

Abstract
Aquaporin 3 (AQP3), a water/glycerol channel protein, has been found to transport hydrogen peroxide (H2O2). Here, we show that H2O2, imported via AQP3, is involved in nuclear factor-κB (NF-κB) signalling in keratinocytes and in the pathogenesis of psoriasis. IL-23-mediated induction of psoriasis is reduced in AQP3 knockout mice (AQP3 −/−), and is accompanied by impaired NF-κB activation and intracellular H2O2 accumulation. In primary keratinocyte cultures, cellular import of H2O2 produced by membrane NADPH oxidase 2 (Nox2) in response to TNF-α is facilitated by AQP3 and required for NF-κB activation by regulation of protein phosphatase 2A. As AQP3 associates with Nox2, we propose that this interplay constitutes H2O2-mediated signalling in response to TNF-α stimulation. Collectively, these data indicate that AQP3-facilitated H2O2 transport is required for NF-κB activation in keratinocytes in the development of psoriasis.
Psoriasis is a chronic inflammatory skin disease characterized by raised plaques, epidermal hyperplasia and infiltration of leukocytes into the skin, though its precise pathogenesis mechanism remains unclear1,2. Recent genetic studies have advanced our understanding of the pathogenesis of psoriasis. Ten genome-wide association studies have identified more than 20 susceptible loci in psoriasis and suggested several pathogenesis mechanisms, including skin barrier functions, IL-17 and IL-23 signalling, TNF-α and NF-κB signalling and HLA-C3–5. IL-23 and TNF-α-targeted therapies, which neutralize subunits of cytokines IL-12/23 or TNF-α respectively, are in clinical use and improve the quality of life for psoriasis patients6. A recent clinical report showed that an IL-17A neutralizing monoclonal antibody also had efficacy in psoriasis7,8.
There have also been recent advances in signalling pathways involved in the pathogenesis of psoriasis. Increasing evidence implicates IL-17 as a centrally important cytokine in psoriatic skin, which stimulates keratinocytes and promotes chronic inflammation3. There is also recent evidence that activation of NF-κB signalling exacerbates psoriatic lesions, potentially by induction of various genes for cytokines, chemokines and growth factors1,9,10. However, the underlying mechanisms of how these pathways produce psoriasis remain unresolved.
Aquaporin-3 (AQP3), a member of the aquaporin water channel family, is a water- and glycerol-transporting protein that is involved in various cellular functions including cell proliferation and migration11. Previous studies have shown the involvement of AQP3 expression in skin diseases, including wound healing, tumorigenesis and atopic dermatitis, in which their pathogenesis was attributed to the AQP3 water or glycerol transport function in epidermal keratinocytes12–14. Building on a study reporting AQP3-facilitated cellular uptake of H2O2 (ref. 15), a reactive oxygen species (ROS), we reported the involvement of AQP3-mediated H2O2 in T-cell signalling in cutaneous contact hypersensitivity16. We showed that extracellular H2O2 produced in response to CXCL12 stimulation was transported into T cells by AQP3, which facilitated CXCL12-dependent cell signalling and T cell migration. Because H2O2 is emerging as an important second messenger in cell signalling17,18, we postulated that AQP3-facilitated intracellular H2O2 might be implicated in a variety of human disease, including the focus of the study here, psoriasis.
We report here that AQP3 is required for TNF-α-induced NF-κB activation in keratinocytes and the development of psoriasis by a mechanism involving TNF-α-induced generation of extracellular H2O2 followed by its intracellular transport by AQP3. An in vivo mouse model of psoriasis showed remarkably reduced pathology in an AQP3-deficient background, in which continuous NF-κB activation and a higher H2O2 level in epidermal keratinocytes were suppressed by AQP3 deficiency.
Results
Reduced IL-23-induced psoriasis-like inflammation in AQP3 −/− mice
Although previous reports showed reduced AQP3 staining or an altered pattern of AQP3 distribution in psoriatic skin, microarray and RNA-seq studies have suggested an upregulation of AQP3 expression19–22. AQP3 immunostaining was done on 18 human psoriatic skin samples. We found strong AQP3 immunostaining, with two patterns of AQP3 localization in keratinocytes: typical plasma membrane staining for eight specimens, and mixed plasma membrane/cytoplasmic staining for nine specimens (Fig. 1a, left, insert). One specimen showed weak AQP3 staining. Immunofluorescence showed AQP3 expression in CD3 + T cells infiltrating in the dermis (Fig. 1a, right). AQP3 transcript expression was more than eight-fold greater in keratinocytes than in CD3 + T cells by PCR with reverse transcription (RT–PCR; Supplementary Fig. 1a).
