

Abstract
Mutation rate in the nuclear genome differs between sexes, with males contributing more mutations than females to their offspring. The male-biased mutation rates in the nuclear genome is most likely to be driven by a higher number of cell divisions in spermatogenesis than in oogenesis, generating more opportunities for DNA replication errors. However, it remains unknown whether male-biased mutation rates are present in mitochondrial DNA (mtDNA). Although mtDNA is maternally inherited and male mtDNA mutation typically does not contribute to genetic variation in offspring, male mtDNA mutations are critical for male reproductive health. In this study, we measured male mtDNA mutation rate using publicly available whole-genome sequences of single sperm of the freshwater microcrustacean Daphnia pulex. Using a stringent mutation detection pipeline, we found that the male mtDNA mutation rate is 3.32 × 10−6 per site per generation. All the detected mutations are heteroplasmic base substitutions, with 57% of mutations converting G/C to A/T nucleotides. Consistent with the male-biased mutation in the nuclear genome, the male mtDNA mutation rate in D. pulex is approximately 20 times higher than the female rate per generation. We propose that the elevated mutation rate per generation in male mtDNA is consistent with an increased number of cell divisions during male gametogenesis.
Keywords: whole-genome amplification, single cell, deleterious mutations
1. Introduction
Substantial evidence across a wide range of taxa supports the idea that germline cells in males contribute many more mutations than females to the nuclear genome of their offspring [1–5]. For example, in mammals the ratio of male-to-female mutation rate ranges from approximately 2 to 20 [6,7]. The male-biased mutation in the nuclear genome has been suggested to be driven by a higher number of cell divisions during spermatogenesis, generating more opportunities for DNA replication errors to occur than in oogenesis [2,8,9].
In contrast to the extensive body of research on the male-biased mutation in the nuclear genome, it remains unknown whether male mitochondrial DNA experiences a higher mutation rate. Because mtDNA in animals is almost always maternally inherited [10,11], mtDNA in males represents an evolutionary dead end and their mutations do not contribute to genetic variation in the offspring. Thus, it has been proposed that mtDNA genetic variants that carry male-specific adverse phenotypic effects may not be effectively removed by natural selection if they are neutral or only slightly deleterious to female fitness [12,13]. On the other hand, maternal mtDNA mutations do contribute to variation in the offspring, and the population frequency of mutations that are deleterious to female fitness is maintained at mutation–selection equilibrium. The asymmetric strength of purifying selection on mtDNA polymorphisms that arise in males versus females may have strong implications for male fertility and disease susceptibility.
Although males do not pass on mtDNA to offspring [14], there are some differences in biological energy requirements between male and female gametes. Unlike eggs, sperm requires a package of mitochondria to provide the energy for motility [15]. Because mitochondria contain an oxidative-rich environment, DNA damage (through oxidation) may be more likely to occur. The direct measurement of mtDNA mutation rates using mutation-accumulation experiments has shown that the mtDNA mutation rate in the female germline is several times higher than nuclear DNA. For example, the mtDNA mutation rate is approximately 7-, 10- and 37-fold higher than nuclear DNA in Drosophila melanogaster [16], Caenorhabditis elegans [17,18] and Saccharomyces cerevisiae [19], respectively. However, to date no direct estimates are available for the mtDNA mutation rates in male germline.
The production of a large number of sperm in the male germline also requires the production of many mitochondria. Additional rounds of mitochondrial replication are required in the male germline, and the number of mutations in the male mtDNA is expected to be higher than the female mtDNA simply due to additional rounds of replication. Moreover, as demonstrated in Drosophila and zebrafish [20], ovary tissue is suppressed for mtDNA transcription, electron transport and free radical production, whereas testis and somatic tissue transcribe mitochondrial genes and produce an excess of reactive oxygen species (ROS) when compared with the ovary. In addition to the DNA damage that can be introduced by ROS, errors that arise during transcription are repaired using transcription-coupled repair, which has been shown to have extremely low levels of repair fidelity [21]. As DNA polymerase γ (pol-γ) is specifically responsible for the replication of mtDNA [22,23] with a high base-substitution fidelity (less than 2 × 10−6 errors per nucleotide) [24], the fidelity of pol-γ may have been optimized for performance in an ovary-specific mitochondrial inner environment. In a more free-radical-rich testis-specific mitochondrial environment, pol-γ may have reduced replication fidelity.
To better understand the mtDNA mutation processes in the male germline, we analyse the mitochondrial genomes of single sperm of the freshwater microcrustacean Daphnia pulex to directly estimate the mutation rate and spectrum of the male mtDNA. The availability of female mtDNA mutation rate in D. pulex from previous studies [25] presents an unprecedented opportunity to draw comparisons between the male and female germline mtDNA mutation rate within the same species.
