

CLL is a highly heritable cancer, with a 7.5-fold increased risk in first-degree relatives1. However, inherited predisposition to CLL remains largely unexplained by traditional linkage or genome-wide association studies. Here, we hypothesized that CLL heritability might arise from rare coding variants not analyzed in previous studies.
We compared rare germline variants (minor allele frequency < 0.01) in coding regions of 516 samples from CLL patients of European descent to those found in 8,920 ethnically matched, normal population controls. This represents the largest and most comprehensive search for risk alleles in CLL exomes to date. To maximize our power to detect significant associations, we combined data from multiple sequencing studies (see Supplementary Methods and Tables S1-S2 for cohort descriptions).
An important consideration when aggregating samples across multiple sequencing studies is controlling for biological and technical heterogeneity. Differences in patient ethnicities, sequencing technologies, depth of coverage, and variant calling methods may give rise to spurious results. Here, we controlled for these factors by: (i) simultaneously processing original sequencing data from all cohorts; (ii) jointly calling variants across all cases and controls; and (iii) analyzing only ethnically matched, unrelated samples over DNA sites with sequencing coverage sufficient to achieve high-confidence genotype calls across the entire sample cohort. We then performed an unbiased, exome-wide rare variant burden test on cases and controls (Figures S1-S2).
We identified two genes significantly associated with CLL (q≤0.05): CDK1 and ATM. CDK1, a gene that encodes a cyclin-dependent kinase critical for cell division, was significantly enriched for rare, non-synonymous germline variants in CLL cases versus controls (p = 5.75 × 10-7, Table 1). One recurrent missense variant, CDK1 p.R59C (rs8755), was observed in 5 cases and 10 controls (Table S3). This missense variant lies in the CDK1 kinase domain (Figure S3) and is predicted to be Possibly Damaging by the PolyPhen2 prediction tool2, suggesting that the variant may affect protein function.
Table 1. Significant hits in gene-based rare variant burden test for CLL.
| Gene | p-value | FDR (q-value) | # of Cases with Rare Variants (%) | # of Controls with Rare Variants (%) | OR (95% CI) | Fisher OR (95% CI) |
|---|---|---|---|---|---|---|
| Discovery Cohort | ||||||
| CDK1 | 5.75 × 10-7 | 0.0091 | 8 (1.6%) | 24 (0.3%) | 5.8 (2.6 – 13.1) | 5.83 (2.25-13.5) |
| ATM | 1.43 × 10-6 | 0.011 | 112 (21.7%) | 1296 (14.5%) | 1.6 (1.3 – 2.0) | 1.66 (1.34-2.06) |
| Extension Cohort* | ||||||
| CDK1 | 2.17 × 10-4 | 0.107 | 8 (1.2%) | 26 (0.3%) | 4.28 (1.93-9.51) | 4.29 (1.67-9.8) |
| ATM | 1.04 × 10-8 | 1.7 × 10-4 | 170 (26.3%) | 1483 (16.6%) | 1.79 (1.49-2.15) | 1.79 (1.49-2.15) |
Due to the inclusion of additional case samples, some variant allele frequencies were altered, leading to an increase in the number of variants that met allele frequency and quality control thresholds. This led to an increase in the number of controls with rare variants in the extension call set.
The second significant gene we identified was ATM (p = 1.43 × 10-6, Table 1), a well-known tumor suppressor gene on chromosome 11q. One of the most enriched recurrent variants is L2307F (2.3% cases, OR = 10.1, 4.9-20.7). Interestingly, L2307F has been previously reported in two CLL cases and a breast cancer case, the latter segregating in a family also affected with hematologic malignancy.3, 4 The L2307F variant lies in the FAT domain of the ATM protein (Figure 1A) and is predicted to be Probably Damaging by PolyPhen2. Subsequent targeted sequencing using Sequenom technology in an independent set of 149 CLL cases revealed a similar frequency of 2.01% (3 out of 149) for the L2307F variant. In 27 cases with available RNASeq data, expression of the rare germline ATM variant was confirmed, and in all but one case, the alternate allele fractions in RNA transcripts were consistent with those in germline genomic DNA (Table S4).
Figure 1. CLL patients harbor multiple ATM lesions: germline variants, somatic mutations, and loss of heterozygosity.
