol (C. kluyveri) or glutamate (A. fermentans) fermentation (6) (Fig. 1). Structural and biochemical analysis of the Etf complex from A. fermentans revealed that it consists of two subunits, with the α subunit coordinating one flavin adenine dinucleotide (FAD) (α-FAD) and the β subunit coordinating a second FAD (β-FAD) (6) (Fig. 2). The β-FAD was established to be the site of hydride acceptance from NADH and electron bifurcation, because NADH binds close to this cofactor (6).
FIG 1.
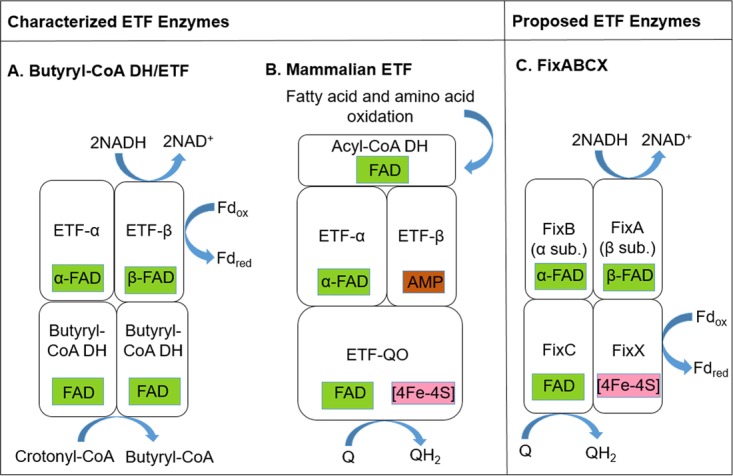
Proposed subunit and cofactor composition and reaction scheme for Etfs. (A) Proposed electron-bifurcating electron transfer mechanism for Etfs from anaerobic fermentative firmicutes (6), in which the β-FAD in Etf-β accepts electrons from NADH and bifurcates them to Fd and to the α-FAD in Etf-α. Etf-α donates electrons to Bcd, which reduces crotonyl-CoA to butyryl-CoA. Notice the absence of a FixX equivalent in this model. (B) Non-Bf electron transfer mechanism in mammalian Etfs. Electrons from the oxidation of fatty acids or amino acids are donated to acyl-CoA dehydrogenase (DH), which passes them to the FAD in Etf-α. Etf-α transfers those electrons to the [4Fe-4S] cluster in Etf-QO, which then reduces a FAD cofactor also in Etf-QO (57). Lastly, this FAD reduces quinone (Q). Note that Etf-β contains a redox-inert AMP cofactor. (C) Electron-bifurcating electron transfer mechanism proposed for FixABCX (2). NADH has been proposed to donate electrons to the β-FAD in FixA, which bifurcates electrons to an [4Fe-4S] cluster in FixX and to the α-FAD in FixB. α-FAD is proposed to subsequently reduce another flavin cofactor (FAD) located in FixC. Lastly, FixX and FixC are proposed to reduce Fd and quinone, respectively.
FIG 2.
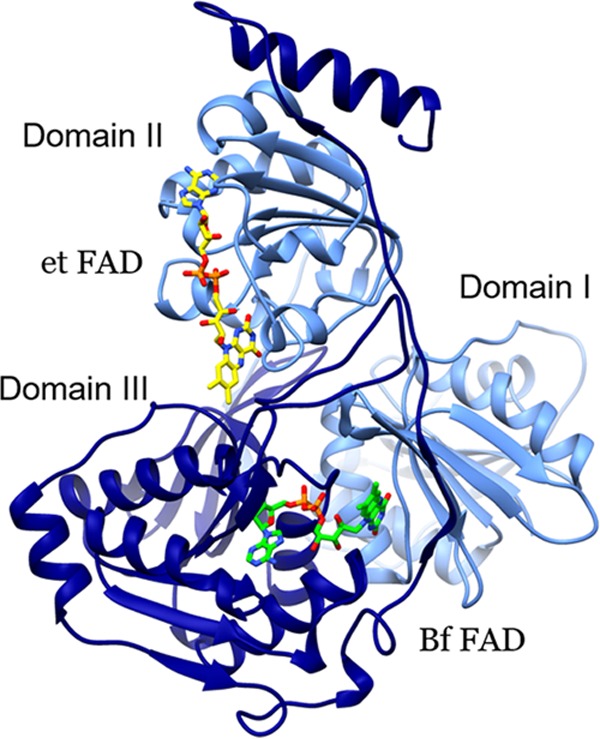
Ribbon diagram of EtfA and EtfB, showing the model of G2 Etfs generated from the consensus sequence based on sequences provided Table S1 in the supplemental material. Light blue indicates Etf-α and dark blue indicates Etf-β; the domains are also labeled. Stick models depict the Bf FAD (green C atoms) and the electron transfer (et) FAD (yellow C atoms). The figure was generated using Chimera (55).
Aside from proposals that a flavin is the site of electron bifurcation, key details of the mechanism of electron bifurcation were lacking until a recent biophysical study of NADP+ oxidoreductase (Nfn) (3), which drives the simultaneous endergonic reduction of Fd and exergonic reduction of NAD+ based on oxidation of NADPH (7). Therein, it was proposed that reduction of Fd is achieved by an unstable flavin anionic semiquinone generated by one-electron oxidation of the doubly reduced flavin hydroquinone formed by hydride transfer from NADPH. Thus, a favorable single-electron transfer from flavin hydroquinone “pays for” production of a much more strongly reducing species that is able to reduce Fd. Moreover, it was proposed that electron transfers from the Bf flavin are mediated by iron-sulfur clusters along two physically separate electron transfer paths whose constituent electron carriers have potentials that span more than 1 V. Given that flavins are common to all known NAD(P)H-utilizing Bf enzymes and that they were proposed (1) and then shown (3) to be the site of bifurcation, it is plausible that unstable flavin anionic semiquinones and additional redox cofactors that differ markedly in their potentials are involved in other Bf systems as well. If so, it is possible that the protein environment of the Bf flavin will be found to produce similar flavin reactivities in diverse bifurcating enzymes, including destabilization of the semiquinone state of flavin and provision of efficient paths of electron transfer as requisites for bifurcation. Thus, the precedent of Nfn suggests that a Bf flavin should be positioned near a binding site for NAD(P)H as well as cofactors or amino acid side chains able to mediate efficient electron transfer (3). Therefore, we hypothesized that such features should be present in Bf Etfs but not necessarily in Etfs in general.
The Bf EtfAB module of the Bf Etf-Bcd complex belongs to a larger group of Etfs, many of which have been biochemically characterized and shown to execute single-electron transfer only, not bifurcation. Members of the larger group include the well-studied mitochondrial EtfAs and EtfBs that are involved in lipid and amino acid metabolism (8). This family of Etfs has been shown to accept electrons from at least nine different acyl-CoA dehydrogenases (9). The acyl-CoA dehydrogenases capture electrons extracted during β-oxidation of CoA-conjugated fatty acids or oxidation of amino acids. Etfs convey those electrons to another flavoprotein, Etf-quinone oxidoreductase (Etf-QO), which in turn passes them into the respiratory electron transport chain via the reduced quinone pool. Hence, Etfs function as an electron funnel in which electrons from a variety of fatty acid or amino acid substrates are channeled into the electron transport chain via Etf-QO (9).
