

ABSTRACT
Many rhodococci are oleaginous and, as such, have considerable potential for the sustainable production of lipid-based commodity chemicals. Herein, we demonstrated that Rhodococcus jostii RHA1, a soil bacterium that catabolizes a wide range of organic compounds, produced wax esters (WEs) up to 0.0002% of its cellular dry weight during exponential growth on glucose. These WEs were fully saturated and contained primarily 31 to 34 carbon atoms. Moreover, they were present at higher levels during exponential growth than under lipid-accumulating conditions. Bioinformatics analyses revealed that RHA1 contains a gene encoding a putative fatty acyl coenzyme A (acyl-CoA) reductase (FcrA). The purified enzyme catalyzed the NADPH-dependent transformation of stearoyl-CoA to stearyl alcohol with a specific activity of 45 ± 3 nmol/mg · min and dodecanal to dodecanol with a specific activity of 5,300 ± 300 nmol/mg · min. Deletion of fcrA did not affect WE accumulation when grown in either carbon- or nitrogen-limited medium. However, the ΔfcrA mutant accumulated less than 20% of the amount of WEs as the wild-type strain under conditions of nitric oxide stress. A strain of RHA1 overproducing FcrA accumulated WEs to ∼13% cellular dry weight under lipid-accumulating conditions, and their acyl moieties had longer average chain lengths than those in wild-type cells (C17 versus C16). The results provide insight into the biosynthesis of WEs in rhodococci and facilitate the development of this genus for the production of high-value neutral lipids.
IMPORTANCE Among the best-studied oleaginous bacteria, rhodococci have considerable potential for the sustainable production of lipid-based commodity chemicals, such as wax esters. However, many aspects of lipid synthesis in these bacteria are poorly understood. The current study identifies a key enzyme in wax ester synthesis in rhodococci and exploits it to significantly improve the yield of wax esters in bacteria. In so doing, this work contributes to the development of novel bioprocesses for an important class of oleochemicals that may ultimately allow us to phase out their unsustainable production from sources such as petroleum and palm oil.
KEYWORDS: fatty acyl-CoA reductase, lipid accumulation, Rhodococcus, wax esters, metabolic engineering
INTRODUCTION
Many bacterial species, particularly the mycolic acid-producing Actinobacteria, synthesize neutral lipids for energy storage. For example, the soil bacterium Rhodococcus jostii RHA1 (referred to as RHA1 here) accumulates neutral lipids up to 70% of cellular dry weight (CDW) in response to environmental stresses, such as nitrogen limitation (1–3). These neutral lipids are primarily triacylglycerides (TAGs) and are stored in lipid bodies within the cytoplasm (4). Proteins associated with these carbon storage organelles (5) facilitate the dynamic shuffling of lipids between utilization and storage, allowing the bacterium to sequester intracellular energy reserves as needed. Due in part to their remarkable biosynthetic capacity, there is considerable interest in using oleaginous microorganisms to sustainably produce neutral lipids to replace oleochemicals currently derived from palm oils and petroleum (6–8). In rhodococci, research has focused on the production of TAGs to manufacture biodiesel (7). However, the poor economics of biodiesel have prevented the development and adoption of industrial-scale processes (9, 10). An alternate approach would be to harness the biosynthetic potential of rhodococci to produce higher-value lipids.
Wax esters (WEs), comprising a fatty alcohol and a fatty acid, are high-value neutral lipids used extensively in cosmetics (11). Found throughout the domains of life, WEs have both functional and structural roles (12–15). In some oleaginous bacteria, WEs are primarily used to store energy (16, 17). In these bacteria, WE synthesis involves the esterification of a fatty alcohol and an acyl coenzyme A (acyl-CoA) in a reaction catalyzed by a bifunctional WE synthase/acyl-CoA:diacylglycerol acyltransferase (WS/DGAT) (Fig. 1) (18, 19). Lipid accumulation is induced by environmental insult or stress, such as nitrogen limitation, in the presence of excess carbon. Under such conditions, fatty acyl-CoA substrates are predominantly provided through lipid biosynthesis. When not available exogenously or generated as intermediates in the degradation of alkanes, fatty alcohols are synthesized de novo from acyl-CoA produced by fatty acid synthases (17, 20). Some oleaginous Actinobacteria have been reported to produce WEs. For example, Mycobacterium tuberculosis accumulates WEs to ∼4% of total neutral lipids alongside TAGs during periods of stress (21), contributing to dormancy and antibiotic resistance (22). Rhodococci also produce WEs under lipid-accumulating conditions when supplied with exogenous fatty alcohols or hydrocarbons (1). Moreover, transient de novo production of WEs was reported in RHA1 by Barney et al. (23). However, because this phenomenon has not been further investigated, the capacity of rhodococci to produce WEs via a de novo route is unclear. Interestingly, oleaginous rhodococci, such as RHA1 and Rhodococcus opacus PD630, harbor over a dozen WS/DGAT homologues, encoded by atf genes (1, 24). Among the characterized rhodococcal WS/DGATs, Atf1PD630 has higher WS than DGAT activity (25).
FIG 1.
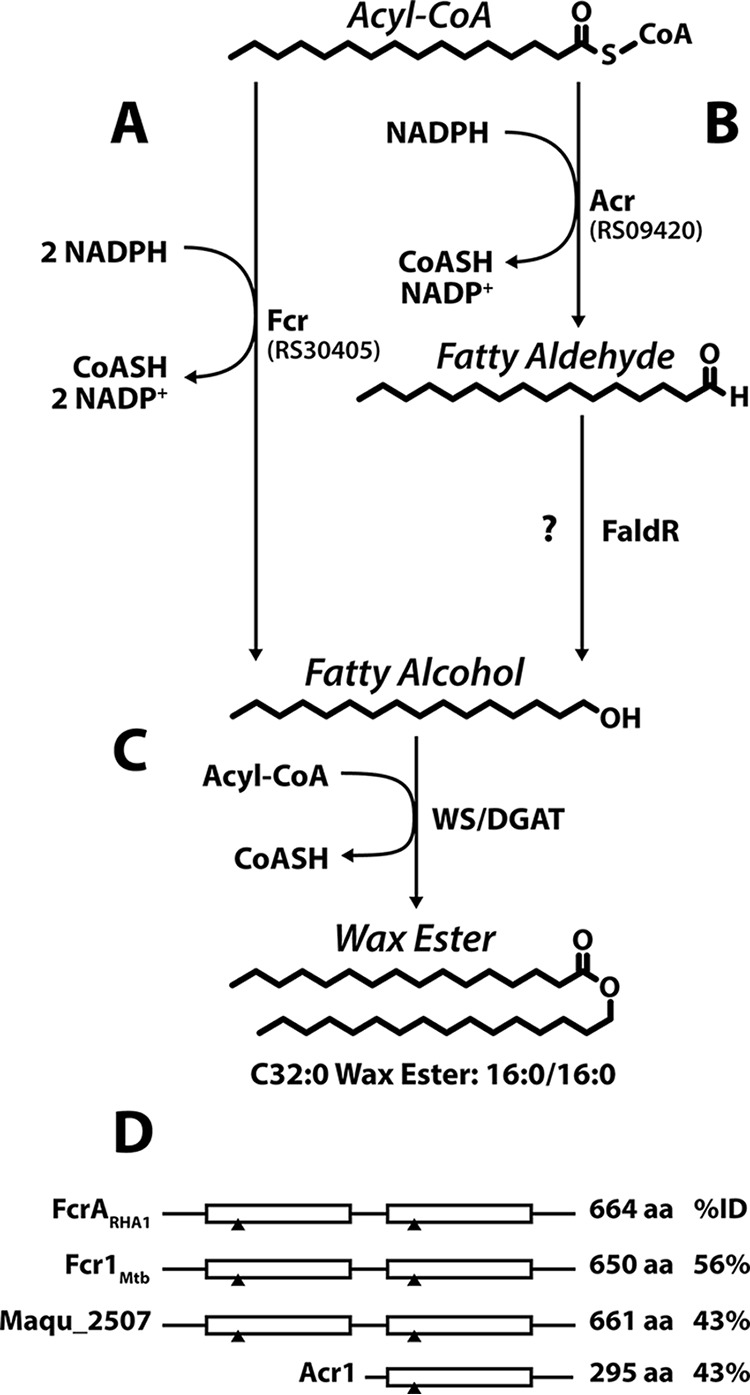
Bacterial WE biosynthesis pathways. Fatty alcohols can be generated through either a single enzyme (A) or consecutive reactions catalyzed by two enzymes (B). (C) Fatty alcohols and an acyl-CoA are esterified to form WEs. The enzymes are as follows: Fcr, alcohol-forming fatty acyl-CoA reductase; Acr, aldehyde-forming fatty acyl-CoA reductase; FaldR, fatty aldehyde reductase; WS/DGAT, WE synthase/acyl-CoA:diacylglycerol acyltransferase. RS30405 and RS09420 are the homologs identified in RHA1. (D) Domain structure of bacterial FARs. White boxes represent NADPH-binding domains. Black triangles indicate regions containing conserved nucleotide-binding residues. Amino acid sequence lengths and percent identity (%ID) to FcrARHA1 are reported.