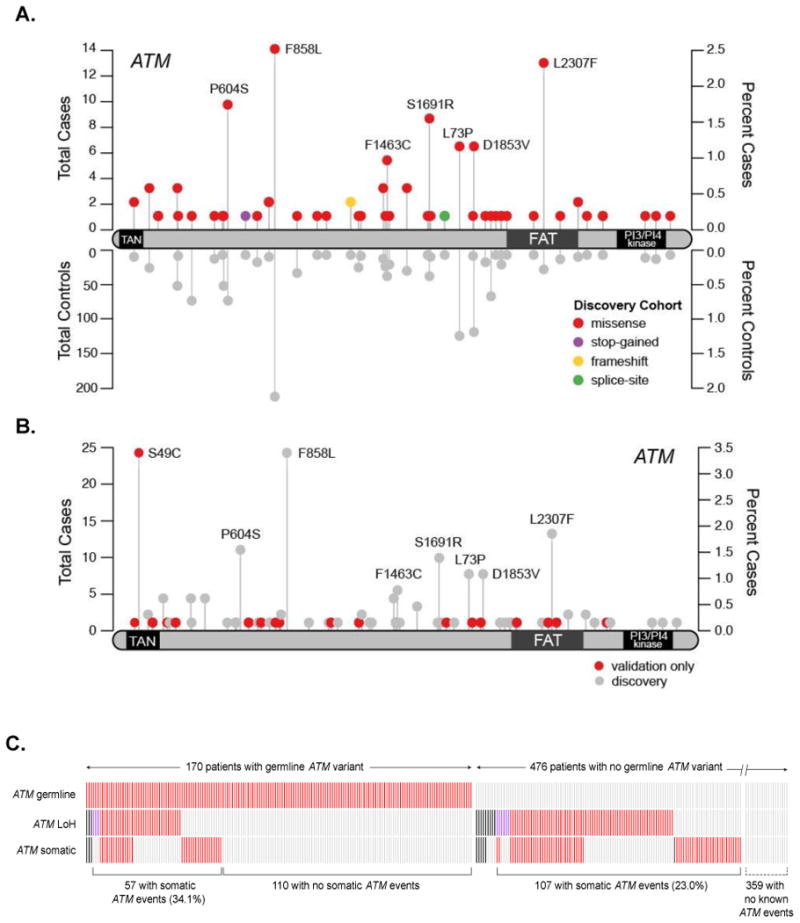
A. Rare germline variants in ATM are enriched in CLL cases (discovery cohort). Variants found in CLL cases are displayed with the total number of cases (above the protein track) and controls (below), along with the corresponding percentages in their respective cohorts. B. Additional rare germline variants in ATM were observed in the extension cohort. C. Co-occurrence of ATM lesions in a total of 646 patient samples: rare germline variants (top row), loss-of-heterozygosity (middle row), and non-synonymous somatic mutations (bottom row). Samples are arranged along the x-axis (unlabeled). Presence of the genetic lesion is indicated in red, absence of the lesion is marked by grey. Missing data are displayed in black. Samples with copy-neutral LOH are indicated by purple boxes; the remaining LOH events are 11q deletions. Percentages of samples with somatic ATM events are labeled in parentheses for the patient cohorts with germline and no germline ATM events, respectively. Data for samples with no known germline or somatic ATM events (359 patients) are not shown to scale due to space limitations.
The majority of the recurrent variants observed in ATM were non-synonymous missense variants (Table S6) in contrast to the predominantly loss-of-function alleles seen in ataxia-telangiectasia, a hereditary disorder associated with increased risk of leukemias and lymphomas. Analysis of the frequency-weighted distribution of PolyPhen2 scores across these missense variants revealed a significant shift toward more damaging scores in the cases versus controls (p=0.0038, one-sided Kolmogorov-Smirnov test; Figure S4). These observations are consistent with recent reports of the potential role of germline missense variants in cancer heritability5, 6. In fact, 22 of the rare missense ATM variants we identified in CLL cases were also associated with breast cancer risk in a meta-analysis of breast cancer studies7.
As an extension to our initial findings, we studied two additional cohorts of CLL cases (n=106 exomes, n=24 genomes). We combined these additional cases with our 516 original cases and compared against our original control cohort. This expanded joint analysis approach has been shown to consistently improve the statistical power for detecting genetic associations8. We found ATM to be the top hit in this combined analysis (p = 1.04 × 10-8, Table 1, Table S5). ATM variants are summarized in Figure 1B and Table S6 while patient characteristics by ATM germline status are summarized in Table S7. For CDK1, no additional rare variants were found in the extension cohort and hence its significance dropped below our significance threshold (q=0.107, Table 1, Table S8). Patient characteristics by CDK1 germline status are summarized in Table S9.
