

ABSTRACT
A central goal of microbial ecology is to identify and quantify the forces that lead to observed population distributions and dynamics. However, these forces, which include environmental selection, dispersal, and organism interactions, are often difficult to assess in natural environments. Here, we present a method that links microbial community structures with selective and stochastic forces through highly replicated subsampling and enrichment of a single environmental inoculum. Specifically, groundwater from a well-studied natural aquifer was serially diluted and inoculated into nearly 1,000 aerobic and anaerobic nitrate-reducing cultures, and the final community structures were evaluated with 16S rRNA gene amplicon sequencing. We analyzed the frequency and abundance of individual operational taxonomic units (OTUs) to understand how probabilistic immigration, relative fitness differences, environmental factors, and organismal interactions contributed to divergent distributions of community structures. We further used a most probable number (MPN) method to estimate the natural condition-dependent cultivable abundance of each of the nearly 400 OTU cultivated in our study and infer the relative fitness of each. Additionally, we infer condition-specific organism interactions and discuss how this high-replicate culturing approach is essential in dissecting the interplay between overlapping ecological forces and taxon-specific attributes that underpin microbial community assembly.
IMPORTANCE Through highly replicated culturing, in which inocula are subsampled from a single environmental sample, we empirically determine how selective forces, interspecific interactions, relative fitness, and probabilistic dispersal shape bacterial communities. These methods offer a novel approach to untangle not only interspecific interactions but also taxon-specific fitness differences that manifest across different cultivation conditions and lead to the selection and enrichment of specific organisms. Additionally, we provide a method for estimating the number of cultivable units of each OTU in the original sample through the MPN approach.
KEYWORDS: 16S RNA, community assembly, enrichment culturing, microbial ecology
INTRODUCTION
Microbial communities are central players in Earth's biogeochemical cycles (1), human health (2), biotechnological processes, such as wastewater treatment (3), and the production of foods (4). Underpinning the structure, function, and evolution of all these communities are the ecological forces of dispersal, drift, selection, and speciation (5, 6). Even on short timescales, in which one can ignore evolutionary mechanisms of diversification, drift, selection, and dispersal interact to turn over populations of organisms in both predictable and unpredictable ways. Unpredictable changes in community structure are rooted in random dispersal and drift, while predictable changes are caused by deterministic fitness differences and environmental selection (6). Capturing the influence of these processes is central to predicting and controlling microbial community structure and function.
Although selective processes can lead to more predictable community compositions (7), the processes themselves are complex and numerous and can stem from biotic sources, abiotic sources, or feedback loops between biotic and abiotic factors (8). Despite there being numerous examples of biotic relationships (e.g., competitive interactions) among microorganisms (9), there is less work exploring how biotic relationships change as a function of the environment in which they are found (6). Moreover, assessment of the impact of selective forces in microbial community structure is hampered by the complexity of natural systems, including the extraordinary diversity of organisms, the numerous uncontrolled (or unmeasured) environmental and historical factors, and large and variegated scales of distance and time. The reduction of these complexities through the use of well-defined experimental platforms (e.g., microcosms) offers a tremendous advantage (10–12). In comparison to studies done in situ, laboratory microcosms allow us to directly evaluate community responses to known and controlled variables while minimizing the influence of unmeasured factors, like resource heterogeneity and historical differences across sites. Furthermore, microcosms allow the preservation of compositional and functional diversity of the seed community (10), and as such, assembly rules garnered from controlled laboratory experiments can be used to better understand the factors that structure microbial communities in the field (13).
In microcosm experiments inoculated with complex and undefined multispecies consortia, there are a number of experiments offering conflicting views regarding the importance of selective forces, and the attendant increase in reproducibility, in the assembly of microbial communities. In some systems, highly reproducible communities formed even from different inocula incubated under similar conditions, which is evidence of niche-based processes and strong selective forces (14–16). On the other hand, some systems exhibit divergent community structures, accounted for by distribution of rare taxa in the inoculum (17), different source communities (18), and stochastic colonization processes (19). Although the results from each of these experiments depend on their own unique source inocula and selective conditions, they highlight the need for a more unified understanding of how both predictable processes (e.g., selection) and unpredictable processes (e.g., random colonization and stochastic drift) interact to shape microbial community assembly.
Here, we leverage the large multiplexing capabilities of Illumina 16S rRNA amplicon sequencing with a highly replicated enrichment experiment in order to examine how selective forces shape community assembly in the presence of random dispersal. Specifically, we aimed to answer the following: (i) how much do community structures vary as a function of probabilistic recruitment from a single regional species pool? (ii) How do abiotic selective factors, such as homogenizing environment (e.g., shaking) and terminal electron-accepting conditions, influence and structure these communities? (iii) How do various taxonomic groups respond to these differentiated selective processes? (iv) Can we detect species interactions, and how do they change as a function of environmental factors? To these ends, we systematically manipulated bacterial diversity by subsampling a single “regional” species pool at several dilutions in order to create many “local” communities that varied in their membership. We carried these experiments out in both an unstructured aerobic environment as well as a structured nitrate-reducing environment to ascertain how these commonly employed cultivation conditions shape community assembly by altering the cultivability, competitive fitness, and interspecific interactions of community members.
RESULTS
Initial sample characterization and estimates of cultivable populations.
The initial inoculum was estimated to contain ∼37,000 cells/ml based on acridine orange direct count (AODC). Based on this initial cell count, the enrichments that received the most concentrated inoculum thus received ∼3,700 cells · ml−1, and those enrichments receiving the most dilute inoculum started with an average of only ∼0.37 cells · ml−1. Following cultivation, all wells that received the two most concentrated inocula (10−1 and 10−2 final inoculum density) showed population growth as measured by optical density at 600 nm (OD600) (see Table S1 in the supplemental material). Anaerobic experiments with initial inoculum densities of 10−3, 10−4, and 10−5, had 69, 12, and 1 positive-growth wells, respectively. Similarly, the aerobic experiments had 79, 13, and 4 positive-growth wells from those same inocula. Using these data, the original sample was calculated to be between 1,400 and 2,200 cultivable cells per milliliter at the 95% confidence level, with 1,700 cells · ml−1 being most probable for aerobic cultivation conditions. Under the anaerobic conditions, the most probable number of initial cultivable cells is estimated to be between 1,000 and 1,600 cultivable cells per milliliter, with 1,400 cells · ml−1 being most probable. Thus, approximately 4% of the total cells counted by the AODC method appear to be cultivable under these conditions (3.8% under nitrate-reducing conditions and 4.6% under aerobic conditions).
In addition to optical density measurements, DNA was extracted from each well and the 16S rRNA gene amplified and sequenced. Across all 960 cultivated communities, OD600 and sequencing data were in agreement in regard to detectable growth in 893 cases (93.0%). There were 23 samples with positive growth by sequencing that did not exceed the OD600 thresholds and 44 samples with growth by optical density that did not exceed read count thresholds. The numbers of positive-growth wells by both methods for each experiment and dilution are shown in Table S1.
Probabilistic immigration and environmental conditions shape microbial community structure.
