

Abstract
The revolution in individualized therapy for patients with advanced NSCLC has seen the emergence of a number of molecularly targeted therapies for distinct patient molecular subgroups. Activating anaplastic lymphoma kinase (ALK)-gene rearrangement has been detected in 3%–7% of NSCLC cases, and the ALK inhibitor crizotinib is now an approved treatment for patients with tumors harboring this event. However, resistance to ALK-targeted therapies is a ubiquitous problem in the management of advanced ALK-positive NSCLC, and can be mediated by secondary kinase mutations or the activation of compensatory alternative oncogenic drivers. New, more potent ALK inhibitors such as ceritinib (LDK378), alectinib (CH5424802), and AP26113, are now emerging, together with an increased knowledge of the molecular basis of resistance. There is therefore a need to evaluate the optimal clinical application of these new agents, either as sequential therapies, and/or in combination with other targeted agents, to combat resistance and prolong survival in patients with ALK-positive NSCLC. The remarkable clinical activity of ALK inhibitors also emphasizes the importance of optimal diagnostic testing algorithms, to ensure that all eligible patients receive these breakthrough therapies.
Keywords: Non-small cell lung cancer, anaplastic lymphoma kinase, ALK inhibitors, diagnostics, resistance
Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide, with a dismal 5-year overall survival of approximately 18% [1]. Non-small cell lung cancer (NSCLC) accounts for 85% of cases, and >70% are diagnosed with advanced disease [2]. The 21st century has witnessed a revolution in treatment for advanced NSCLC from a one-size-fits-all to a personalized approach. Traditionally, histologic subtypes have dictated the choice of chemotherapy, but now key oncogenic driver mutations are known, and there are increasing data regarding genetic alterations allowing adenocarcinomas to be further classified into clinically relevant molecular subtypes which predict response to novel agents. The first clinically relevant molecular alterations to be characterized were epidermal growth factor receptor (EGFR) mutations that respond to tyrosine kinase inhibitors (TKIs). Recently, the discovery of translocations, involving the anaplastic lymphoma kinase (ALK) gene has driven the investigation of novel treatment options for the 3%–7% of patients with NSCLC whose tumors harbor this event [3]. Crizotinib, the first-in-class small molecule ALK inhibitor, gained US Food and Drug Administration (FDA) approval for the treatment of ALK-rearranged (ALK-positive) NSCLC in 2011. In this review, we discuss the discovery of and testing for ALK rearrangements in NSCLC, and review data on crizotinib, upcoming agents, and trials for this patient population.
Discovery of ALK fusion genes
Oncogenic addiction is the phenomenon whereby tumor cells depend on an oncogene for survival and proliferation, making them attractive therapeutic targets [4]. The rearrangement of the ALK gene represents such a dependency in NSCLC. First reported as a fusion gene in a small proportion of anaplastic large cell lymphomas (ALCLs) [5], ALK rearrangement was subsequently discovered in NSCLC [6, 7], mostly in adenocarcinomas [8-11]. The most common alteration involves inversion on chromosome 2, leading to fusion of the protein encoded by the echinoderm microtubule–associated protein-like 4 (EML4) gene with the intracellular portion of the receptor tyrosine kinase encoded by the ALK gene [6, 12]. This EML4–ALK fusion protein constitutively activates a number of signaling cascades (Figure 1) [13]. These pathways promote initiation, progression, and survival of NSCLC [13, 14]. Four other ALK fusion proteins are also associated with NSCLC [15-18]. Several studies suggest that ALK rearrangements are largely independent of EGFR and KRAS mutations [19-24].
Figure 1.
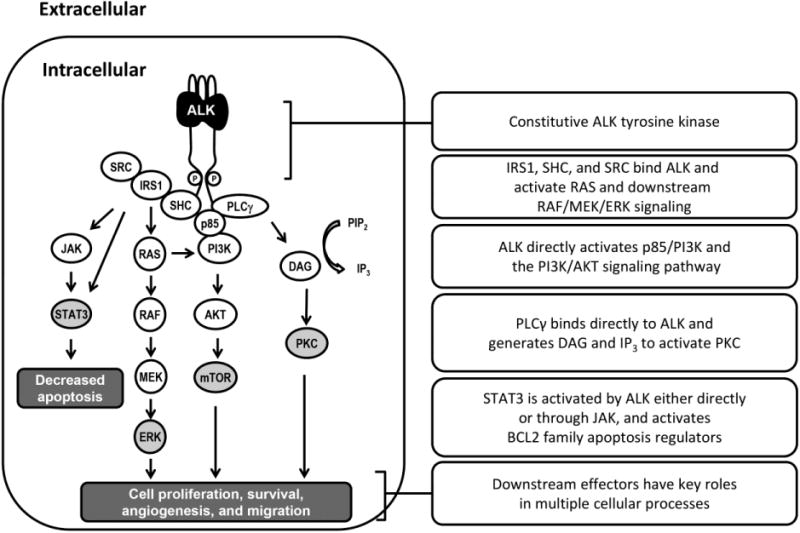
Signaling cascades activated by the EML4-ALK fusion protein.
Optimal screening strategy
A subset of patients with NSCLC may possess clinicopathologic features that predict ALK-positivity. Most studies quoting a higher incidence of ALK-positivity involved patients who were light/never smokers (chance of carrying mutation 20% vs. 2% in smokers), were younger in age (median age 54 vs. 64 years for the ALK-negative [ALK wild-type] population), had acinar/signet ring histology, and had transcription termination factor 1 (TTF-1)-positive histology [8, 10, 25-29]. Furthermore, if we understand EGFR and ALK alterations to be mutually exclusive, the presence of an activating EGFR mutation or response to EGFR TKIs may predict for ALK-negative status[30]. However, ALK rearrangements are not entirely restricted to non-smokers or certain age groups. In the absence of strong data suggesting predictive factors, current National Comprehensive Cancer Network (NCCN) guidelines suggest screening all patients with advanced non-squamous NSCLC and patients with squamous disease if they are never smokers or were diagnosed based on small biopsy specimens [31]. The remarkable clinical activity of ALK inhibitors emphasizes the importance of testing for these mutations and ensuring that eligible patients receive appropriate targeted therapy.
Optimal testing modality
The discovery of ALK rearrangement and its potential as a therapeutic target triggered the co-development of diagnostic assays. The current FDA-approved break-apart fluorescence in situ hybridization (FISH) assay (AbbVie, Inc.), was clinically validated in Phase I/II trials involving crizotinib [10, 32]. The cut-off point for a positive result is >15% of tumor cells positive in ≥50 cell nuclei [10, 21]. The test can be performed on formalin-fixed paraffin-embedded specimens, and detects novel ALK fusion genes by targeting the tyrosine kinase domain of ALK, independent of the fusion partner [33]. Disadvantages include the need for specialized expertise to both perform the test and interpret the results, a risk of false negatives due to subtle splitting of colored signals, and associated costs [30, 33].