In addition to the Etfs that work with Etf-QO, some bacterial genomes encode Etfs that are not involved in fatty acid or amino acid oxidation or quinone reduction (10–13). For example, Etfs involved in trimethylamine oxidation by a methylotrophic bacterium (10, 14, 15) and carnitine oxidation by Escherichia coli (11, 16) have been described. These Etfs are not associated with Etf-QO but rather are associated with a substrate-specific dehydrogenase (16, 17). Other Etf homologs were identified in the nitrogen-fixing bacterium Sinorhizobium meliloti; the genes were termed fixA and fixB because mutations in them rendered S. meliloti unable to fix nitrogen (12, 18, 19). Regrettably, FixA and FixB correspond to Etf-β and Etf-α, respectively. These nitrogen fixation-associated Etfs have a conserved gene organization in which the genes coding for Etf-α and Etf-β are immediately followed by the genes fixC and fixX, encoding an oxidoreductase and a Fd-like protein, respectively. FixC and FixX, when concatenated, are homologous to Etf-QO and have been shown to be essential for nitrogen fixation in Rhodopseudomonas palustris (20), where they presumably accept electrons from FixA and FixB (12) (Fig. 1). FixABCX was proposed (12, 21) and then shown (22) to oxidize NADH to generate reduced quinone, which feeds the respiratory chain, and reduced Fd, which is the reductant of nitrogenase for N2 fixation.
Structural characterization of Etfs from human mitochondria as well as from the bacterium Paracoccus denitrificans revealed that the two Etf subunits, Etf-α and Etf-β, are tightly associated in a heterodimer composed of three different domains (Fig. 2) (23, 24). Domain I is formed by the N-terminal portion of the α subunit and does not appear to be involved in cofactor binding. Domain II is formed mostly by the C-terminal region of the α subunit, with some contribution from the C-terminal region of the β subunit. A single FAD cofactor is coordinated by residues found primarily within the C terminus of the α subunit, in a region that is highly conserved among all known Etfs. Domain III is formed by most of the β subunit and, in non-Bf Etfs, it coordinates an AMP moiety thought to stabilize the protein but not to play a redox function (10, 24). The Bf Etf-Bcd differs structurally and mechanistically from non-Bf Etfs (1), in that the bound AMP is replaced by a FAD that binds to domain III with its flavin at the interface, where it also interacts with residues in domain I (4–6, 25) (Fig. 1 and 2). Indeed, a previous analysis suggested that a peptide motif from domain III near the binding site of the Bf FAD might differentiate Bf and non-Bf Etfs (26).
Although it is clear from work carried out over the past 30 years that Etf-associated enzymes are widely prevalent in biology and play diverse roles in metabolism, a comprehensive perspective on the diversity of these enzymes is lacking. Here we analyze the phylogenetic and structural variations among Etf proteins in genome sequence databases to discern putative functional variations within this group of enzymes and to identify sequence motifs that potentially signify and even underlie the ability of members of this class of enzymes to bifurcate electrons. A new scheme that classifies Etfs into five phylogenetic and functionally coherent groups is presented. Structural analyses of representatives of these groups reveal amino acid motifs that may be diagnostic and predictive of the ability of Etfs to catalyze electron bifurcation reactions.
RESULTS AND DISCUSSION
Diverse archaea and bacteria encode Etf-α and Etf-β homologs.
Identified Etf-α and Etf-β homologs were first examined at the level of the automated annotations specified in the gene ontology. In most groups, automated or manual annotation during genome curation correctly identified the sequences as Etfs. In some cases, however, Etf gene sequences were incorrectly identified or annotated as fixA or fixB. For example, the α subunit of Etfs (Etf-α) encoded by E. coli genomes and other enterobacteria was consistently annotated as “nitrogen fixation protein FixB” in NCBI databases, although these enterobacteria do not encode the other component needed to fix nitrogen. The annotated fixA and fixB genes in these taxa were often flanked by genes annotated as fixC or fixX. Homologs in actinobacteria were often annotated as fixA or fixB although homologs of fixC and fixX were not identified in the genome. Based on numerous instances of inconsistency in genome annotation, we subjected homologs identified in genome sequence databases to thorough taxonomic and phylogenetic analyses, to develop a more consistent framework for describing and annotating Etf proteins.
A total of 1,314 homologous Etf sequences were identified from 890 bacterial and archaeal genomes (see Table S2 in the supplemental material). Of the 51 archaeal genomes that encoded Etfs, 45% (i.e., n = 23) belonged to the phylum Crenarchaeota, while 55% (n = 28) belonged to the phylum Euryarchaeota. Similarly, a total of 839 bacterial genomes encoded Etfs. The majority of Etfs were found in the phyla Proteobacteria (i.e., 52% of the total of 839 bacterial genomes), Firmicutes (i.e., 17% of the total bacterial genomes), and Actinobacteria (i.e., 15% of the total bacterial genomes) (Table S2). However, the identification of genes encoding Etf-α, Etf-β, FixA, and FixB homologs in numerous phyla previously not known to possess those genes points to their likely widespread importance in cellular metabolism and suggests that they may have a broader array of functions than currently known. In support of this notion, the numbers of copies of etfα and etfβ genes per genome varied from one in about one-third of the genomes to as many as eight or nine in the genomes of some species of Azoarcus, Desulfitobacterium, Geobacter, and Burkholderia (Table S2).
Phylogenetic analyses support five distinct Etf groups.
Maximum-likelihood phylogenetic reconstruction of 1,314 concatenated Etf-α/β sequences and analysis of their relatedness resulted in five well-defined clades, which were designated group 1 (G1) to G5 (Fig. 3A and Table 1). G1 sequences include Etf-α and Etf-β from P. denitrificans and human mitochondria that have been biochemically characterized; these largely correspond to the canonical housekeeping Etfs (23, 24). This group of proteins encompasses the largest number of sequences (473 of 1,314 sequences) and contains representatives of over 300 different Alphaproteobacteria, Betaproteobacteria, Deltaproteobacteria, and Gammaproteobacteria species. About 70 organisms in this group possess multiple G1 Etfs. These organisms also have genes corresponding to etfQO, but they are often separate from the etf genes. Furthermore, there is no biochemical evidence to suggest involvement of any of these enzymes in electron bifurcation; informatic analyses (presented below) support this proposal.
FIG 3.

Maximum-likelihood phylogenetic tree depicting relationships among 1,314 concatenated Etf-α/β sequences. (A) Five distinct phylogenetic groups (shown in distinct colors) that emerged from phylogenetic reconstruction of Etf-β and Etf-α. Sequences that did not belong to any of those groups were left black and are not discussed further (see Table S2 in the supplemental material). Sequences in G2 and G3 were further classified into subgroups (denoted by uppercase letters). (B) Closeup of G2 subgroups, showing further diversity among lineages putatively involved in electron bifurcation; stars denote the presence of biochemically characterized bifurcating Etfs. Sequences in G2D2 have been annotated as FixAB because of their proposed role in nitrogen fixation.
TABLE 1.
