

Abstract
The kidneys receive 20-25 % of cardiac output and play a main role in the control of cardiovascular homeostasis. It is an endocrine organ that regulates and produces many substances, scavenger particles and immune complexes. Cytokines, growth factors, reactive oxygen metabolites, bioactive lipids, proteases, vasoactive substances such as nitric oxide (NO), adrenomedullin (AM), urotensin-II (U-II), have been released in several diseases, and kidney is one of mostly affected organs in body. Some of these mediators act in a paracrine fashion while some act in autocrine. They play important roles in modulating the cardiovascular responses, renal hemodynamics, and probably in mediating the clinical and laboratory manifestations of several renal diseases.
These mediators are like “a double edged sword.” While small amounts of them mediate many physiological events, little excess may cause the damage to the healthy cells. Many investigators have searched the role(s) of mediators in several diseases. However, the findings are mostly like the model of “chicken and egg”, and indistinguishable as to whether they are the causes of, or results of the diseases.
We will discuss mainly the possible roles of NO, AM and U-II in children with several renal diseases and summarize what is known, and what must be known about these mediators.
Keywords: adrenomedullin, children, glomerulonephritis, nephrotic syndrome, nitric oxide, urotensin-II
Kidney is in trouble with mediators
Nitric oxide (NO), a molecule of the millennium, is synthesized from L-arginine by the enzyme nitric oxide synthase (NOS). It is a highly reactive, free radical gas and serves many functions within the kidney, including regulation of afferent arteriolar tone and proliferation of mesangial cells (1-3). Although small amounts of NO mediate many physiological events such as vasodilation, memory, neuroprotection, and immune defense, in fact, it is like a double edged sword. It has a complex role in immune functions, and mediates immunological injury to kidney mesangium in experimental glomerulonephritis (4). Immunologic and inflammatory stimuli induce the production of NO over longer periods, and it exerts cytotoxic and cytostatic effects not only against invading cells, but also against healthy cells (5). Therefore, it has been the recent focus on the role of endothelium in the inflammation. Adrenomedullin (AM) is originally isolated from human pheochromocytoma, and has potent vasorelaxing and natriuretic properties (6). It has been reported to be present in normal adrenal medulla, heart, lung, and kidney as well as in plasma and urine. Adrenomedullin may function as a circulating hormone and an autocrine/paracrine mediator involved in the regulation of the cardiovascular system, blood pressure, and renal function (6,7). There is also a close interaction between AM and NO. Adrenomedullin stimulates NO production by endothelial cells, and inhibits potassium and angiotensin II-stimulated aldosterone secretion in the adrenal gland. Natriuretic and diuretic actions of AM reflect unique actions of this peptide on renal blood flow and tubular function (8). Considering the important influences of NO and AM on kidney, we investigated their possible roles on several important childhood diesases.
Nitric oxide and adrenomedullin in Henoch -Schönlein purpura
Henoch-Schönlein purpura (HSP) is one of the most common types of vasculitis seen in children. It is characterized by non-thrombocytopenic purpura, arthritis, abdominal pain, and renal involvement (9). Its clinical manifestations are due to a generalized vasculitis involving capillaries, arteries and venules. Considering the endothelial synthesis of AM and NO, and endothelial injury in HSP, we measured their levels in children with HSP (10). This was the first report on AM levels in HSP, while a study has been previously demonstrated that serum and urinary nitrite levels are elevated in children with HSP (11).
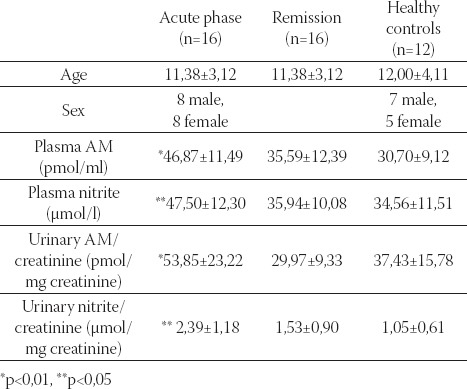
Sixteen children with HSP evaluated during the acute and remission phases of disease, and compared with 12 healthy controls. This study demonstrated that plasma and urinary AM and nitrite levels were elevated during acute phase in children with HSP, and returned to normal levels during remission (Table 1) (10).
TABLE 1.