Fatty alcohols are produced through the reduction of an activated fatty acid. In bacteria, the latter is usually a fatty acyl-CoA, and its reduction is accomplished by either a single enzyme or consecutive reactions catalyzed by two enzymes (Fig. 1). The two-enzyme pathway was first described in the WE-accumulating bacterium Acinetobacter calcoaceticus BD413. In this bacterium, an NADPH-dependent fatty acyl-CoA reductase (FAR), encoded by acr1, performs the two-electron reduction of the acyl-CoA to a fatty aldehyde. The second enzyme, responsible for reducing the fatty aldehyde to a fatty alcohol, has yet to be identified (26). Marinobacter aquaeolei VT8 accomplishes the same process using a FAR that catalyzes two successive two-electron reductions of the acyl-CoA to the corresponding fatty alcohol. M. aquaeolei VT8 contains two FAR-encoding genes with this activity, maqu_2220 and maqu_2507 (27, 28). M. tuberculosis contains both enzyme types; fcr1 (Rv3391) is a homolog of maqu_2507, while fcr2 (Rv1543) is a homolog of acr1 (22). Maqu_2507 and Fcr1 contain two NADPH-binding domains. Interestingly, the C-terminal domain of these enzymes is homologous to the NADPH-binding domain of Acr1 and Fcr2 (Fig. 1D).
Herein, we studied the production of WEs in RHA1. WE levels were investigated in different growth media. A fatty acyl-CoA reductase (FcrA) encoded by the RHA1 genome was purified and characterized with respect to its ability to transform various substrates. A deletion mutant and an FcrA overproduction strain were generated to examine the role of this fatty acyl-CoA reductase in WE synthesis. The results are discussed in terms of the potential of rhodococcal species for the production of high-value oleochemicals.
RESULTS
Production of WEs in RHA1.
Based on the ability of mycobacteria to produce WEs (21, 22) and the similar physiologies of mycobacteria and rhodococci, we hypothesized that the latter also produce WEs when supplied with a growth substrate other than a fatty alcohol or hydrocarbon (1). To test this hypothesis, we grew RHA1 on M9 Goodies minimal medium containing 4 g/liter glucose (defined here as carbon-limiting or C− medium, as glucose is completely consumed before nitrogen is depleted), extracted the neutral lipids from exponentially growing cells, and fractionated them using flash chromatography. Initial gas chromatography-mass spectrometry (GC/MS) analyses revealed that cells contained saturated WEs ranging from 31 to 34 carbons in length, with C32 species being the most abundant (Fig. 2A). Using electron ionization mass spectrometry, we analyzed the WEs using the RCOOH2+ and RCO-H+· ions of their saturated and unsaturated fatty acid components, respectively (29). For example, the length of the fatty acyl component of the C32 WEs varied from 14 to 18 carbons as seen from the RCOOH2+ ions with m/z values of 229, 243, 257, 271, and 285, respectively (Fig. 2B). These ions indicate that ∼70% of the C32 WEs were C16:C16 species, while the remainder were a mixture of species varying from C14:C18 to C18:C14. Finally, using the highly abundant C16 ion, which is more abundant than the corresponding WE molecular ion, we were able to identify additional WEs with 29, 30, and 35 carbon atoms that were not apparent in the total ion trace (Fig. 2C). In summary, the most abundant WEs in exponentially growing cells contained 32 carbons (Fig. 3A). Moreover, C16 was the most abundant WE fatty acyl moiety in the detected WEs (Fig. 3B). No unsaturated fatty acids were detected under these conditions. Quantification by GC/MS revealed that the cells contained 2.2 ± 0.2 μg of WEs/g cell dry weight (CDW). Interestingly, the cells contained smaller amounts of WEs (0.6 ± 0.2 μg/g CDW) when grown in nitrogen-limiting (N−) medium (i.e., medium in which nitrogen is depleted before glucose is consumed) and examined in stationary phase (Table 1), conditions under which they accumulate TAGs to over 50% of their CDW (1). WEs produced in N− medium were similar in length and composition to those in C− medium (Fig. 3).
FIG 2.

Analyses of WEs in RHA1. (A) GC/MS chromatogram of WE-containing lipid fraction. Identified WEs are labeled according to total number of carbon atoms. (B) Mass spectrum of the detected C32 WEs. Molecular ion and major fatty acyl fragments are labeled. (C) WEs detected using the C16 RCOOH2+ ion (m/z = 257), the most abundant ion fragment associated with the WEs. Cells were grown on glucose, and neutral lipids were extracted and fractionated using flash chromatography.
FIG 3.