Designation, taxonomic distribution, abundance, and proposed functions of Etf groups
| Group and subgroup | Dominant phylum (% of total)a | No. homologs/no. genomesb | Gene name | Proposed functionc |
|---|---|---|---|---|
| Group 1 | Proteobacteria (100) | 473/360 | eftAB | Fatty acid oxidation |
| Group 2 | ||||
| 2A | Firmicutes (85) | 81/63 | etfAB | Fatty acid oxidation, fermentation (butyrate)d |
| 2B | Firmicutes (56) | 53/49 | etfAB | Fatty acid oxidation, fermentation (lactate)d |
| 2C1 | Crenarchaeota (100) | 15/15 | etfABCXe | Mo/W cofactor biosynthesis, vitamin B synthesis |
| 2C2 | Bacteroidetes (50) | 50/48 | etfABCX/etfAB | Unknown |
| 2D1 | Firmicutes (66) | 35/35 | etfABCX | CO2/CO metabolism |
| 2D2 | Proteobacteria (98) | 55/53 | fixABCX | Nitrogen fixationd |
| 2E | Proteobacteria (100) | 23/10 | etfABCXe | Nitrogen fixation, fatty acid aldehyde metabolism |
| Group 3 | ||||
| 3A | Proteobacteria (100) | 76/76 | etfAB | Amino acid metabolism, redox metabolism |
| 3B | Proteobacteria (71) | 66/51 | Carnitine metabolism,d anaerobic fatty acid metabolism | |
| 3C | Euryarchaeota (54) | 76/61 | etfAB | Fatty acid oxidation, menaquinone metabolism |
| 3D | Actinobacteria (100) | 137/130 | etfAB | Amino acid metabolism |
| Group 4 | Bacteroidetes (100) | 51/51 | etfAB | Pyruvate metabolism, nucleotide metabolism |
| Group 5 | Firmicutes (100) | 83/70 | etfAB | Fatty acid oxidation, amino acid metabolism |
| Unclassified | Deinococcus-Thermus (45) | 40/39 | etfAB | Not analyzed |
The number in parentheses represents the percentage of the total sequences within a designated group or subgroup that is within the specified phylum. For simplicity, only the dominant phyla are shown. See Table S2 in the supplemental material for the remaining phyla that encode Etfs in each group.
A given genome may encode multiple Etf homologs and thus be counted multiple times in this table. The total number of genomes encoding Etf homologs was 890.
Based on the annotated function of genes colocalized (±20 open reading frames) with etfAB, etfABCX, or fixABCX.
The link of Etf to this metabolic function is supported by genetic and/or biochemical studies.
etfA and etfB are fused.
G2 sequences are of interest since they include Etf-α and Etf-β from A. fermentans, Megasphaera elsdenii, Acetobacterium woodii, and C. kluyveri, all of which have been shown to function in electron bifurcation and are proposed to coordinate two flavins (4, 6, 26, 27). All of the Etfs known to bifurcate electrons cluster in this group, which nonetheless exhibits substantial phylogenetic diversity, as its 311 included Etf sequences are derived from 15 different phyla (Table 1; also see Table S2).
Functional diversity among G2 enzymes is indicated by the emergence of five subgroups based on phylogenetic clustering and gene neighborhood analyses (described below), with several subgroups being further divided into subsets (Fig. 3B and Table 1; also see Table S2). G2A and G2B consist primarily of Etfs from Firmicutes (i.e., 85% and 56%, respectively, of the total homologous sequences) and include all of the biochemically characterized Etfs that are known to bifurcate. G2C1 consists exclusively of archaeal Etfs, all affiliated with the phylum Crenarchaeota. Interestingly, genes coding for the Etf-α and Etf-β homologs in the genus Sulfolobus are fused and form a single open reading frame, which may indicate that these proteins form a functional complex in a manner similar to that described for several maturase proteins involved in nitrogen fixation (28) and hydrogen metabolism (29). G2C2 Etf genes were identified in 52% of the Bacteroidetes genomes; these proteins represent 50% of the total G2C2 sequences. G2D1 genes primarily include sequences from Firmicutes genomes (i.e., 66% of the total G2D1 sequences), and many of the organisms in this group also encode G1 Etfs. G2D2 Etfs are predominantly found in phylum Proteobacteria (i.e., 98% of the total G2D2 sequences). These G2D2 Etfs are classified as Fix and were proposed (12, 21) and then shown (22) to function in coupling the oxidation of NADH to the formation of quinol and reduced Fd as a reductant for nitrogen fixation through the process of electron bifurcation (21). In support of a role for G2D2 Etfs (Fix) in delivering reduced Fd to nitrogenase, all of the genomes that encode G2D2 Etfs also encode the minimal set of proteins required to fix nitrogen via molybdenum-dependent nitrogenase (i.e., NifHDKENB) (28, 30). Lastly, all G2E Etfs are affiliated with the phylum Proteobacteria. A representative of G2E Etf (i.e., Gmet_2067/2066 from Geobacter metallireducens) has been proposed to be capable of electron bifurcation (31). These Etfs also harbor a peptide motif in the β-FAD cofactor environment that has been proposed to allow for bifurcation (26), which is described in more detail below. A small number of genes encoding G2E ETF-α and Etf-β homologs are flanked by genes encoding homologs of FixC and FixX (described below), suggesting that they may be involved in nitrogen fixation, based on the observation that several of these genomes also encode NifHDKENB.
G3 Etfs include sequences belonging to a variety of bacterial and archaeal phyla (Table 1; also see Table S2). This group forms four subgroups (i.e., G3A to G3D). G3A and G3B Etfs are primarily found in members of Proteobacteria (100% and 70%, respectively, of the total sequences in each group). G3B includes the characterized Etf involved in carnitine oxidation in E. coli (10, 14, 15). Interestingly, many enterobacteria have a G3B Etf coding sequence that has been annotated as ydiQRST (represented as etfABCX in Table 1) and is associated with fadK (ydiD). Genetic studies have shown that ydiQRST and ydiD are required for the capacity to oxidize fatty acids under anoxic conditions in E. coli (11, 16). G3C-like Etfs were enriched in members of the phylum Euryarchaeota (i.e., 54% of the total G3C sequences), although they were also identified in bacteria; this group includes a protein from a methylotrophic bacterium that is involved in trimethylamine oxidation (10, 14, 15). Lastly, all G3D Etfs belonged to members of the phylum Actinobacteria. Based on a lack of conserved residues correlated with the capability to bifurcate in other groups (described in more detail below), we surmise that these enzymes are unlikely to bifurcate.
G4 Etfs and G5 Etfs are confined to the bacterial phyla Bacteroidetes and Firmicutes, respectively. Representative enzymes from G4 and G5 have yet to be biochemically characterized. However, based on the similarity of the proteins encoded in their gene neighborhoods (described below), it is possible that their functions are analogous to those of Etfs in G1 (Table 1). Moreover, 40 Etf homologs could not be readily classified into any of these phylogenetic groups and hence were left unclassified (Table S2). These unclassified Etfs primarily belonged to the phylum Deinococcus-Thermus.
Gene neighborhood analyses associate metabolic processes with Etf function.
To investigate the potential functions of the diverse Etfs uncovered here and to identify potential determinants that define Bf and non-Bf enzymes, we examined the gene neighborhoods flanking Etf genes for each group. Unfortunately, no genes were consistently identified in the flanking regions of G1 Etf genes, precluding a prediction of the functionality of these Etfs based on gene neighborhood analysis. However, all G1 Etf-encoding genomes encoded ubiquinone oxidoreductase (QO) elsewhere in the genome (data not shown); Etf-QO is involved in fatty acid metabolism (32). Therefore, we speculate that G1 Etfs are involved in fatty acid metabolism.