Plasma/urinary adrenomedullin(AM) and nitrite levels in children with Henoch-Schönlein purpura (10)
Considering the important roles of NO in inflammation, its production is expected to be increased in patients with HSP, especially in the acute inflammatory stage of the disease. Because the Ca2+-independent isoform of NOS (iNOS) can be activated by inflammatory cytokines, interferon gamma, bacterial lipopolysaccharides, and endotoxins during inflammatory and infectious processes, and generates large amounts of NO (12,13). It has been suggested that proinflammatory cytokines including IL-1, IL-6, and tumor necrosis factor (TNF) may play a role as a mediator of inflammation in HSP (14). This observation was possibly due to NO-generating cells such as endothelial, polymorphonuclear cells, and macrophages found in inflammation. It could also derive from vascular endothelial cells (ECs) through the endothelial, calcium-dependent isoforms of NOS (15). Alternatively, increased urinary excretion of nitrite may reflect enhanced NO production by glomerular mesangial or renal tubular epithelial cells that contain calmodulin and calcium-independent-inducible NOS (16,17). It has been shown that AM inhibits platelet-derived growth factor-induced endothelin-1 (ET-1) production in cultured VSMCs probably through a cAMP-dependent process (18). It is also known that TNF, IL-1, and lipopolysaccharide stimulate AM production and secretion from ECs and VSMCs and enhance AM secretion by macrophages and monocytes (19). Considering the increased levels of proinflammatory cytokines in HSP (14), we suggest that AM production and secretion may be activated by these inflammatory cytokines. Since AM has cytoprotective (20,21) and antiproliferative (renoprotective) effects on mesangial cells (22), the increased levels of plasma AM may also be compensatory against inflammatory reactions in HSP. Based on these results, it is possible that AM participates (as a cytoprotective peptide) in the pathophysiology of vascular lesions in HSP, although without having a disease-specific character. This study indicated that AM and NO may have a role in the immunoinflammatory process of HSP, especially in the active stage, although whether this perpetuates, or protects against, further vascular injury is not clear (10).
Nitric oxide and adrenomedullin: A suspicious pair, responsible from vascular hyporeactivity, in Bartter syndrome
Bartter syndrome includes hypokalemia, decreased sensitivity of the distal nephron to vasopressin, hyperreninemia, a high plasma level of angiotensin II (ATII), hyperaldosteronism, and reduced vascular response to pressor action of ATII and norepinephrine which causes its persistent normotension in spite of biochemical and hormonal abnormalities typical of hypertension (23). These events are not caused by hypokalemia, because they are still present even after the patients are made normokalemic (24), nor by prostaglandin overproduction, because they do not disappear after treatment with inhibitors of prostaglandin synthesis (25). Recent evidence in adult Bartter syndrome suggests that increased nitric oxide (NO) synthesis may account for the reduced vascular response of the disease (26). We have firstly investigated the possible roles of NO and AM in maintaining this reduced vascular reactivity by studying plasma and urinary nitrite, a stable metabolite of NO, and AM levels in 10 children with Bartter syndrome, 10 healthy controls, and 5 children with hypokalemia of causes other than Bartter syndrome (pseudo-Bartter) (27). Urinary excretion of nitrite (pmol/mg urinary creatinine) was 8,9±1,2 in children with Bartter syndrome, 4,7±0,9 in healthy controls, and 2,9±0,8 in pseudo-Bartter (P<0,05). Plasma nitrite levels (pmol/l) were 101,9±23,4, 59,9±14,7, and 65,0±29,7, respectively (P>0,05), in the three groups. Urinary excretion of AM (pmol/mg urinary creatinine) was 187±40, 65±10, and 160±50, respectively (P<0,05), in the three groups. Plasma AM levels were 47,4±1,8; 39,9±5,9, and 42,4±3,9, respectively (P>0,05), in the three groups. The same parameters were repeated in the two groups of controls and in the Bartter patients in the 6th month of therapy. Urinary nitrite and AM levels were still higher in the Bartter patients than in the other groups. We concluded that increased NO production may be responsible for the reduced vascular response in Bartter syndrome (27). Increased NO may originate from vascular ECs (15), or may be produced by glomerular mesangial or renal tubular epithelial cells (16,17,26). Hypokalemia did not seem to play a role in the increased NO production, since there was no significant difference in urinary nitrite excretion between pseudo-Bartter and healthy controls. Adrenomedullin induces vasorelaxation by at least two mechanisms: a direct action on vascular smooth muscle to increase intracellular cAMP, and stimulation of the release of NO from ECs (7). We found higher urinary AM levels, in children with Bartter syndrome and pseudo-Bartter at the pretreatment period. Two possible sources of high urinary AM can be considered. One may be glomerular filtration of circulating AM since plasma AM levels are increased. Another possible explanation is that the kidney may be one of the major sites of AM synthesis. Cultured rat mesangial cells and renal tubular cells have been shown to produce AM, indicating that AM was produced and secreted in the kidney (7,28,29), and suggesting that urinary excretion of AM may be partly derived from renal tissues. The initial increased urinary levels of AM, both in children with Bartter syndrome and in those with pseudo-Bartter, may support a role of acute hypokalemia in the increased AM production. Hypokalemia may induce the synthesis of AM from the adrenal medulla by stimulation of sympathoadrenal activity. However, we observed that increased AM production was still going on at the 6th month of therapy in Bartter syndrome when compared to pseudo-Bartter, after normalization of potassium, and healthy controls. Considering the potent hypotensive and vasodilatory effect of AM, we speculate that it may also be responsible for vascular hyporeactivity in children with Bartter syndrome (27).