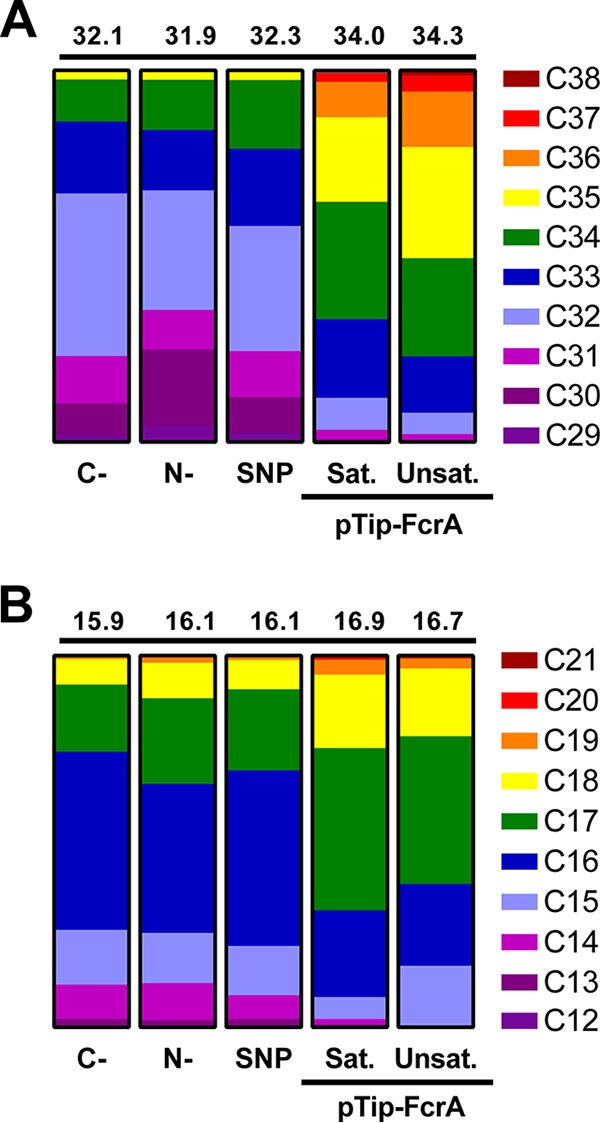
WE content of RHA1 under different conditions. (A) Distribution of WE chain lengths detected. (B) Distribution of acyl-chain lengths of detected WEs. Weighted mean chain lengths were calculated and are displayed above each column. Values were obtained from biological duplicates. Errors ranged from 0.01 to 0.17. Conditions were as follows: C-, carbon limited (exponential); N-, nitrogen limited (stationary); SNP, SNP-treated RHA1; pTip-FcrA, RHA1 overproducing FcrA in N− medium. Sat. and Unsat. indicate saturated and unsaturated WEs/acyl-chains, respectively.
TABLE 1.
WE levels in RHA1 strains grown on different media and at different growth stagesa
| RHA1 strain | WE level (μg/g) for growth condition |
||
|---|---|---|---|
| N− (stationary) | C− (exponential) | C− and SNP-treated (exponential) | |
| WT | 0.6 ± 0.2 | 2.2 ± 0.3 | 3.0 ± 0.3 |
| ΔfcrA mutant | 0.7 ± 0.1 | 3.2 ± 0.5 | 0.5 ± 0.1 |
Experimental values represent micrograms WE per gram CDW. Values represent the mean of biological duplicates. Errors indicate the standard deviation.
Bioinformatic identification of FcrA.
Having identified WEs in RHA1, we searched the strain's genome for homologs of fcr1 or fcr2 of M. tuberculosis, which encode fatty acyl-CoA reductases (22). Using BLAST, we identified RHA1_RS30405 and RHA1_RS09420 as reciprocal best hits of Fcr1 and Fcr2, respectively. RS30405 encodes a protein of 664 amino acid residues, identified here as FcrA, that shares 56% and 43% amino acid sequence identity with Fcr1 and Maqu_2507 from M. aquaeolei VT8, respectively. The three enzymes share a similar two-domain structure in which each domain contains a predicted nucleotide-binding site (Fig. 1D). RS30405 is conserved in all rhodococci whose genomes have been sequenced. RS09420 encodes a protein of 280 amino acids that shares 48% and 46% amino acid sequence identity with Fcr2 and Acr1 from A. calcoaceticus, respectively, as well as 46% identity with the second nucleotide-binding domain of FcrA.
To gain further insight into the physiological role of the identified FARs in RHA1, we considered their transcript levels in previous whole-transcriptome shotgun sequencing (RNA-seq) studies aimed at understanding TAG biosynthesis (3) (GEO accession number GSE77158). In N− medium, the transcript levels of both fcrA and RS09420 were low, with reads per kilobase per million mapped (RPKM) values between 6 and 11 in the exponential and stationary phases. Interestingly, fcrA appears to be more highly expressed in C− medium, with RPKMs of 19 ± 9 and 87 ± 6 in the exponential and stationary phases, respectively. In contrast, RS09420 transcript levels were also low in C− medium; the RPKM value was 9 ± 5 in exponential phase, and no transcripts were detected in stationary phase.
The RNA-seq data also provide insight into the operon structure of fcrA. Thus, RS30410, located immediately upstream of fcrA and encoding a putative membrane protein, was not cotranscribed with fcrA. Interestingly, RS30410 is conserved upstream of fcrA across the diverse phylogenetic clades of rhodococci (30), while the surrounding genomic context is not. In contrast, a homolog of RS30410 is not present upstream of fcr1 in mycobacterial species, suggesting that RS30410 is not essential for the activity of fcrA. The low transcription levels for RS09420 did not allow us to make meaningful observations about the structure of its transcript.
Production, purification, and characterization of recombinant FcrA.
We focused further efforts on exploring the function of FcrA, as related two-domain enzymes completely reduce acyl-CoAs to fatty alcohols (28) and have been used in biotechnology applications (31, 32). To investigate the activity of FcrA, we produced it as a C-terminally His-tagged protein in RHA1 and purified it to greater than 95% apparent homogeneity (Fig. 4A). To check for posttranslational modification, purified FcrA was subjected to mass spectrometry. The molecular mass of FcrA-His6 was 73,627 Da, which corresponds to the mass of the protein (73,758 Da) less the N-terminal methionine (131 Da). This result indicates that the heterologously produced enzyme was not subjected to other posttranslational modifications.
FIG 4.

Purification and activity of recombinant FcrA. (A) SDS-PAGE of 8 μg His6-tagged FcrA purified from RHA1. (B) GC/MS trace of a reaction mixture containing 1.4 μM FcrA-His6, 100 μM oleoyl-CoA, and 400 μM NADPH (20 mM Tris-HCl, pH 7.0, 50 mM NaCl) incubated for 20 h.
To evaluate the ability of FcrA to catalyze the formation of fatty alcohols, 1.4 μM FcrA was incubated with 400 μM NADPH and 100 μM oleoyl-CoA in 1 ml of 20 mM Tris-HCl (pH 7.0) and 50 mM NaCl for 20 h at room temperature. The reaction was quenched with 1 ml saturated NaCl, and 100 μM tetradecanol was added as a standard. Analysis of the extracted and derivatized reaction products by GC/MS revealed that FcrA converted oleoyl-CoA to oleyl alcohol (Fig. 4B). No alcohol was detected in control reactions in which FcrA was omitted.
Using a spectrophotometric assay following either the production of 2-nitro-5-thiobenzoate (NTB2−) or the consumption of NADPH, FcrA catalyzed the reduction of various acyl-CoAs. As with previously characterized FARs (22, 28), activity was maximal at pH 7.0. Moreover, no activity was detected when NADPH was substituted with NADH. Similar to Maqu_2507 (28), the rate of NADPH consumption was twice that of NTB2− production, indicating that FcrA oxidizes two equivalents of NADPH for every molecule of CoA released. NTB2− production was used to query a range of acyl-CoA substrates. Among the tested substrates, FcrA had the highest specific activity for stearoyl-CoA (C18-CoA), reducing it at a rate of 45 ± 3 nmol/mg · min. The specific activity of FcrA dropped off with decreasing chain length of the acyl-CoA substrate (Table 2). In contrast, oleoyl-CoA (C18:1-CoA), a monounsaturated acyl-CoA, was reduced at a similar rate to stearoyl-CoA.
TABLE 2.