G2 Etfs include characterized bifurcating Etfs (3, 5, 19, 33). Each G2 subgroup was accompanied by a unique suite of proteins encoded by genes in its gene neighborhood (Fig. 4). The gene for acyl-CoA dehydrogenase, which is involved in fatty acid oxidation (9), was identified in the flanking region of 76% and 26% of the genomes that encode G2A and G2B Etfs, respectively. This indicates potential involvement of these Etfs in electron transfer reactions involving fatty acid metabolism. In support of this hypothesized role, G2A and G2B Etfs have been found to be involved in electron transfer reactions during the oxidation of butyrate and lactate (4, 25). The neighborhoods of G2C1 Etf genes included genes for homologs of FixX, FixC-like, and MoaD-like enzymes. MoaD homologs have been shown to be involved in molybdenum/tungsten cofactor biosynthesis or thiamine biosynthesis (34, 35). Thus, it is possible that G2C1 Etfs participate in electron transfer reactions contributing to cofactor or vitamin biosynthesis. All genes for subgroups of G2D Etfs encoded FixC and FixX-like enzymes in their gene neighborhoods. The neighborhoods of 27% of the G2D1 Etf genes included genes for homologs of carbon monoxide dehydrogenase and acetyl-CoA synthase, which raises the intriguing possibility of involvement in CO or CO2 metabolism (36). G2D2 Etfs were all derived from putatively diazotrophic genomes (i.e., the genomes also encoded NifHDKENB). Mutations in Etf gene subunits suppressed growth under nitrogen-fixing conditions, which led to the Etf genes being named fix (fixABCX) in nitrogen-fixing microbes (12, 21, 33, 37, 38). Lastly, the neighborhoods of 26% of the G2E Etf genes encoded acetyl-CoA dehydrogenase, acetyl-CoA acetyltransferase, the nitrogenase cofactor biosynthesis proteins NifE and NifN, and aldehyde oxidoreductase. It is not clear from gene neighborhood analysis what functional role these Etfs may have.
FIG 4.
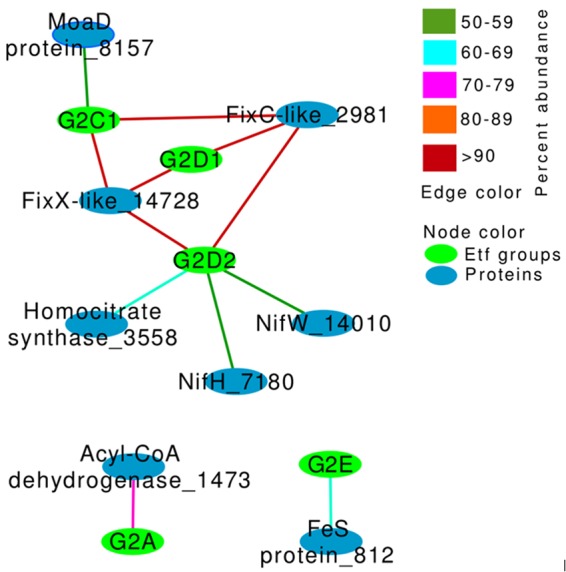
Network depiction of the covariation of proteins encoded in the flanking regions (±20 genes) of etf and specific group designations. Only proteins (n = 8) encoded by ≥50% of the G2 Etf-encoding genomes (i.e., relative frequency of ≥50%) were considered in this analysis. Here, nodes represent either the Etf group designation (green) or proteins in the flanking gene environment of Etfs (blue), while the edge color represents the abundance of the proteins in the group.
G3 Etfs were divided into four groups, with G3A deriving from aerobic and facultatively anaerobic organisms. The gene environments for these Etfs included genes for NADH oxidase and multiple proteins containing iron-sulfur clusters (Fig. S1). G3B Etf genes were found to be accompanied by genes for FixC-like and FixX-like enzymes, carnitine dehydratase, and acyl-CoA dehydrogenase in the gene neighborhood, supporting the idea that they are involved in carnitine metabolism (11, 14). The neighborhoods of 32% of G3C Etf genes encoded homologs of acetyl-CoA dehydrogenase and acetyl-CoA acetyltransferase, both of which are involved in fatty acid oxidation. In addition, the neighborhoods of 29% of G3C Etf genes encoded homologs of ubiquinone methyltransferase, which is involved in menaquinone biosynthesis (39); thus, these Etfs may support biosynthesis of menaquinone. Genes that flanked G3D Etf genes commonly encoded cysteine desulfurase (73% of G3D Etfs), Asn/Gln amidotransferase (62% of G3D Etfs), and Gln amidotransferase (60% of G3D Etfs), suggesting a role for these flavoproteins in electron transfer during amino acid biosynthesis (Fig. S1).
The neighborhoods of G4 Etf genes did not reveal any conserved (>50% relative frequency) genes. However, among the more commonly observed genes flanking G4 Etf genes were those encoding pyruvate dehydrogenase (41% of G4 Etfs) and thymidylate synthase (41% of G4 Etfs). Lastly, the neighborhood of G5 Etf genes included homologs of genes for fumarate reductase (52% of G5 Etfs) and the cytochrome b558 subunit of succinate dehydrogenase (52% of G5 Etfs) (Fig. S1).
Structural modeling reveals key differences between bifurcating and nonbifurcating Etfs.
Regardless of their putative roles in metabolism (Table 1), Etf proteins participate in two mechanistically distinct sets of redox reactions, namely, reactions that bifurcate electrons and reactions that do not bifurcate electrons (4–6, 26, 40). From the recent analysis of the mechanism of the flavoenzyme Nfn (3), it was established that FAD was the site of bifurcation, and we refer to this as the Bf FAD. This flavin accepts a pair of electron equivalents from NADPH and donates one to each of two different electron acceptors.
We interrogated our sequence alignments for motifs and individual residues that could differentiate Bf from non-Bf enzymes. Toward these ends, we carried out structural homology analyses with sequences belonging to G1 and G2 Etfs. Available evidence from biochemical characterization of selected members of these two groups suggests that G1 Etfs do not bifurcate (23, 24), whereas several G2 Etfs do (4, 6, 26, 27), making comparisons between these two types of Etfs especially attractive. A crucial difference is that the Etfs that bifurcate have been demonstrated to contain two FADs, whereas Etfs that do not bifurcate have been found to contain only one. Moreover, bifurcating Etfs can accept electrons from NADH, whereas those that do not bifurcate cannot. Indeed, our analyses revealed conserved amino acid differences affecting residues that interact with NAD(H), based on a recent crystal structure (6) (motif 1), and a motif that coordinates with β-FAD (motif 2), which is the proposed site of bifurcation in Etfs (1, 4).