Other screening methods being evaluated include reverse transcription polymerase chain reaction (RT-PCR) assays and immunohistochemistry (IHC). Sanders et al. used multiplexed RT-PCR to detect 5 known EML–ALK variants, identified in 9% of specimens [34]. RT-PCR is highly sensitive and specific but requires high-quality RNA (unobtainable from many archived samples), and only detects known fusion variants, with the consequent potential of false negative results (in the setting of novel fusion genes), and lacks clinical validation.
ALK-directed IHC is an attractive alternative to FISH and may soon become an established diagnostic algorithm. IHC is quick, affordable, can be performed on a variety of tumor specimens, and also facilitates histologic comparison. Currently, the low degree of ALK expression in NSCLC makes the use of this technique challenging. More sensitive techniques using ALK monoclonal antibodies are being investigated. Yi et al. correlated IHC with FISH using the ALK1 antibody, and found >90% sensitivity and specificity when 2/3+ scores were considered IHC positive [35]. However, the poor transcriptional activity of EML–ALK in NSCLC leads to low staining intensity, and may impact the reliability of this assay [17, 36]. Results obtained using 5A4 and D5F3 antibodies have been more encouraging, and studies have suggested a sensitivity and specificity of 95%-100% for IHC using the 5A4 antibody [37-39]. One study supported a scoring algorithm in which ALK IHC scores of 0, 1, and 3+ were highly compatible with FISH results, while a score of 2+ was variable [37]. In another study, correlation between an IHC score of 0 with negative FISH status and between an IHC score of 1+ with positive FISH status were observed [39]. Both of these studies propose a two-tier system for evaluating ALK with an initial IHC screening followed by FISH assay for IHC 1+ and/or 2+ specimens. The D5F3 monoclonal antibody provided a sensitivity and specificity of 100% and 99%, respectively, using ALK FISH as gold standard [36]. The lack of clinical validation of these techniques prevents their widespread approval; nonetheless, an automated IHC companion diagnostic ALK assay (Ventana Medical Systems, Inc.) has now been launched in Europe [40]. The break-apart FISH assay remains the US standard and recommended testing strategy.
Treatment of ALK-positive lung cancer
The majority of ongoing trials involving ALK-positive patients are in the metastatic/advanced setting and this is therefore the focus of this review. Future trials will be needed in order to evaluate ALK inhibitors and other novel agents for early stage lung cancer treatment.
Chemotherapy
Retrospective analyses indicate that ALK status does not predict chemotherapy response [8, 27, 29]. Patients with ALK-positive NSCLC do not benefit from EGFR TKI therapy [29, 41, 42], and this decreased responsiveness highlights the mutual exclusivity of ALK rearrangements and EGFR mutations. Gandara et al. evaluated the expression of thymidylate synthase (TS) in 63 patients with ALK-positive lung adenocarcinoma and 1698 patients with ALK-negative disease. TS gene expression was low in ALK-positive tumors compared with ALK-negative tumors, supporting a rationale for pemetrexed therapy for ALK-positive NSCLC [43]. Retrospective analyses have evaluated the differential activity of pemetrexed in patients with ALK-positive NSCLC [44, 45]. In one study, multivariate analysis, adjusting for age, sex,smoking status, histology, line and type of therapy, ALK-positivity was associated with prolonged PFS on pemetrexed (hazard ratio 0.36) [45]. In contrast, a large multicenter, retrospective analysis did not support these findings, and the median PFS of ALK-positive patients treated with single-agent pemetrexed or non-platinum/pemetrexed combination was similar to that of ALK-negative patients. In the same series, among patients undergoing first-line platinum/pemetrexed therapy, the median PFS in patients with ALK-positive tumors was 7.3 months compared with 5.4 months for wild-type tumors. However, patients who were never/light smokers had a similar PFS to the ALK-positive group [46].The retrospective nature of the analysis, and the finding of improved sensitivity to chemotherapy among non-smoking patients [47], make interpretation of the data difficult.
ALK-targeted therapy
Preclinical studies have shown that ALK fusion gene products are oncogenic drivers of transformation, and ALK has therefore been extensively explored as a therapeutic target. Clinical investigation of crizotinib began as a c-Met inhibitor in patients with various malignancies. The subsequent discovery of ALK gene rearrangement in NSCLC, and promising results in patients with NSCLC, led to the addition of an expansion cohort to include this population, in which a response rate of 61% was seen, with a median PFS of 9.7 months [10, 48]. The singe-arm Phase II study PROFILE 1005 showed a comparable overall response rate (ORR) of 60% and median PFS of 8.1 months [32].A Phase III trial, PROFILE 1007, comparing crizotinib with standard chemotherapy in the second-line setting resulted in an improved ORR (65% vs. 20%), a shorter time to response (6.3 vs. 12.6 weeks), and an improved median PFS (7.7 vs. 3.0 months) with crizotinib. Overall survival benefit was not demonstrated on interim analysis, and this was likely related to crossover (64% of patients on chemotherapy crossed over to crizotinib after progression) [49]. In all these studies, toxicities were acceptable, with some visual disturbances, gastrointestinal side effects, fatigue, and edema. The PROFILE 1014 study is designed to answer the question of the superiority of crizotinib over front-line platinum/pemetrexed combination chemotherapy (NCT01154140). Despite the absence of mature randomized data, the NCCN panel recommends crizotinib in a front-line setting in advanced ALK-positive NSCLC [31]. However, in clinical practice, systemic chemotherapy may be started before genotyping results are available. Berge et al. reported that PFS benefit from crizotinib appears higher than with pemetrexed in patients with advanced ALK-positive NSCLC [50]. Pemetrexed exposure did not affect outcome with crizotinib; however, PFS benefit from pemetrexed was less after crizotinib use (4.5 months) compared with before crizotinib use (6 months).
Progression on crizotinib and emergence of resistance
Unfortunately, about 40% of ALK-positive patients show intrinsic resistance to crizotinib [10, 32, 48], while others derive dramatic responses initially but develop resistance within 12 months [51, 52]. Proposed mechanisms of acquired resistance include target gene alteration/amplification (≈30% of cases) and up-regulation of alternate cell-signaling pathways. One study describes four different mutations in the ALK tyrosine kinase (ALK-TK) domain that confer various degrees of resistance to crizotinib [51]. Most common is the L1196M amino acid substitution, similar to the gatekeeper mutations observed in EGFR (T790M) and BCR–ABL genes (T315I), originally identified as an independent mutation in a tumor from a patient with ALK-positive NSCLC [53]. Some other mutations, such as G1202R and S1206Y, are located close to the crizotinib-binding site on the ALK-TK domain and decrease the affinity of crizotinib for ALK, while the 1151T insertion may affect the affinity of ALK for ATP, conferring strong crizotinib resistance [51]. Additional studies have also identified novel mutations in the ALK-TK domain that predict for crizotinib resistance [52, 54]. Other mechanisms implicated in resistance include target gene amplification, with increase in ALK gene rearrangement copy numbers without a documented mutation [52] and up-regulation of alternate pathways including EGFR activation, and c-KIT amplification [51]. A recent study also suggests EGF-mediated HER family activation as a mechanism of ALK-TKI resistance [55]. There may be diverse and multiple mechanisms involved in resistance even within an individual patient, and these factors have emerged as major roadblocks in the transformative clinical impact of the ALK inhibitors.