As expected, based on 16S rRNA gene amplicon sequencing data, enrichment cultures started with the highest inoculum concentrations had the highest operational taxonomic unit (OTU) richness. The communities receiving the most concentrated inoculum had statistically similar numbers of OTUs under nitrate-reducing and aerobic conditions (t test, P = 0.10), with the nitrate-reducing communities averaging 26.5 OTUs (n = 94; standard deviation [SD], 11.27 OTUs) and the aerobic communities averaging 29.2 (n = 96; SD, 10.53 OTUs). OTU richness declined in experiments that received less concentrated inocula (Fig. 1). In the 10−2 dilutions, the aerobic communities tended to have higher species richness than the nitrate-reducing communities (t test, P = 2.09e−06), with nitrate-reducing cultures having on average 9.3 OTUs (n = 96; SD, 5.7 OTUs) and the aerobically cultivated communities with 13.5 OTUs (n = 96; SD, 6.4 OTUs). Aerobic communities that received the most diluted inoculum had on average only 2.3 OTUs (n = 3; SD, 2.31 OTUs), and only a single OTU in a single sample was detected in the nitrate-reducing communities begun with the most dilute inoculum. In addition to species richness, we quantified how evenly communities were structured with Pielou's index. At all dilutions, the anaerobic communities showed significantly reduced evenness (Fig. S2), despite being seeded from the same populations that seeded the aerobic communities. These results indicate that the anaerobic cultivation conditions favor the outgrowth of a smaller number of taxa, results consistent with stronger selective forces under the anaerobic conditions.
FIG 1.
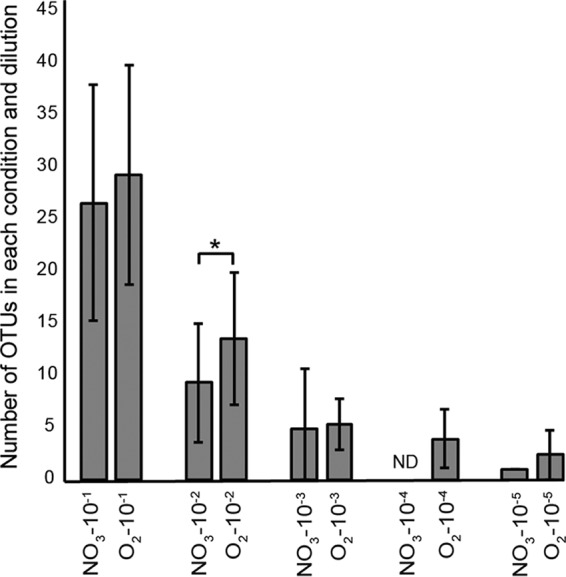
For each experiment and dilution, the mean number of OTUs assigned. Error bars represent standard deviations. Statistical significance between means was tested using Student's t test for the first three dilutions (10−1 to 10−3). Significance (P < 0.05) is marked with an asterisk. ND, no data acquired for that set of samples.
Overall, there were 399 unique OTUs identified across all cultures. Of these, 197 OTUs were found only in nitrate-reducing cultures, 99 OTUs only in aerobic cultures, and 103 OTUs in both aerobic and nitrate-reducing samples (Fig. 2). Some families, like the Pseudomonadaceae, had fewer OTUs unique to anaerobic samples (n = 8) than OTUs unique to aerobic samples (n = 40). Other families, like the Paenibacillaceae, had a larger number of OTUs uniquely identified in anaerobic samples (n = 44) than identified in aerobic samples (n = 4).
FIG 2.
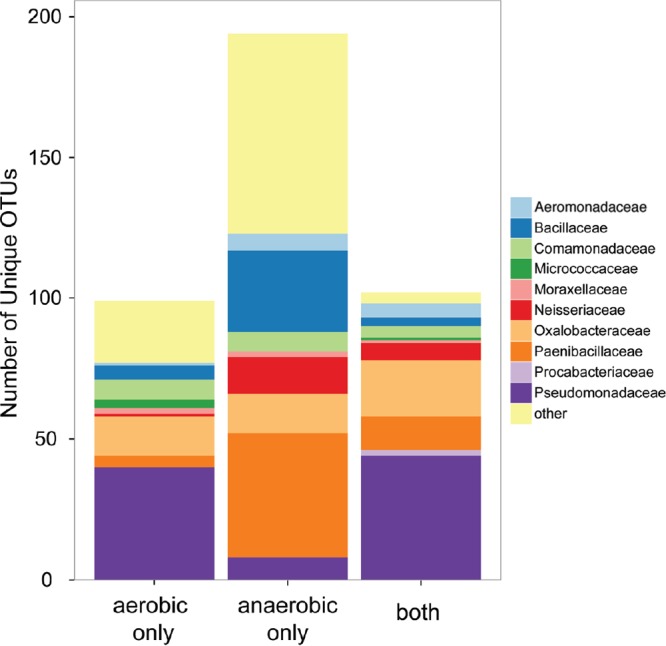
Number of OTUs found uniquely in anaerobic enrichments or aerobic enrichments, as well as OTUs identified in both.
Many cultures started from more dilute inocula were dominated by a single OTU (“New.ReferenceOTU30,” Pseudomonas sp.; Fig. 3). The abundance of this OTU in cultures started from more dilute inocula is indicative of its higher cultivable abundance in the initial sample, precluding it from being removed by successive dilutions. Most OTUs (69.3% in anaerobic samples and 64.4% in aerobic samples) were identified only in communities started from the two most concentrated inocula, reflecting their low cultivable abundance in the groundwater inoculum and resultant extinction upon dilution. Conversely, only 13.3% of the OTUs in anaerobic samples were limited to communities cultivated from more dilute inocula (NO3-10−3 through NO3-10−5), and only 3.9% of aerobically identified OTUs were limited to those communities from the more dilute inocula (O2-10−3 through O2-10−5).
FIG 3.

Relative abundance of OTUs from (y axes) across all communities (x axes) in the first four dilutions of aerobic enrichments and first three dilutions of anaerobic nitrate-reducing enrichments. Only the most abundant 11 OTU are shown for clarity.
We quantified the dispersion of community structures in each dilution and under each condition in order to examine how probabilistic processes and environmental selection interact and contribute to stabilizing or destabilizing the range of community structure outcomes. Stochastic recruitment drives variation among replicate communities of a condition and dilution, and we expect that communities formed from fewer taxa, either because of selective filtering or removal by dilution, will tend to be more similar to each other. Among communities formed from the most concentrated inocula, the aerobically cultivated communities were typically more similar to each other than the nitrate-reducing communities (Fig. 4). The dominance of one or several of a small subset of organisms in the anaerobic communities drives the divergence in community structure outcomes (Fig. 3). Conversely, in the communities formed from the next inoculum dilutions (NO3-10−2 and O2-10−2), the nitrate-reducing communities are actually more similar to each other than the aerobic communities are (Fig. 4). At this dilution, the selective pressures of the nitrate-reducing conditions prevented a number of OTU populations from growing as they did in the aerobic cultures. By the third dilution (10−3), most communities under either condition are very similar to each other (i.e., the median of the distances are low); however, there is a larger range of community dispersions. These data reflect that fact that most communities at these dilutions are dominated by a single OTU, precluding significant dissimilarities between them.
FIG 4.