2. Material and methods
(a). Sample collection and sequencing
The whole-genome sequence data of Daphnia single sperm have been previously published [26]. We briefly describe the experimental procedure here. Daphnia females are capable of parthenogenetic production of males under environmental stress. Taking advantage of this, sperm samples were collected from fifteen 20-day-old males of the Daphnia pulex genotype PA42, which was a natural isolate sampled from a vernal pond in Portland Arch Nature Reserve, Indiana. The pooled sperm sample was stained using a double-stranded DNA dye Hoechst 33 342 (Sigma-Aldrich) at room temperature. The AriaII cell sorter (BD Biosciences) was used to isolate single sperm cells into 96-well plates that contained cell lysis buffer. Based on cell sorting, we estimated that the total number of sperm from 15 males is approximately 50 000, with each male carrying about 3300 sperm. We performed MALBAC whole-genome amplification [27] procedure on the lysed single sperm cell. The MALBAC method introduces a quasi-linear pre-amplification stage that avoids bias associated with nonlinear amplification such as uneven coverage across the genome [27]. Details regarding cell sorting, cell lysis and MALBAC amplification can be found in [26]. The whole-genome amplified DNA for each sperm cell (approx. 1 µg) was used for preparing Illumina short-read sequencing library following standard protocols. Each library was sequenced using 150 bp paired-end reads on a HiSeq 2500 Illumina sequencer. All the raw reads were downloaded from NCBI Sequence Read Archive (SRA) study no. SRP058678.
(b). Mutation detection
The raw reads for each sample were mapped to the Daphnia pulex (GenBank accession no. 117817.1) mitochondrial reference sequence using the software Novoalign version 3.06.05. Because of the presence of potential nuclear sequences of mitochondrial origin, we removed any putative mtDNA mutations that are covered by raw reads that can be mapped to the Daphnia pulex nuclear genome reference [28], which was derived from the same PA42 clone as used in this study.
An initial set of mutations was generated using the mpileup function and bcftools in Samtools. In this case, false mutation calls can result from PCR errors in whole-genome amplification, PCR artefacts in library construction, sequencing errors, mismapped reads and misalignment caused by indels. To filter out the false-positive mutations, we employed the following set of stringent criteria: Phred-scaled quality score greater than 30 (i.e. less than 0.1% chance of a wrong base call); no signs of strand bias (i.e. covered by both forward and reverse reads); and no signs of tail distance bias (i.e. not enriched at the ends of reads that are prone to sequencing errors). Most importantly, all the mutant SNP alleles need to have at least 10× coverage, with a minimum of 5× coverage by forward and reverse reads, respectively, which translates to a heteroplasmic mutation frequency between 0.15 and 0.5 (see Results). This threshold is set based on the empirically estimated mean error rate across all samples is 0.006 per site (see below), with the most extreme error rate at approximately 0.1.
As all the mutations that we detected are heteroplasmic, the mutation rate (μ) is calculated using the equation
where Ri is the number of reads supporting mutant nucleotide at a given site for the ith sample and Di is the total number of reads at the same site, L is the number of single sperm samples, n is the number of mtDNA nucleotide sites examined in each sample and T (=1) is the number of generations.
(c). Estimation of copy number of mtDNA in Daphnia sperm
We can obtain a relative estimate of the copy number of mtDNA in each sperm by calculating the ratio of mtDNA average sequence coverage per site to that of nuclear sites at known single-copy genes (1 copy in a single sperm) in the Daphnia genome [28]. The raw reads of the nuclear genome were mapped to the Daphnia pulex reference assembly [28] using Novoalign. Raw reads mapped to multiple locations of the nuclear genome were removed from the final estimation of average coverage.
(d). Simulation of PCR errors in whole-genome amplification
Polymerase replication errors are a major concern in detecting mutations in genomic sequences that have undergone whole-genome amplification. To assess the likelihood that mutant alleles due to amplification errors could be falsely identified as true mutations by our mutation detection method, we performed computer simulation of the MALBAC whole-genome amplification procedure to measure the frequency of mutants in the final pool of amplicons and evaluate the likelihood that these errors could pass our computational filtering procedure. The MALBAC whole-genome amplification achieves a quasi-linear amplification of DNA templates using strand-displacement polymerase (Bst Large Fragment) and randomly annealing primers [27]. The MALBAC procedure consists of a pre-amplification stage followed by a regular PCR stage. The pre-amplification stage consists of five amplification cycles, where newly formed full amplicons that possess complementary 5′ and 3′ ends form a DNA loop structure and do not get amplified in subsequent round of pre-amplifications. This means that even if a polymerase error occurred in the very first round of amplification, its frequency in the pool of generated molecules would decrease significantly (at least fivefold) by the end of the pre-amplification stage. The presence of multiple mtDNA and multiple amplification of a single site by strand-displacement polymerase in each round of pre-amplification work in concert to further reduce the frequency of replication errors. At the end of pre-amplification, all the amplicons including the mutants will be used for regular PCR amplification.