Specific activities of FcrA with various acyl-CoA and fatty aldehyde substrates
| Substrate | Chain length | Specific activitya,b (nmol/mg [protein] · min) |
|---|---|---|
| Stearoyl-CoA | C18 | 45 ± 3 |
| Oleoyl-CoA | C18:1 | 41 ± 1 |
| Palmitoyl-CoA | C16 | 7.0 ± 0.1 |
| Lauroyl-CoA | C12 | 1.5 ± 0.3 |
| Decanal | C10 | 3,300 ± 200 |
| Dodecanal | C12 | 5,300 ± 300 |
| cis-11-Hexadecanal | C16:1 | 2,800 ± 300 |
Specific activity for acyl-CoA substrates was determined from NTB2− ion formation as detected at 412 nm.
Specific activity for aldehyde substrates was determined from NADP+ formation as detected at 340 nm. Values represent means determined from triplicate experiments. Errors represent the standard deviations.
As previously characterized FARs also reduce fatty aldehydes, we used a spectrophotometric assay to investigate whether FcrA also catalyzes this transformation. Like Maqu_2507 (28), FcrA reduced fatty aldehydes at a rate ∼100-fold greater than that of fatty acyl-CoAs. Among the three tested aldehydes, FcrA had the highest specific activity for dodecanal, reducing it at a rate of 5,300 ± 300 nmol/mg · min (Table 2).
Wax ester production in a ΔfcrA mutant.
Having established the identity of RS30405 as a fatty acyl-CoA reductase, we further investigated the physiological role of fcrA by deleting the gene in RHA1. Interestingly, the ΔfcrA mutant contained similar amounts of WEs as wild-type (WT) RHA1 when grown in both C− and N− media in exponential and stationary phase, respectively (Table 1).
In M. tuberculosis, a Δfcr1 mutant did not show a decrease in WEs when starved for both carbon and nitrogen but did so under conditions of nitric oxide (NO) stress (22). Therefore, we sought to investigate whether WE production by fcrA is linked to the presence of reactive nitrogen species in RHA1 by using sodium nitroprusside (SNP) to generate NO. When stressed with NO, WT cells growing exponentially on glucose minimal medium produced similar levels of WEs as nonstressed cells. The majority of the WE species observed in WT RHA1 are similar in chain length and composition to those seen under other growth conditions (Fig. 3). However, trace amounts of unsaturated C32 to C34 WEs were detected in the SNP-treated cells. The production of WEs in response to NO stress was strikingly different in the ΔfcrA mutant (see Fig. S1 in the supplemental material), with a 6-fold reduction in accumulated WEs (Table 1).
Overexpression of fcrA.
To investigate the potential of FcrA to promote WE accumulation, we overproduced a tagless form of the enzyme in RHA1 using a pTip vector. We performed this experiment in N− medium, which promotes neutral lipid accumulation (1). Indeed, thin-layer chromatography (TLC) analyses of cells overproducing FcrA indicated that they contained significant amounts of WEs under these conditions (Fig. 5A). Thus, overproduction of FcrA resulted in the appearance of a large band that ran similarly to stearyl stearate. Gravimetric analysis indicated that the FcrA-overproducing strain accumulated WEs to 13% ± 5% of CDW in N− medium. No significant accumulation of WEs was observed in cells growing exponentially or at stationary phase in C− medium. These WEs ranged from 30 to 38 C atoms in length (Fig. 5B). The WEs were on average a couple of carbon atoms longer than those in WT cells (Fig. 3A). Moreover, a significant portion (∼20%) of the WEs was unsaturated. Analysis of the fragmentation ions further revealed that the most abundant saturated and unsaturated fatty acyl chain lengths were C17 (Fig. 3B). Finally, only trace amounts (<1% of total) of unsaturated fatty alcohols were detected, signifying that the unsaturation was primarily located within the acyl moiety.
FIG 5.

WE content of RHA1 overproducing FcrA. (A) Total lipid extracts as visualized by TLC. Lanes (from left to right) were loaded with the following: 1, extract of RHA1 carrying an empty vector (pTipQC2); 2, extract of RHA1 overproducing FcrA (pTip-fcrA); and 3, stearyl stearate and glyceryl tripalmitate. (B) GC/MS analysis of WE fraction isolated from RHA1 overproducing FcrA from pTip-fcrA.
DISCUSSION
This study presents evidence for the de novo synthesis of WEs in rhodococci from nonalkane substrates and establishes that FcrA is an alcohol-forming fatty acyl-CoA reductase that contributes to the biosynthesis of WEs in RHA1. More specifically, purified FcrA catalyzed the reduction of various fatty acyl-CoAs to the corresponding fatty alcohol. Although the enzyme did not appear to contribute significantly to the synthesis of WEs under normal growth conditions, deletion of fcrA resulted in significantly lower production of WEs under NO stress. Finally, overproduction of FcrA in RHA1 resulted in the bacterium accumulating WEs to greater than 10% of CDW under conditions that promote TAG production.
FcrA of RHA1 had comparable activity to the other two-domain FARs that have been characterized to date but had a slightly different substrate preference. For example, FcrA showed a strong preference for C18-CoAs in contrast to Maqu_2507, which had the highest activity for C16-CoA (28). Both enzymes showed decreased activity with shorter-chain acyl-CoAs. Monounsaturation of the acyl-CoA substrate appears to have no significant effect on the activity of either enzyme. In contrast, Fcr1 of M. tuberculosis had a strong preference for C18:1-CoA over saturated acyl-CoAs (22). Maqu_2507 and FcrA reduced fatty aldehydes at comparable rates. However, while Maqu_2507 had maximal activity with decanal (C10), FcrA had maximal activity with dodecanal (C12). Nevertheless, additional studies are required to properly compare the substrate preferences of the two enzymes.
Our data indicate that, like M. tuberculosis and M. aquaeolei VT8, RHA1 has more than one WE biosynthesis pathway. Moreover, FcrA only contributes to WE biosynthesis under specific stresses. Thus, deletion of fcrA had no effect on WE synthesis during nutrient-limited growth, similar to the phenotypes of the fcr1 mutant in M. tuberculosis (22) and the maqu_2507 mutant in M. aquaeolei VT8 (16), a Gram-negative WE-accumulating bacterium isolated from an offshore oil-producing well. In further similarity to the phenotype of the fcr1 mutant in M. tuberculosis (22), deletion of fcrA impacted WE synthesis in the presence of reactive nitrogen species. Consistent with the apparently minor role of FcrA in WE biosynthesis during exponential growth, the WE composition of RHA1 under these conditions did not reflect the substrate preference of FcrA. More specifically, FcrA had a preference for longer acyl-CoAs than the average chain length of the WE alcohols under these conditions. In contrast, the WE content of RHA1 cells was more reflective of FcrA substrate preference in cells overproducing the enzyme in N− medium. That is, the WEs contained alcohols with longer chain lengths. Overall, this suggests that the two-domain FARs may be responsible for producing WEs only in response to specific stresses (22).