Figure 1. Impaired IL-23-induced psoriasis-like skin in AQP3 −/− mice.

(a) Left: representative immunohistochemical staining for AQP3 in the skin from five healthy volunteers and 18 psoriatic patients. Right: immunostaining with anti-AQP3 (cy3, red) and anti-CD3 (FITC, green) in psoriatic skin. Scale bar, 100 μm. Epi, epidermis; der, dermis. (b–f) IL-23 (500 ng) or vehicle control (PBS) was intradermally injected daily into the ear skin of WT and AQP3 −/− mice for 4 days. Skin samples were excised at 24 h after the final IL-23 injection. (b) Left: haematoxylin and eosin staining of ears from WT and AQP3 −/− mice. Scale bar, 100 μm. Arrow head, infiltrating lymphocytes. Right: whole skin and epidermal thickness determined from haematoxylin and eosin staining (s.e., n =5, *P<0.01 by t-test).(c) Immunostaining with anti-AQP3 (FITC, green) and anti-CD3 (cy3, red) in WT mouse skin. Scale bar, 100 μm (left), 20 μm (right). (d) Left: representative immunohistochemical staining for Ki67. Right: the ratio of Ki67 positive cells in the epidermis (s.e., n =3, *P<0.01 by t-test). (e) CD3 + γδTCR + cell numbers in epidermis and dermis from WT and AQP3 −/− mice analysed by flow cytometry (s.e., n =4–5, *P<0.01 by t-test). (f) mRNA expression of IL-17A, IL-17F, IL-22 and IL-19 in skin tissues determined by real-time RT–PCR (s.e., n =4–5,*P<0.01 by t-test). Data are expressed as the ratio to GAPDH.
We used an experimental mouse model of IL-23-induced psoriasis to study the potential role of AQP3 in the pathogenesis of psoriasis23,24. IL-23 was injected into the skin of the ear of wild-type (WT) and AQP3 knockout (AQP3 −/−) mice. Consistent with previous reports, injection of IL-23 into WT skin induced marked ear swelling, accompanied by pronounced acanthosis, hyperparakeratosis and lymphocytic infiltration (Fig. 1b, left). The skin of AQP3 −/− mice showed remarkably less swelling and inflammation after injection of IL-23. Whole skin and epidermal thickness in haematoxylin and eosin-stained sections showed significantly reduced swelling and hyperkeratosis in AQP3 −/− mice (Fig. 1b, right). Immunostaining showed AQP3 expression in keratinocytes and dermal CD3 + T cells in the psoriasis-induced mouse skin (Fig. 1c), similar to observations in human psoriatic skin. The AQP3 messenger RNA (mRNA) expression level in isolated epidermal cells was similar in control and IL-23-treated WT skin (Supplementary Fig. 1b). Immunoblot analysis showed that the increased AQP3 expression after IL-23 injection was associated with greater keratin 5 and 14 expression in epidermis compared with control skin (Supplementary Fig. 1c). Immunohistochemical analysis showed that IL-23 application remarkably increased Ki67 + keratinocytes in WT but not in AQP3 −/− epidermis, suggesting that IL-23 stimulation induced keratinocytes proliferation in WT skin (Fig. 1d). These data indicate that the higher AQP3 expression by immunoblotting is due to an increase in proliferative keratinocytes.
Previous studies showed that IL-23 injection increased the number of CD3 + γδTCR + cells (γδ T cells) in the dermis, which contributes to the development of psoriatic skin by IL-17 production25. We confirmed that CD3 + γδTCR + cells specifically accumulated in IL-23-treated WT skin (Supplementary Fig. 1d). Fluorescence-activated cell sorting (FACS) analysis showed greater numbers of γδ T cells in the dermis after IL-23 treatment in WT than in AQP3 −/− mice (Fig. 1e). Quantitative RT–PCR analysis revealed that continuous IL-23 injection increased mRNAs encoding IL-17A, IL-17F, IL-22 and IL-19 in WT epidermis, as previously reported, with reduced elevations in AQP3 −/− epidermis (Fig. 1f). These data indicate the involvement of AQP3 in the development of psoriasis in the IL-23 mouse model.