Therapeutic advances in the setting of resistance
The identification of resistance mechanisms provides groundwork for the development of new ALK inhibitors to combat crizotinib resistance, including the development of combination therapies to attack bypass track pathways.
Novel ALK inhibitors
Next-generation ALK inhibitors currently under clinical evaluation include ceritinib (LDK378; Novartis), AP26113 (ARIAD), alectinib (CH5424802/RO5424802; Chugai/Hoffmann-La Roche), and ASP3026 (Astellas). Other agents are in earlier stages of development (Table 1).
Table 1. Anaplastic lymphoma kinase inhibitors in clinical and preclinical development.
| Agent [references] | Targets (IC50) | Clinical trial phase/status | Common AEs | Response rate (%) | Median PFS (months) | Other targets |
|---|---|---|---|---|---|---|
| Crizotinib (Pfizer) [10, 32, 80] | WT-ALK (24 nM) | Approved | Visual disorders (59%), nausea (57%), vomiting (44%) | 60 (ALK+ NSCLC [n=259]) | 8.1 | c-MET, ROS-1 |
| Ceritinib (LDK378; Novartis)[57, 58, 80] | WT-ALK (0.15 nM) G1269S, F1245C |
Phase II/III | Nausea (82%), diarrhea (75%), vomiting (65%) | 58 (ALK+ NSCLC [n=114]) 62 (crizotinib-naïve ALK+ NSCLC [n=34]) 56 (crizotinib-refractory ALK+ NSCLC [n=80]) |
10.4a 6.9b | IGF-1R, ROS1 |
| Alectinib (CH5424802/RO54 24802; Chugai/Roche) [61, 62] |
WT-ALK (1.9 nM) L1196M, F1174L |
Phase I/II | Dysgeusia (30%), AST increased (28%), blood bilirubin increased (24%) | 93 (crizotinib-naïve ALK+ NSCLC [Japan; n=46]) 59 (crizotinib-refractory ALK+ NSCLC [n=45]) |
– | GAK, LTK |
| AP26113 (ARIAD) [66, 80] | WT-ALK (0.62 nM) L1196M, F1171T |
Phase I/II | Nausea (43%), fatigue (41%), diarrhea (35%) | 62 (ALK+ tumors [n=21]) 71 (crizotinib-refractory ALK+ NSCLC [n=14]) |
– | EGFR, ROS1 |
| TSR-011 (Tesaro) [81, 82] | WT-ALK (0.7 nM) L1196M, R1275Q |
Phase I/IIa | QTc prolongation (20%; DLT), fatigue/asthenia (10%) | 67 (ALK+ NSCLC [n=3]) | – | TRKA, TRKB, TRKC |
| ASP3026 (Astellas) [80] | WT-ALK L1196M |
Phase I | – | – | – | ROS1 |
| X-396 (Xcovery) [83] | WT-ALK (<0.4 nM) L1196M, C1156Y |
Phase I | – | – | – | – |
| EP-28122 (Teva/Cephalon) [80] | WT-ALK (1.9 nM) F1174L, R1275Q |
Preclinical | – | – | – | – |
| AZD3463 (AstraZeneca) [84] | WT-ALK (Ki 0.75 nM) L1196M, T115Ins |
Preclinical | – | – | – | IGF-1R |
Crizotinib-naive patients;
Crizotinib-refractory patients.
AE, adverse event; ALK, anaplastic lymphoma kinase; ALK+, ALK-positive; AST, aspartate transaminase; DLT, dose-limiting toxicity; EGFR, epidermal growth factor receptor; GAK, cyclin G-associated kinase; IGF-1R, insulin-like growth factor-1 receptor; LTK, leukocyte receptor tyrosine kinase; NSCLC, non-small cell lung cancer; PFS, progression-free survival; ROS1, c-ros oncogene 1; TRK, tropomycin receptor kinase; WT-ALK, wild-type ALK.
Ceritinib is an oral ALK inhibitor with 20-fold greater preclinical potency than crizotinib, and activity against crizotinib-resistant mutations [56]. Ceritinib shows marked antitumor activity against both crizotinib-sensitive and crizotinib-resistant ALK-rearranged xenograft tumors [57]. An ongoing Phase I trial includes 130 patients with advanced cancers harboring genetic alterations in ALK [58]. Preliminary results have shown that in 114 patients with ALK-positive NSCLC treated with ceritinib ≥400 mg/day, the ORR and median PFS were 58% and 7.0 months, respectively. Significant clinical benefit was noted even in the crizotinib-pretreated group (n=80), including an ORR of 56%. Ceritinib was tolerated up to the maximum tolerated dose of 750 mg/day with primarily gastrointestinal side effects such as nausea, diarrhea, and vomiting (Table 1). Based on the encouraging results observed with this agent, the FDA granted it Breakthrough Therapy designation for the treatment of patients with ALK-positive metastatic NSCLC who have progressed on or are intolerant to crizotinib [59]. This status is intended to help expedite the drug's development and review, with Phase II trials currently underway [60]. Two Phase III trials comparing ceritinib with single-agent chemotherapy after progression on a platinum-based doublet and crizotinib (NCT01828112), and with a pemetrexed-platinum doublet in a first-line setting (NCT01828099) are currently recruiting patients.
Alectinib (CH5424802/RO5424802) is a potent ALK inhibitor that also targets the ALK L1196M gatekeeper mutation in vitro [61]. In a Phase II study in 46 Japanese patients with ALK-positive, crizotinib-naïve NSCLC, the objective response rate was 93.5%, including 2 (4%) complete responses and 41 (89%) partial responses; 40 of 46 patients continued to be on trial at the time of data reporting [62]. Adverse events included dysgeusia and increased aspartate aminotransferase (Table 1): visual disorders were rare and gastrointestinal toxicities were mild. Preliminary data from a Phase I study (n=45) in crizotinib-refractory patients indicated an ORR of 59% [63].