Aldehyde-forming fatty acyl-CoA reductases may play a larger role in WE synthesis in mycolic acid-producing Actinobacteria. For example, the fcr2 mutant of M. tuberculosis contained smaller amounts of WEs under all conditions examined. It seems reasonable that in RHA1, RS09420 plays a similar role. Fcr2 and RS09420 are homologs of Acr1 (26), the aldehyde-forming fatty acyl-CoA reductase of A. calcoaceticus BD413, and may therefore function as such. Sirakova et al. reported that Fcr2 generated fatty alcohols from acyl-CoA substrates (22). However, Fcr2 was characterized in Escherichia coli lysate, which contains an unidentified enzyme that reduces fatty aldehydes to fatty alcohols (26). Indeed, the presence of this enzyme has been exploited to produce fatty alcohols in E. coli expressing acr1 (8). Genetic and biochemical characterizations of RS09420 are required to definitively assign this gene's function.
The amount of WEs in RHA1 is significantly less than what has been reported in M. tuberculosis. More specifically, WEs comprised a larger portion of the neutral lipids in M. tuberculosis than in WT RHA1 under all of the conditions that we tested and, as quantified by [1-14C]oleate incorporation into neutral lipid pools after 6 h, were approximately 10,000-fold higher than in RHA1 under stress conditions (21, 22). However, it is unclear whether the WE content of M. tuberculosis is representative of mycolic acid-containing bacteria. For example, WEs were not detected in Mycobacterium smegmatis under nitrogen-limited conditions by TLC (18), an observation that does not preclude their presence in the trace amounts reported here for RHA1. WEs have been suggested to contribute to dormancy and membrane permeability in M. tuberculosis (22). Interestingly, M. tuberculosis is one of the few mycobacterial species that does not produce mycolate-derived WEs (33), a class of lipids synthesized from keto-mycolic acids by a Baeyer-Villiger monooxygenase (34). These mycolate-derived WEs are integral components of the mycolic acid layer of mycobacteria (35) and appear to be replaced by methoxy-mycolates in M. tuberculosis. More studies are required to elucidate the role of WEs in mycolic acid-containing bacteria.
The current data highlight the potential of rhodococci for the sustainable production of WEs. More specifically, simply overproducing FcrA in RHA1 yielded a system that accumulated ∼13% of its CDW in WEs. This exceeds the levels of any natural or engineered strains reported to date, including A. calcoaceticus and M. aquaeolei VT8, which accumulated de novo WEs to ∼6% and ∼10% CDW, respectively (16, 36). Similarly, engineered strains of E. coli and Acinetobacter accumulated WEs to 1% and 3% CDW, respectively (37, 38), or resulted in large reductions to growth yields (39). Further metabolic engineering should significantly increase WE yields in RHA1 strains. This may be achieved by increasing carbon flow into WEs, modulating redox cofactors—analogously to what was accomplished in the oleaginous yeast Yarrowia lipolytica (40), and/or the simultaneous overproduction of a WE synthase. Interestingly, the WEs accumulated by our engineered strain are remarkably similar to spermaceti WEs, which were historically valued as lubricants and cosmetics due to their excellent physicochemical properties (41). The rhodococcal and spermaceti WEs are comparable with respect to several characteristics, including range of chain lengths (with C34 WEs being the majority species), lengths of acyl and alcohol components, and degree of unsaturation (14). The similarity of rhodococcal and spermaceti WEs enhances the former's industrial relevance and justifies further research into the production of rhodococcal WEs.
MATERIALS AND METHODS
Strains and culture conditions.
Escherichia coli DH5α was used to propagate DNA. E. coli S17.1 was used to conjugate pK18-derived plasmids into RHA1. E. coli strains were grown in LB broth at 37°C and 200 rpm. RHA1 was grown at 30°C while shaking at 200 rpm. For protein production, RHA1 was grown in LB. For lipid production, RHA1 was grown in M9 minimal medium supplemented with trace elements plus thiamine (Goodies mix) and 4 g/liter glucose as the growth substrate (42, 43). In carbon-limited (C−) medium (i.e., medium in which the carbon concentration limits maximum cell growth), the M9 medium contained 1 g/liter ammonium chloride. In nitrogen-limiting (N−) medium (i.e., medium in which the nitrogen concentration limits maximum cell growth), this concentration was 0.05 g/liter as previously described (3). For solid medium, LB broth was supplemented with Bacto agar (1.5% [wt/vol]; Difco). Media were further supplemented with 100 μg/ml ampicillin (E. coli carrying pTip-derived plasmids), 50 μg/ml kanamycin (E. coli carrying pK18-derived plasmids), 34 μg/ml chloramphenicol (RHA1 carrying pTip-derived plasmids), or 10 μg/ml neomycin (RHA1 carrying pK18-derived plasmids) as appropriate.
Reagents.
Enzymes for cloning were purchased from New England BioLabs unless otherwise noted. Primers were ordered from Integrated DNA Technologies. Chemicals were of at least reagent grade unless otherwise noted. Buffers were prepared using water purified on a Barnstead Nanopure UV apparatus to a resistivity of greater than 17 MΩ cm.
DNA manipulation, plasmid construction, and gene deletion.
DNA was isolated, manipulated, and analyzed using standard protocols (43). E. coli and RHA1 were transformed with DNA by electroporation using a MicroPulser with Gene Pulser cuvettes (Bio-Rad). To produce a C-terminal His6-tagged FcrA, RHA1_RS30405 was amplified from RHA1 genomic DNA using Phusion polymerase with the primers 5′-TTTAAGAAGGAGATATACATATGGCCACCTACCTCGTCACCG-3′ and 5′-ATGGTGATGGTGATGCTCGAGGCTGCTGCCCTGGAAATACAAGTTTTCTCTGCCGCTGCTCCA-GTGGGTGCCGGGCAC-3′. The resulting amplicon was inserted into pTip-QC2 linearized with Nde1 and Xho1 using Gibson assembly. Tagless FcrA was produced as above using the primers 5′-TTTAAGAAGGAGATATACATATGGCCACCTACCTCGTCACC-3′ and 5′-GTGCCGGTGGGTCGACTAGTCACCAGTGGGTGCCGGG-3′. The nucleotide sequence of the cloned genes was verified. The ΔfcrA mutant was constructed using a sacB counter selection system (44). Two 500-bp flanking regions of RHA1_RS30405 were amplified from RHA1 genomic DNA using the upstream primers 5′-TGAGGTGCGAGGACGTGGATCTCGGCTG-3′ and 5′-CGGGTACCGAGCTCGAATTCGTCTCCGCGCC-ATCACGC-3′ and the downstream primers 5′-GCTATGACATGATTACGAATTCGGCCGGGGTGCGG-GTCA-3′ and 5′-ACGTCCTCGCACCTCAGCACACACCGGAACCC-3′. The resulting amplicons were inserted into pK18mobsacB linearized with EcoR1 using Gibson assembly. The nucleotide sequence of the resulting construct was verified. Kanamycin-sensitive/sucrose-resistant colonies were screened using PCR, and the gene deletion was confirmed by sequencing.
FcrA production and purification.
RHA1 freshly transformed with pTip-fcrA was grown overnight in LB. These cultures were used to inoculate 1 liter of fresh LB medium to an optical density at 600 nm (OD600) of 0.05. Cultures were grown to an OD600 of ∼0.8, the expression of fcrA was induced with 10 μg/ml of thiostrepton, and cultures were incubated for a further 24 h. Cells were harvested by centrifugation and stored at −80°C.