Impaired psoriasis is dependent on AQP3 expression
Previous studies implicated a critical role for both T cells and keratinocytes in the pathogenesis of psoriasis as well as in the psoriatic mouse model induced by IL-23 application2,3,23–25. AQP3 was found to be expressed in epidermal keratinocytes and T cells in psoriatic skin (Fig. 1a,c). To investigate the potential significance of AQP3 expression in these cell types individually, lethally irradiated WT and AQP3 −/− mice (recipients) were reconstituted with bone marrow (BM) cells from WT and AQP3 −/− mice (donors; Supplementary Fig. 2a). When these mice were injected with IL-23 to induce psoriatic skin changes, ear swelling measured with a micrometer was increased in chimeric mice produced by transferring WT BM cells into WT recipients (Fig. 2a,b). In contrast, chimeric mice produced by transferring WT BM cells into AQP3 −/− recipients showed reduced IL-23-induced ear swelling, indicating that the development of psoriasis requires the expression of AQP3 in keratinocytes. Transfer of AQP3 −/− BM into WT recipients also suppressed IL-23-induced ear swelling, suggesting the involvement of AQP3 expression in haematopoietic cells, probably in T cells (Fig. 2a,b).
Figure 2. Impaired IL-23-induced psoriasis is dependent on AQP3 expression in both keratinocytes and T cells.

(a,b) IL-23-induced psoriatic model using bone marrow (BM) cell-transferred mice. Recipient WT and AQP3 −/− mice (Rec) received transplants of BM cells from donor WT and AQP3 −/− mice (Don). (a) Ear thickness at 24 h after final injections (s.e., n =5, *P<0.01 by t-test). (b) Representative haematoxylin and eosin staining. Scale bar, 100 μm. (c) Chemotaxis assay. The migration efficiency of CD3 + T cells toward the ligands CCL20 (100 ng ml−1) or CXCL9 (100 ng ml −1) was determined using a transwell chamber with 5 μm pores. Data are expressed as the percentage of applied cells (s.e., n =5, *P<0.01 by t-test). (d) mRNA expression levels of CCR6 in epidermis (left) and CXCR3 in dermis (right) by real-time RT–PCR (s.e., n =5, *P<0.01 by t-test). (e) mRNA expression levels of IL-17A and IL-17F by real-time RT–PCR in sorted T cells incubated with IL-23 in the presence of CD3/28 for 3 days (s.e., n =5, *P<0.01 by t-test). Data are expressed as the IL-17/GAPDH ratio. (f) IL-22 level by ELISA in the culture medium with IL-23 in the presence of anti-IL-4 and anti-IFNγ for 3 days (s.e., n =5, *P<0.01 by t-test).
We next determined the contribution of AQP3-facilitated T-cell chemotaxis to the development of psoriasis, based on our recent demonstration that AQP3 is required for T-cell chemotaxis16. The mRNA levels of CCL20, CXCL9 and CXCL10, key chemokines to recruit T cells or dendritic cells during the development of psoriasis26, were remarkably increased in IL-23-treated WT skin (Supplementary Fig. 2b). The chemotaxis of CD3 + T cells to CCL20 or CXCL9 was significantly reduced in AQP3 −/− cells (Fig. 2c). We also found that expression of CCR6 and CXCR3, the receptors of CCL20 and CXCL9 respectively, was decreased in IL-23-treated AQP3 −/− skin compared with WT skin (Fig. 2d). These results suggest that the impaired T-cell chemotaxis leads to less accumulation of CCR6 + and CXCR3 + T cells in AQP3 −/− skin.
During the development of psoriasis induced by IL-23 application, IL-17 and IL-22 production by T cells in response to IL-23 stimulation is crucial for the following steps23–25. We found comparable expression of mRNA encoding IL-17A/F in WT and AQP3 −/− primary cultured T cells, as well as comparable IL-22 production (Fig. 2e,f). FACS analysis also showed a comparable ratio of IL-17 expressing cells among γδT cells in skin-draining lymph nodes in WT and AQP3 −/− mice following IL-23 application (Supplementary Fig. 2c). From these results, we propose that AQP3 deficiency results in the impaired T-cell migration into the dermal and epidermal layers, which reduces downstream immune response during the development of psoriasis.