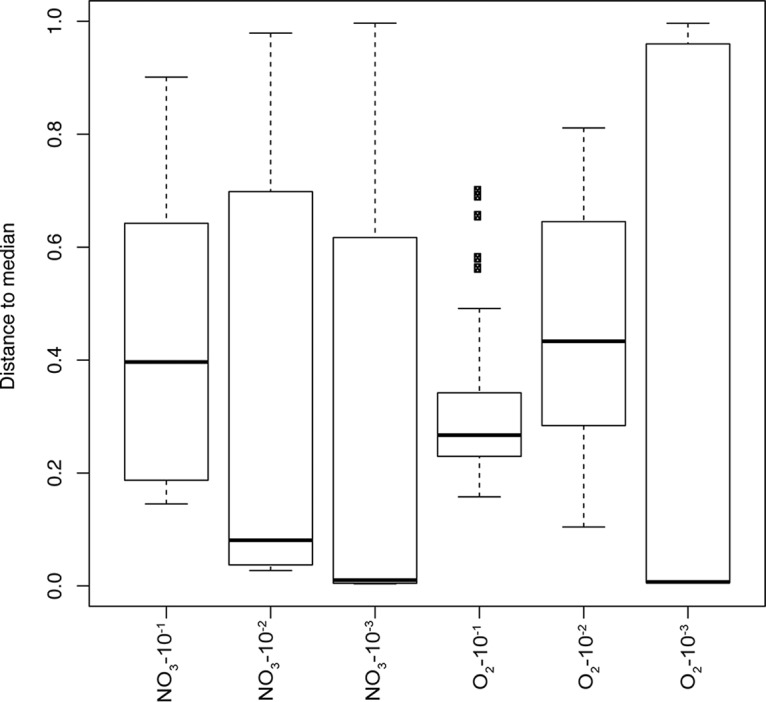
Analysis of group dispersions calculated by measuring each community's distance from a median point in multivariate space using Bray-Curtis dissimilarity. Higher median values indicate more within-group variation, and lower values indicate more homogeneous communities.
Environmental selection shapes cultivable fraction of inoculum.
For each OTU, we used the frequency at which the OTU was identified (i.e., the number of wells in which it was found) at multiple dilution levels under each condition to estimate the most probable number of cultivable units in the original inoculum sample. Since cultivability is condition dependent, we compared how these numbers varied between aerobic and anaerobic samples (Fig. 5). Notably, members of the Pseudomonadaceae, Comamonadaceae, and Micrococcaceae tended to be more cultivable in aerobic cultures, while OTUs assigned to the Paenibacillaceae and Bacillaceae tended to be more frequently found in the anaerobic cultures. Members of the Oxalobacteraceae, on the other hand, could be more cultivable under either aerobic or anaerobic conditions.
FIG 5.

Most probable number estimates of cultivable units per ml for each OTU, colored by family, under both anaerobic and aerobic conditions. Line of perfect concordance shown to clarify OTUs that are more cultivable under aerobic versus anaerobic conditions.
Most probable number (MPN) calculations are built upon several key assumptions, including that each OTU is randomly mixed, and different OTUs do not repel each other (20), assumptions that are unlikely to hold for natural bacterial communities. We calculated rarity values for each MPN as a means of assessing the extent to which these assumptions hold. Rarity values assess the probability that our observed detections for each OTU were likely to have occurred given the calculated MPN, and they are calculated by dividing the likelihood of the observed outcome by the largest likelihood of any outcome at that same MPN (21). We found that 38.6% and 32.8% of OTUs from aerobic and anaerobic cultures, respectively, had distribution frequencies categorized as unlikely or extremely unlikely (rarity values, <0.05). Of those MPN estimations with unlikely or extremely unlikely distributions, nearly all had a lower-than-expected number of positive observations from high-inoculum cultures and a concomitant higher-than-expected number of positive observations in low-inoculum cultures (Fig. S3). Explanations for this behavior include competitive mechanisms in high-inoculum cultures preventing growth and detection of these OTUs, or clumps of colocalized OTUs in the initial inoculum being broken up upon dilution, leading to a higher-than-expected number of observations in low-dilution cultures (22).
Identifying organism relative fitness.
We sought to classify OTUs as strong or weak competitors under each condition by comparing measured organism abundance with predicted organism abundance in a null model of community assembly in which all organisms have identical growth properties (no net positive or negative growth differences, and no interaction between OTUs). Using the estimated initial cultivable abundances of each OTU, we simulated the seeding and cultivation of 10,000 replicate communities from the lowest dilution inoculum into the aerobic and anaerobic environments. We focused on the lowest dilution cultures, since these cultures represent the greatest inclusion of taxa and thus overall highest expected frequency of competition. We then compared these estimated average abundances to the measured average abundance of each OTU and identified OTUs whose measured relative abundances were higher or lower than the predicted abundances at a 99% confidence level (Fig. 6). In essence, we are using only the frequency at which each OTU is identified to create expectations of how abundant taxa are during inoculation. We then compare these expected values to observed postcultivation average abundances. Most organisms tended to be poor competitors, including the most abundant OTU in our experiment, Pseudomonadaceae New.ReferenceOTU30. Using its estimated cultivable units per milliliter, the model predicts that this OTU should be an average of 19.5% of the NO3-10−1 communities and 32.4% of the O2-10−1 communities. The measured average relative abundances, however, were only 9.4% and 12.1%, respectively, reflecting the poor relative fitness of this taxon (Fig. 6). Put more simply, this OTU is expected to be very abundant in the neutral model of cultivation because it was estimated to be very abundant in the inoculum (i.e., was found in many cultures). At the end of cultivation, however, its relative abundance is lower than that expectation.
FIG 6.

Each OTU's final average percent abundance plotted against initial estimated percent abundance for the nitrate-reducing (left) and anaerobic (right) enrichments begun with the most concentrated inoculum. Red and blue lines indicate the upper and lower boundaries, respectively, of the 99% confidence interval of expected average abundance in 10,000 communities simulated in the null model of community assembly. Note the log scale. The right-most point in both graphs represents the Pseudomonas OTU New.ReferenceOTU30.
Some OTUs, such as those belonging to the Neisseriaceae and Aeromonadaceae, tended to be strong competitors under both aerobic and nitrate-reducing conditions (Fig. 7). Others, like the Pseudomonadaceae and Paenibacillaceae, tended to have strong competitors under only one condition (the Paenibacillaceae under anaerobic conditions and the Pseudomonadaceae under aerobic conditions). On the other hand, the Oxalobacteraceae had only a few, if any, strong competitors under either aerobic conditions or nitrate-reducing conditions. In some cases, rare taxa dominate cultures, including OTUs 581021 and 922761 (family Enterobacteriaceae), which are both predicted to be less than 0.008% of the cultivable inoculum and yet come to represent 33.2 and 62.1% of the anaerobic cultures in which they are found, respectively (Table S2). In the aerobic cultures, a single taxon of Aeromonadaceae (778059), representing only 0.002% of the initial cultivable inoculum, came to represent 66.0% of a single community.
FIG 7.

OTUs binned as having high, low, or nonsignificant relative fitness advantages in the anaerobic nitrate-reducing (left) and aerobic (right) communities at the lowest dilution.
As it makes the unrealistic assumption of no fitness differences between taxa, the null model simulation of community assembly did not predict true final organism abundances (Fig. 6). The true average abundances for the vast majority of taxa fell below the 99% confidence threshold of their expected abundances. Nearly all of these were predicted to be low-abundance taxa in the inoculum (i.e., <1%) that were driven to even lower relative abundances during cultivation. In addition to extraction and amplification biases, fitness differences and competition likely contribute to the lower-than-predicted abundances for many of these OTUs.
Predicting organism interactions.
Given the probabilistic nature of how we seeded each replicate, we sought to identify pairs of taxa that may be interacting by observing if they were found more or less frequently together than one would expect by chance. For each condition and dilution, the total number of pairwise comparisons, the number of significant positive and negative associations, and the median strength of the associations for each condition and dilution are shown in Table 1. Overall, we identified 115 putative interactions (56 negative and 59 positive) among 34 OTU in the nitrate-reducing samples and 34 putative interactions (23 positive and 11 negative) among 15 OTU in the aerobic samples (Fig. 8). There was very little overlap between interaction predictions across conditions, with only 14 OTU and 5 predicted interactions shared in the aerobic and anaerobic communities. Of those five shared interactions, all were positive associations among pairs of closely related OTUs.