The possible roles of NO and AM in minimal change nephrotic syndrome
The mediator(s) of glomerular hyperpermeability is (are) not well understood in minimal change nephrotic syndrome (MCNS) (30), and the relationship between immunological alterations and glomerular changes related to proteinuria is not clear. Many investigators have investigated the roles of several mediators which may induce proteinuria in glomerulonephritis. Kubo et al. (31) showed that plasma AM concentrations correlated with the extent of proteinuria in chronic glomerulonephritis, including MCNS, in adults. We have examined the possible roles of NO and AM in children with MCNS (32). In comparison with healthy controls, children with MCNS had increased urinary nitrite excretion (μmol urinary creatinine), irrespective of whether the disease was in relapse or remission (3,2±0,2 in relapse, n=13 1,9±3 in remission, n=12; 1,0±0,2 in controls, n=10; P<0,05). Plasma nitrite levels (pmol/l) were high in relapse compared with controls (53,2±8,7 vs. 32±4,0, P<0,05). This finding indicate that NO may play a role in mediating the clinical manifestations of MCNS in children (32). As we mentioned above, increased NO could derive from vascular ECs, glomerular mesangial or renal tubular epithelial cells (15-17). In the absence of circulating or in situ immune complexes, a circulating factor, such as the lymphokine produced by activated suppressor T cells (33) or other cytokines (34), may be responsible for stimulating NO synthesis in children with MCNS. The role of increased urinary nitrite excretion in mediating the manifestations of MCNS is unknown. Recently, Trachtman et al. (35) showed that increased urinary nitrite excretion in MCNS cannot be attributed to the nephrotic syndrome itself, because they found no difference between control subjects and patients with focal segmental glomerulosclerosis (FSGS). In our study, the elevated nitrite excretion in relapse and remission suggests that renal NO synthesis does not influence proteinuria in MCNS. However, it may play a role in mediating other manifestations of the disease (32). Recently, two important studies showed an antimitogenic effect of AM on glomerular mesangial cells (20), and migration inhibition by this peptide (21). These studies suggested that AM may exert an important beneficial effect, tending to lessen the severity of glomerular diseases involving mainly the mesangium. We demonstrated that AM levels were significantly lowered in plasma and elevated in urine of patients with MCNS in relapse (32). The increased urinary AM observed in glomerular disease may be compensatory to reduce the renal damage of high proteinuria, or it could result from tubular protein overload saturating protein reabsorptive mechanisms. Urinary excretion of AM showed a significant correlation with urinary excretion of sodium during relapse, suggesting that tubular delivery of AM may have a natriuretic effect in MCNS, probably by decreasing distal tubular sodium reabsorption, since tubular reabsorption of phosphorus was not impaired in our patients (32). Further investigations will clarify the role(s) of AM in children with MCNS.
Urotensin-H in children with renal diseases: new messages from an ancient peptide
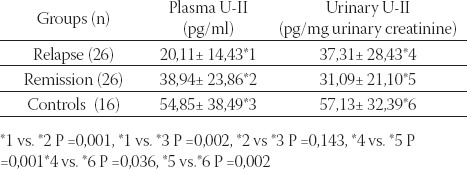
Although urotensin-II has been recognized for over 30 years, only recently it has become a major focus of clinical researches (36). Human urotensin-II (hU-II) is a cyclic peptide of 11 amino acids cleaved from a larger prepro-U-II precursor peptide of about 130 amino acids (36,37). It is a ligand for the orphan G-protein-coupled receptor, GPR14 (36). Although human prepro-U-II mRNA is expressed mainly in the brain and spinal cord, it is also detected in other tissues, such as kidney, spleen, small intestine, thymus, prostate, pituitary, and adrenal gland (37). It is the most potent mammalian vasoconstrictor identified to date (36), being ten-fold more potent than endothelin-I. It circulates in the plasma of healthy individuals, and acts as a circulating vasoactive hormone and as a locally acting paracrine or autocrine factor in cardiovascular regulation (38,39). It also has a vasodilatory effect on the small arteries of rats (40) and on the resistance arteries of humans (41), through release of endothelium-derived hyperpolarizing factor and NO. Although some studies have been searched the circulating levels of U-II in several diseases, such as hypertension (42), congestive heart failure (43), renal failure (36), MCNS (44), preeclampsia-eclampsia (45), little is known about the actions of this peptide within the kidney. Some studies have shown that renal dysfunction affected the U-II levels. The plasma U-II level has been found to be elevated in renal failure (36), congestive heart failure(43), and systemic hypertension (42). However, there are no sufucient data on the level of this vasoactive peptide in glomerular diseases of childhood. Recently, we firstly demonstrated that U-II was present in plasma and urine samples of 26 children with minimal change nephrotic syndrome (MCNS) (44). It showed important changes in relapse and remission periods. Plasma U-II concentrations during relapse were significantly lower than in remission and in controls, whereas urinary U-II levels were higher in relapse than in remission (Table 2) (44).
TABLE 2.