AQP3 deficiency suppresses NF-κB activation and H2O2 induction
We next sought to elucidate the role of AQP3 in keratinocytes in the pathogenesis of psoriasis, since the reduced psoriasis in AQP3 −/− mice cannot be accounted for by functions of AQP3 reported previously13,14. Several studies suggest an important role of NF-κB in the pathogenesis of psoriasis1,9,10, including overexpression of active phosphorylated NF-κB in psoriatic epidermis27,28. Immunostaining showed a much greater number of positive keratinocytes for phospho-p65 and phospho-IKKα/β in IL-23-treated WT skin than control skin, as found in humans27,28 (Fig. 3a, Supplementary Fig. 3a). AQP3 −/− epidermis showed fewer cells positive for phospho-p65 and phospho-IKKα/β compared with WT skin. Immunoblot also showed reduced IL-23-induced phosphorylation of Iκβα and its degradation in AQP3 −/− epidermis (Fig. 3b, Supplementary Fig. 7), indicating that NF-κB was activated by IL-23 application in WT but not in AQP3 −/− skin.
Figure 3. AQP3 deficiency impairs H2O2 elevation and NF-κB activation in IL-23-treated skin.

(a,b) Mice were injected with IL-23 into the ear as described in Fig. 1. (a) Immunostaining of phospho-p65 in PBS (control) or IL-23 injected WT and AQP3 −/− skin. Scale bar, 100 μm. (b) Representative immunoblot analysis using antibodies against phospho-Iκβα, Iκβα and β-actin. Experiments were performed in two other independent experiments with similar results. (c–e) Haematoxylin and eosin staining (c), ear thickness (d) and phospho-p65 immunostaining (e) of ears from WT mice treated daily with IL-23 or PBS (control) in the presence of anti-TNF-α blocking monoclonal antibody or isotype control. (f) H2DCFDA fluorescence in the skin treated with IL-23 (three continuous applications) with or without anti-TNF-α antibody. Anti-TNF-α antibody was injected 1 h before sampling. (g) H2DCFDA fluorescence in WT and AQP3 −/− skin treated daily with PBS (control) or IL-23 by fluorescence microscopy. (h) The mean fluorescence intensity (MFI) of H2DCFDA by FACS analysis in epidermal cells (s.e., n = 5, *P<0.05, **P<0.01 by t-test).
Reported evidence suggests that TNF-α-dependent NF-κB signalling is required for the development of psoriasis1,9,10. A previous study suggested that IL-23-induced psoriatic changes are dependent on TNF-α-mediated cell signalling in mice, as pretreatment with a blocking monoclonal TNF-α antibody reduced psoriatic changes in 129SvEv mice24. We confirmed that administration of TNF-α antibody attenuated IL-23-induced skin swelling in WT mice (C57B/6 background; Fig. 3c,d). NF-κB activation was suppressed with TNF-α antibody, with significantly fewer phospho-p65-positive cells than with an IgG-isotype control (Fig. 3e, Supplementary Fig. 3c), which is consistent with the data in human psoriasis27.
It has been also suggested that TNF-α-dependent NF-κB activation is H2O2-regulated29,30. To test our hypothesis that AQP3-mediated intracellular H2O2 is a central signalling event in the development of psoriasis, we measured cellular H2O2 levels in skin using a previously described method31. Injections of IL-23 increased intracellular H2O2 in epidermal keratinocytes and in some dermal cells in WT mice (Fig. 3f). The administration of TNF-α antibody before IL-23 application reduced the increase in intracellular in H2O2 (Fig. 3f), suggesting IL-23-induced H2O2 accumulation was partially dependent on TNF-α cell signalling. In addition, the H2O2 level was greater in WT epidermal cells than in AQP3 −/− cells after IL-23 application (Fig. 3g). Finally, we quantified cellular H2O2 in epidermal cells by FACS analysis. After trypsinization of the epidermal sheet, single-cell suspensions were incubated with H2DCFDA, as validated for measurement of intracellular H2O2 (Supplementary Fig. 3d). The H2DCFDA fluorescence in epidermal cells showed significantly higher H2O2 in WT than in AQP3 −/− epidermal cells after IL-23 application (Fig. 3h).