We examined conserved differences at each position, looking for instances in which sequences from G1 or G2 share a unique residue within their group that is distinct from the type present in the other group (Fig. 5). Figure 5A depicts the functional distinctions between nonbifurcating (G1) and bifurcating (G2) Etfs. The structural comparison reveals that the motifs identified previously are clustered near the β-FAD binding site and indeed are differentially conserved in G1 versus G2 Etfs (Fig. S2). For example, β-Gly127 (using R. palustris numbering) represents a conserved difference in amino acid type; it is a small residue that minimizes steric conflict with the flavin in motif 2 in all G2 proteins, but it is replaced by a larger and generally anionic residue in G1. This negative charge would be expected to repel the phosphate of β-FAD (Bf FAD in G2 enzymes) and thus disfavor binding. In addition, G1 proteins tend to have bulkier residues overall around this residue, further predicting a diminished capacity to bind the flavin mononucleotide (FMN) portion of β-FAD. Thus, our approach reproduces previous findings but adds residues that are nearby in the 3-dimensional protein structure but not necessarily in the amino acid sequence.
FIG 5.

Structural analyses of Bf and non-Bf Etfs showing differences near the β-FAD site. (A) Homology model of Etf-α and Etf-β with mapped conserved differences in amino acid type between G1 Etfs, with representatives that do not bifurcate and contain AMP in the β subunit, and G2 Etfs, with representatives that have been demonstrated to catalyze electron-bifurcating reactions. Red indicates equivalent residues at a position, whereas blue indicates a position at which the distribution of residues characterizing one group differs from the distribution of residues found in the other group. The thickness of the worms indicates whether amino acids at a position share the same functional property (wide worm) or not (thin worm) within G2. (B) Homology model of Etf-α/β with mapped conserved differences in amino acid use between Etfs in G2A and G2B combined, which have representatives known to bifurcate, and Etfs in G2D2, which includes the FixAB Etfs. Note the high degree of conservation (thick red worms) around the putative β-FAD binding site, leading us to predict that G2D2 Etfs also bifurcate. Thick worms indicate high levels of conservation within G2D2. (C) Closeup depiction of the NADH-binding (blue green) and FAD-binding (bright green) residues in bifurcating Etfs. Key residues proposed to coordinate these two molecules are numbered with their positions in the sequence of R. palustris Etf (GenBank accession no. YP_783418). Only a portion of the NADH molecule, where the quality of the electron density was good enough to permit unambiguous refinement in the structure upon which our model is based (PDB accession no. 4L2I), is shown. Only residues associated with the proposed ability to bifurcate are shown. (D) Alignment of residues predicted to coordinate NADH and FAD in bifurcating Etfs. Groups in the first column correspond to the groups mentioned in Table 1. Unc indicates the uncharacterized group. Residues highlighted in panel C are shown in red boxes.
Other conserved distinctions in the region in which the β-FAD binds to G2 Etfs are observed in the Etf-α subunit, where two strands of the α chain interface with the β chain and are predicted to participate in the binding of the flavin moiety of β-FAD (Fig. 5C). Thus, a structurally coherent pattern of amino acid conservation supports a tendency for FAD binding in G2 versus AMP binding in G1. Figure 5B depicts a second comparison, in which bifurcating G2 Etfs are compared with predicted bifurcating G2D2 Etfs (also known as Fix). This structural comparison shows that G2D2 Etfs share the motif identified in bifurcating Etfs in G2A and G2B (Fig. 5B), strongly suggesting that G2D2 Etfs (or Fix), like the rest of the G2 subgroups, are capable of electron bifurcation. The region displaying the most blue-colored residues and the most residues with intermediate constancy (shades of purple) coincides with the recognition loop that has been found to interact with partner proteins (41). This is consistent with G2D2 Etfs interacting with FixC and FixX while the genes of some other bifurcating Etfs (G2A and G2B) are not accompanied by genes for FixC or Etf-QO homologs, suggesting that these Etfs have different partners.
A region found to interact with NAD+ in the crystal structure of the A. fermentans Bf Etf (6) (motif 1) also exhibits a contiguous string of conserved differences between Bf and non-Bf Etfs (Fig. 5A). This region includes the peptide bond between β-Phe89 and β-Ala90, which π stacks with the NAD+ adenine ring, the backbone amide of β-Asp86, which forms hydrogen bonds with the adenine ring, and the backbone amide of β-Gly91, which forms hydrogen bonds with the α-phosphate of NAD+ (Fig. 5C). These combined conserved differences in the region of the β-FAD flavin-binding pocket all suggest that NADH is a substrate for G2 but not G1 Etfs.
To investigate whether motifs 1 and 2 that are conserved in G2A and G2B Etfs are conserved in other Etf groups, we aligned the corresponding consensus sequences from each of the subgroups that emerged from our database (Fig. 5D). As expected, nearly all of the G2 sequences exhibited conservation in the residues that were identified as being important in distinguishing Bf G2 sequences from non-Bf G1 sequences, whereas the same residues were not conserved in the sequences belonging to the other groups. Importantly, our informatic analyses suggested that more members of G2A and G2B may be found to be capable of electron bifurcation, providing the impetus for biochemical studies of Etfs from G2C (primarily found in Archaea) and G2D (including the enzymes that were hypothesized [12, 21] and then shown [22] to be involved in providing reductant for nitrogen fixation), to test for bifurcation activity. Importantly, there are likely exceptions or limitations to the predictions of which enzymes can bifurcate, based on conservation of the aforementioned amino acid sequence motifs. For example, Clostridium propionicum has been shown to have an insertion in Etf-α that prevents bifurcation, although the sequence for this Etf has the identified conserved motifs that predict bifurcation capability (26).
Our structural comparison predicts that only G2 Etfs have the capacity to bifurcate, which raises the questions of how and when bifurcating Etfs evolved. Since our phylogenetic tree is not rooted (i.e., it is a protein family tree consisting of all paralogs), we cannot use it to predict the origin of bifurcating Etfs. However, the Etf protein family phylogeny does suggest that, once the ability to bifurcate electrons arose (as either an ancestral or derived characteristic), this activity was maintained (i.e., G2 Etf all share an apparent ability to bifurcate). Both archaeal and bacterial genomes encode putative bifurcating G2 Etfs; however, the archaeal versions known so far all occur in G2B and G2C. These lineages branch closer to the crown of the G2 lineage, suggesting that G2 Etfs diverged from a bacterial ancestor. Furthermore, the majority of G2C and G2D Etfs, which have genes encoding FixC- and FixX-like proteins in their neighborhoods, diverged after G2A and G2B. Parsimony would suggest that fixC- and fixX-like genes were acquired during the evolution of G2 Etfs. Importantly, the genomes of several members of G3B also encode FixC- and FixX-like subunits, and these are nested among G3 sequences that lack these subunits; this suggests that these genes were acquired independently multiple times during the evolution of Etfs. These collective observations, including those showing that Etf-α and Etf-β can form complexes with other enzymes, such butyryl-CoA dehydrogenase (4), crotonyl-CoA dehydrogenase (4), and pyruvate dehydrogenase (42), underscore the versatility of the Etf-α/β protein “chassis” in the emergence and evolution of new metabolic processes in microorganisms.
Overall, the Etfs provide a unique opportunity to compare many amino acid sequences for proteins that are structural homologs but represent two distinct functional capacities, i.e., single-electron transfer via a single FAD versus electron bifurcation within a system containing two FADs in the Etf plus additional cofactors in the associated enzyme complex. The diverse genomic contexts in which Etf genes are found may reflect their versatility and the essential nature of the energy and electron transactions they enable. However, the distinction between single-electron transfer and electron bifurcation is critical in energy conservation. Therefore, the identified protein characteristics that demarcate Bf and non-Bf groups of Etfs provide exciting predictions regarding which Etfs should be tested for their ability to bifurcate and which residues and types of residues provide the protein environment allowing for this capability.