Plasma and urinary urotensin (U)-II levels in children with minimal change nephrotic syndrome and controls (44)
The plasma U-II level showed a significant positive correlation with the plasma albumin concentration during remission (r=0,52, P=0,006). However, there was no correlation between the extent of proteinuria and plasma/urinary U-II levels, and we could not detect any relationship between U-II levels and other clinical and laboratory parameters such as the age at onset of disease, number of relapses, time to remission, blood pressure, serum creatinine, and hematological parameters. We speculated that there were a few possible reasons for the high urinary excretion of U-II. One was the loss of U-II in urine since the plasma U-II levels were markedly decreased in relapse and increased in remission for the same patients. Another possible explanation is that the kidney may be one of the major sites for U-II synthesis. Since the distal convoluted tubules and endothelial cells of renal capillaries have been shown to produce U-II (46), urinary excretion of U-II may be partly derived from renal tissues. In that study (44), all of our patients were diagnosed with MCNS, the most benign form of childhood glomerular diseases, we suggested that the important changes in plasma and urinary U-II levels during relapse may be the result of heavy proteinuria rather than playing a role in mediating the clinical and laboratory manifestations of MCNS. After this, it would be interesting to search the possible role(s) of this peptide in children with glomerular diseases other than MCNS. Therefore, we examined renal biopsy specimens of children with several renal diseases, including 24 with membranoproliferative (MPGN), and 6 with membranous nephropathy (MGN), 5 with IgA nephropathy, 5 with HSP, and 3 with focal segmental glomerulosclerosis (FSGS) (47,48). The positivities in proximal, distal and collecting tubules, renal capillaries, Bowman capsules, glomerular capillaries and basal membranes, mesangium and interstisium were revealed as weak (+), moderate (++) and severe(+++). In normal human kidney, there was weak expression of human U-II in glomerulus, while abundant expressions were seen in proximal, distal tubules, and collecting ducts (Figure 1) (47). In kidneys of children with MPGN, as opposed to the normal kidneys, more dense U-II immunoreactivity was seen in the glomerular basement membrane(GBM), glomerular mesangium, Bowman capsule, and tubules (Figure 2) (47). Interestingly, we also observed U-II immunoreactivity in crescents (Figure 3) (47). Systolic BP was positively correlated with mesangial expression of U-II (p=0,042, r=0,418), while diastolic BP was correlated with endothelial U-II expression (p=0,021, r=0,469) in MPGN (47). In children with MGN, U-II was mostly seen in GBM and Bowman capsule. We observed more dense U-II immunoreactivity in distal tubules (p = 0,030), endothelium (p= 0,009), and mesangium (p= 0,002) in children with MPGN than in MGN. Diastolic BP was positively correlated with the expression of U-II in Bowman capsule (p=0,000, r=1) in children with MGN (47).
FIGURE 1.
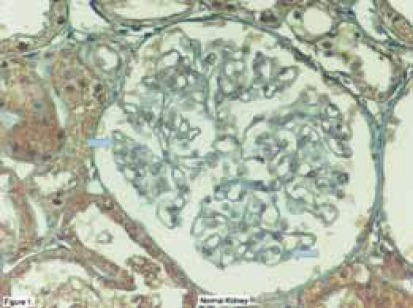
Localization of urotensin-II İmmunoreacüvity (brown color) in the normal human kidney. Weak immunos-taining in glomerulus, abundant expression of U-II in tubules. Thick arrows: weak immunostaining, black arrow: strong immunostaining (47).
FIGURE 2.
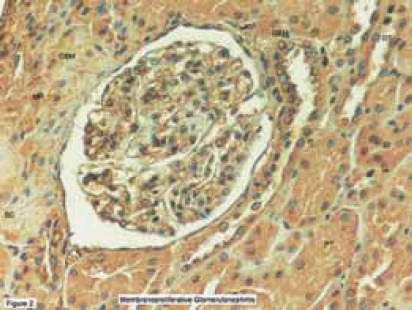
Localization of urotensin-II immunoreactivity in membranoproliferative glomerulonephritis. Immunostaining in glomerular basement membrane, mesangium, Bowman capsule, and tubules.
FIGURE 3.
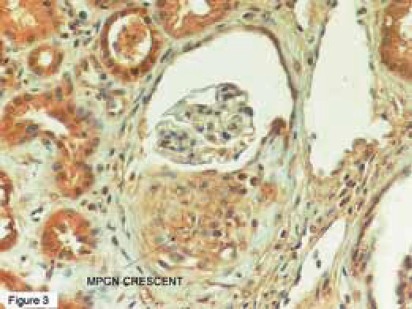
Localization of urotensin-II immunoreactivity in a crescent (47). MPGN: Membranoproliferative Glomerulo-nephritis
Abbreviations: GBM=glomerular basement membrane, BC=Bowman capsule, M=mesangium, PT=proximal tubule, DT=distal tubule (47).