As further evidence for the involvement of AQP3 in the development of psoriasis, we performed an imiquimod (IMQ)-induced psoriasis model32. The IMQ-treated AQP3 −/− mice showed reduced ear swelling and psoriasis-like phenotype (acanthosis and lymphocytic infiltration) compared with WT skin (Supplementary Fig. 4a,b). AQP3 was expressed in a wide range of hyperplastic psoriatic epidermis (Supplementary Fig. 4c). The IMQ-treated WT epidermal cells showed an increased number of phospho-p65-positive and a higher ROS level, as seen in IL-23-treated WT skin (Supplementary Fig. 4d,e).
Together, these findings provide evidence for involvement of AQP3 in H2O2 induction and NF-κB activation in IL-23-induced psoriatic changes in skin, which are dependent on TNF-α signalling.
Impaired TNF-α-induced NF-κB activation in AQP3 −/−
We next investigated the involvement of AQP3 in TNF-α-induced NF-κB activation in primary keratinocytes from WT and AQP3 −/− mice. Immunoblot analysis showed that 1-h TNF-α stimulation induced the phosphorylation of p65/NF-κB in WT cells, which was reduced in AQP3 −/− cells (Fig. 4a, Supplementary Fig. 6a and 7). TNF-α-induced signalling is mediated by one of the two cell-surface receptors, TNFR1 and TNFR2 (ref. 33). Classical NF-κB activation following TNF-α stimulation is initiated by recruitment of the death domain-containing proteins RIP1 and TRADD, after binding of TNF receptor (TNFR)-associated factor 2 (TRAF2) to TNFR1 (refs 33,34). A previous study showed that the recruitment of TRAF2 to TNFR1 in response to TNF-α was dependent on the ROS level35. Immunoprecipitation with anti-TNFR1 showed that AQP3 deficiency did not affect the recruitment of TRAF2 and RIP1 to TNFR1 upon TNF-α stimulation (Fig. 4b, Supplementary Fig. 7). In contrast, AQP3 −/− keratinocytes showed reduced TNF-α-induced phosphorylation of IKKβ and Iκβα and degradation of Iκβα (Fig. 4a, Supplementary Figs 6a and 7).
Figure 4. Impaired TNF-α-induced NF-κB activation in AQP3-deficient keratinocytes.

(a,b) Primary keratinocytes from WT and AQP3 −/− mice were incubated with TNF-α (100 ng ml −1) for 5 min to 1 h. (a) Representative immunoblot using antibodies against phospho-IKKβ, IKKβ, phospho-Iκβα, Iκβα, phospho-p65, p65 and β-actin at indicated times. (b) Cell lysate (TNF-α, 100 ng ml −1, 5 min) was immunoprecipitated with anti-TNFR1. Immunoblot was performed with anti-Traf2, anti-RIP1 and anti-TNFR1. Ct, control; T, TNF-α. Experiments in a and b were performed in two other independent experiments with similar results. (c) mRNA expression in primary keratinocytes by real-time RT–PCR. Cells from WT and AQP3−/− mice were incubated with TNF-α (100 ng ml −1) for 24 h. Data are expressed as the ratio to GAPDH (s.e., n =5, *P<0.05, **P<0.01 by t-test). (d) Primary keratinocytes from WT and AQP3 −/− mice were incubated with IL-22 (100 ng ml −1, 10 min) or IFNγ (100 ng ml −1, 10 min). Representative immunoblot analysis with anti-phospho-Stat3, -Jak2, Stat3 or Jak2. A second set of experiments gave similar results.
TNF-α-dependent NF-κB signalling regulates inflammation and apoptosis during the development of psoriasis36. Quantitative RT–PCR of primary keratinocytes showed that TNF-α stimulation increased levels of IL-17c, IL-6 and S100A8, which are related to inflammation in response to NF-κB activation, with lesser increases in AQP3 −/− keratinocytes (Fig. 4c). AQP3 deficiency also attenuated the increase in the response to TNF-α of caspase 3, a key enzyme in the apoptotic pathway.
To verify the specific role of AQP3 in NF-κB signalling, we examined Jak2/Stat3 activation in response to IL-22 or interferon gamma (INFγ), which are also thought to be key cell signalling molecules in the development of psoriasis37,38. We found comparable phosphorylation of Jak2 and Stat3 with IL-22 or INFγ stimulation (Fig. 4d, Supplementary Fig. 7). These results suggest that AQP3 expression in keratinocytes is required for NF-κB activation in response to TNF-α stimulation.