TABLE 1.
Summary table of pairwise cooccurrence analyses for each environment and dilution
| Dilution | No. of samples | No. of species | Total no. of species pair combinations | No. of analyzed combinationsa | No. of positive interactionsb | No. of negative interactionsb | Median powerc | FDR (%)d |
|---|---|---|---|---|---|---|---|---|
| NO3-10−1 | 94 | 230 | 26,335 | 802 | 58 | 47 | 6.2 | 1.53 |
| NO3-10−2 | 96 | 124 | 7,626 | 317 | 12 | 9 | 7.5 | 3.02 |
| NO3-10−3 | 54 | 91 | 4,095 | 37 | 1 | 0 | 8.2 | 7.40 |
| NO3-10−4 | 0 | 0 | NAe | NA | NA | NA | NA | NA |
| NO3-10−5 | 1 | 1 | NA | NA | NA | NA | NA | NA |
| O2-10−1 | 96 | 164 | 13,366 | 1,303 | 8 | 8 | 9.75 | 16.29 |
| O2-10−2 | 96 | 109 | 5,886 | 564 | 15 | 3 | 9.85 | 6.27 |
| O2-10−3 | 79 | 65 | 2,080 | 74 | 2 | 0 | 7.7 | 7.40 |
| O2-10−4 | 22 | 37 | 666 | 12 | 0 | 0 | NA | NA |
| O2-10−5 | 3 | 6 | NA | NA | NA | NA | NA | NA |
Analyzed combinations represent only those species pairs expected to have 1 or more cooccurrences.
Significance at the threshold of a P value of <0.001.
Calculated as |observed interactions − expected interactions|.
FDR, false-discovery rate.
NA, not applicable.
FIG 8.
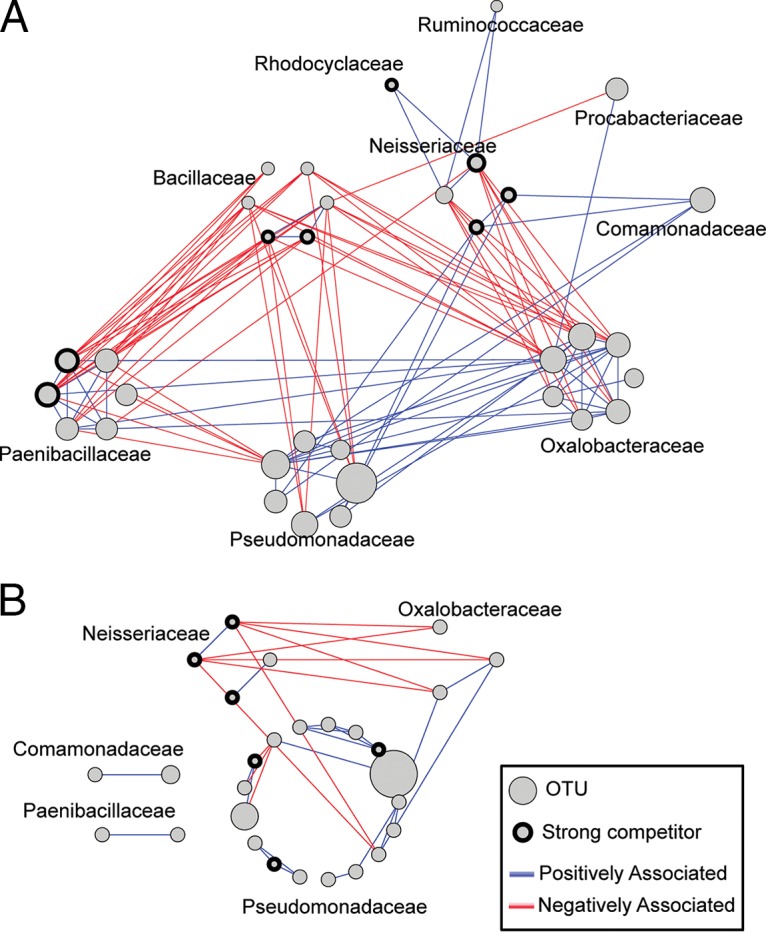
Networks depicting positive and negative associations between pairs of taxa in anaerobic nitrate-reducing communities (A) and aerobic communities (B). Graphs were made by the union of interaction graphs at each dilution for aerobic and anaerobic samples, respectively. Positive associations are shown in blue and negative associations in red. OTUs predicted to be strong competitors (Fig. 6 and 7) are indicated with a bold outline. The size of the node for each OTU scales with the estimated number of cultivable units of that OTU in the initial inoculum (Fig. 5).
In the anaerobic samples, OTUs of the Pseudomonadaceae were positively associated with members of the Oxalobacteraceae and negatively associated with members of the Bacillaceae and Paenibacillaceae. Oxalobacteraceae, on the other hand, were positively associated with the Paenibacillaceae and negatively associated with members of the Neisseriaceae and Bacillaceae. The Bacillaceae had no positive connections to other families and were negatively associated with members of the Pseudomonadaceae, Oxalobacteraceae, and Paenibacillaceae. In aerobic samples, some positive associations between the Pseudomonadaceae and Oxalobacteraceae were identified, and the Neisseriaceae share negative associations with members of both the Oxalobacteraceae and Pseudomonadaceae families.
DISCUSSION
We utilized multiple dilutions in a highly replicated enrichment experiment to understand how probabilistic recruitment and selection shape community assembly. We show that probabilistic subsampling can produce a range of community structure outcomes constrained by environmental selection.
Divergence among replicate communities formed from a single inoculum dilution and under a single selective pressure is rooted in varied recruitment. Together with this probabilistic process, selective forces act by winnowing down the types and sizes of populations that will thrive. This effect, for example, is seen when comparing communities in the anaerobic versus aerobic enrichments of the first dilutions (NO3-10−1 and O2-10−1). The anaerobic cultivations, despite being seeded with the same numbers and populations of cells as the aerobic enrichments, favored the outgrowth and dominance of a smaller number of taxa, as indicated by Pielou's evenness index (Fig. S2). In other words, the NO3-10−1 communities are more varied because fewer organisms are fit and emerge as “winners,” creating distinct sets of reproducible outcomes. The communities under the O2-10−1 condition are more cohesive because many organisms are fit.
As with strong selective pressures, dilution can create variance in community structures by bottlenecking the number of cultivable organisms. For example, the communities of the O2-10−1 enrichments tend to be more similar to each other than the communities of the O2-10−2 enrichments. Additionally, the O2-10−1 enrichments are more evenly structured than the communities of the O2-10−2 enrichments, which are often dominated by a single organism. These findings are consistent with stochastic recruitment creating fewer “winning” organisms and ultimately more divergent community structures in the O2-10−2 enrichments. Continuing to inoculate with more and more dilute inocula, however, ultimately reduces variance in community structure outcomes, because a single OTU comes to dominate. Under aerobic conditions, this organism's relative cultivable abundance means it dominated the 10−3 dilutions, while the overall reduced cultivability of other organisms in the stark selective pressures of the anaerobic environment led to this OTU's dominance in the 10−2 dilutions.