There are no sufficient data about the precise role of hU-II in renal diseases, and that was the first report demonstrating the presence of U-II by IHC in children with several renal diseases. Shenouda et al (46) demonstrated that U-II was mostly present in the epithelial cells of tubules and ducts, with greater intensity in the distal convoluted tubules in normal human kidneys. Moderate U-II immunoreactivity was seen in the endothelial cells of renal capillaries, but only focal immunoreactivity was found in the endothelial cells of the glomeruli. We observed similar results in normal kidneys, and these findings suggest that hU-II may contribute to the pathophysiology of human kidneys. It has been speculated that U-II might be an important physiological mediator of vascular tone and blood pressure in hums (40), and is also an extremely potent constrictor of renal blood vessels from primates, more potent than endothelin-I (49). Considering the positive correlation between BP and intensity of U-II expression in endothelium, and mesangium, it is reasonable to suggest that U-II may play an important role in the regulation of BP in MPGN. As shown previously, mainly two basic mechanisms are possible in primary mechanisms of GNs: antibody interaction with antigens in situ within the glomerulus and antibody binding to soluble antigens in the circulation, followed by immune-complex deposition within the glomeruli (50). The secondary immune mechanisms of glomerular injury are the cascade of inflammatory mediators that are recruited to propagate renal damage following the primary glomerular attack. Some of these mediators play essential roles, whereas others may aggravate the glomerular lesion (50) . Most of the secondary mediators include cytokines and growth factors, reactive oxygen metabolites, bioactive lipids (PAF and eicosanoids), proteases, and vasoactive substances, such as ET and NO (50). Since U-II is abundantly expressed in the glomeruli in MPGN and MGN, it is reasonable to suggest that U-II may play a role in this mechanism, probably in the secondary immune mechanisms of glomerular injury, by a paracrine or an endocrine action. The abundant expression of hU-II in the proximal and distal tubules of children (patients and controls), may also suggest a possible endocrine role for U-II in electrolyte handling (47,48). Interestingly, we observed abundant U-II immunoreactivity in crescents. Crescents are composed of large, swollen cells arising from both macrophages of hematogenous origin and native parietal epithelial cells (51). As time elapses, the cellular crescents are progressively replaced by fibroblasts, and in more advanced stages, the fibroblastic component is entirely replaced by collagenous lamellar materials with a few remnant cells (52). Recent reports have shown a mitogenic role for U-II through induction of smooth muscle cell proliferation (53,54), and additionally, it has been shown to induce collagen deposition by fibroblasts (55). Although we could not detect the cellular source(s) of U-II in the crescents from that study, we speculate that it may play an important role in crescent formation (47). MGN is an antibody-mediated disease of uncertain and imprecise pathogenesis. However, the hypothesis that it is an autoimmune disease of the kidney, and that the subepithelial immune deposits are formed in-situ with an endogenous glomerular antigen are attractive (56). The electron-dense deposits are generally located at the site of the slit diaphragm, and subepithelial space, while no electron-dense deposits are seen in the subendothelial space or in the mesangium, and hypertension at onset is associated with a less favorable outcome in MGN (56). In our study, hU-II expression was mostly seen in GBM and Bowman capsule, and there was a strong positive correlation between diastolic BP and intensity of U-II expression in Bowman capsule. These findings may raise two interesting questions: May U-II play a role in the formation of these deposits as a mitogenic factor, and may it have any role in the clinical course of MGN by regulating the BP ? However, it is difficult to answer these questions with that study and these hypotheses must be clarified by further studies. In kidneys of 5 children with Henoch-Schönlein nephritis (HSN) and 5 with IgA nephropathy (IgAN), similarly to each other, more dense U-II immunoreactivity was seen in GBM, glomerular mesangium, Bowman capsule, proximal and distal tubules. We also observed U-II immunoreactivity in crescents (48). The pathogenesis of IgAN and HSN is unknown (57). Animal studies have shown the key role of cytokines and growth factors (particularly PDGF and TGFg) in the induction and resolution of mesangial injury and there is some evidence that these are also involved in IgAN (57). The similar expression pattern of U-II in HSN and IgAN has been considered that U-II may have a role in mesangial inflammation and crescent formation, as we mentioned above. In 3 patients with FSGS, deposition of U-II was similar to MPGN patients, but interestingly there was also abundant expression of U-II in sclerotic areas (48). The pathogenesis of glomerulosclerosis is still unknown. Several factors, cytokines and growth factors, hyperlipidemia and platelet activation lead to an increase of mesangial matrix production by resident cells. Several data demonstrate that abnormal glomerular growth is associated with glomerular sclerosis (58). Considering the abundant expression of U-II in sclerotic areas, we may suggest that U-II may also play a role in the progression of glomerular sclerosis, probably as a growth factor or as an inflammatory peptide. This hypothesis must be investigated in the future.
In summary, NO, AM, and U-II may be important mediators in kidney diseases. Whether the observed findings are primary or secondary to these pathological conditions still remains unclear. They suggest a possible role of these peptides in the pathophysiology of childhood glomerulonephritis. Further, detailed studies are needed to address the exact roles of them in children with renal diseases. I think that kidney will always be in trouble with mediators and, “Every mediator is like a bullet for kidney. Some of them miss it, some glance off but some hit it”.