AP26113 is a dual ALK/EGFR inhibitor that also overcomes crizotinib resistance mediated by L1196M and other mutations in preclinical models [64, 65]. In a Phase I/II study in patients with advanced malignancies, preliminary responses have been reported in 13/21 (62%) patients with ALK-positive NSCLC, including responses in both crizotinib-naive and crizotinib-pretreated patients [66]. Phase II expansion cohorts will enroll both crizotinib-naïve and crizotinib-resistant patients [67].
These data indicate that new ALK inhibitors improve responses in patients who have progressed on crizotinib. For secondary mutations, knowledge of the precise resistance-inducing mutation may be important in selecting future salvage therapies since some crizotinib-resistance mutations have been found to show cross-resistance to other ALK inhibitors [68].
Alternative targets and combination therapies
With regards to alternative signaling pathways, ALK regulates downstream signaling such as the RAF/MEK/ERK and PI3K/AKT/mTOR pathways [13]. Combining targeted therapy against these pathways may help overcome crizotinib resistance; for example, combining an ALK inhibitor with a MEK, mTOR or EGFR inhibitor upfront may be explored.
Heat-shock protein 90 (Hsp90) is a molecular chaperone that facilitates correct folding and maturation of oncogenic client proteins, including ALK [69].The Hsp90 inhibitor ganetespib (Synta), exhibits single-agent activity against ALK-positive tumors in preclinical and clinical studies, with activity in resistant cells [54, 70]. In a Phase II study, the Hsp90 inhibitor retaspimycin (Infinity) demonstrated clinical activity in three heavily pretreated patients with ALK-positive NSCLC, two of whom had partial responses and the third had prolonged stable disease (7.2 months) [71]. Thus, Hsp90 inhibitors may represent an alternative strategy to overcome crizotinib resistance. Differential sensitivity of ALK-fusion variants to ALK inhibitors correlates with fusion protein stability [72], and combining Hsp90 and ALK inhibitors has provided synergistic cytotoxicity.[72] Clinical studies are underway involving AT13387 (Astex) plus crizotinib (NCT01712217) and AUY922 (Novartis) plus ceritinib (NCT01772797).
Although ALK-positive tumors are unresponsive to EGFR inhibitors, activation of a secondary pathway such as EGFR is a recognized resistance mechanism. Therefore, EGFR TKIs may improve sensitivity to crizotinib in combination regimens by targeting signaling pathways that contribute to resistance [51].
Clinical challenges in ALK inhibitor therapy – central nervous system (CNS) metastasis
Although some current data does not support an inherent propensity of ALK-positive NSCLC for CNS spread, [73] it has been encountered in the setting of progression after crizotinib therapy, and likely related to poor cerebrospinal fluid (CSF) penetration of the drug despite good systemic control [74]. In one reported case of CNS metastasis, the CSF concentration of crizotinib was 0.62 ng/mL, compared with a serum concentration of 237 ng/mL [75]. A retrospective study evaluating ALK inhibition after therapy for oligoprogressive NSCLC showed CNS to be the first site of progression in 46% of patients with ALK-positive disease [76]. In such patients, continuation of crizotinib after local therapy provided ongoing benefit. Otterson et al. showed that patients may be able to continue with crizotinib for a period of time following clinically documented progression [77]. Another retrospective analysis found that 30% of patients with ALK-positive NSCLC with isolated CNS failure on crizotinib were able to resume therapy after completion of radiotherapy, and continued to receive crizotinib for ≥4 more months without disease progression [78]. Thus, continuing ALK inhibitor therapy in such patients may be a valid option. Notably, favorable effects on brain metastases have been reported for alectinib [62, 63], ceritinib,[58] and AP26113 [66]. Combined high-dose pemetrexed and crizotinib also showed activity in an isolated case with miliary CNS metastases, suggesting that the synergistic effect of this combination may be beneficial in treating patients with ALK-positive NSCLC and brain metastases [79].
Conclusions
The emergence of targeted treatment options for ALK-positive NSCLC has revolutionized the care of patients with this disease. However, resistance to approved treatment often develops, and more research is required to further understand the molecular events associated with ALK-positive NSCLC as well as mechanisms of resistance. Future work will not only focus on optimal diagnosis and treatment at earlier stages of disease, but also on rational combinations of effective agents and the ideal sequence of therapy, particularly as more next-generation agents obtain regulatory approval. In addition, optimal supportive care and toxicity management is essential for patients who may hopefully live longer on sequential treatment.
Acknowledgments
This manuscript was written by the authors. Medical editorial assistance was provided by Matthew Naylor PhD, funded by Novartis Pharmaceuticals.
Footnotes
Conflict of interest: Dr. Ranee Mehra: Consulting (Novartis, Bristol Myers Squibb), Honoraria (Pfizer). Dr. Namrata Vijayvergia: None.
References
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.NCI Surveillance Epidemiology and End Results. [Accessed March 2014]; From http://seer.cancer.gov/statfacts/html/lungb.html#survival.