Our simulation algorithm explores a range of parameter values for the copy number of mtDNA (e.g. 2, 10, 50, 100, 500) in a single sperm, the polymerase error rates (1 × 10−5 to 1 × 10−3) and the number of times a single site amplified by strand-displacement polymerase in one round of pre-amplification. At the end of the simulation, we used the average mutant frequency across the entire mtDNA to estimate the likelihood that these mutants would achieve the same sequencing profile (i.e. depth of coverage) as the identified mutations under a binomial sampling distribution.
Furthermore, to validate the predications from our simulation, we calculated the base-substitution error rate for each site, excluding sites where heteroplasmic mutations and indels are found across all the samples.
3. Results
(a). Overview of single sperm sequencing data
We analysed whole-genome sequence data of 96 single Daphnia sperm from a previous study [26]. Raw sequence reads were mapped to the D. pulex mtDNA reference sequence [29]. Across the 96 sperm genomes, the sperm mitochondrial genome has a coverage of 176 (s.d. = 130.7) reads per site on average (electronic supplementary material, figure S1), with a median value of 159.
(b). Number of copies of mtDNA in a Daphnia sperm
In mammals, a typical sperm cell contains approximately 50–75 mitochondria with one copy of mtDNA in each [15]. Although we do not know the exact copy number of mtDNA in a single Daphnia sperm, we obtained the relative copy number of mtDNA by using the ratio of the average sequence coverage at a site in the mtDNA compared to the average sequence coverage of single-copy gene sites in the nuclear genome. This normalizes the sperm mtDNA to a single copy of sperm nuclear DNA, but this estimate may be slightly upwardly biased. Because the animal mtDNA and nuclear genome have different genomic characteristics (e.g. repetitive regions), it is possible that the mtDNA can amplify at a higher efficiency than the nuclear genome, leading to the slight overestimation of mtDNA copy number per sperm. After mapping raw reads of sperm samples to the D. pulex mtDNA reference sequence and nuclear genome reference assembly [28], we calculated the ratio of average depth of coverage per site of mtDNA to that for single-copy nuclear gene for each sperm sample. The mean value of this ratio is 19 (s.d. = 8), indicating that each sperm on average contains 19 copies of mtDNA.
The likely overestimation of mtDNA copy number should not have an effect on the estimated mutation rate and spectrum in this study. However, because of the uncertainty in mtDNA copy number, it is extremely difficult to detect low-frequency (e.g. 0.001–0.01) heteroplasmic mutations, whose genomic signature could be similar to PCR errors. This is verified by our computer simulation of PCR errors incorporating a wide range of mtDNA copy numbers (see below).
(c). Mutation rate and spectrum
We used a stringent mutation detection pipeline based on sequence read depth to detect and quantify heteroplasmic mtDNA mutations in the Daphnia sperm samples. A total of 19 heteroplasmic single-base substitutions were detected with a mutant allele frequency (MAF) ranging from 0.14 to 0.4. No indels (insertion/deletion) were detected. Based on the number of heteroplasmic mutations, the base-substitution mutation rate in Daphnia is 3.32 × 10−6 (s.e. = 1.77 × 10−8) per site per generation.
Among the base-substitution events, 16 are transition events, whereas only 3 are transversion events. We examined the effects of these heteroplasmic mutations, and found 7 synonymous substitutions, 10 non-synonymous substitutions and 2 non-coding changes. We note that 2 non-synonymous substitutions generated new stop codons in the genes NDII and COIII (table 1).
Table 1.
Summary of identified Daphnia mtDNA mutations. Total depth represents the number of sequence coverage at a site. syn: synonymous substitution. non-syn: non-synonymous substitution.