Concluding remarks.
Our analysis shows that Etf enzymes are phylogenetically diverse and widely distributed in archaea and bacteria, consistent with the essential roles of these enzymes in the metabolism and fitness of numerous strains (43, 44). Many strains encode more than one Etf, which is also consistent with the roles of Etfs in diverse metabolic processes. These Etf homologs (or paralogs, if they are not functionally redundant) sometimes belong to the same phylogenetic group, e.g., multiple G3B Etfs in E. coli and multiple G2E ETFs in Geobacter spp., but in other instances belong to distinct groups, e.g., many proteobacteria with G2D2 Etfs also encode G1 Etfs and crenarchaeota with G2C1 Etfs also encode G3B Etfs. The vast phylogenetic diversity and numerous copies of Etfs in microbial genomes suggest that they have unexplored functional diversity, which could shed light on the metabolism of these organisms. In several cases, predictions can be made regarding the potential functions of these uncharacterized Etfs based on proteins encoded by adjacent genes, as discussed above. These predictions provide a roadmap for future biochemical studies aimed at characterizing the functional diversity of this enzyme family.
The demonstration of bifurcation for Etfs of anaerobic bacteria (4–6) significantly advanced our understanding of the interplay among processes that contribute to energy conservation in fermentative metabolism. Perhaps our most intriguing findings here are the identification of residues that apparently enable bifurcation capability in Etfs in phylogenetically and physiologically diverse organisms that extend beyond those recognized previously (26) and residues dispersed in the sequences of both subunits. Indeed, ours are the first such analyses to integrate information from putative Bf Etfs obtained from a comprehensive database of sequences from nonfermentative archaeal and bacterial taxa as well as those putatively associated with nitrogen fixation.
The identification of bifurcation-associated residues provides a foundation for future biochemical studies aimed at elucidating the specific mechanism by which FAD transfers electrons to acceptors with differing midpoint potentials. Bifurcating Etfs need to provide a protein environment that potentiates the states of FAD likely required during bifurcation, such as the proposed unstable anionic semiquinone. Our study identifies residues coordinating FAD that could play such a role.
MATERIALS AND METHODS
Generation of an Etf/Fix database.
Homologs of Etf-α were identified using a BLASTp search (cutoff E value of 10−5) against completed bacterial and archaeal genomes available in the Department of Energy Integrated Microbial Genomes (IMG) database (45), with FixB (Etf-α) from Azotobacter vinelandii (STRING network identifier: Avin_10530) serving as the query. A total of 1,314 Etf-α homologs across 890 genomes were identified. Homologs of Etf-β (compiled from the IMG database for each Etf-α sequence) and Etf-α were aligned individually using Clustal Omega (46), and the two resultant alignment blocks were concatenated.
Protein clustering and phylogenetic analyses.
The concatenated Etf-α and Etf-β sequences were subjected to maximum-likelihood phylogenetic reconstruction with RaxML (version 7.3.0) (47), specifying the LG substitution matrix, the PROTGAMMA option, and 100 bootstrap iterations. The tree was projected with FigTree. In addition, a custom Python (version 2.7.6) script was used to extract gene sequences (20 upstream and 20 downstream) that flanked each Etf-β. The 40 genes extracted were translated in silico, and the protein sequences were clustered using identity thresholds of 90%, 60%, and 30% while holding the pairwise sequence coverage threshold constant at >60%. The clusters generated by the three-step clustering method were later combined to yield a final “averaged” cluster identity.
Network analysis.
Protein sequence clusters obtained from the earlier step were used to generate a binary matrix describing the presence or absence of genes. The binary matrix generated was then used to identify the abundant proteins (i.e., present in ≥50% of the genomes) in each group to create a network plot, with Cytoscape specifying the force-directed organic layout (58). Each unique protein or group identified was denoted as a node, and the edges between the nodes represent the abundance of the genes in the group.
Structural modeling.
Amino acid diversity was scored as the number of different amino acids found in an alignment position. Functional diversity at each aligned position was described in terms of five defined types of residues, i.e., negatively charged residues, including Asp and Glu; positively charged residues, including Lys and Arg; hydrophobic residues, including Ile, Leu, Val, Met, and Phe; and polar uncharged residues, including Gln, Asn, Ser, His, Thr, Cys, Ala, and Gly (48). Trp, Tyr, and Pro were considered to be special, based on their ability to mediate electron transfer within proteins (49) and their ability to influence protein dynamics and folding (50).
Structural models were generated by using the consensus sequence and online tools provided by the SwissModeler suite (51, 52) (see the supplemental material for the consensus sequences used in this study). Etf-α and Etf-β protein templates were chosen from the same complex to optimize preservation of the interface between monomers, based on metrics of structure quality captured by Qmean4 (53, 54). Monomer coordinates were combined with cofactor coordinates from the template, and the resulting holoproteins were assessed on the basis of the number of clashes between protein and FAD molecules and between monomers, using tools provided within the Chimera modeling software package (55). Holoprotein dimers with the fewest clashes and clashes derived mainly from side chain interactions were subjected to energy minimization after the addition of H atoms and the assignment of charges (56), as implemented in Chimera (55). The resulting minimized holoprotein dimer models were again evaluated with respect to clashes, and only models free of clashes were used for further work. The templates used for each model are reported in Table S1 in the supplemental material, along with metrics of the quality of the models.
To identify conserved differences between groups of sequences, we compared the prevalence of amino acid types at analogous positions. At each position, we constructed a vector in the 5-dimensional space defined by the five functional types of amino acids. The five components of the vector represented the prevalence of each of the five functional types of amino acids, as outlined above. To determine the extent of similarity at each position of the two groups being compared, we calculated the scalar product of the corresponding vectors. Thus, conservation of the same functional type at an alignment position (parallel vectors) yields the maximum value of 1, whereas conservation of different functional types in each of the two groups yields a minimum value of 0. Positions characterized by similar variations among functional types in the two groups of sequences yield high values for the scalar product because the two vectors point in the same direction in the 5-dimensional space, even though no one type is conserved. To highlight positions where a single functional type is conserved in the reference group, we displayed the degree of type conservation using worm width, with wide worm segments for positions at which the type is conserved and thinner worm segments for positions at which the type varies.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported as part of the Biological Electron Transfer and Catalysis Energy Frontier Research Center, funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under award DE-SC0012518.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00440-17.