- 3.Li Y, Ye X, Liu J, Zha J, Pei L. Evaluation of EML4-ALK fusion proteins in non-small cell lung cancer using small molecule inhibitors. Neoplasia. 2011;13:1–11. doi: 10.1593/neo.101120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma SV, Fischbach MA, Haber DA, Settleman J. Oncogenic shock: explaining oncogene addiction through differential signal attenuation. Clin Cancer Res. 2006;12:4392s–4395s. doi: 10.1158/1078-0432.CCR-06-0096. [DOI] [PubMed] [Google Scholar]
- 5.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 6.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 7.Mano H. Non-solid oncogenes in solid tumors: EML4-ALK fusion genes in lung cancer. Cancer Sci. 2008;99:2349–2355. doi: 10.1111/j.1349-7006.2008.00972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeda M, Okamoto I, Sakai K, Kawakami H, Nishio K, Nakagawa K. Clinical outcome for EML4-ALK-positive patients with advanced non-small-cell lung cancer treated with first-line platinum-based chemotherapy. Ann Oncol. 2012;23:2931–2936. doi: 10.1093/annonc/mds124. [DOI] [PubMed] [Google Scholar]
- 9.Takeuchi K, Choi YL, Soda M, Inamura K, Togashi Y, Hatano S, Enomoto M, Takada S, Yamashita Y, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y, Mano H. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res. 2008;14:6618–6624. doi: 10.1158/1078-0432.CCR-08-1018. [DOI] [PubMed] [Google Scholar]
- 10.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, Fidias P, Stubbs H, Engelman JA, Sequist LV, Tan W, Gandhi L, Mino-Kenudson M, Wei GC, Shreeve SM, Ratain MJ, Settleman J, Christensen JG, Haber DA, Wilner K, Salgia R, Shapiro GI, Clark JW, Iafrate AJ. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ, Choi HG, Kim J, Chiang D, Thomas R, Lee J, Richards WG, Sugarbaker DJ, Ducko C, Lindeman N, Marcoux JP, Engelman JA, Gray NS, Lee C, Meyerson M, Janne PA. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14:4275–4283. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8:11–23. doi: 10.1038/nrc2291. [DOI] [PubMed] [Google Scholar]
- 13.Shaw AT, Solomon B. Targeting anaplastic lymphoma kinase in lung cancer. Clin Cancer Res. 2011;17:2081–2086. doi: 10.1158/1078-0432.CCR-10-1591. [DOI] [PubMed] [Google Scholar]
- 14.Mosse YP, Balis FM, Lim MS, Laliberte J, Voss SD, Fox E, Bagatell R, Weigel B, Adamson PC, Ingle AM, Ahem CH, Blaney S. Efficacy of crizotinib in children with relapsed/refractory ALK-driven tumorsincluding anaplastic large cell lymphoma and neuroblastoma: a Children's Oncology Group phase I consortium study. J Clin Oncol. 2012;30(suppl) abstract 9500. [Google Scholar]
- 15.Solomon B, Varella-Garcia M, Camidge DR. ALK gene rearrangements: a new therapeutic target in a molecularly defined subset of non-small cell lung cancer. J Thorac Oncol. 2009;4:1450–1454. doi: 10.1097/JTO.0b013e3181c4dedb. [DOI] [PubMed] [Google Scholar]
- 16.Choi YL, Takeuchi K, Soda M, Inamura K, Togashi Y, Hatano S, Enomoto M, Hamada T, Haruta H, Watanabe H, Kurashina K, Hatanaka H, Ueno T, Takada S, Yamashita Y, Sugiyama Y, Ishikawa Y, Mano H. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res. 2008;68:4971–4976. doi: 10.1158/0008-5472.CAN-07-6158. [DOI] [PubMed] [Google Scholar]
- 17.Takeuchi K, Choi YL, Togashi Y, Soda M, Hatano S, Inamura K, Takada S, Ueno T, Yamashita Y, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y, Mano H. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009;15:3143–3149. doi: 10.1158/1078-0432.CCR-08-3248. [DOI] [PubMed] [Google Scholar]
- 18.Weickhardt AJ, Camidge DR. The therapeutic potential of eneplastic lymphoma kinase inhibitors in lung cancer: rationale and clinical evidence. Clinical Investigation. 2011;1:1119–1126. [Google Scholar]
- 19.Wong DW, Leung EL, So KK, Tam IY, Sihoe AD, Cheng LC, Ho KK, Au JS, Chung LP, Pik Wong M University of Hong Kong Lung Cancer Study Group. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 2009;115:1723–1733. doi: 10.1002/cncr.24181. [DOI] [PubMed] [Google Scholar]
- 20.Paik JH, Choi CM, Kim H, Jang SJ, Choe G, Kim DK, Kim HJ, Yoon H, Lee CT, Jheon S, Choe JY, Chung JH. Clinicopathologic implication of ALK rearrangement in surgically resected lung cancer: a proposal of diagnostic algorithm for ALK-rearranged adenocarcinoma. Lung Cancer. 2012;76:403–409. doi: 10.1016/j.lungcan.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 21.Camidge DR, Kono SA, Flacco A, Tan AC, Doebele RC, Zhou Q, Crino L, Franklin WA, Varella-Garcia M. Optimizing the detection of lung cancer patients harboring anaplastic lymphoma kinase (ALK) gene rearrangements potentially suitable for ALK inhibitor treatment. Clin Cancer Res. 2010;16:5581–5590. doi: 10.1158/1078-0432.CCR-10-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Varella-Garcia M, Cho Y, Lu X, Baron AE, Terracciano L, Camidge DR, Bunn PA, Jr, Franklin WA, Cappuzzo F, Doebele RC. ALK gene rearrangements in unselected caucasians with non-small cell lng carcinoma (NSCLC) J Clin Oncol. 2010;28(15s) abstract 10533. [Google Scholar]
- 23.Kris MG, Johnson BE, Kwatkowski DJ, Iafrate AJ, Wistuba II, Aronson SL, Engelman JA, Shyr Y, Khuri FR, Rudin CM, Garon EB, Pao W, Schiller JH, Haura EB, Shirai K, Giaccone G, Berry LD, Kugler K, Minna JD, Bunn PA. Identification of driver mutations in tumor specimens from 1,000 patients with adenocarcinoma: The NCI's Cancer Mutation Consortium (LCMC) J Clin Oncol. 2011;29(suppl) abstract CRA7506. [Google Scholar]
- 24.Gainor JF, Varghese AM, Ou SH, Kabraji S, Awad MM, Katayama R, Pawlak A, Mino-Kenudson M, Yeap BY, Riely GJ, Iafrate AJ, Arcila ME, Ladanyi M, Engelman JA, Dias-Santagata D, Shaw AT. ALK Rearrangements Are Mutually Exclusive with Mutations in EGFR or KRAS: An Analysis of 1,683 Patients with Non-Small Cell Lung Cancer. Clin Cancer Res. 2013;19:4273–4281. doi: 10.1158/1078-0432.CCR-13-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takahashi T, Sonobe M, Kobayashi M, Yoshizawa A, Menju T, Nakayama E, Mino N, Iwakiri S, Sato K, Miyahara R, Okubo K, Manabe T, Date H. Clinicopathologic features of non-small-cell lung cancer with EML4-ALK fusion gene. Ann Surg Oncol. 2010;17:889–897. doi: 10.1245/s10434-009-0808-7. [DOI] [PubMed] [Google Scholar]