REFERENCES
- 1.Neveu H, Ito S, Ren Y.L. Evidence for the role of nitric oxide in macula densa control of glomerular hemodynamics. J. Clin. Invest. 1993;92:1093–1098. doi: 10.1172/JCI116615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garg U.C, Hassid A. Inhibition of rat mesangial cell mitogenesis by nitric oxide-generating vasodilators. Am. J. Physiol. 1989;257:F60–F66. doi: 10.1152/ajprenal.1989.257.1.F60. [DOI] [PubMed] [Google Scholar]
- 3.Shinde U.A, Mehta A.A, Goyal R.K. Nitric oxide: a molecule of the millennium. Indian J. Exp. Biol. 2000;38(3):201–210. [PubMed] [Google Scholar]
- 4.Narita I, Border W, Ketteler M, Noble N.A. Nitric oxide mediates immunologic injury to kidney mesangium in experimental glomerulonephritis. Lab. Invest. 1995;72:17–24. [PubMed] [Google Scholar]
- 5.Nathan C, Xie Q.W. Regulation of biosynthesis of nitric oxide. J. Biol. Chem. 1994;269:13725–13728. [PubMed] [Google Scholar]
- 6.Kitamura K, Kangawa K, Kawamoto M, Ichiki Y, Nakamura S, Matsuo H, Eto T. Adrenomedullin: a novel hypotension peptide isolated from human pheochromocytoma. Biochem. Biophys. Res. Commun. 1993;192:553–560. doi: 10.1006/bbrc.1993.1451. [DOI] [PubMed] [Google Scholar]
- 7.Kangawa K, Kitamura K, Minamino N, Eto T, Matsuo H. Adrenomedullin: a new hypotensive peptide 1996. J. Hypertension. 1996;14:S105–S110. [PubMed] [Google Scholar]
- 8.Samson W.K. Adrenomedullin and the control of fluid and electrolyte homeostasis. Annu. Rev. Physiol. 1999;61:363–389. doi: 10.1146/annurev.physiol.61.1.363. [DOI] [PubMed] [Google Scholar]
- 9.Miller M.L, Pachman L.M. Vasculitis syndromes. In: Behrman R.E, Kliegman R.M, Jenson H.B, editors. Nelson textbook of pediatrics. 16th edn. Philadelphia: WB Saunders; 2000. pp. 727–735. [Google Scholar]
- 10.Islek I, Balat A, Cekmen M, Yurekli M, Muslu A, Sahinoz S, Sivasli E. Adrenomedullin and total nitrite levels in children with Henoch-Schonlein purpura. Pediatr. Nephrol. 2003;18(11):1132–1137. doi: 10.1007/s00467-003-1193-2. [DOI] [PubMed] [Google Scholar]
- 11.Soylemezoglu O, Ozkaya O, Erbas D, Atok N, Buyan N, Hasanoglu E. Nitric oxide in Henoch-Schonlein purpura. Scand. J. Rheum. 2002;31:271–274. doi: 10.1080/030097402760375151. [DOI] [PubMed] [Google Scholar]
- 12.Farrell A.J, Blake D.R. Nitric oxide. Ann. Rheum. 1996;55:7–20. doi: 10.1136/ard.55.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lincoln J, Hoyie C.H, Burnstock G. Nitric oxide in health and disease. Cambridge: Cambridge University Press; 1997. pp. 22–41. [Google Scholar]
- 14.Besbas N, Saatci U, Ruacan S, Ozen S, Sungur A, Bakkaloglu A, Elnahas A.M. The role of cytokines in Henoch Schonlein purpura. Scand. J. Rheumatol. 1997;26:456–460. doi: 10.3109/03009749709065719. [DOI] [PubMed] [Google Scholar]
- 15.Nathan C, Xie Q.W. Nitric oxide synthases: roles, tolls, and controls. Cell. 1994;78:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 16.Shultz P.J, Archer S.L, Rosenberg M.E. Inducible nitric oxide synthase mRNA and activity in glomerular mesangial cells. Kidney Int. 1994;46:683–689. doi: 10.1038/ki.1994.321. [DOI] [PubMed] [Google Scholar]
- 17.Markewitz B.A, Michael J.R, Kohan D.E. Cytokine-induced expression of nitric oxide synthase in rat renal tubule cells. J. Clin. Invest. 1993;91:2138–2143. doi: 10.1172/JCI116439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kohno M, Yasunari Y, Yokokawa K, et al. Interaction of adrenomedullin and platelet-derived growth factor on the production of endothelin. Hypertension. 1996;27:663–667. doi: 10.1161/01.hyp.27.3.663. [DOI] [PubMed] [Google Scholar]
- 19.Sugo S, Minamino N, Shoji H, et al. Interleukin-1, tumor necrosis factor and lipopolysaccharide additively stimulate production of adrenomedullin in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1995;207:25–32. doi: 10.1006/bbrc.1995.1148. [DOI] [PubMed] [Google Scholar]
- 20.Chini E.N, Choi E, Grande J.P, Burnett J.C, Dousa T.P. Adrenomedullin suppresses mitogenesis in rat mesangial cells via cAMP pathway. Biochem. Biophys. Res. Commun. 1995;215:868–873. doi: 10.1006/bbrc.1995.2544. [DOI] [PubMed] [Google Scholar]
- 21.Kohno M, Yasunari K, Minami M, et al. Regulation of rat mesangial cell migration by platelet-derived growth factor, angiotensin II, and adrenomedullin. J. Am. Soc. Nephrol. 1999;10:2495–2502. doi: 10.1681/ASN.V10122495. [DOI] [PubMed] [Google Scholar]