REFERENCES
- 1.Buckel W, Thauer RK. 2013. Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochim Biophys Acta Bioenerg 1827:94–113. doi: 10.1016/j.bbabio.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Peters JW, Miller AF, Jones AK, King PW, Adams MW. 2016. Electron bifurcation. Curr Opin Chem Biol 31:146–152. doi: 10.1016/j.cbpa.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Lubner CE, Jennings DP, Mulder DW, Schut GJ, Zadvornyy OA, Hoben JP, Tokmina-Lukaszewska M, Berry L, Nguyen DM, Lipscomb GL, Bothner B, Jones AK, Miller AF, King PW, Adams MWW, Peters JW. 2017. Mechanistic insights into energy conservation by flavin-based electron bifurcation. Nat Chem Biol 13:655–659. doi: 10.1038/nchembio.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herrmann G, Jayamani E, Mai G, Buckel W. 2008. Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J Bacteriol 190:784–791. doi: 10.1128/JB.01422-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li F, Hinderberger J, Seedorf H, Zhang J, Buckel W, Thauer RK. 2008. Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J Bacteriol 190:843–850. doi: 10.1128/JB.01417-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chowdhury NP, Mowafy AM, Demmer JK, Upadhyay V, Koelzer S, Jayamani E, Kahnt J, Hornung M, Demmer U, Ermler U, Buckel W. 2014. Studies on the mechanism of electron bifurcation catalyzed by electron transferring flavoprotein (Etf) and butyryl-CoA dehydrogenase (Bcd) of Acidaminococcus fermentans. J Biol Chem 289:5145–5157. doi: 10.1074/jbc.M113.521013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demmer JK, Huang H, Wang S, Demmer U, Thauer RK, Ermler U. 2015. Insights into flavin-based electron bifurcation via the NADH-dependent reduced ferredoxin:NADP oxidoreductase structure. J Biol Chem 290:21985–21995. doi: 10.1074/jbc.M115.656520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crane KL, Beinert H. 1956. On the mechanism of dehydrogenation of fatty acyl derivatives of coenzyme A. II. The electron-transferring flavoprotein. J Biol Chem 218:717–731. [PubMed] [Google Scholar]
- 9.Ghisla S, Thorpe C. 2004. Acyl-CoA dehydrogenases: a mechanistic overview. Eur J Biochem 271:494–508. doi: 10.1046/j.1432-1033.2003.03946.x. [DOI] [PubMed] [Google Scholar]
- 10.Chen D, Swenson RP. 1994. Cloning, sequence analysis, and expression of the genes encoding the two subunits of the methylotrophic bacterium W3A1 electron transfer flavoprotein. J Biol Chem 269:32120–32130. [PubMed] [Google Scholar]
- 11.Eichler K, Buchet A, Bourgis F, Kleber HP, Mandrand-Berthelot MA. 1995. The fix Escherichia coli region contains four genes related to carnitine metabolism. J Basic Microbiol 35:217–227. doi: 10.1002/jobm.3620350404. [DOI] [PubMed] [Google Scholar]
- 12.Earl CD, Ronson CW, Ausubel FM. 1987. Genetic and structural analysis of the Rhizobium meliloti fixA, fixB, fixC, and fixX genes. J Bacteriol 169:1127–1136. doi: 10.1128/jb.169.3.1127-1136.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsai MH, Saier MH Jr. 1995. Phylogenetic characterization of the ubiquitous electron transfer flavoprotein families ETF-α and ETF-β. Res Microbiol 146:397–404. doi: 10.1016/0923-2508(96)80285-3. [DOI] [PubMed] [Google Scholar]
- 14.Davidson VL, Husain M, Neher JW. 1986. Electron transfer flavoprotein from Methylophilus methylotrophus: properties, comparison with other electron transfer flavoproteins, and regulation of expression by carbon source. J Bacteriol 166:812–817. doi: 10.1128/jb.166.3.812-817.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DuPlessis ER, Rohlfs RJ, Hille R, Thorpe C. 1994. Electron-transferring flavoprotein from pig and the methylotrophic bacterium W3A1 contains AMP as well as FAD. Biochem Mol Biol Int 32:195–199. [PubMed] [Google Scholar]
- 16.Walt A, Kahn ML. 2002. The fixA and fixB genes are necessary for anaerobic carnitine reduction in Escherichia coli. J Bacteriol 184:4044–4047. doi: 10.1128/JB.184.14.4044-4047.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang L, Rohlfs RJ, Hille R. 1995. The reaction of trimethylamine dehydrogenase with electron transferring flavoprotein. J Biol Chem 270:23958–23965. doi: 10.1074/jbc.270.41.23958. [DOI] [PubMed] [Google Scholar]
- 18.Pühler A, Aguilar MO, Hynes M, Müller P, Klipp W, Priefer U, Simon R, Weber G. 1984. Advances in the genetics of free-living and symbiotic nitrogen fixing bacteria, p 609–619. In Veeger C, Newton WE (ed), Advances in nitrogen fixation research: proceedings of the 5th International Symposium on Nitrogen Fixation, Noordwijkerhout, The Netherlands, August 28–September 3, 1983. Springer Netherlands, Dordrecht, Netherlands. [Google Scholar]
- 19.Ruvkun GB, Sundaresan V, Ausubel FM. 1982. Directed transposon Tn5 mutagenesis and complementation analysis of Rhizobium meliloti symbiotic nitrogen fixation genes. Cell 29:551–559. doi: 10.1016/0092-8674(82)90171-4. [DOI] [PubMed] [Google Scholar]
- 20.Huang JJ, Heiniger EK, McKinlay JB, Harwood CS. 2010. Production of hydrogen gas from light and the inorganic electron donor thiosulfate by Rhodopseudomonas palustris. Appl Environ Microbiol 76:7717–7722. doi: 10.1128/AEM.01143-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edgren T, Nordlund S. 2004. The fixABCX genes in Rhodospirillum rubrum encode a putative membrane complex participating in electron transfer to nitrogenase. J Bacteriol 186:2052–2060. doi: 10.1128/JB.186.7.2052-2060.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ledbetter RN, Garcia Costas AM, Lubner CE, Mulder DW, Tokmina-Lukaszewska M, Artz JH, Patterson A, Magnuson TS, Jay ZJ, Duan HD, Miller J, Plunkett MH, Hoben JP, Barney BM, Carlson RP, Miller A-F, Bothner B, King PW, Peters JW, Seefeldt LC. 2017. The electron bifurcating FixABCX protein complex from Azotobacter vinelandii: generation of low-potential reducing equivalents for nitrogenase catalysis. Biochemistry doi: 10.1021/acs.biochem.7b00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts DL, Frerman FE, Kim JJ. 1996. Three-dimensional structure of human electron transfer flavoprotein to 2.1-Å resolution. Proc Natl Acad Sci U S A 93:14355–14360. doi: 10.1073/pnas.93.25.14355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roberts DL, Salazar D, Fulmer JP, Frerman FE, Kim JJ. 1999. Crystal structure of Paracoccus denitrificans electron transfer flavoprotein: structural and electrostatic analysis of a conserved flavin binding domain. Biochemistry 38:1977–1989. doi: 10.1021/bi9820917. [DOI] [PubMed] [Google Scholar]
- 25.Sato K, Nishina Y, Shiga K. 2003. Purification of electron-transferring flavoprotein from Megasphaera elsdenii and binding of additional FAD with an unusual absorption spectrum. J Biochem 134:719–729. doi: 10.1093/jb/mvg199. [DOI] [PubMed] [Google Scholar]
- 26.Chowdhury NP, Kahnt J, Buckel W. 2015. Reduction of ferredoxin or oxygen by flavin-based electron bifurcation in Megasphaera elsdenii. FEBS J 282:3149–3160. doi: 10.1111/febs.13308. [DOI] [PubMed] [Google Scholar]