- 26.Inamura K, Takeuchi K, Togashi Y, Hatano S, Ninomiya H, Motoi N, Mun MY, Sakao Y, Okumura S, Nakagawa K, Soda M, Choi YL, Mano H, Ishikawa Y. EML4-ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod Pathol. 2009;22:508–515. doi: 10.1038/modpathol.2009.2. [DOI] [PubMed] [Google Scholar]
- 27.Koh Y, Kim DW, Kim TM, Lee SH, Jeon YK, Chung DH, Kim YW, Heo DS, Kim WH, Bang YJ. Clinicopathologic characteristics and outcomes of patients with anaplastic lymphoma kinase-positive advanced pulmonary adenocarcinoma: suggestion for an effective screening strategy for these tumors. J Thorac Oncol. 2011;6:905–912. doi: 10.1097/JTO.0b013e3182111461. [DOI] [PubMed] [Google Scholar]
- 28.Shaw AT, Yeap B, Costa DB, Solomon BJ, Kwak EL, Nguyen AT, Bergethon K, Engelman JA, Iafrate AJ. Prognostic versus predictive value of EML4-ALK translocation in metastatic non-small cell lung cancer. J Clin Oncol. 2010;28(15s) abstract 7606. [Google Scholar]
- 29.Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, Solomon B, Stubbs H, Admane S, McDermott U, Settleman J, Kobayashi S, Mark EJ, Rodig SJ, Chirieac LR, Kwak EL, Lynch TJ, Iafrate AJ. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247–4253. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Atherly AJ, Camidge DR. The cost-effectiveness of screening lung cancer patients for targeted drug sensitivity markers. Br J Cancer. 2012;106:1100–1106. doi: 10.1038/bjc.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.NCCN Guidelines: NSCLC. 2014;3 [Google Scholar]
- 32.Kim D, Ahn M, Shi Y, Yang P, Liu X, De Pas TM, Crino L, Lanzalone S, Polli A, Shaw AT. Updated results of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancer. European Society for Medical Oncology Congress. 2012 abstract 1230PD. [Google Scholar]
- 33.Shaw AT, Solomon B, Kenudson MM. Crizotinib and testing for ALK. J Natl Compr Canc Netw. 2011;9:1335–1341. doi: 10.6004/jnccn.2011.0115. [DOI] [PubMed] [Google Scholar]
- 34.Sanders HR, Li HR, Bruey JM, Scheerle JA, Meloni-Ehrig AM, Kelly JC, Novick C, Albitar M. Exon scanning by reverse transcriptase-polymerase chain reaction for detection of known and novel EML4-ALK fusion variants in non-small cell lung cancer. Cancer Genet. 2011;204:45–52. doi: 10.1016/j.cancergencyto.2010.08.024. [DOI] [PubMed] [Google Scholar]
- 35.Yi ES, Boland JM, Maleszewski JJ, Roden AC, Oliveira AM, Aubry MC, Erickson-Johnson MR, Caron BL, Li Y, Tang H, Stoddard S, Wampfler J, Kulig K, Yang P. Correlation of IHC and FISH for ALK gene rearrangement in non-small cell lung carcinoma: IHC score algorithm for FISH. J Thorac Oncol. 2011;6:459–465. doi: 10.1097/JTO.0b013e318209edb9. [DOI] [PubMed] [Google Scholar]
- 36.Mino-Kenudson M, Chirieac LR, Law K, Hornick JL, Lindeman N, Mark EJ, Cohen DW, Johnson BE, Janne PA, Iafrate AJ, Rodig SJ. A novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistry. Clin Cancer Res. 2010;16:1561–1571. doi: 10.1158/1078-0432.CCR-09-2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paik JH, Choe G, Kim H, Choe JY, Lee HJ, Lee CT, Lee JS, Jheon S, Chung JH. Screening of anaplastic lymphoma kinase rearrangement by immunohistochemistry in non-small cell lung cancer: correlation with fluorescence in situ hybridization. J Thorac Oncol. 2011;6:466–472. doi: 10.1097/JTO.0b013e31820b82e8. [DOI] [PubMed] [Google Scholar]
- 38.McLeer-Florin A, Moro-Sibilot D, Melis A, Salameire D, Lefebvre C, Ceccaldi F, de Fraipont F, Brambilla E, Lantuejoul S. Dual IHC and FISH testing for ALK gene rearrangement in lung adenocarcinomas in a routine practice: a French study. J Thorac Oncol. 2012;7:348–354. doi: 10.1097/JTO.0b013e3182381535. [DOI] [PubMed] [Google Scholar]
- 39.Park HS, Lee JK, Kim DW, Kulig K, Kim TM, Lee SH, Jeon YK, Chung DH, Heo DS. Immunohistochemical screening for anaplastic lymphoma kinase (ALK) rearrangement in advanced non-small cell lung cancer patients. Lung Cancer. 2012;77:288–292. doi: 10.1016/j.lungcan.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 40.Ventana Medical Systems, Inc. [Accessed March 2014]; From http://www.ventana.com/site/page?view=press-release-oct25-2012.
- 41.Lee JK, Park HS, Kim DW, Kulig K, Kim TM, Lee SH, Jeon YK, Chung DH, Heo DS, Kim WH, Bang YJ. Comparative analyses of overall survival in patients with anaplastic lymphoma kinase-positive and matched wild-type advanced nonsmall cell lung cancer. Cancer. 2012;118:3579–3586. doi: 10.1002/cncr.26668. [DOI] [PubMed] [Google Scholar]
- 42.Kim HR, Shim HS, Chung JH, Lee YJ, Hong YK, Rha SY, Kim SH, Ha SJ, Kim SK, Chung KY, Soo R, Kim JH, Cho BC. Distinct clinical features and outcomes in never-smokers with nonsmall cell lung cancer who harbor EGFR or KRAS mutations or ALK rearrangement. Cancer. 2012;118:729–739. doi: 10.1002/cncr.26311. [DOI] [PubMed] [Google Scholar]
- 43.Gandara DR, Huang E, Desai S, Mack PC, Beckett L, Stephens C, Zeger G, Denenberg KD, Maus MKH, Li T. Thymidylate synthase (TS) gene expression in patients with ALK positive (ALK+) non-small cell lung cancer (NSCLC): Implications for therapy. J Clin Oncol. 2012;30(suppl) abstract 7582. [Google Scholar]
- 44.Lee HY, Ahn HK, Jeong JY, Kwon MJ, Han JH, Sun JM, Ahn JS, Park K, Choi YL, Ahn MJ. Favorable clinical outcomes of pemetrexed treatment in anaplastic lymphoma kinase positive non-small-cell lung cancer. Lung Cancer. 2013;79:40–45. doi: 10.1016/j.lungcan.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 45.Camidge DR, Kono SA, Lu X, Okuyama S, Baron AE, Oton AB, Davies AM, Varella-Garcia M, Franklin W, Doebele RC. Anaplastic lymphoma kinase gene rearrangements in non-small cell lung cancer are associated with prolonged progression-free survival on pemetrexed. J Thorac Oncol. 2011;6:774–780. doi: 10.1097/JTO.0b013e31820cf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaw AT, Varghese AM, Solomon BJ, Costa DB, Novello S, Mino-Kenudson M, Awad MM, Engelman JA, Riely GJ, Monica V, Yeap BY, Scagliotti GV. Pemetrexed-based chemotherapy in patients with advanced, ALK-positive non-small cell lung cancer. Ann Oncol. 2013;24:59–66. doi: 10.1093/annonc/mds242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsao AS, Liu D, Lee JJ, Spitz M, Hong WK. Smoking affects treatment outcome in patients with advanced nonsmall cell lung cancer. Cancer. 2006;106:2428–2436. doi: 10.1002/cncr.21884. [DOI] [PubMed] [Google Scholar]