Strong selective pressures are also evident when examining how different phylogenetic groups are enriched under the different cultivation conditions. For example, the majority of Paenibacillaceae OTUs were unique to anaerobic samples (Fig. 2). Overall, the dominant detected families, including the Pseudomonadaceae, Bacillaceae, Paenibacillaceae, Comamonadaceae, and Neisseriaceae, are commonly found in the ground waters of the Oak Ridge field site and represent frequently identified heterotrophic members of bacterial soil and groundwater communities (23, 24).
An organism's initial abundance in any given local community, indeed, the chance it arrives in that community at all, is a function of its abundance in the inoculum. In agreement with that expectation, species richness declined with increasing dilution of the inoculum, as did the number of wells with positive detectable growth (Table S1). Similar dilution-to-extinction approaches have been used previously to examine the link between biodiversity and ecosystem functioning (25–27). Here, however, the high replication at each dilution allows us to extrapolate the abundance of each OTU in the initial inoculum by examining the number of communities in which each OTU is found at each dilution. We estimate, using an MPN technique, the absolute cultivable abundance of each taxon in the inoculum, data unobtainable from 16S rRNA amplicon sequencing of the inoculum alone. We estimated that the most abundant Pseudomonas OTU (New.ReferenceOTU30), for instance, had approximately 840 cultivable units per ml under anaerobic conditions and 2,590 cultivable units per ml under aerobic conditions (Table S2). Although MPN techniques are commonly used for estimation of bacterial abundance in a wide variety of applications (28), the application of 16S rRNA amplicon sequencing to the approach is, to our knowledge, novel, and offers the advantage of estimating the cultivability of a large number of taxa simultaneously. Many taxa had extremely small cultivable populations in the inoculum. In fact, 66.8% of OTUs cultivable under aerobic conditions and 78.3% of those cultivable under anaerobic conditions are estimated to have less than one cultivable unit per milliliter. These results reflect the diversity and high number of low-abundance species in the inoculum, consistent with previous results (23). Importantly, these results also highlight the need for careful consideration of experimental design, volume of inoculum used, and microbial density and diversity in the inoculum when evaluating reproducibility across any enrichment experiment. It is worth noting that having the 16S rRNA amplicon sequencing of the inoculum would add an exciting dimension to this analysis, including the extent to which detected taxa in the inoculum were cultivable and how well cultivable abundances align with OTU abundances. However, insufficient biomass for adequate extraction and sequencing was obtained from the inoculum, and these data were not collected. We further want to highlight that although the inoculum was submitted to two different selective regimes, they share a cultivation medium, R2A, which may select against large fractions of the inoculum community (e.g., approximately 4% of the cells counted by microscopy were cultivated). The use of other cultivation media would not only offer opportunities to recover different fractions of the inoculum but could also be used to dissect how specific selective factors impact the fitness of different populations.
We assessed how the relative fitness of individual OTUs differed across environmental conditions by predicting the relative abundance of each OTU in a null model of community assembly devoid of fitness differences, and we compared this to actual measured relative abundance (Fig. 6). In this way, we were able to identify OTUs as having either high, low, or no competitive fitness advantage in both the NO3-10−1 and O2-10−1 communities (Fig. 7). Again, we see some family-level differences in competitive abilities as a function of the enrichment conditions. For example, some OTUs of Pseudomonadaceae were strong competitors in aerobic environments, yet none were identified as strongly competitive under nitrate-reducing conditions. This is somewhat surprising, as members of the Pseudomonadaceae are frequent nitrate reducers (29, 30) and had many representatives capable of growth under anaerobic nitrate-reducing conditions (Fig. 2). The dominance of these Pseudomonadaceae in predominantly aerobic samples may be a reflection of an aerobic or facultatively aerobic ecological strategy in the natural environment of the Field Research Center (FRC) groundwater. On the other hand, representatives of the Paenibacillaceae are likely better adapted to conditions of low oxygen concentrations, as evidenced by their higher relative fitness under only anaerobic conditions (Fig. 7). Furthermore, despite their overall preference for anaerobic conditions, some Bacillaceae were strong competitors even in aerobic environments (Fig. 7), reflecting the broad metabolic versatility of these organisms (31). In both aerobic and anaerobic environments, some of the most competitive taxa belonged to members of the Neisseriaceae, especially the genus Chromobacterium (Table S2).
In addition to revealing how abiotic factors and probabilistic immigration shape community assembly, we sought to identify the roles of organism interactions in structuring communities. To that end, pairs of taxa were identified as potentially interacting if they were found more or less frequently together than expected by random chance. Given that every local environment is initially identical, cooccurrence patterns are not linked to initial abiotic conditions and “habitat-filtering,” a common problem for studies done in situ (32). Overall, we observed a larger number of interactions in the anaerobic samples than in the aerobic samples (Table 1 and Fig. 8), although the reasons for this remain unclear. In general, negative interactions could be explained by antibiotic production or resource competition. Paenibacillaceae, Pseudomonadaceae, Bacillaceae, Neisseriaceae, and Oxalobacteraceae all harbor species capable of producing antibiotics (33, 34). The higher number of negative interactions in the anaerobic samples may be linked to the regulation of antibiotic production by oxygen availability, as has been shown in species of Pseudomonas (35). Alternatively, anaerobic negative interactions might be linked to accumulations of by-products of fermentative metabolisms that inhibit competing organisms. Finally, negative interactions could be linked to the structured (i.e., unshaken) environment of the anaerobic cultures, with physical proximity possibly being an important factor. Members of the family Neisseriaceae and Oxalobacteraceae were unique in that they showed negative interaction patterns in both aerobic and anaerobic samples, even though no individual OTUs and interacting pairs were preserved in the two interaction networks.
Positive interactions can be more difficult to interpret, as in some cases, cooccurring OTUs may be ultimately caused by sequence variation among copies of the 16S rRNA gene cooccurring within cells (36). For this reason, we focus predominately on associations across broader phylogenetic distances. Intriguingly, members of the Oxalobacteraceae were positively associated with members of the Pseudomonadaceae and the Paenibacillaceae in anaerobic samples and with the Pseudomonadaceae alone in aerobic samples. Associations between Oxalobacteraceae and Pseudomonadaceae have been reported previously in human-associated samples (37). One possibility is that the Oxalobacteraceae are supported by CO2 released from the oxidation of organic carbon in the medium, as these organisms exhibit capnophilic physiologies (38).
Nonrandom positive cooccurrences might also be caused by colocalization on the same particle in the environment and subsequent coseeding in each enrichment community. These types of positive cooccurrences would be of particular interest since these organisms are more likely to be in close association in their natural environments. However, the poor overlap in positive cooccurrences between aerobic and anaerobic communities suggests that this may not be the case. Some positive interactions may also be a case of being the common target of another organism. In this case, negative interactions stemming from a broad-spectrum “killer” (e.g., members of the Bacillaceae) may eliminate multiple taxa from certain communities, leading to increased incidence of cooccurrence of those taxa in communities where the killer strain is not found.
Conclusion.
Here, we show that the combination of random dispersal with abiotic and biotic selections gives rise to numerous and variegated communities. We examine how random variation in community outcome is strongly throttled by selective pressures and dissect how those selective pressures alter the structure of the cultivable inoculum, as well as the competitive hierarchy of specific taxa. Ultimately, this approach offers a method to simultaneously explore the “niche” parameters of many coexisting populations, identify organism interactions, and explore processes of community assembly for ecological or biotechnological applications.
MATERIALS AND METHODS
Sampling and cell counting.