Cells from 2 liters of culture were suspended in 40 ml of lysis buffer (50 mM Na-phosphate, pH 8.5, 500 mM NaCl, 2.5 mM imidazole) containing cOmplete mini EDTA-free protease inhibitors (Roche Diagnostics). The cell suspension was split between four 15-ml conical tubes, each containing ∼1 ml of 0.1-mm zirconium/silica beads and ∼1 ml of 0.5-mm glass beads (BioSpec Products), and subjected to three rounds of bead beating at 5 to 6 m/s using a FastPrep-24 (MP Biomedicals) with 5 min on ice between rounds. The lysate was centrifuged (4,000 × g for 5 min) to remove unbroken cells. The supernatant lysate was removed and stored on ice. The pellets were suspended in 5 ml lysis buffer and subjected to another 3 rounds of bead beating. The supernatants were combined and further clarified by ultracentrifugation (40,000 × g for 60 min) and passage through a 0.22-μm filter.
The clarified lysate was incubated with 5 ml Ni Sepharose 6 fast flow resin (GE Healthcare Bio-Sciences) for 3 h. After incubation, the resin was poured into a column and then washed with 50 ml lysis buffer, 50 ml lysis buffer containing 50 mM imidazole, and 15 ml lysis buffer containing 75 mM imidazole. FcrA was eluted with 15 ml lysis buffer containing 250 mM imidazole. FcrA-containing fractions, as judged by SDS-PAGE, were pooled, exchanged into 50 mM Na-phosphate, pH 8.5, and 500 mM NaCl, and concentrated to ∼10 mg/ml using an Amicon Ultra-15 centrifugal filtration unit (Merck KGaA) equipped with a 30-kDa cutoff membrane. Protein was flash frozen as beads in liquid nitrogen and stored at −80°C. Protein concentration was determined by Micro BCA (Thermo Fisher Scientific, Rockford, IL) and by absorbance at 280 nm.
Mass spectrometry.
Purified FcrA (∼4 μg/ml in 5% acetonitrile, 0.1% formic acid [vol/vol]) was injected onto a 5-mm C4 column connected to a Waters Xevo GS-2 QTof mass spectrometer via a NanoAcquity ultraperformance liquid chromatography (UPLC) system operated at 20 μl/min. Samples were eluted in a 40-μl gradient of 5% to 100% acetonitrile at 20 μl/min. MS spectra were summed and deconvoluted using Waters' MaxEnt algorithm.
Activity assays.
FcrA activity was evaluated using two spectrophotometric assays (28). Assays were performed at 25°C in 1 ml of 20 mM morpholinepropanesulfonic acid (MOPS), 80 mM NaCl, pH 7.0 (I = 0.1 M) containing 5 μM acyl-CoA or 60 μM aldehyde, and 200 μM NADPH. Reactions were initiated by the addition of FcrA to a final concentration of 0.1 to 2 μM. In one assay, the oxidation of NADPH was followed at 340 nm (ε = 6.3 mM−1 cm−1 [45]). In the second assay, the reaction mixture also contained 0.1 mg/ml 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB), and the formation of NTB2− was followed at 412 nm (ε = 14.15 mM−1 cm−1 for NTB2− [46]). Reaction rates were calculated from progress curves using Cary WinUV Kinetics Application (Agilent). The pH-dependence of the reaction was evaluated using the DTNB assay and a series of 20 mM Good's buffers {morpholineethanesulfonic acid [MES], MOPS, 3-[4-(2-hydroxyethyl)-1-piperazinyl]propanesulfonic acid [HEPPS] or [tris(hydroxymethyl)methylamino]propanesulfonic acid [TAPS]} and 80 mM NaCl (I = 0.1 M), pH 6 to 9.
Production of neutral lipids and WEs.
Fresh media were inoculated to an OD600 of 0.1 using washed RHA1 cells harvested from cultures grown overnight in C− medium. For analyses of C− cultures, cells were harvested during exponential growth, approximately 24 h after inoculation. For analyses of N− cultures, cells were harvested after approximately 72 h (3). To induce nitric oxide (NO) stress, cells were grown in C− medium to an OD600 of 0.5 to 0.6, at which point sodium nitroprusside (SNP) was added to the cultures to a concentration of 1 mM at 2-h intervals for 6 h. NO-stressed cells were harvested 2 h after the final addition of SNP. For RHA1 transformed with pTip-fcrA or empty pTipQC2, cultures were grown as above, except that when they reached an OD600 of between 0.6 and 1.0, thiostrepton was added to 10 μg/ml. Cells were pelleted at 4,000 × g for 30 min at 4°C, washed with distilled water, and stored at −80°C.
Lipid extraction and thin-layer chromatography.
Frozen cell pellets were lyophilized for 24 to 48 h using a FreeZone 2.5 (Labconco). Dried cells were suspended in water and sonicated to lyse cells. Myristyl myristate (Nu-Chek Prep) and/or ethyl myristate (Sigma) was added as a standard. Total lipids were extracted from lysate using 2:1 chloroform/methanol with 1% acetic acid (47). The organic phase was collected and dried using a rotary evaporator (Buchi) and/or nitrogen gas. Dried extracts were suspended in chloroform and stored at −20°C. Lipid extracts were analyzed using silica-TLC and a mobile phase of 90:8:1 (vol/vol/vol) of hexane/diethyl ether/acetic acid. Lipids were visualized by staining with 10% cupric sulfate in 8% aqueous phosphoric acid (vol/vol) and charring for 5 min at 200°C.
Isolation, fractionation, and gravimetric quantification of neutral lipids.
Neutral cellular lipids were purified using flash chromatography. Total lipid extracts were applied to a column of silica resin (10 mg lipids per 300 mg resin [48]) equilibrated with 5 column volumes (CV) of chloroform. Neutral lipids eluted with 5 CV of chloroform were dried as described above and quantified by weight using an AT200 analytical balance (METTLER). The neutral lipids were suspended in hexanes and fractionated using silica equilibrated with 5 CV of hexanes. Neutral lipid extracts were applied to the column, and the hydrocarbon fraction was eluted with 10 CV of hexanes, the WE fraction with 10 CV of 98:2 (vol/vol) hexanes/diethyl ether, and the remaining TAGs and neutral lipids with 10 CV of chloroform. The fractions were dried, weighed, and suspended in chloroform for storage at −20°C.
To analyze fatty alcohols produced in vitro, quenched reactions of 2 ml were extracted with 5 ml of 2:1 chloroform/methanol (vol/vol). The organic phase was collected and washed once with 1:1 H2O/methanol and three times with H2O. The organic phase was removed and dried under nitrogen stream. The extract was suspended in pyridine and derivatized with tetramethylsilane for 1 h at 60°C. Derivatized extracts were analyzed by gas chromatography-mass spectrometry (GC/MS) as described below.
GC/MS analyses.
Analyses were performed using an Agilent 6890n gas chromatograph system fitted with an HP-5 MS 30-m by 0.25-mm capillary column (Hewlett-Packard) and an Agilent 5973n mass-selective detector. The GC was operated at an injector temperature of 300°C, a transfer line temperature of 320°C, a quad temperature of 150°C, a source temperature of 230°C, and a helium flow rate of 1 ml/min. Samples of 1 μl were injected in splitless mode. For fatty alcohol analyses, the temperature program of the oven was 40°C for 2 min, increased to 160°C at a rate of 40°C per min, then increased to 240°C at a rate of 5°C per min, and finally increased to 300°C at a rate of 60°C per min and held for 5 min. The mass spectrometer was operated in electron emission scanning mode at 40 to 800 m/z and 1.97 scans per second. For WE analysis, the temperature program of the oven was 40°C for 2 min, increased to 180°C at a rate of 40°C per min, and then increased to 320°C at a rate of 2.5°C per min and held for 20 min. Derivatized fatty alcohols and WEs were identified using ChemStation E.02.02.1431 (Agilent) and the NIST08 Library. The identity of fatty alcohols was further verified using similarly derivatized authentic fatty alcohols. WEs were further identified by comparison to an authentic stearyl stearate standard and were quantified using a standard curve generated using palmityl myristate (Nu-Chek Prep).