- 48.Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, Riely GJ, Solomon B, Ou SH, Kim DW, Salgia R, Fidias P, Engelman JA, Gandhi L, Janne PA, Costa DB, Shapiro GI, Lorusso P, Ruffner K, Stephenson P, Tang Y, Wilner K, Clark JW, Shaw AT. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011–1019. doi: 10.1016/S1470-2045(12)70344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, Wu YL, Thomas M, O'Byrne KJ, Moro-Sibilot D, Camidge DR, Mok T, Hirsh V, Riely GJ, Iyer S, Tassell V, Polli A, Wilner KD, Janne PA. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 50.Berge EM, Lu X, Maxson D, Baron AE, Gadgeel SM, Solomon BJ, Doebele RC, Varella-Garcia M, Camidge DR. Clinical Benefit From Pemetrexed Before and After Crizotinib Exposure and From Crizotinib Before and After Pemetrexed Exposure in Patients With Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer. Clin Lung Cancer. 2013;14:636–643. doi: 10.1016/j.cllc.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, Drew L, Saeh JC, Crosby K, Sequist LV, Iafrate AJ, Engelman JA. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012;4:120ra17. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WA, Varella-Garcia M, Camidge DR. Mechanisms of Resistance to Crizotinib in Patients with ALK Gene Rearranged Non-Small Cell Lung Cancer. Clin Cancer Res. 2012;18:1472–1482. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, Yatabe Y, Takeuchi K, Hamada T, Haruta H, Ishikawa Y, Kimura H, Mitsudomi T, Tanio Y, Mano H ALK Lung Cancer Study Group. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734–1739. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- 54.Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, Shakespeare WC, Iafrate AJ, Engelman JA, Shaw AT. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108:7535–7540. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanizaki J, Okamoto I, Okabe T, Sakai K, Tanaka K, Hayashi H, Kaneda H, Takezawa K, Kuwata K, Yamaguchi H, Hatashita E, Nishio K, Nakagawa K. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clin Cancer Res. 2012;18:6219–6226. doi: 10.1158/1078-0432.CCR-12-0392. [DOI] [PubMed] [Google Scholar]
- 56.Marsilje TH, Pei W, Chen B, Lu W, Uno T, Jin Y, Jiang T, Kim S, Li N, Warmuth M, Sarkisova Y, Sun F, Steffy A, Pferdekamper AC, Li AG, Joseph SB, Kim Y, Liu B, Tuntland T, Cui X, Gray NS, Steensma R, Wan Y, Jiang J, Chopiuk G, Li J, Gordon WP, Richmond W, Johnson K, Chang J, Groessl T, He YQ, Phimister A, Aycinena A, Lee CC, Bursulaya B, Karanewsky DS, Seidel HM, Harris JL, Michellys PY. Synthesis, Structure-Activity Relationships, and in Vivo Efficacy of the Novel Potent and Selective Anaplastic Lymphoma Kinase (ALK) Inhibitor 5-Chloro-N2-(2-isopropoxy-5-methyl-4-(piperidin-4-yl)phenyl)-N4-(2-(isopropylsulf onyl)phenyl)pyrimidine-2,4-diamine (LDK378) Currently in Phase 1 and Phase 2 Clinical Trials. J Med Chem. 2013;56:5675–5690. doi: 10.1021/jm400402q. [DOI] [PubMed] [Google Scholar]
- 57.Li N, Michellys P, Kim S, Culazzo Pferdekamper A, Li J, Kasibhatla S, Tompkins CS, Steffy A, Li A, Sun F, Sun X, Hua S, Tiedt R, Sarkisova Y, Marsilje TH, McNamara P, Harris J. Activity of a potent and selective phase I ALK inhibitor LDK378 in naive and crizotinib-resistant preclinical tumor models. Mol Cancer Ther. 2011;10 abstract B232. [Google Scholar]
- 58.Shaw AT, Kim D, Mehra R, Tan DS, Felip E, Chow LQM, Camidge DR, Vansteenkiste JF, Sharma S, De Pas T, Riely GJ, Solomon BJ, Wolf J, Thomas M, Schuler M, Liu G, Santoro A, Lau Y, Goldwasser M, BoralAL, Engelman JA. LDK378 in anaplastic lymphoma kinase-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370:1189–1197. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. [Accessed March 2014];LDK378 Breakthrough Therapy designation. From www.novartis.com/newsroom/media-releases/en/2013/1685517.shtml.
- 60.Shaw AT, Camidge DR, Felip E, Sharma S, Tan SW, Kim D, De Pas T, Vansteenkiste JF, Santoro A, Liu G, Chow LQM, Goldwasser M, Lau Y, Boral AL, Mehra R. Results of the first-in-human phase I study of the ALK inhibitor LDK378 in advanced malignancies. European Society for Medical Oncology Congress. 2012 abstract 440O. [Google Scholar]
- 61.Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, Oikawa N, Tsukuda T, Ishii N, Aoki Y. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19:679–690. doi: 10.1016/j.ccr.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 62.Seto T, Kiura K, Nishio M, Nakagawa K, Maemondo M, Inoue A, Hida T, Yamamoto N, Yoshioka H, Harada M, Ohe Y, Nogami N, Takeuchi K, Shimada T, Tanaka T, Tamura T. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1-2 study. Lancet Oncol. 2013;14:590–598. doi: 10.1016/S1470-2045(13)70142-6. [DOI] [PubMed] [Google Scholar]
- 63.Ou S, Gadgeel SM, Chiappori A, Riely G, Lee R, Garcia L, Kikuchi H, Tatsuno M, Tanaka T, Gandhi L. Safety and efficacy analysis of RO5424802/CH5424802 in anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer (NSCLC) patients who have failed crizotinib in a dose-finding phase I study (AF-002JG, NCT01588028) European Society for Medical Oncology Congress. 2013 abstract LAB44. [Google Scholar]
- 64.Zhang S, Wang F, Keats J, Ning Y, Wardwell SD, Moran L, Mohemmad QK, Ye E, Anjum R, Wang Y, Zhu X, Miret JJ, Delgarno D, Narasimhan I, Clackson T, Shakespeare WC, Rivera VM. AP26113, a potent ALK inhibitor, overcomes mutations in EML4-ALK that confer resistance to PF-02341066. Cancer Res. 2010;70(suppl) abstract LB-298. [Google Scholar]
- 65.Camidge DR, Bazhenova L, Salgia R, Weiss GJ, Langer CJ, Shaw AT, Narasimhan I, Dorer DJ, Rivera VM, Zhang J, Clackson T, Haluska FG, Gettinger SN. First-in-human dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignancies: Updated results. J Clin Oncol. 2013;31(suppl) abstract 8031. [Google Scholar]
- 66.Camidge DR, Bazhenova L, Salgia R, Weiss GJ, Langer CJ, Shaw AT, Narasimhan I, Dorer DJ, Zhang J, Gettinger SN. Updated results of a first-in-human dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignancies. European Society for Medical Oncology Congress. 2013 abstract 3401. [Google Scholar]
- 67.Clinicaltrials.gov. [Accessed March 2014]; From http://www.clinicaltrials.gov.