Groundwater was collected from an uncontaminated well (FW301: N35.94106884 and W84.33618124) at the Oak Ridge Field Research Site on 5 May 2015. The well is considered uncontaminated because, unlike many other wells at the Oak Ridge Field Research Site, it does not sample groundwater from the radioactive and hazardous contaminant plume emanating from the former waste disposal ponds (23). Prior to the collection of samples, approximately 10 liters of groundwater was pumped until pH, conductivity, and oxidation-reduction (redox) values were stabilized. Following this purge, approximately 50 ml was pumped from the midscreen level into a sterilized serum vial, minimizing residual headspace. The vial was sealed and shipped overnight at 4°C to the laboratory for cultivation. An additional ∼40 ml of water sample was taken immediately following the first, preserved with 4% formaldehyde, and stored at 4°C for cell counting. Initial inoculum cell counts were determined using the acridine orange direct count (AODC) method (39). A 20-ml volume was filtered through a 0.2-μm-pore-size black polycarbonate membrane (Whatman International Ltd., Piscataway, NJ), supported by a vacuum filtration sampling manifold (Millipore Corp., Billerica, MA). Filtered cells were stained with 25 mg/ml acridine orange for 2 min in the dark. Unbound stain was rinsed through the membrane with 10 ml of filter-sterilized 1× phosphate-buffered saline (PBS; Sigma-Aldrich Corp., St. Louis, MO). The rinsed membrane was mounted onto a slide and cells were imaged with a fluorescein isothiocyanate (FITC) filter on a Zeiss Axioskop (Carl Zeiss, Inc., Germany).
Inoculation and culturing.
Five milliliters of the groundwater sample was diluted serially four times into a 4 mM phosphate-buffered saline solution (pH 7.4) at a 1:10 ratio. For aerobic experiments, 100 μl of the original undiluted sample and the four serially diluted samples (1:10, 1:100, 1:1,000, and 1:10,000) were each inoculated into deep-well 96-well plates, with each well containing 900 μl of autoclaved R2A medium (HiMedia, Mumbai, India). Thus, each dilution was inoculated into 96 replicates. Plates were sealed with breathable plate seals and placed on a 30°C shaking incubator for 24 h (Infors HT, Switzerland) at 750 rpm. All experiments are designated by the incubation condition (e.g., O2) and the dilution with respect to original sample (e.g., 10−1, 10−2, etc.), giving five sets of incubations: O2-10−1, O2-10−2, O2-10−3, O2-10−4, and O2-10−5. Anaerobic experiments were inoculated from the same dilutions but into R2A that had been supplemented with 20 mM sodium nitrate (Sigma-Aldrich, St. Louis, MO, USA). The anaerobic experiments were immediately transferred into an anaerobic glove bag (Coy, Grass Lake, MI, USA) containing a N2:H2:CO2 atmosphere (85:10:5) and cultivated, unshaken, at 30°C for ∼96 h. The aerobic and anaerobic experiments were both cultivated until visible growth had occurred in some wells, and the anaerobic experiments thus necessitated a longer incubation. These experiments are referred to as NO3-10−1, NO3-10−2, NO3-10−3, NO3-10−4, and NO3-10−5. In addition to plates inoculated with the groundwater, two additional plates were inoculated with 100 μl of PBS solution and served as a negative control for growth under both aerobic and anaerobic conditions.
DNA extraction and PCR.
Two-hundred microliter aliquots of culture were extracted using the Wizard SV 96 Genomic DNA purification system (Promega, Madison, WI, USA), as per the manufacturer's specifications. In addition to the samples, we extracted 36 no-inoculum control samples and 24 extraction blanks. The extraction blanks are DNA extractions carried out solely on the extraction reagents themselves and thus serve as a control for contaminating DNA both in the extraction and the downstream PCR. DNA was quantified with the Quant-iT double-stranded DNA (dsDNA) assay kit (Life Technologies, Eugene, OR, USA). Samples were normalized so that ∼5 ng of each sample was input into each 20-μl PCR. Some samples, especially extraction blanks, received less than 5 ng, as they were limited by the concentrations of extracted DNA. Primers used in the PCRs amplified the V3-V4 hypervariable regions of the 16S gene (341F [5′-CCTACGGGAGGCAGCAG] and 806R [5′-GGACTACHVGGGTWTCTAAT]). Both forward and reverse primers contained TruSeq Illumina adapters, barcodes, phasing, and linker sequences and are similar to previously described designs (40, 41), with the exception that the barcodes here were included so as to be part of a sequencing read instead of a separate indexing read. Each PCR mixture contained 4 μl of 5× Phusion high-fidelity (HF) buffer, 0.2 μl of Phusion high-fidelity DNA polymerase, 200 μM dinucleoside triphosphates (dNTPs), 3% dimethyl sulfoxide (DMSO), and each primer at a concentration of 0.05 μM. All PCR reagents were obtained from NEB (Ipswich, MA, USA), except for primers, which were synthesized and PAGE purified by IDT (Coralville, IA, USA). The thermal cycling conditions were as follows: an initial denaturation at 98°C for 30 s, followed by 30 cycles at 98°C for 10 s, 50°C for 30 s, and 72°C for 30 s, with a final extension at 72°C for 7 min. Following PCR, samples from the same experiment and dilution (i.e., plate) were pooled and purified with Zymo Clean and Concentrator kits (Irvine, CA, USA) and quantified with quantitative PCR (qPCR; Kapa Biosystems, Wilmington, MA, USA). Each of the 11 pooled PCR products (each representing 96 samples) was then normalized and combined.
Sequencing and OTU calling.
The single aliquot of all combined PCRs was diluted and denatured according to the MiSeq reagent kit preparation guide (Illumina, San Diego, CA, USA). A sample concentration of 6 pM was loaded and sequenced on a 600-cycle (2 × 300 paired ends) MiSeq kit without PhiX. Paired-end reads overlapped and were merged with PEAR (42) under default parameters (minimum overlap of 10 bases and P = 0.01). Merged reads were quality filtered with custom scripts in which each read was matched to both forward and reverse barcodes allowing for zero mismatches and kept only if the maximum expected errors in the whole read were ≤2 (https://github.com/polyatail/arkin). Additional trimming removed reads that did not contain both forward and reverse primer sequences or were less than 420 bp. Finally, the remaining reads were trimmed of chimeric sequences using UCHIME against the Greengenes database (43), resulting in 9,026,027 high-quality reads across all samples. Reads were clustered with QIIME 1.9.0, using the pick_open_references.py script and a 97% clustering threshold (44). Taxonomic calls were made against the Greengenes database version 13_5 (45), with a minimum cluster size of 2.
Data processing and analysis.
OTU tables from QIIME were imported into R with Ruby scripts that assigned each well to the corresponding experiment (i.e., condition and dilution). As not all wells had positive growth but were extracted and sequenced anyway, it was vital to separate reads accumulated from either barcode sequencing errors or reagent contamination from true-positive detected OTUs. We controlled for these potential sources of error by sequencing and analyzing no-inoculum cultures and extraction-only blanks. First, R scripts were used to identify all OTUs that were found in the no-inoculum controls and the extraction blank samples. OTUs that represented more than 0.1% of summed reads in the no-inoculum controls and the extraction blank samples were called contaminants and excluded from the analysis. Next, in any given sample, any OTU with fewer reads than the summed read count of all contaminant OTUs in that sample was excluded from the analysis. Overall, contaminant reads were high (e.g., >0.5%) only in samples with few sequencing reads (<500) and with no detected growth by OD600 (<0.055 absorbance). Finally, any sample with fewer than 500 total reads was excluded from the analysis. The median and mean read counts of samples kept in the analysis were 9,177 and 14,529, respectively. The read count data for each sample are depicted in Fig. S1.