The classical model of tumor suppressor inactivation involves the loss of both wild-type alleles of the tumor suppressor gene. In CLL, ATM is frequently lost through somatic deletion of the chromosome 11q region that spans the ATM locus9 and through inactivating somatic mutations10, 11. We observed an enrichment of these somatic “second hits” in patient samples harboring rare ATM germline variants. Among the 112 patients in the discovery cohort with ATM germline variants, we found 23 with somatic ATM mutations, 29 with 11q deletions, and 2 with copy-neutral loss of heterozygosity (LOH) in the ATM locus (Figure 1C). The presence of a germline ATM variant was significantly associated with the occurrence of an ATM somatic mutation (OR=1.79, 95% CI 0.98-3.16; p=0.047, two-tailed Fisher exact test), as well as with LOH, either copy-neutral or via 11q deletion (OR=1.68, 95% CI 0.98-2.82; p=0.042, two-tailed Fisher exact test). Overall, the presence of a rare germline ATM variant was significantly associated with the presence of at least one of these somatic events (OR=2.17, 95% CI 1.35-3.48; p=9.1×10-4, two-tailed Fisher exact test). The association remained significant in the extension cohort (OR=1.74, 95% CI 1.16-2.60; p=5.5×10-3, two-tailed Fisher exact test). The observation that patients with rare coding germline variants in ATM were significantly more likely to harbor a second inactivating somatic lesion in ATM suggests that rare germline variants in ATM behave as tumor suppressor alleles.
To test further whether the rare germline ATM variants are likely to be functional, we examined which ATM allele is lost when 11q is deleted. If the variants had no effect on the development of CLL, we would expect an equal likelihood of their loss or retention in 11q deleted cases. Strikingly, we found that patients with rare germline ATM variants who acquired an 11q deletion more often lost the wild-type ATM allele. Specifically, 80% carried only the rare variant germline allele in their tumor samples (16 out of 20 with clonal or near-clonal 11qdel). This rate of wild-type allele loss is significantly greater than expected by chance (p=0.012, two-tailed binomial test). Furthermore, in the four cases that lost the variant allele, none had Probably Damaging PolyPhen2 scores, suggesting that their effect on protein function was less severe. These results suggest that many rare germline ATM alleles may confer selective advantage to malignant B-cells.
Given the association of somatic 11q deletion9 or ATM mutation12 with worse clinical outcomes in CLL, we investigated whether the presence of a rare germline variant in ATM would be associated with worse clinical outcomes. In a Cox regression analysis adjusting for treatment arm, somatic del(11q), but not germline ATM variant, was a significant predictor of outcome (Tables S10-S11, Figure S5). When we added two other CLL prognostic factors, del(17p) and IGHV status, to the Cox regression analysis (Table S12), del(11q) was no longer significantly associated with PFS. We also investigated the effect of a rare germline ATM variant and/or 11q deletion on overall survival, and saw no effect, including in a Cox regression analysis adjusting for treatment arm (Table S13, Figure S6).
Taken together, our results show that rare, protein-coding germline variants in ATM are frequent events in CLL, with ATM behaving as a classic tumor suppressor gene, showing preferential somatic loss of the wild-type allele. Although previous research has hinted at a role for specific alleles of ATM in CLL risk, these studies either involved relatively small sample sizes, resulting in low statistical power, or were targeted approaches that did not evaluate the entire ATM gene or did not evaluate ATM against other potential risk genes in an unbiased, exome-wide manner13-15. In contrast, we have applied consistent technical processing to a large cohort of jointly-called, ethnically matched CLL and normal population controls, with a focus on rare coding variants. This, along with careful quality control, variant filtering, and accounting for population substructure in an unbiased, exome-wide association analysis, allowed us to identify ATM as a CLL risk gene.
Because CLL predominantly affects individuals of European descent, we chose to focus our study on patients of European ethnicity. We expect, however, that further studies examining patients of different ethnic backgrounds may uncover additional germline risk genes not detectable in the European study cohort. Indeed, the presence of residual population substructure within the cohort of European subjects in this study suggests that there may be germline predisposition genes affecting different European sub-populations that we have not yet identified. Another area requiring further exploration is the search for germline risk factors in familial CLL cases. Our study included many patients for whom the familial or sporadic disease status was not available (n=387), and among those with known status, most were sporadic or without living affected or available relatives (n=195). Larger studies of whole exomes and whole genomes with a focus on familial cases and underrepresented ethnic populations will be needed to further increase power to detect additional risk genes and alleles for CLL. The approach we describe here, combining rare variant association with somatic sequencing analysis, can be applied to any type of heritable cancer and holds great promise for identifying new germline cancer predisposition alleles as progressively larger cohorts of germline cancer cohorts are sequenced.
Supplementary Material
Acknowledgments
The authors thank all patients and physicians for trial participation and sample donation. We would also like to thank Myriam Mendila, Nancy Valente, Stephan Zurfluh, Michael Wenger, and Jamie Wingate for their support in the CLL8 trial conception and conduct.