- 68.Ceccon M, Mologni L, Bisson W, Scapozza L, Gambacorti-Passerini C. Crizotinib-resistant NPM-ALK mutants confer differential sensitivity to unrelated Alk inhibitors. Mol Cancer Res. 2013;11:122–132. doi: 10.1158/1541-7786.MCR-12-0569. [DOI] [PubMed] [Google Scholar]
- 69.Bonvini P, Gastaldi T, Falini B, Rosolen A. Nucleophosmin-anaplastic lymphoma kinase (NPM-ALK), a novel Hsp90-client tyrosine kinase: down-regulation of NPM-ALK expression and tyrosine phosphorylation in ALK(+) CD30(+) lymphoma cells by the Hsp90 antagonist 17-allylamino,17-demethoxygeldanamycin. Cancer Res. 2002;62:1559–1566. [PubMed] [Google Scholar]
- 70.Socinski MA, Goldman J, El-Hariry I, Koczywas M, Vukovic V, Horn L, Paschold E, Salgia R, West H, Sequist LV, Bonomi P, Brahmer J, Chen LC, Sandler A, Belani CP, Webb T, Harper H, Huberman M, Ramalingam S, Wong KK, Teofilovici F, Guo W, Shapiro GI. A multicenter phase II study of ganetespib monotherapy in patients with genotypically defined advanced non-small cell lung cancer. Clin Cancer Res. 2013;19:3068–3077. doi: 10.1158/1078-0432.CCR-12-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sequist LV, Gettinger S, Senzer NN, Martins RG, Janne PA, Lilenbaum R, Gray JE, Iafrate AJ, Katayama R, Hafeez N, Sweeney J, Walker JR, Fritz C, Ross RW, Grayzel D, Engelman JA, Borger DR, Paez G, Natale R. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol. 2010;28:4953–4960. doi: 10.1200/JCO.2010.30.8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heuckmann JM, Balke-Want H, Malchers F, Peifer M, Sos ML, Koker M, Meder L, Lovly CM, Heukamp LC, Pao W, Kuppers R, Thomas RK. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin Cancer Res. 2012;18:4682–4690. doi: 10.1158/1078-0432.CCR-11-3260. [DOI] [PubMed] [Google Scholar]
- 73.Doebele RC, Lu X, Sumey C, Maxson DA, Weickhardt AJ, Oton AB, Bunn PA, Jr, Baron AE, Franklin WA, Aisner DL, Varella-Garcia M, Camidge DR. Oncogene status predicts patterns of metastatic spread in treatment-naive nonsmall cell lung cancer. Cancer. 2012;118:4502–4511. doi: 10.1002/cncr.27409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chun SG, Choe KS, Iyengar P, Yordy JS, Timmerman RD. Isolated central nervous system progression on Crizotinib: an Achilles heel of non-small cell lung cancer with EML4-ALK translocation? Cancer Biol Ther. 2012;13:1376–1383. doi: 10.4161/cbt.22255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Costa DB, Kobayashi S, Pandya SS, Yeo WL, Shen Z, Tan W, Wilner KD. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol. 2011;29:e443–5. doi: 10.1200/JCO.2010.34.1313. [DOI] [PubMed] [Google Scholar]
- 76.Weickhardt AJ, Scheler B, Burke JM, Gan G, Doebele RC, Bunn PA, Gaspar LE, Kavanagh BD, Camidge DR. Continuation of EGFR/ALK inhibition after local therapy of oligoprogressive disease in EGFR mutant (Mt) and ALK+ non-small cell lung cancer (NSCLC) J Clin Oncol. 2012;30(suppl) doi: 10.1097/JTO.0b013e3182745948. abstract 7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Otterson GA, Riely GJ, Shaw AT, Crino L, Kim D, Martins R, Salgia R, Zhou C, Solomon BJ, Wilner KD, Poli A, Tang Y, Bartlett CH, Ou SI. Clinical characteristics of ALK+ NSCLC patients (pts) treated with crizotinib beyond disease progression (PD): Potential implications for management. J Clin Oncol. 2012;30(suppl) abstract 7600. [Google Scholar]
- 78.Takeda M, Okamoto I, Nakagawa K. Clinical impact of continued crizotinib administration after isolated central nervous system progression in patients with lung cancer positive for ALK rearrangement. J Thorac Oncol. 2013;8:654–657. doi: 10.1097/JTO.0b013e31828c28e7. [DOI] [PubMed] [Google Scholar]
- 79.Gandhi L, Drappatz J, Ramaiya NH, Otterson GA. High-dose pemetrexed in combination with high-dose crizotinib for the treatment of refractory CNS metastases in ALK-rearranged non-small-cell lung cancer. J Thorac Oncol. 2013;8:e3–5. doi: 10.1097/JTO.0b013e3182762d20. [DOI] [PubMed] [Google Scholar]
- 80.Puig de la Bellacasa R, Karachaliou N, Estrada-Tejedor R, Teixido J, Costa C, Borrell JL. ALK and ROS1 as a joint target for the treatment of lung cancer: a review. Transl Lung Cancer. 2013;2:72–86. doi: 10.3978/j.issn.2218-6751.2013.03.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wilcoxen KM, Brake RL, Saffran D, Taffera Y, Choquette D, Whittington D, Yu V, Romero K, Bode C, Stellwagen J, Potashman M, Emkey R, Andrews P, Drew AE, Xu M, Szilvassy S, Al-Assad S, Lewis RT. Characterization of a novel series of potent, selective inhibitors of wild-type and mutant/fusion anaplastic lymphoma kinase. Cancer Res. 2012;72(suppl) abstract 1795. [Google Scholar]
- 82.Weiss G, Sachdev JC, Infante JR, Mita M, Wilcoxen KM, Kansra V, Brooks DG, Martell RE, Anthony SP. TSR-011: A potent inhibitor of ALK with activity in crizotinib-resistant tumor models in phase 1-2 development for ALK+ NSCLC. E European Society for Medical Oncology Congress. 2013 abstract 417. [Google Scholar]
- 83.Lovly CM, Heuckmann JM, de Stanchina E, Chen H, Thomas RK, Liang C, Pao W. Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res. 2011;71:4920–4931. doi: 10.1158/0008-5472.CAN-10-3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Drew L, Cheng J, Engelman J, Ferguson D, Katayama R, McDermott B, Saeh J, Shaw A, Shen M, Widzowski D, Wu A, Smith G. AZD3463, a novel ALK/IGF1R inhibitor, overcomes multiple mechanisms of acquired resistance to crizotinib. Cancer Res. 2013;73(suppl) abstract 919. [Google Scholar]