The variance in community structures within samples and dilutions was calculated using the “betadispers” function in the R package vegan (46). The multivariate analyses of group dispersions were done by calculating each community's distance from a median point in multivariate space using Bray-Curtis dissimilarity.
We used the MPN technique to calculate the cultivable abundance of every taxon in the inoculum. This widely employed technique provides the most probable number of cultivable units of an organism in an inoculum sample given a distribution of positive and negative outgrowths at several dilutions (24). The cultivable abundance is thus a function of both the number of cells of that organism in the inoculum as well as their ability to replicate under the prescribed cultivation condition. First, we calculated an overall estimated number of cultivable cells using OD600 data (24). To obtain the OTU-specific cultivable units per ml, we used the same technique coded into the statistical package R on the sequencing data of our cultivations. Data from the last two anaerobic dilutions were excluded in the MPN calculations, given that there were no samples with detectable OTUs in the NO3-10−4 dilution and only a single sample with a single OTU in the NO3-10−5 dilution. Rarity values for each OTU's MPN-estimated cultivable abundance were calculated by dividing the likelihood of the observed outcome by the largest likelihood of any outcome at that same estimated inoculum concentration (21). All data, including raw reads, and processed and demultiplexed reads, as well as code for calculating most probable number and rarity values for each OTU were calculated in R with scripts available at http://genomics.lbl.gov/supplemental/enrichments.
Null model analysis.
In order to determine which OTUs were the strongest competitors and which were the weakest competitors, we compared, across replicates, the average relative abundance of each OTU with its average expected abundance. Expected abundances are derived by simulating the assembly of many communities using the cultivable units per ml for each OTU estimated from MPN analyses. The communities are assembled in a null model in which no organism interactions or fitness differences were allowed. As such, this model is not meant to accurately predict outcomes, only to serve as a metric against which to measure and compare the strength of nonrandom forces (i.e., relative fitness in light of environmental selection). For each dilution and experimental condition, 10,000 communities were simulated. In each simulation, the number of seeded cells for a given OTU was randomly sampled from a Poisson distribution, with a mean value equal to the expected number of cells for that OTU under the condition/dilution. To account for potential error in the MPN-estimated cell abundances, both the mean number of cells for each OTU and the total number of cells (sum of all OTU abundances) were allowed to vary 2-fold. A 99% confidence interval was calculated for the percent relative abundance of each OTU in all simulated communities for the condition/dilution.
Predicting organism interactions.
OTU cooccurrence patterns were examined for each dilution under each experimental condition using the R package cooccur (47). Briefly, we identified, within all replicates of a condition and dilution, the number of times two taxa occur in the same cultivation well (i.e., replicate) and the number of times they occur apart. The model provides the probability that cooccurrences would occur more or less often than the observed cooccurrences assuming random and independent distribution of OTUs. Only OTUs with a relative abundance greater than 0.1% were counted in order to focus on only the most abundant taxa as well as to reduce false-positive associations from artifacts of OTU sequencing and clustering. Significant positive and negative associations (α = 0.001) were visualized as networks in Cytoscape by taking the union of all aerobic and nitrate-reducing experiments, respectively (48).
Accession number(s).
Raw data can be downloaded from the Sequence Read Archive under project accession no. PRJNA387349.
Supplementary Material
ACKNOWLEDGMENTS
We thank Morgan Price and Ryan Melnyk for their helpful discussions, and Dominique Joyner for her assistance with sampling.
This research was funded by Ecosystems and Networks Integrated with Genes and Molecular Assemblies (ENIGMA; http://enigma.lbl.gov), a Scientific Focus Area Program at Lawrence Berkeley National Laboratory supported by the U.S. Department of Energy, Office of Science, Office of Biological & Environmental Research under contract number DE-AC02-05CH11231. N. B. Justice was supported by the National Institutes of Health under Ruth L. Kirschstein National Research Service Award F32GM113547 from the National Institute of General Medical Sciences.
We declare no competing conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01253-17.
REFERENCES
- 1.Falkowski PG, Fenchel T, DeLong EF. 2008. The microbial engines that drive Earth's biogeochemical cycles. Science 320:1034–1039. doi: 10.1126/science.1153213. [DOI] [PubMed] [Google Scholar]
- 2.Cho I, Blaser MJ. 2012. The human microbiome: at the interface of health and disease. Nat Rev Genet 13:260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Daims H, Taylor MW, Wagner M. 2006. Wastewater treatment: a model system for microbial ecology. Trends Biotechnol 24:483–489. doi: 10.1016/j.tibtech.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Beresford TP, Fitzsimons NA, Brennan NL, Cogan TM. 2001. Recent advances in cheese microbiology. Int Dairy J 11:259–274. doi: 10.1016/S0958-6946(01)00056-5. [DOI] [Google Scholar]
- 5.Vellend M. 2010. Conceptual synthesis in community ecology. Q Rev Biol 85:183–206. doi: 10.1086/652373. [DOI] [PubMed] [Google Scholar]
- 6.Nemergut DR, Schmidt SK, Fukami T, O'Neill SP, Bilinski TM, Stanish LF, Knelman JE, Darcy JL, Lynch RC, Wickey P, Ferrenberg S. 2013. Patterns and processes of microbial community assembly. Microbiol Mol Biol Rev 77:342–356. doi: 10.1128/MMBR.00051-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chase JM. 2007. Drought mediates the importance of stochastic community assembly. Proc Natl Acad Sci U S A 104:17430–17434. doi: 10.1073/pnas.0704350104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.HilleRisLambers J, Adler PB, Harpole WS, Levine JM, Mayfield MM. 2012. Rethinking community assembly through the lens of coexistence theory. Annu Rev Ecol Evol Syst 43:227–248. doi: 10.1146/annurev-ecolsys-110411-160411. [DOI] [Google Scholar]
- 9.Hibbing ME, Fuqua C, Parsek MR, Peterson SB. 2010. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol 8:15–25. doi: 10.1038/nrmicro2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Widder S, Allen RJ, Pfeiffer T, Curtis TP, Wiuf C, Sloan WT, Cordero OX, Brown SP, Momeni B, Shou W, Kettle H, Flint HJ, Haas AF, Laroche B, Kreft J-U, Rainey PB, Freilich S, Schuster S, Milferstedt K, van der Meer JR, Groszkopf T, Huisman J, Free A, Picioreanu C, Quince C, Klapper I, Labarthe S, Smets BF, Wang H, Isaac Newton Institute Fellows, Soyer OS. 2016. Challenges in microbial ecology: building predictive understanding of community function and dynamics. ISME J 10:2557–2568. doi: 10.1038/ismej.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drake JA, Huxel GR, Hewitt CL. 1996. Microcosms as models for generating and testing community theory. Ecology 77:670–677. doi: 10.2307/2265489. [DOI] [Google Scholar]