- 22.Kubo A, Nishitani Y, Minamino N, et al. Adrenomedullin gene transcription is decreased in peripheral blood mononuclear cells of patients with IgA nephropathy. Nephron. 2000;85:201–206. doi: 10.1159/000045662. [DOI] [PubMed] [Google Scholar]
- 23.Bartter F.C. Bartter's syndrome: a disorder of vascular reactivity. Hypertension. 1981;3:69–73. doi: 10.1161/01.hyp.3.3_pt_2.i69. [DOI] [PubMed] [Google Scholar]
- 24.Zipser R.D, Rude R.K, Zia P.K, Fichman MP. Regulation of urinary prostaglandins in Bartter's syndrome. Am. J. Med. 1979;67:263–267. doi: 10.1016/0002-9343(79)90401-7. [DOI] [PubMed] [Google Scholar]
- 25.Delaney V.B, Oliver J.F, Simms M, Costello J, Bourke E. Bartter's syndrome: physiological and pharmacological studies. J. Med. 1981;198:213–232. [PubMed] [Google Scholar]
- 26.Calo L, D'Angelo A, Cantaro S, et al. Increased urinary NO2-/NO3- and cyclic guanosine monophosphate levels in patients with Bartter's syndrome: relationship to vascular reactivity. Am. J. Kidney Dis. 1996;27:784–789. doi: 10.1016/s0272-6386(96)90514-4. [DOI] [PubMed] [Google Scholar]
- 27.Balat A, Çekmen M, Yürekli M, et al. Adrenomedullin and total nitrite levels in children with Bartter's syndrome. Pediatr. Nephrol. 2000;15:266–270. doi: 10.1007/s004670000474. [DOI] [PubMed] [Google Scholar]
- 28.Chini E.N, Chini C.C.S, Bolliger C, et al. Cytoprotective effects of adrenomedullin in glomerular cell injury: central role of cAMP signaling pathway. Kidney Int. 1997;52:917–925. doi: 10.1038/ki.1997.413. [DOI] [PubMed] [Google Scholar]
- 29.Sato K, Imai T, Iwashina M, Marumo F, Hirata Y. Secretion of adrenomedullin by renal tubular cell lines. Nephron. 1998;78:914. doi: 10.1159/000044875. [DOI] [PubMed] [Google Scholar]
- 30.Shaloub R.J. Pathogenesis of lipoid nephrosis: a disorder of T cell function. Lancet. 1974;II:556–569. doi: 10.1016/s0140-6736(74)91880-7. [DOI] [PubMed] [Google Scholar]
- 31.Kubo A, Kurioka H, Minamino N, et al. Plasma and urinary levels of adrenomedullin in chronic glomerulonephritis patients with proteinuria. Nephron. 1998;80:227–230. doi: 10.1159/000045172. [DOI] [PubMed] [Google Scholar]
- 32.Balat A, Çekmen M, Yürekli M, et al. Adrenomedullin and total nitrite levels in children with minimal change nephrotic syndrome. Pediatr. Nephrol. 2000;15:70–73. doi: 10.1007/s004670000440. [DOI] [PubMed] [Google Scholar]
- 33.Schnaper H.W, Aune T.M. Identification of the lympho^kinesoluble immune response suppressor in urine of nephrotic children. J. Clin. Invest. 1985;76:341–349. doi: 10.1172/JCI111967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saxena S, Mittal A, Andal A. Pattern of interleukins in minimal change nephrotic syndrome of childhood. Nephron. 1993;65:5661. doi: 10.1159/000187441. [DOI] [PubMed] [Google Scholar]
- 35.Trachtman H, Gauthier B, Frank R, Futterweit S, Goldstein A, Tomczak J. Increased urinary nitrite excretion in children with minimal change nephrotic syndrome. J. Pediatr. 1996;128:173–176. doi: 10.1016/s0022-3476(96)70385-2. [DOI] [PubMed] [Google Scholar]
- 36.Totsune 1С, Takahashi 1С, Arihara Z, et al. Role of urotensin II in patients on dialysis. Lancet. 2000;358:81–811. doi: 10.1016/S0140-6736(01)06002-0. [DOI] [PubMed] [Google Scholar]
- 37.Pearson D, Shively J.E, Clark B.R, et al. Urotensin-II: a somatostatin like-peptide in the caudal neurosecretory system of fishes. Proc. Natl. Acad. Sci USA. 1980;77:5021–5024. doi: 10.1073/pnas.77.8.5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coulouarn Y, Lihrmann I, Jegou S, et al. Cloning of the cDNA encoding the urotensin II precursor in frog and human reveals intense expression of the urotensin II gene in motorneurons of the spinal cord. Proc. Natl. Acad. Sci. USA. 1998;95:15803–15808. doi: 10.1073/pnas.95.26.15803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maguire J.J, Kuc R.E, Wiley ICE, Kleinz M.J, Davenport A.P. Cellular distribution of immunoreactive urotensin-II in human tissues with evidence of increased expression in atherosclerosis and a greater constrictor response of small compared to large coronary arteries. Peptides. 2004;25(10):1767–1774. doi: 10.1016/j.peptides.2004.01.028. [DOI] [PubMed] [Google Scholar]