| position | sample | reference | mutation | total depth | mutation frequency | gene | effect | amino acid changes |
|---|---|---|---|---|---|---|---|---|
| 839 | 57 | C | T | 28 | 0.357 | ND2 | syn | AAC(N) → AAU (N) |
| 997 | 19 | T | A | 45 | 0.400 | ND2 | non-syn | UUA (L) → UAA (STOP) |
| 1775 | 26 | C | T | 77 | 0.247 | COI | syn | ACC(T) → ACU (T) |
| 3594 | 37 | C | A | 140 | 0.164 | COII | syn | UCC(S) → UCA (S) |
| 3898 | 46 | C | T | 120 | 0.275 | ATPase8 | syn | UUC(F) → UUU (F) |
| 5090 | 47 | G | A | 133 | 0.321 | COIII | non-syn | UGG (W) → UGA (STOP) |
| 7482 | 52 | G | A | 109 | 0.288 | ND5 | non-syn | GCU (A) → GUU (V) |
| 8317 | 49 | A | G | 69 | 0.145 | ND4 | syn | ACU(T) → ACC (T) |
| 8709 | 90 | C | T | 49 | 0.551 | ND4 | non-syn | GCA (A) → ACA (T) |
| 8734 | 7 | A | G | 76 | 0.211 | ND4 | syn | CCU (P) → CCC (P) |
| 9072 | 51 | C | T | 58 | 0.254 | ND4 | non-syn | GUG (V) → AUG (Start) |
| 9141 | 33 | T | C | 66 | 0.182 | ND4 | non-syn | AUG (M) → GUG (V) |
| 10 087 | 40 | T | C | 85 | 0.235 | ND6 | non-syn | UCC (S) → CCC (P) |
| 10 217 | 96 | T | C | 76 | 0.171 | ND6 | non-syn | CUU (L) → CCU (P) |
| 10 625 | 77 | T | A | 61 | 0.442 | CytB | non-syn | UUA (L) → AUA (I) |
| 11 775 | 60 | T | C | 121 | 0.147 | ND1 | non-syn | AGU (S) → GGU (G) |
| 11 920 | 87 | A | G | 76 | 0.145 | ND1 | syn | UUU (F) → UUC (F) |
| 12 801 | 55 | T | C | 88 | 0.159 | rRNA-Large | non-coding | n.a. |
| 13 533 | 87 | C | T | 94 | 0.191 | rRNA-Large | non-coding | n.a. |
(d). Mutation spectrum
The majority of Daphnia male (57%) mitochondrial mutations are towards A/T nucleotide (figure 1), which is consistent with the A/T-biased mutation in Drosophila melanogaster [16,30]. There is also substantial proportion of mutations are towards G/C nucleotide, with T : A → C : G mutations constituting 42% of the mutations in Daphnia males (figure 1).
Figure 1.
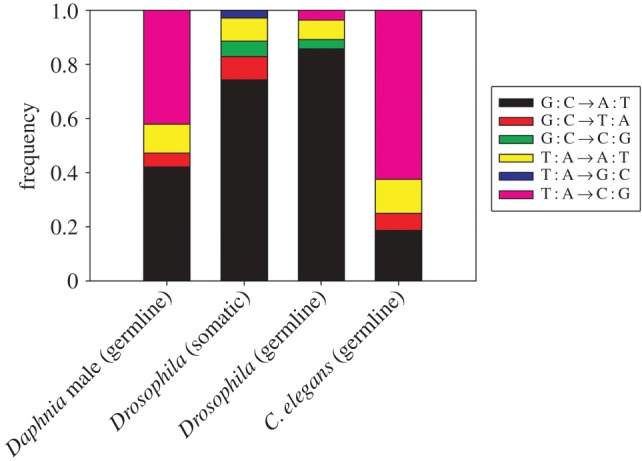
Mitochondrial mutation spectra of Daphnia pulex male (present study), Drosophila melanogaster somatic tissue [30], D. melanogaster mitochondrial mutation-accumulation experiment [16] and Caenorhabditis elegans mitochondrial mutation-accumulation experiment [18].
(e). PCR errors and mutation detection
A primary concern with detecting mutations from whole-genome sequencing of single cells that have undergone whole-genome amplification is whether the mutation rate is inflated by false positives that result from PCR amplification errors. PCR errors that arise early in genome amplification can be driven to high frequencies after repeated rounds of replication. The MALBAC procedure employs quasi-linear amplification instead of exponential amplification in the first few rounds of amplification to ensure that the frequency of PCR errors in the final pool of DNA molecules that are subject to short-read sequencing in this study ranges between 0.001 and 0.01 (see below). Nevertheless, to verify whether PCR errors from MALBAC whole-genome amplification could be mistakenly identified as mutations, we simulated whole-genome amplification using various parameters, including a wide range of PCR error rates (1 × 10−5 to 1 × 10−3) and mtDNA copy number (e.g. 2, 10, 50, 100, 500), and found that the final error rate is between 0.001 and 0.01. Similarly, the average base-substitution error rate per site estimated using all sperm samples (excluding mutant and indel sites) is 0.0065 (s.d. = 0.0065). Given that the frequency of PCR errors in the final pool of amplicons is between 0.001 and 0.01, the probability for PCR errors to pass our filtering procedure is orders of magnitude lower (10−70–10−9) than the estimated mutation rate in both species (approx. 10−6). Thus, amplification errors are unlikely to contribute substantially to the overall mtDNA mutation rate.
4. Discussion
In this study, we analysed the mtDNA sequences derived from single-sperm whole-genome sequencing to examine the rate and spectrum of mtDNA mutations in the male germline of the microcrustacean Daphnia pulex. To the best of our knowledge, this is the first estimation of male mtDNA mutation rate in eukaryotic species.