Supplementary Material
ACKNOWLEDGMENTS
William Mohn and Gordon Stewart provided access to, and assistance with, the GC/MS. Protein MS was performed at the UBC Proteomics Core Facility.
This research was supported by Large Scale Applied Research Project 2108 from Genome Canada and Discovery grant 171359 from the Natural Sciences and Engineering Research Council (NSERC) of Canada to L.D.E.
L.D.E. is the recipient of a Canada Research Chair.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00902-17.
REFERENCES
- 1.Hernandez MA, Mohn WW, Martinez E, Rost E, Alvarez AF, Alvarez HM. 2008. Biosynthesis of storage compounds by Rhodococcus jostii RHA1 and global identification of genes involved in their metabolism. BMC Genomics 9:600. doi: 10.1186/1471-2164-9-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dávila Costa JS, Herrero OM, Alvarez HM, Leichert L. 2015. Label-free and redox proteomic analyses of the triacylglycerol-accumulating Rhodococcus jostii RHA1. Microbiology 161:593–610. doi: 10.1099/mic.0.000028. [DOI] [PubMed] [Google Scholar]
- 3.Amara S, Seghezzi N, Otani H, Diaz-Salazar C, Liu J, Eltis LD. 2016. Characterization of key triacylglycerol biosynthesis processes in rhodococci. Sci Rep 6:24985. doi: 10.1038/srep24985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waltermann M, Steinbuchel A. 2005. Neutral lipid bodies in prokaryotes: recent insights into structure, formation, and relationship to eukaryotic lipid depots. J Bacteriol 187:3607–3619. doi: 10.1128/JB.187.11.3607-3619.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding Y, Yang L, Zhang S, Wang Y, Du Y, Pu J, Peng G, Chen Y, Zhang H, Yu J, Hang H, Wu P, Yang F, Yang H, Steinbüchel A, Liu P. 2012. Identification of the major functional proteins of prokaryotic lipid droplets. J Lipid Res 53:399–411. doi: 10.1194/jlr.M021899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang M-H, Jiang J-G. 2013. Advancing oleaginous microorganisms to produce lipid via metabolic engineering technology. Prog Lipid Res 52:395–408. doi: 10.1016/j.plipres.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Alvarez HM. 2010. Biotechnological production and significance of triacylglycerols and wax esters, p 2995–3002. In Timmis KN. (ed), Handbook of hydrocarbon and lipid microbiology. Springer, Berlin, Germany. [Google Scholar]
- 8.Steen EJ, Kang Y, Bokinsky G, Hu Z, Schirmer A, McClure A, Del Cardayre SB, Keasling JD. 2010. Microbial production of fatty-acid-derived fuels and chemicals from plant biomass. Nature 463:559–562. doi: 10.1038/nature08721. [DOI] [PubMed] [Google Scholar]
- 9.Vlysidis A, Binns M, Webb C, Theodoropoulos C. 2011. A techno-economic analysis of biodiesel biorefineries: assessment of integrated designs for the co-production of fuels and chemicals. Energy 36:4671–4683. doi: 10.1016/j.energy.2011.04.046. [DOI] [Google Scholar]
- 10.Balan V. 2014. Current challenges in commercially producing biofuels from lignocellulosic biomass. ISRN Biotechnol 2014:463074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Keng PS, Basri M, Zakaria MRS, Rahman MBA, Ariff AB, Rahman RNZA, Salleh AB. 2009. Newly synthesized palm esters for cosmetics industry. Ind Crops Prod 29:37–44. doi: 10.1016/j.indcrop.2008.04.002. [DOI] [Google Scholar]
- 12.Post-Beittenmiller D. 1996. Biochemistry and molecular biology of wax production in plants. Annu Rev Plant Physiol Plant Mol Biol 47:405–430. doi: 10.1146/annurev.arplant.47.1.405. [DOI] [PubMed] [Google Scholar]
- 13.Jacobsen E, Billings JK, Frantz RA, Kinney CK, Stewart ME, Downing DT. 1985. Age-related changes in sebaceous wax ester secretion rates in men and women. J Investig Dermatol 85:483–485. doi: 10.1111/1523-1747.ep12277224. [DOI] [PubMed] [Google Scholar]
- 14.Nevenzel JC. 1970. Occurrence, function and biosynthesis of wax esters in marine organisms. Lipids 5:308–319. doi: 10.1007/BF02531462. [DOI] [PubMed] [Google Scholar]
- 15.Kaiser J-P, Hanselmann KW. 1982. Fermentative metabolism of substituted monoaromatic compounds by a bacterial community from anaerobic sediments. Arch Microbiol 133:185–194. doi: 10.1007/BF00414999. [DOI] [Google Scholar]
- 16.Lenneman EM, Ohlert JM, Palani NP, Barney BM. 2013. Fatty alcohols for wax esters in Marinobacter aquaeolei VT8: two optional routes in the wax biosynthesis pathway. Appl Environ Microbiol 79:7055–7062. doi: 10.1128/AEM.02420-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishige T, Tani A, Sakai Y, Kato N. 2003. Wax ester production by bacteria. Curr Opin Microbiol 6:244–250. doi: 10.1016/S1369-5274(03)00053-5. [DOI] [PubMed] [Google Scholar]
- 18.Kalscheuer R, Steinbuchel A. 2003. A novel bifunctional wax ester synthase/acyl-CoA:diacylglycerol acyltransferase mediates wax ester and triacylglycerol biosynthesis in Acinetobacter calcoaceticus ADP1. J Biol Chem 278:8075–8082. doi: 10.1074/jbc.M210533200. [DOI] [PubMed] [Google Scholar]
- 19.Stoveken T, Kalscheuer R, Malkus U, Reichelt R, Steinbuchel A. 2005. The wax ester synthase/acyl coenzyme A:diacylglycerol acyltransferase from Acinetobacter sp. strain ADP1: characterization of a novel type of acyltransferase. J Bacteriol 187:1369–1376. doi: 10.1128/JB.187.4.1369-1376.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rontani J-F. 2010. Production of wax esters by bacteria, p 459–470. In Timmis KN. (ed), Handbook of hydrocarbon and lipid microbiology. Springer, Berlin, Germany. [Google Scholar]
- 21.Deb C, Lee C-M, Dubey VS, Daniel J, Abomoelak B, Sirakova TD, Pawar S, Rogers L, Kolattukudy PE. 2009. A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen. PLoS One 4:e6077. doi: 10.1371/journal.pone.0006077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sirakova TD, Deb C, Daniel J, Singh HD, Maamar H, Dubey VS, Kolattukudy PE. 2012. Wax ester synthesis is required for Mycobacterium tuberculosis to enter in vitro dormancy. PLoS One 7:e51641. doi: 10.1371/journal.pone.0051641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barney BM, Wahlen BD, Garner E, Wei J, Seefeldt LC. 2012. Differences in substrate specificities of five bacterial wax ester synthases. Appl Environ Microbiol 78:5734–5745. doi: 10.1128/AEM.00534-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holder JW, Ulrich JC, DeBono AC, Godfrey PA, Desjardins CA, Zucker J, Zeng Q, Leach AL, Ghiviriga I, Dancel C, Abeel T, Gevers D, Kodira CD, Desany B, Affourtit JP, Birren BW, Sinskey AJ. 2011. Comparative and functional genomics of Rhodococcus opacus PD630 for biofuels development. PLoS Genet 7:e1002219. doi: 10.1371/journal.pgen.1002219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alvarez AF, Alvarez HM, Kalscheuer R, Waltermann M, Steinbuchel A. 2008. Cloning and characterization of a gene involved in triacylglycerol biosynthesis and identification of additional homologous genes in the oleaginous bacterium Rhodococcus opacus PD630. Microbiology 154:2327–2335. doi: 10.1099/mic.0.2008/016568-0. [DOI] [PubMed] [Google Scholar]