- 12.Jessup CM, Kassen R, Forde SE, Kerr B, Buckling A, Rainey PB, Bohannan BJM. 2004. Big questions, small worlds: microbial model systems in ecology. Trends Ecol Evol 19:189–197. doi: 10.1016/j.tree.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 13.Cadotte MW, Drake JA, Fukami T. 2005. Constructing nature: laboratory models as necessary tools for investigating complex ecological communities. Adv Ecol Res 37:333–353. doi: 10.1016/S0065-2504(04)37011-X. [DOI] [Google Scholar]
- 14.Vanwonterghem I, Jensen PD, Dennis PG, Hugenholtz P, Rabaey K, Tyson GW. 2014. Deterministic processes guide long-term synchronised population dynamics in replicate anaerobic digesters. ISME J 8:2015–2028. doi: 10.1038/ismej.2014.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falk MW, Song K-G, Matiasek MG, Wuertz S. 2009. Microbial community dynamics in replicate membrane bioreactors–natural reproducible fluctuations. Water Res 43:842–852. doi: 10.1016/j.watres.2008.11.021. [DOI] [PubMed] [Google Scholar]
- 16.Ofiţeru ID, Lunn M, Curtis TP, Wells GF, Criddle CS, Francis CA, Sloan WT. 2010. Combined niche and neutral effects in a microbial wastewater treatment community. Proc Natl Acad Sci U S A 107:15345–15350. doi: 10.1073/pnas.1000604107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pagaling E, Strathdee F, Spears BM, Cates ME, Allen RJ, Free A. 2014. Community history affects the predictability of microbial ecosystem development. ISME J 8:19–30. doi: 10.1038/ismej.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langenheder S, Lindstrom ES, Tranvik LJ. 2006. Structure and function of bacterial communities emerging from different sources under identical conditions. Appl Environ Microbiol 72:212–220. doi: 10.1128/AEM.72.1.212-220.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou J, Liu W, Deng Y, Jiang Y-H, Xue K, He Z, Van Nostrand JD, Wu L, Yang Y, Wang A. 2013. Stochastic assembly leads to alternative communities with distinct functions in a bioreactor microbial community. mBio 4(2):e00584-12. doi: 10.1128/mBio.00584-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jarvis B, Wilrich C, Wilrich PT. 2010. Reconsideration of the derivation of most probable numbers, their standard deviations, confidence bounds and rarity values. J Appl Microbiol 109:1660–1667. [DOI] [PubMed] [Google Scholar]
- 21.Blodgett RJ. 2002. Measuring Improbability of Outcomes from a Serial Dilution Test. Commun Stat Theory Methods 31:2209–2223. doi: 10.1081/STA-120017222. [DOI] [Google Scholar]
- 22.Blodgett RJ, Garthright WE. 1998. Several MPN models for serial dilutions with suppressed growth at low dilutions. Food Microbiol 15:91–99. doi: 10.1006/fmic.1997.0144. [DOI] [Google Scholar]
- 23.Hemme CL, Tu Q, Shi Z, Qin Y, Gao W, Deng Y, Van Nostrand JD, Wu L, He Z, Chain PSG, Tringe SG, Fields MW, Rubin EM, Tiedje JM, Hazen TC, Arkin AP, Zhou J. 2015. Comparative metagenomics reveals impact of contaminants on groundwater microbiomes. Front Microbiol 6:1205. doi: 10.3389/fmicb.2015.01205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spain AM, Krumholz LR. 2011. Nitrate-reducing bacteria at the nitrate and radionuclide Oak Ridge Integrated Field Research Challenge site: a review. Geomicrobiol J 28:418–429. doi: 10.1080/01490451.2010.507642. [DOI] [Google Scholar]
- 25.Szabo KE, Itor POB, Bertilsson S, Tranvik L, Eiler A. 2007. Importance of rare and abundant populations for the structure and functional potential of freshwater bacterial communities. Aquatic Microb Ecol 47:1–10. doi: 10.3354/ame047001. [DOI] [Google Scholar]
- 26.Peter H, Beier S, Bertilsson S, Lindstrom ES, Langenheder S, Tranvik LJ. 2011. Function-specific response to depletion of microbial diversity. ISME J 5:351–361. doi: 10.1038/ismej.2010.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Philippot L, Spor A, Henault C, Bru D, Bizouard F, Jones CM, Sarr A, Maron P-A. 2013. Loss in microbial diversity affects nitrogen cycling in soil. ISME J 7:1609–1619. doi: 10.1038/ismej.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garthright WE, Blodgett RJ. 2003. FDA's preferred MPN methods for standard, large or unusual tests, with a spreadsheet. Food Microbiol 20:439–445. doi: 10.1016/S0740-0020(02)00144-2. [DOI] [Google Scholar]
- 29.Heylen K, Vanparys B, Wittebolle L, Verstraete W, Boon N, De Vos P. 2006. Cultivation of denitrifying bacteria: optimization of isolation conditions and diversity study. Appl Environ Microbiol 72:2637–2643. doi: 10.1128/AEM.72.4.2637-2643.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones CM, Stres B, Rosenquist M, Hallin S. 2008. Phylogenetic analysis of nitrite, nitric oxide, and nitrous oxide respiratory enzymes reveal a complex evolutionary history for denitrification. Mol Biol Evol 25:1955–1966. doi: 10.1093/molbev/msn146. [DOI] [PubMed] [Google Scholar]
- 31.Vos P, Garrity G, Jones D, Krieg NR, Ludwig W. 2011. Bergey's manual of systematic bacteriology. Volume 3: the Firmicutes. Springer Science & Business Media, New York, NY. [Google Scholar]
- 32.Bar-Massada A. 2015. Complex relationships between species niches and environmental heterogeneity affect species co-occurrence patterns in modelled and real communities. Proc Biol Sci 282:20150927. doi: 10.1098/rspb.2015.0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E. 2006. The Prokaryotes: a handbook on the biology of bacteria. Springer-Verlag, New York, NY. [Google Scholar]
- 34.Krieg NR, Parte A, Ludwig W, Whitman WB, Hedlund BP, Paster BJ, Staley JT, Ward N, Brown D. 2011. Bergey's manual of systematic bacteriology, vol 2 Springer Science & Business Media, New York, NY. [Google Scholar]
- 35.Haas D, Keel C. 2003. Regulation of antibiotic production in root-colonizing Pseudomonas spp. and relevance for biological control of plant disease. Annu Rev Phytopathol 41:117–153. doi: 10.1146/annurev.phyto.41.052002.095656. [DOI] [PubMed] [Google Scholar]
- 36.Vĕtrovsk T, Baldrian P. 2013. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One 8:e57923. doi: 10.1371/journal.pone.0057923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Price LB, Liu CM, Johnson KE, Aziz M, Lau MK, Bowers J, Ravel J, Keim PS, Serwadda D, Wawer MJ, Gray RH. 2010. The effects of circumcision on the penis microbiome. PLoS One 5:e8422. doi: 10.1371/journal.pone.0008422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valdes N, Soto P, Cottet L, Alarcon P, Gonzalez A, Castillo A, Corsini G, Tello M. 2015. Draft genome sequence of Janthinobacterium lividum strain MTR reveals its mechanism of capnophilic behavior. Stand Genomic Sci 10:110. doi: 10.1186/s40793-015-0104-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Francisco DE, Mah RA, Rabin AC. 1973. Acridine orange-epifluorescence technique for counting bacteria in natural waters. Trans Am Microsc Soc 92:416–421. doi: 10.2307/3225245. [DOI] [PubMed] [Google Scholar]
- 40.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. 2013. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, Ravel J. 2014. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2:6. doi: 10.1186/2049-2618-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Kobert K, Flouri T, Stamatakis A. 2014. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30:614–620. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens H, Szoecs E, Wagner H. 2017. vegan: community ecology package. R package version 2.4-3. R Core Development Team, Vienna, Austria: https://CRAN.R-project.org/package=vegan. [Google Scholar]
- 47.Veech JA. 2012. A probabilistic model for analysing species co-occurrence. Glob Ecol Biogeogr 22:252–260. [Google Scholar]
- 48.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.