The Daphnia male mtDNA mutation rate estimated from single sperm is 3.32 × 10−6 per site per generation. This estimate is at least an order of magnitude higher than mtDNA mutation rates derived from mutation-accumulation experiments for other species such as D. melanogaster (7.8 × 10−8 per site per generation) and C. elegans (1.6 × 10−7 per site per generation). The Daphnia male mtDNA mutation rate is approximately 20 times higher than the Daphnia female mtDNA mutation rate estimated in a prior mutation-accumulation experiment (approx. 1.5 × 10−7 per site per generation). Because all the detected mutations are base substitutions, the base-substitution rate in male Daphnia mtDNA is approximately 100-fold higher than that in female Daphnia mtDNA (approx. 3 × 10−8 per site per generation), which is much higher than the 20-fold overall mutation rate difference between sexes. Although the mutation rate per cell division may be identical, male germline cells undergo additional rounds of mtDNA amplification when compared with their female counterparts, thus leading to a higher mutation rate per generation. It should be noted that the difference in Daphnia male and female mtDNA mutation rates are likely to be even greater, as estimates from the mutation-accumulation experiment could be downwardly biased because only effectively neutral or slightly deleterious mutations can persist in long-term mutation-accumulation experiments [31]. While the sampled sperm cells in this study are subject to selection pressures that ensure sperm viability, they are not exposed to any selection pressures that may arise at fertilization and development, providing an unbiased estimate of the male mtDNA mutation rate and spectrum. This view is supported by the detection of mutations causing stop codons in the functionally important NDII and COIII genes.
A main concern with detecting mutations from single cells that have undergone whole-genome amplification is whether mutation rate is inflated by false positives that result from amplification errors. In general, an amplification error at a single nucleotide site in the very first few amplification cycles of a PCR reaction could be exponentially propagated in subsequent cycles. In a simple scenario where only a double-stranded DNA molecule is the original template for whole-genome amplification, a replication error at a site in the first cycle would be one of the four templates for the next round of amplification and would constitute 25% of the final pool of sequence reads for this site. Technically speaking, tagging single DNA molecule with unique molecular identifiers (UMIs) can help identify PCR errors during amplification [32]. Although not employed by the current study, UMIs should be considered for future single-cell mutation studies.
Nonetheless, two factors in our experiment guard our results against false positives due to polymerase errors. First, each sperm probably contains multiple mitochondria, with each mitochondrion possibly containing multiple copies of mtDNA. Our estimates show that each Daphnia sperm on average contains approximately 19 copy of mtDNA. Assuming a polymerase error rate (ɛ) on the order of 10−4–10−3 per base, occurrence of polymerase error at the same site on multiple copies of mtDNA (n) in one round of amplification is an extremely small possibility (ɛn). Because of the dilution effect of multiple amplicons, amplification errors are unlikely to produce the sequencing read profile that would pass our criteria to identify mutations. Second, the MALBAC whole-genome amplification technique achieves quasi-linear amplification with five pre-amplification cycles [27]. During these pre-amplification cycles, newly formed amplicons do not engage in subsequent pre-amplification cycles, which limits the amplification of early PCR errors. In essence, the pre-amplification cycles reduce the frequency of newly arisen errors in the amplicons because the possibility for DNA polymerase to make the same mistake in multiple rounds of amplification is very small (see above). Our computer simulation of MALBAC whole-genome amplification confirms this logic and shows that amplification errors only constitute about 0.001–0.01 per site in the final pool of molecules subject to short-read sequencing, which is also validated by our empirical estimate of per-site error rate at non-mutation sites across the samples (0.0065 per site). The likelihood of amplification errors passing our mutation detection filters is extremely small (see Results) under a binomial sampling scheme [33].
Mitochondrial mutation spectrum varies greatly across eukaryotic species [34]. The A/T-biased mutation spectrum in Daphnia male is consistent with that of Drosophila melanogaster somatic and germline mitochondrial mutations (figure 1) [16,30]. Moreover, transitions (G : C → A : T and T : A → C : G) dominate the mutation spectrum in Daphnia male (figure 1), which is consistent with the dominance of transitions in Drosophila (figure 1), Caenorhabditis elegans [18] and Saccharomyces cerevisiae [19]. A potential source of false-positive mutations that could bias the mutation spectrum detected from the single-cell genomic dataset is DNA damage occurring during cell lysis. During single-cell genomic lysis, high levels of cytosine deamination may lead to a substantial increase of C-to-T mutations in the mutation spectrum of nuclear DNA [35], leading to the elevated mutation rate observed in male mtDNA. However, we argue that this factor is unlikely to create false positives in the current dataset. Since multiple copies of mtDNA exist (approx. 19 in Daphnia) in a single sperm cell and DNA damage during lysis only affects a single strand of a DNA molecule [36], the frequency of false positive mutations should be around 3% (1/30) in the Daphnia male mtDNA, far below the threshold used to detect mutations detected in our dataset.