Grant Support: J.R. Brown is a clinical scholar of the Leukemia and Lymphoma Society and is supported by the Leukemia and Lymphoma Society (TRP# 6289-13) and the American Cancer Society (RSG-13-002-01-CCE). J.R. Brown would also like to acknowledge the support of the Melton Family Fund for CLL Research, the Susan and Gary Rosenbach Fund for Lymphoma Research, and the Okonow Lipton Family Lymphoma Research Fund. C.J. Wu acknowledges support from the Blavatnik Family Foundation and NIH/NCI (1R01CA182461-02, 1R01CA184922-01, 1U10CA180861-01). C.J. Wu is a Scholar of the Leukemia and Lymphoma Society. S.S. and E.T. are supported by the Else Kröner-Fresenius-Stiftung (2010_Kolleg24, 2012_A146) and Deutsche Forschungsgemeinschaft (SFB 1074 projects B1, B2). Genetic analyses in CLL8 were supported by Roche.
Footnotes
Conflict of Interest: No potential conflicts of interest were disclosed.
Author Contributions: Conception and design: GT, MRI, AK, GG, JRB; Development of methodology: GT, MRI, SK, WP, DAL, ATW, AK, GG, JRB. Acquisition of data: GT, MRI, SK, WP, AK, DAL, MH, SS, CJW, GG, JRB; Data analysis and interpretation: GT, MRI, SK, WP, AK, ET, HTK, ER, JB, SR, KF, AK, GG, JRB. Writing, review, and/or revision of the manuscript: GT, MRI, DAL, ESL, GG, JRB; Administrative, technical, or material support: GT, MRI, SK, CC, SB, SMF, KH, SG; Study supervision: GG, JRB.
References
- 1.Goldin LR, Pfeiffer RM, Li X, Hemminki K. Familial risk of lymphoproliferative tumors in families of patients with chronic lymphocytic leukemia: results from the Swedish Family-Cancer Database. Blood. 2004 Sep 15;104(6):1850–1854. doi: 10.1182/blood-2004-01-0341. [DOI] [PubMed] [Google Scholar]
- 2.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010 Apr;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paglia LL, Lauge A, Weber J, Champ J, Cavaciuti E, Russo A, et al. ATM germline mutations in women with familial breast cancer and a relative with haematological malignancy. Breast Cancer Res Treat. 2010 Jan;119(2):443–452. doi: 10.1007/s10549-009-0396-z. [DOI] [PubMed] [Google Scholar]
- 4.Lahdesmaki A, Kimby E, Duke V, Foroni L, Hammarstrom L. ATM mutations in B-cell chronic lymphocytic leukemia. Haematologica. 2004 Jan;89(1):109–110. [PubMed] [Google Scholar]
- 5.Young EL, Feng BJ, Stark AW, Damiola F, Durand G, Forey N, et al. Multigene testing of moderate-risk genes: be mindful of the missense. J Med Genet. 2016 Jan 19; doi: 10.1136/jmedgenet-2015-103398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jhuraney A, Velkova A, Johnson RC, Kessing B, Carvalho RS, Whiley P, et al. BRCA1 Circos: a visualisation resource for functional analysis of missense variants. J Med Genet. 2015 Apr;52(4):224–230. doi: 10.1136/jmedgenet-2014-102766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tavtigian SV, Oefner PJ, Babikyan D, Hartmann A, Healey S, Le Calvez-Kelm F, et al. Rare, evolutionarily unlikely missense substitutions in ATM confer increased risk of breast cancer. American journal of human genetics. 2009 Oct;85(4):427–446. doi: 10.1016/j.ajhg.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nature genetics. 2006 Feb;38(2):209–213. doi: 10.1038/ng1706. [DOI] [PubMed] [Google Scholar]
- 9.Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. The New England journal of medicine. 2000 Dec;343(26):28. 1910–1916. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 10.Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013 Feb 14;152(4):714–726. doi: 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011 Jul 7;475(7354):101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. The New England journal of medicine. 2011 Dec 29;365(26):2497–2506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rudd MF, Sellick GS, Webb EL, Catovsky D, Houlston RS. Variants in the ATM-BRCA2 CHEK2 axis predispose to chronic lymphocytic leukemia. Blood. 2006 Jul 15;108(2):638–644. doi: 10.1182/blood-2005-12-5022. [DOI] [PubMed] [Google Scholar]
- 14.Stankovic T, Weber P, Stewart G, Bedenham T, Murray J, Byrd PJ, et al. Inactivation of ataxia telangiectasia mutated gene in B-cell chronic lymphocytic leukaemia. Lancet. 1999 Jan 2;353(9146):26–29. doi: 10.1016/S0140-6736(98)10117-4. [DOI] [PubMed] [Google Scholar]
- 15.Boultwood J. Ataxia telangiectasia gene mutations in leukaemia and lymphoma. J Clin Pathol. 2001 Jul;54(7):512–516. doi: 10.1136/jcp.54.7.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.