- 40.Affolter J, Webb D.J. Urotensin II: a new mediator in cardiopulmonary regulation? Lancet. 2001;358:774–775. doi: 10.1016/S0140-6736(01)06005-6. [DOI] [PubMed] [Google Scholar]
- 41.Bottrill F.E, Douglas S.A, Hiley C.R, White R. Human urotensin-II is an endothelium-dependent vasodilator in rat small arteries. Br. J. Pharmacol. 2000;130:1865–1870. doi: 10.1038/sj.bjp.0703513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheung B.M, Leung R, Man Y.B, Wong L.Y. Plasma concentration of urotensin II is raised in hypertension. J. Hypertens. 2004;22(7):1341–1344. doi: 10.1097/01.hjh.0000125452.28861.f1. [DOI] [PubMed] [Google Scholar]
- 43.Russell F.D, Meyers D, Galbraith A.J, et al. Elevated plasma levels of human urotensin-II immunoreactivity in congestive heart failure. Am. J. Physiol. Heart Circ Physiol. 2003;285:H1576–H1581. doi: 10.1152/ajpheart.00217.2003. [DOI] [PubMed] [Google Scholar]
- 44.Balat A, Pakır H.İ, Gök F, Anarat R, Sahinoz S. Urotensin-II levels in children with minimal change nephrotic syndrome. Pediatr. Nephrol. 2005(20):42–45. doi: 10.1007/s00467-004-1716-5. [DOI] [PubMed] [Google Scholar]
- 45.Balat O, Aksoy F, Kutlar I, Ugur M.G, Iyikosker H, Balat A, Anarat R. Increased plasma levels of Urotensin-II in preeclampsia-eclampsia: a new mediator in pathogenesis?Eur. J. Obstet. Gynecol. Reprod. Biol. 2005;120(1):33–38. doi: 10.1016/j.ejogrb.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 46.Shenouda A, Douglas S.A, Ohlstein E.H, Giaid A. Localization of urotensin-II immunoreactivity in normal human kidneys and renal carcinoma. J. Histochem. Cytochem. 2002;50:885–889. doi: 10.1177/002215540205000702. [DOI] [PubMed] [Google Scholar]
- 47.Balat A, Karakok M, Yilmaz K, Kibar Y. Urotensin-II immunoreactivity in children with chronic glomerulonephritis. Ren. Fail. 2007;29(5):573–578. doi: 10.1080/08860220701392108. [DOI] [PubMed] [Google Scholar]
- 48.Balat A. Urotensin-II in children with renal diseases: New messages from an ancient peptide. Columnista Experta de SIIC. 2007. www.siicsalud.com/des/des0s3/0760s001.hhtm .
- 49.Ames R.S, Sarau H.M, Chambers J.K, et al. Human urotensin-II is a potent vasoconstrictor and agonist for the orphan receptor GPR 14. Nature. 1999;401:282–286. doi: 10.1038/45809. [DOI] [PubMed] [Google Scholar]
- 50.Eddy A.A. Immune mechanisms of glomerular injury. In: Barratt TM, Avner ED, Harmon WE, editors. Pediatric Nephrology. 4th Ed. Baltimore: Lippincott Williams&Wilkins; 1999. pp. 641–668. [Google Scholar]
- 51.Rosen S. Crescentic glomerulonephritis. Occurrence, mechanisms and prognosis. Pathol Annu. 1975;10:37–61. [PubMed] [Google Scholar]
- 52.Cole B.R, Salinas-Madrigal L. Acute proliferativeglomerulonephritis and crescentic glomerulonephritis. In: Barratt TM, Avner ED, Harmon WE, editors. Pediatric Nephrology. 4th Ed. Baltimore: Lippincott Williams&Wilkins; 1999. pp. 669–689. [Google Scholar]
- 53.Sauzeau V, Le M.E, Bertoglio J, Scalbert E, Pacaud Loirand G. Human urotensin-II induced contraction and arterial smooth muscle cell prolipheration are mediated by RhoA and Rho kinase. Circ. Res. 2001;88:1102–1104. doi: 10.1161/hh1101.092034. [DOI] [PubMed] [Google Scholar]
- 54.Watanebe T, Pakala R, Katagiri T, Benedict C.R. Synergistic effect of urotensin-II with mildly oxidized LDL on DNA synthesis in vascular smooth muscle cells. Circulation. 2001;104:16–18. doi: 10.1161/hc2601.092848. [DOI] [PubMed] [Google Scholar]
- 55.Tzanidis A, Hannan R.D, Thomas W.G, et al. Direct actions of urotensin II on the heart: implications for cardiac fibrosis and hypertrophy. Circ. Res. 2003;93:246–253. doi: 10.1161/01.RES.0000084382.64418.BC. [DOI] [PubMed] [Google Scholar]
- 56.Makker S.P. Membranous glomerulonephropathy. In: Barratt T.M, Avner E.D, Harmon W.E, editors. Pediatric Nephrology. 4th Ed. Baltimore: Lippincott Williams&Wilkins; 1999. pp. 719–730. [Google Scholar]
- 57.White R.H.R, Yoshikawa N, Feehally J. IgA nephropathy and Henoch-Schönlein nephritis. In: Barratt TM, Avner ED, Harmon WE, editors. Pediatric Nephrology. 4th Ed. Baltimore: Lippincott Williams&Wilkins; 1999. pp. 691–706. [Google Scholar]
- 58.Niaudet P. Steroid-resistant idiopathic nephrotic syndrome. In: Barratt TM, Avner ED, Harmon WE, editors. Pediatric Nephrology. 4th Ed. Baltimore: Lippincott Williams&Wilkins; 1999. pp. 749–763. [Google Scholar]