The substantially elevated mutation rate in Daphnia male mtDNA with respect to the female mtDNA is an intriguing observation that merits further discussion. As discussed earlier, there are a few factors that could collectively increase the male mtDNA mutation rate relative to that of females. First of all, mitochondria contain large amounts of reactive oxidative species (ROS) as a result of ATP production. Considering the substantially higher level of mtDNA transcription, electron transport and free radical production that occur in the testis, and not in ovary tissue, the excessive amount of reactive oxidative species (ROS) in male gametogenesis could be a potential source of elevated mutation rate in male mtDNA. The most common signature for DNA damage caused by ROS is the oxidation of guanine to 8-oxoguanine [37], which allows G-to-A mispairings that generate a high frequency of G : C → T : A transversions. We examined the types of mutations that existed in a heteroplasmic state in the Daphnia dataset and only found a single case of G : C → T : A transversions in each dataset, suggesting that the oxidative condition in the male environment is not a large factor in driving the elevation of the male-specific mtDNA mutation rate. This finding is consistent with the observation of a relatively low rate of G : C → T : A mutations in somatic mitochondrial DNA of Drosophila melanogaster when oxidative damage repair enzyme (Ogg1) was removed [30]. Together, these data support the idea that the environmental conditions in the testis do not drive the elevation of mitochondrial mutation rate in the male germline.
Alternatively, the number of germline cell division and mtDNA replications could be a driving force for the elevated male mutation rate. It is important to keep in mind that while germline cell division certainly involves mitochondria replication and mtDNA replication, these replications can be independent of germline cell division [38,39]. Unfortunately, little is known about how many rounds of independent replication mitochondria go through in the Daphnia male germline.
The high depth coverage of sequencing of the Daphnia sperm mtDNA allows an unbiased estimate of the mtDNA mutation rate in the male germline. However, no sex-specific germline cell division numbers are available for Daphnia. The 20-fold higher mutation rate in male mtDNA compared with female mtDNA probably results from the joint effect of the different number of germline cell divisions and independent mtDNA replication. Furthermore, given that the mtDNA mutation rate in somatic tissue of Drosophila appears to increase with age [30], it is expected that the male Daphnia mtDNA mutation rate is substantially higher than the female mtDNA at any age.
The third (and also the least tangible) explanation for the male-biased mutation rate lies in the fact that mitochondria are maternally inherited, so that any mutations that arise in the male germline mtDNA are not under selection in the offspring. As a result, natural selection on the enzymes that maintain high-replication fidelity in mtDNA may be relaxed in the male-specific environment. It has been argued that ovary tissue has suppressed mtDNA transcription, electron transport and free radical production when compared with testis tissue and somatic tissue in both sexes [20]. Thus, if the suppressed mitochondrial metabolic environment in the ovary is a major selective force on the maintenance of polymerase fidelity, we would expect a substantially lower fidelity of mtDNA replication in testis and somatic tissues. If this hypothesis holds true, it would be an inevitable outcome that sperm and somatic cells will be subject to an increased influx of deleterious mutations. In fact, some of the detected mutations in the current study certainly have strong deleterious effects, such as creating new stop codons in important genes (i.e. NDII and COIII) within the male Daphnia mtDNA. However, it should be noted that all the mutations in the present dataset are heteroplasmic, thus inflicting deleterious effects on only a fraction of the mitochondria in a sperm cell.
In summary, we find an elevated rate of mutation in the mitochondrial genome of sperm in Daphnia, consistent with a general pattern of male-biased mutation rate in the nuclear genome. We find little evidence that the male-specific mitochondrial inner environment contributes to the male mtDNA mutation spectrum, suggesting that other factors are responsible for a male-biased mutation rate. Although the mutation rate per round of mitochondrial replication may not differ between males and females, male mtDNA may undergo many more rounds of mtDNA replication per generation when compared with their female counterparts, which is probably a key factor in the male-biased mtDNA mutation rate.
Supplementary Material
Acknowledgements
We thank Drs H. Long, W. Hao and Mavrine Neiman, and two anonymous reviewers, for constructive comments on an early draft of this manuscript.
Data accessibility
NCBI Sequence Read Archive (SRA) study no. SRP058678.
Authors' contributions
S.X. conceived the study, performed data analyses and wrote the manuscript. K.V.T. and S.N. performed genomic data analyses and mutation annotation. M.S. and T.V.H. performed computer simulation and analyses. W.S. conceived the study, performed data analyses and wrote the manuscript. All authors gave final approval for publication.