- 26.Reiser S, Somerville C. 1997. Isolation of mutants of Acinetobacter calcoaceticus deficient in wax ester synthesis and complementation of one mutation with a gene encoding a fatty acyl coenzyme A reductase. J Bacteriol 179:2969–2975. doi: 10.1128/jb.179.9.2969-2975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hofvander P, Doan TT, Hamberg M. 2011. A prokaryotic acyl-CoA reductase performing reduction of fatty acyl-CoA to fatty alcohol. FEBS Lett 585:3538–3543. doi: 10.1016/j.febslet.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 28.Willis RM, Wahlen BD, Seefeldt LC, Barney BM. 2011. Characterization of a fatty acyl-CoA reductase from Marinobacter aquaeolei VT8: a bacterial enzyme catalyzing the reduction of fatty acyl-CoA to fatty alcohol. Biochemistry 50:10550–10558. doi: 10.1021/bi2008646. [DOI] [PubMed] [Google Scholar]
- 29.Urbanová K, Vrkoslav V, Valterová I, Háková M, Cvačka J. 2012. Structural characterization of wax esters by electron ionization mass spectrometry. J Lipid Res 53:204–213. doi: 10.1194/jlr.D020834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Creason AL, Davis EW, Putnam ML, Vandeputte OM, Chang JH. 2014. Use of whole genome sequences to develop a molecular phylogenetic framework for Rhodococcus fascians and the Rhodococcus genus. Front Plant Sci 5:406. doi: 10.3389/fpls.2014.00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Youngquist JT, Schumacher MH, Rose JP, Raines TC, Politz MC, Copeland MF, Pfleger BF. 2013. Production of medium chain length fatty alcohols from glucose in Escherichia coli. Metab Eng 20:177–186. doi: 10.1016/j.ymben.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aslan S, Sun C, Leonova S, Dutta P, Dörmann P, Domergue F, Stymne S, Hofvander P. 2014. Wax esters of different compositions produced via engineering of leaf chloroplast metabolism in Nicotiana benthamiana. Metab Eng 25:103–112. doi: 10.1016/j.ymben.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 33.Marrakchi H, Lanéelle M-A, Daffé M. 2014. Mycolic acids: structures, biosynthesis, and beyond. Chem Biol 21:67–85. doi: 10.1016/j.chembiol.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 34.Toriyama S, Imaizumi S, Tomiyasu I, Masui M, Yano I. 1982. Incorporation of 18O into long-chain, secondary alcohols derived from ester mycolic acids in Mycobacterium phlei. BBA-Lipid Lipid Metab 712:427–429. doi: 10.1016/0005-2760(82)90363-0. [DOI] [Google Scholar]
- 35.Lacave C, Laneelle M-A, Daffe M, Montrozier H, Laneelle G. 1989. Mycolic acid metabolic filiation and location in Mycobacterium aurum and Mycobacterium phlei. Eur J Biochem 181:459–466. doi: 10.1111/j.1432-1033.1989.tb14747.x. [DOI] [PubMed] [Google Scholar]
- 36.Fixter LM, Nagi MN, Mccormack JG, Fewson CA. 1986. Structure, distribution and function of wax esters in Acinetobacter calcoaceticus. J Gen Microbiol 132:3147–3157. [Google Scholar]
- 37.Kalscheuer R, Stoveken T, Luftmann H, Malkus U, Reichelt R, Steinbuchel A. 2006. Neutral lipid biosynthesis in engineered Escherichia coli: jojoba oil-like wax esters and fatty acid butyl esters. Appl Environ Microbiol 72:1373–1379. doi: 10.1128/AEM.72.2.1373-1379.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santala S, Efimova E, Koskinen P, Karp MT, Santala V. 2014. Rewiring the wax ester production pathway of Acinetobacter baylyi ADP1. ACS Synth Biol 3:145–151. doi: 10.1021/sb4000788. [DOI] [PubMed] [Google Scholar]
- 39.Kannisto M, Efimova E, Karp M, Santala V. 2017. Growth and wax ester production of an Acinetobacter baylyi ADP1 mutant deficient in exopolysaccharide capsule synthesis. J Ind Microbiol Biotechnol 44:99–105. doi: 10.1007/s10295-016-1872-1. [DOI] [PubMed] [Google Scholar]
- 40.Qiao K, Wasylenko TM, Zhou K, Xu P, Stephanopoulos G. 2017. Lipid production in Yarrowia lipolytica is maximized by engineering cytosolic redox metabolism. Nat Biotech 35:173–177. doi: 10.1038/nbt.3763. [DOI] [PubMed] [Google Scholar]
- 41.Wolfmeier U, Schmidt H, Heinrichs F-L, Michalczyk G, Payer W, Dietsche W, Boehlke K, Hohner G, Wildgruber J. 2000. Waxes. Ullmann's encyclopedia of industrial chemistry. Wiley, Hoboken, New Jersey. [Google Scholar]
- 42.Bauchop T, Elsden SR. 1960. The growth of micro-organisms in relation to their energy supply. J Gen Microbiol 23:457–469. [DOI] [PubMed] [Google Scholar]
- 43.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 44.van der Geize R, Hessels GI, van Gerwen R, van der Meijden P, Dijkhuizen L. 2001. Unmarked gene deletion mutagenesis of kstD, encoding 3-ketosteroid delta1-dehydrogenase, in Rhodococcus erythropolis SQ1 using sacB as counter-selectable marker. FEMS Microbiol Lett 205:197–202. doi: 10.1111/j.1574-6968.2001.tb10947.x. [DOI] [PubMed] [Google Scholar]
- 45.Bergmeyer HU. 1975. New values for the molar extinction coefficients of NADH and NADPH for the use in routine laboratories. Z Klin Chem Klin Biochem 13:507–508. [PubMed] [Google Scholar]
- 46.Eyer P, Worek F, Kiderlen D, Sinko G, Stuglin A, Simeon-Rudolf V, Reiner E. 2003. Molar absorption coefficients for the reduced Ellman reagent: reassessment. Anal Biochem 312:224–227. doi: 10.1016/S0003-2697(02)00506-7. [DOI] [PubMed] [Google Scholar]
- 47.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 48.Pernet F, Pelletier CJ, Milley J. 2006. Comparison of three solid-phase extraction methods for fatty acid analysis of lipid fractions in tissues of marine bivalves. J Chromatogr A 1137:127–137. doi: 10.1016/j.chroma.2006.10.059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.