- 27.Weghoff MC, Bertsch J, Muller V. 2015. A novel mode of lactate metabolism in strictly anaerobic bacteria. Environ Microbiol 17:670–677. doi: 10.1111/1462-2920.12493. [DOI] [PubMed] [Google Scholar]
- 28.Boyd ES, Anbar AD, Miller S, Hamilton TL, Lavin M, Peters JW. 2011. A late methanogen origin for molybdenum-dependent nitrogenase. Geobiology 9:221–232. doi: 10.1111/j.1472-4669.2011.00278.x. [DOI] [PubMed] [Google Scholar]
- 29.Posewitz MC, King PW, Smolinski SL, Zhang L, Seibert M, Ghirardi ML. 2004. Discovery of two novel radical S-adenosylmethionine proteins required for the assembly of an active [Fe] hydrogenase. J Biol Chem 279:25711–25720. doi: 10.1074/jbc.M403206200. [DOI] [PubMed] [Google Scholar]
- 30.Dos Santos PC, Fang Z, Mason SW, Setubal JC, Dixon R. 2012. Distribution of nitrogen fixation and nitrogenase-like sequences amongst microbial genomes. BMC Genomics 13:162. doi: 10.1186/1471-2164-13-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Estelmann S, Boll M. 2014. Glutaryl-coenzyme A dehydrogenase from Geobacter metallireducens: interaction with electron transferring flavoprotein and kinetic basis of unidirectional catalysis. FEBS J 281:5120–5131. doi: 10.1111/febs.13051. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Frerman FE, Kim JJ. 2006. Structure of electron transfer flavoprotein-ubiquinone oxidoreductase and electron transfer to the mitochondrial ubiquinone pool. Proc Natl Acad Sci U S A 103:16212–16217. doi: 10.1073/pnas.0604567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gubler M, Zürcher T, Hennecke H. 1989. The Bradyrhizobium japonicum fixBCX operon: identification of fixX and of a 5′mRNA region affecting the level of the fixSCX transcript. Mol Microbiol 3:141–148. doi: 10.1111/j.1365-2958.1989.tb01803.x. [DOI] [PubMed] [Google Scholar]
- 34.Hochstrasser M. 2009. Origin and function of ubiquitin-like protein conjugation. Nature 458:422–429. doi: 10.1038/nature07958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makarova KS, Koonin EV. 2010. Archaeal ubiquitin-like proteins: functional versatility and putative ancestral involvement in tRNA modification revealed by comparative genomic analysis. Archaea 2010:710303. doi: 10.1155/2010/710303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tan X, Volbeda A, Fontecilla-Camps JC, Lindahl PA. 2006. Function of the tunnel in acetylcoenzyme A synthase/carbon monoxide dehydrogenase. J Biol Inorg Chem 11:371–378. doi: 10.1007/s00775-006-0086-9. [DOI] [PubMed] [Google Scholar]
- 37.Grönger P, Manian SS, Reiländer H, O'Connell M, Priefer UB, Pühler A. 1987. Organization and partial sequence of a DNA region of the Rhizobium leguminosarum symbiotic plasmid pRL6JI containing the genes fixABC, nifA, nifB and a novel open reading frame. Nucleic Acids Res 15:31–49. doi: 10.1093/nar/15.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaminski AP, Norel F, Desnoues N, Kush A, Salzano G, Elmerich C. 1988. Characterization of the fixABC region of Azorhizobium caulinodans ORS571 and identification of a new nitrogen fixation gene. Mol Gen Genet 214:496–502. doi: 10.1007/BF00330486. [DOI] [PubMed] [Google Scholar]
- 39.Lee PT, Hsu AY, Ha HT, Clarke CF. 1997. A C-methyltransferase involved in both ubiquinone and menaquinone biosynthesis: isolation and identification of the Escherichia coli ubiE gene. J Bacteriol 179:1748–1754. doi: 10.1128/jb.179.5.1748-1754.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jang M-H, Scrutton NS, Hille R. 2000. Formation of W3A1 electron-transferring flavoprotein (ETF) hydroquinone in the trimethylamine dehydrogenase·ETF protein complex. J Biol Chem 275:12546–12552. doi: 10.1074/jbc.275.17.12546. [DOI] [PubMed] [Google Scholar]
- 41.Toogood HS, Leys D, Scrutton NS. 2007. Dynamics driving function: new insights from electron transferring flavoproteins and partner complexes. FEBS J 274:5481–5504. doi: 10.1111/j.1742-4658.2007.06107.x. [DOI] [PubMed] [Google Scholar]
- 42.Scott JD, Ludwig RA. 2004. Azorhizobium caulinodans electron-transferring flavoprotein N electrochemically couples pyruvate dehydrogenase complex activity to N2 fixation. Microbiology 150:117–126. doi: 10.1099/mic.0.26603-0. [DOI] [PubMed] [Google Scholar]
- 43.Luo H, Lin Y, Gao F, Zhang C-T, Zhang R. 2014. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res 42:D574–D580. doi: 10.1093/nar/gkt1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pechter KB, Gallagher L, Pyles H, Manoil CS, Harwood CS. 2015. Essential genome of the metabolically versatile alphaproteobacterium Rhodopseudomonas palustris. J Bacteriol 198:867–876. doi: 10.1128/JB.00771-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, Huntemann M, Anderson I, Mavromatis K, Ivanova NN, Kyrpides NC. 2012. IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucleic Acids Res 40:D115–D122. doi: 10.1093/nar/gkr1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silvestro D, Michalak I. 2012. raxmlGUI: a graphical front-end for RAxML. Org Divers Evol 12:335–337. doi: 10.1007/s13127-011-0056-0. [DOI] [Google Scholar]
- 48.Wimley WC, Creamer TP, White SH. 1996. Solvation energies of amino acid side chains and backbone in a family of host-guest pentapeptides. Biochemistry 35:5109–5124. doi: 10.1021/bi9600153. [DOI] [PubMed] [Google Scholar]
- 49.Gray HB, Winkler JR. 2015. Hole hopping through tyrosine/tryptophan chains protects proteins from oxidative damage. Proc Natl Acad Sci U S A 112:10920–10925. doi: 10.1073/pnas.1512704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pradeep L, Shin HC, Scheraga HA. 2006. Correlation of folding kinetics with the number and isomerization states of prolines in three homologous proteins of the RNase family. FEBS Lett 580:5029–5032. doi: 10.1016/j.febslet.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 51.Arnold K, Bordoli L, Kopp J, Schwede T. 2006. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 52.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, Schwede T. 2014. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benkert P, Biasini M, Schwede T. 2011. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27:343–350. doi: 10.1093/bioinformatics/btq662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benkert P, Tosatto SC, Schomburg D. 2008. QMEAN: a comprehensive scoring function for model quality assessment. Proteins 71:261–277. doi: 10.1002/prot.21715. [DOI] [PubMed] [Google Scholar]
- 55.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera: a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 56.Wang J, Wang W, Kollman PA, Case DA. 2006. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 57.Watmough NJ, Kiss J, Frerman FE. 1992. Structural and redox relationships between Paracoccus denitrificans, porcine and human electron-transferring flavoproteins. Eur J Biochem 205:1089–1097. doi: 10.1111/j.1432-1033.1992.tb16877.x. [DOI] [PubMed] [Google Scholar]
- 58.Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. 2011. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27:431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.