Competing interests
We have no competing interests.
Funding
This work is supported by start-up funds from University of Texas at Arlington to S.X. and start-up funds from the University of North Carolina at Charlotte to W.S. The funding for sequencing single Daphnia sperm was provided to Michael Lynch at Indiana University by NIH grant R01GM101672.
References
- 1.Ellegren H, Fridolfsson AK. 1997. Male-driven evolution of DNA sequences in birds. Nat. Genet. 17, 182–184. ( 10.1038/Ng1097-182) [DOI] [PubMed] [Google Scholar]
- 2.Hurst LD, Ellegren H. 1998. Sex biases in the mutation rate. Trends Genet. 14, 446–452. ( 10.1016/S0168-9525(98)01577-7) [DOI] [PubMed] [Google Scholar]
- 3.Li WH, Yi SJ, Makova K. 2002. Male-driven evolution. Curr. Opin. Genet. Dev. 12, 650–656. ( 10.1016/S0959-437x(02)00354-4) [DOI] [PubMed] [Google Scholar]
- 4.Makova KD, Li WH. 2002. Strong male-driven evolution of DNA sequences in humans and apes. Nature 416, 624–626. ( 10.1038/416624a) [DOI] [PubMed] [Google Scholar]
- 5.Miyata T, Hayashida H, Kuma K, Mitsuyasu K, Yasunaga T. 1987. Male-driven molecular evolution—a model and nucleotide-sequence analysis. Cold Spring Harb. Symp. Quant. Biol. 52, 863–867. ( 10.1101/SQB.1987.052.01.094) [DOI] [PubMed] [Google Scholar]
- 6.Sayres MAW, Makova KD. 2011. Genome analyses substantiate male mutation bias in many species. BioEssays 33, 938–945. ( 10.1002/bies.201100091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sayres MAW, Venditti C, Pagel M, Makova KD. 2011. Do variations in substitution rates and male mutation bias correlate with life-history traits? A study of 32 mammalian genomes. Evolution 65, 2800–2815. ( 10.1111/j.1558-5646.2011.01337.x) [DOI] [PubMed] [Google Scholar]
- 8.Ellegren H. 2007. Characteristics, causes and evolutionary consequences of male-biased mutation. Proc. R. Soc. B 274, 1–10. ( 10.1098/rspb.2006.3720) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kong A, et al. 2012. Rate of de novo mutations and the importance of father's age to disease risk. Nature 488, 471–475. ( 10.1038/nature11396). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ballard JWO, Rand DM. 2005. The population biology of mitochondrial DNA and its phylogenetic implications. Annu. Rev. Ecol. Evol. Syst. 36, 621–642. ( 10.1146/annurev.ecolsys.36.091704.175513). [DOI] [Google Scholar]
- 11.Avise JC. 2004. Molecular markers, natural history, and evolution, 2nd edn Sunderland, MA: Sinauer Associates. [Google Scholar]
- 12.Frank SA, Hurst LD. 1996. Mitochondria and male disease. Nature 383, 224 ( 10.1038/383224a0) [DOI] [PubMed] [Google Scholar]
- 13.Beekman M, Dowling DK, Aanen DK. 2014. The costs of being male: are there sex-specific effects of uniparental mitochondrial inheritance? Phil. Trans. R. Soc. B 369, 20130440 ( 10.1098/rstb.2013.0440) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birky CW., Jr 1995. Uniparental inheritance of mitochondrial and chloroplast genes: mechanisms and evolution. Proc. Natl Acad. Sci. USA 92, 11 331–11 338. ( 10.1073/pnas.92.25.11331) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ankel-Simons F, Cummins JM. 1996. Misconceptions about mitochondria and mammalian fertilization: implications for theories on human evolution. Proc. Natl Acad. Sci. USA 93, 13 859–13 863. ( 10.1073/pnas.93.24.13859) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haag-Liautard C, Coffey N, Houle D, Lynch M, Charlesworth B, Keightley PD. 2008. Direct estimation of the mitochondrial DNA mutation rate in Drosophila melanogaster. PLoS Biol. 6, 1706–1714. ( 10.1371/journal.pbio.0060204) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denver DR, Morris K, Lynch M, Thomas WK. 2004. High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature 430, 679–682. ( 10.1038/nature02697) [DOI] [PubMed] [Google Scholar]
- 18.Denver DR, Morris K, Lynch M, Vassilieva LL, Thomas WK. 2000. High direct estimate of the mutation rate in the mitochondrial genome of Caenorhabditis elegans. Science 289, 2342–2344. ( 10.1126/science.289.5488.2342) [DOI] [PubMed] [Google Scholar]
- 19.Lynch M, et al. 2008. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc. Natl Acad. Sci. USA 105, 9272–9277. ( 10.1073/pnas.0803466105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Paula WBM, Agip ANA, Missirlis F, Ashworth R, Vizcay-Barrena G, Lucas CH, Allen JF. 2013. Female and male gamete mitochondria are distinct and complementary in transcription, structure, and genome function. Genome Biol. Evol. 5, 1969–1977. ( 10.1093/gbe/evt147) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kunkel TA. 2009. Evolving views of DNA replication (in)fidelity. Cold Spring Harb. Symp. Quant. Biol. 74, 91–101. ( 10.1101/sqb.2009.74.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hudson G, Chinnery PF. 2006. Mitochondrial DNA polymerase-gamma and human disease. Hum. Mol. Genet. 15, R244-R252. ( 10.1093/hmg/ddl233) [DOI] [PubMed] [Google Scholar]
- 23.Kaguni LS. 2004. DNA polymerase gamma, the mitochondrial replicase. Annu. Rev. Biochem. 73, 293–320. ( 10.1146/annurev.biochem.72.121801.161455) [DOI] [PubMed] [Google Scholar]
- 24.Kunkel TA, Soni A. 1988. Exonucleolytic proofreading enhances the fidelity of DNA synthesis by chick embryo DNA polymerase-gamma. J. Biol. Chem. 263, 4450–4459. [PubMed] [Google Scholar]
- 25.Xu S, Schaack S, Seyfert A, Choi E, Lynch M, Cristescu ME. 2012. High mutation rates in the mitochondrial genomes of Daphnia pulex. Mol. Biol. Evol. 29, 763–769. ( 10.1093/molbev/msr243) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu S, Ackerman MS, Long H, Bright L, Spitze K, Ramsdell JS, Thomas WK, Lynch M. 2015. A male-specific genetic map of the microcrustacean Daphnia pulex based on single-sperm whole-genome sequencing. Genetics 201, 31–38. ( 10.1534/genetics.115.179028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zong C, Lu S, Chapman AR, Xie XS. 2012. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 338, 1622–1626. ( 10.1126/science.1229164) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye Z, et al. 2017. A new reference genome assembly for the microcrustacean Daphnia pulex. G3: Genes|Genomes|Genetics 7, 1405–1416. ( 10.1534/g3.116.038638) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crease TJ. 1999. The complete sequence of the mitochondrial genome of Daphnia pulex (Cladocera: Crustacea). Gene 233, 89–99. ( 10.1016/S0378-1119(99)00151-1) [DOI] [PubMed] [Google Scholar]
- 30.Itsara LS, Kennedy SR, Fox EJ, Yu S, Hewitt JJ, Sanchez-Contreras M, Cardozo-Pelaez F, Pallanck LJ. 2014. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet. 10, e1003974 ( 10.1371/journal.pgen.1003974) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keightley PD, Caballero A. 1997. Genomic mutation rates for lifetime reproductive output and lifespan in Caenorhabditis elegans. Proc. Natl Acad. Sci. USA 94, 3823–3827. ( 10.1073/pnas.94.8.3823) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, Lonnerberg P, Linnarsson S. 2014. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 11, 163–166. ( 10.1038/Nmeth.2772) [DOI] [PubMed] [Google Scholar]
- 33.Lynch M. 2008. Estimation of nucleotide diversity, disequilibrium coefficients, and mutation rates from high-coverage genome sequencing projects. Mol. Biol. Evol. 25, 2409–2419. ( 10.1093/molbev/msn185) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montooth KL, Rand DM. 2008. The spectrum of mitochondrial mutation differs across species. PLoS Biol. 6, 1634–1637. ( 10.1371/journal.pbio.0060213) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen C, Xing D, Tan L, Li H, Zhou G, Huang L, Xie XS. 2017. Single-cell whole-genome analyses by linear amplification via transposon insertion (LIANTI). Science 356, 189–194. ( 10.1126/science.aak9787) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hofreiter M, Jaenicke V, Serre D, von Haeseler A, Paabo S. 2001. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 29, 4793–4799. ( 10.1093/nar/29.23.4793) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Bont R, van Larebeke N. 2004. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 19, 169–185. ( 10.1093/mutage/geh025) [DOI] [PubMed] [Google Scholar]
- 38.Bogenhagen D, Clayton DA. 1977. Mouse L cell mitochondrial DNA molecules are selected randomly for replication throughout cell-cycle. Cell 11, 719–727. ( 10.1016/0092-8674(77)90286-0) [DOI] [PubMed] [Google Scholar]
- 39.Birky CW. 1994. Relaxed and stringent genomes—why cytoplasmic genes don't obey Mendel's laws. J. Hered. 85, 355–365. ( 10.1093/oxfordjournals.jhered.a111480) [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
NCBI Sequence Read Archive (SRA) study no. SRP058678.