TNF-α-mediated increase in intracellular H2O2 depends on AQP3
There is increasing evidence that H2O2 plays an important role in the regulation of NF-κB29,30,39. We hypothesized that AQP3-facilitated intracellular H2O2 accumulation is involved in NF-κB activation in the presence of TNF-α. First, we investigated whether AQP3 can transport extracellular H2O2 into keratinocytes. Intracellular H2O2 was measured in primary keratinocyte cultures from WT and AQP3 −/− mice after extracellular addition of 10–300 μM H2O2 using the fluorescent dye CM-H2DCFDA, which reacts with ROS including H2O2. We found that cellular H2O2 increased to a significantly greater extent in WT than in AQP3 −/− keratinocytes, demonstrating the involvement of AQP3 in H2O2 transport in keratinocytes (Fig. 5a,b). Cell morphology and apoptosis after incubation with H2O2 was comparable in WT and AQP3 −/− cells (Supplementary Fig. 5a,b).
Figure 5. AQP3-dependent H2O2 permeability in keratinocytes.

(a, b) H2O2 uptake into primary cultured keratinocytes. Keratinocytes were incubated with H2O2 (10 to 300 μM), and cellular H2O2 was detected using CM-H2DCFDA fluorescence using a plate reader. (a) Representative fluorescence intensity of CM-H2DCFDA. (b) Increased fluorescence intensity at 15 s after addition of H2O2 (s.e., n = 5, *P<0.01, H2O2 added versus control cells by t-test). (c–d) Intracellular H2O2 was monitored by CM-H2DCFDA fluorescence with TNF-α stimulation (100 ng ml− 1). (c) Representative fluorescence intensity in WT and AQP3 −/− cells. (d) Cells were incubated with DPI (5 μM), catalase (2,000 U ml −1) or vehicle (PBS) for 30 min, and followed by TNF-α (100 ng ml −1) for 30 s (s.e., n =5, *P<0.01 by t-test). (e) WT and AQP3 −/− cells transfected with HyPer were incubated with TNF-α (100 ng ml −1) or H2O2 (100 μM) for 3 min. Representative immunofluorescence. Scale bar, 20 μm. (f) Cellular H2O2 after stimulation by TNF-α (100 ng ml −1) or H2O2 (100 μM) in primary keratinocytes from WT, AQP3−/−, or Nox2−/− mice using CM-H2DCFDA fluorescence (1 min, s.e., n = 7, *P<0.01 by t-test). (g) Left: immunoblot of AQP3 and Nox2 in membrane-rich fraction from WT, AQP3 −/− and Nox2 −/− keratinocytes. Right: cell lysates by RIPA were immunoprecipitated with anti-AQP3 or anti-Nox2 showing the interaction between endogenous AQP3 and Nox2.
Extracellular H2O2, which is mainly produced by the activation of plasma membrane NADPH oxidase (Nox) in response to various stimulation including TNF-α, is believed to be highly diffusible across the membrane and to act as a second messenger17,18,30,40,41. TNF-α stimulation markedly increased intracellular H2O2 in WT mouse keratinocytes within 30 s (Fig. 5c). Pretreatment with diphenyleneiodonium (DPI), a general Nox inhibitor, or incubation with catalase, which removes any extracellular H2O2, greatly reduced the TNF-α-induced increase intracellular H2O2 in WT cells, suggesting that TNF-α stimulation activates Nox for extracellular production of H2O2 (Fig. 5d). The TNF-α-induced increase in intracellular H2O2 level was remarkably reduced in AQP3 −/− keratinocytes, and neither DPI nor catalase treatment affected cellular H2O2 (Fig. 5d). To verify the TNF-α-mediated increase in the intracellular H2O2 by an independent method, cells were transfected with HyPer, a genetically encoded ratiometric sensor that is selective to H2O2 and allows dynamic monitoring of intracellular H2O2 concentration42. Confocal microscopy showed increased HyPer fluorescence in the cytosol and around the cell membrane after the addition of TNF-α (100 ng ml −1) or H2O2 (100 μM) in WT cells, with much less fluorescence in AQP3 −/− cells (Fig. 5e).
It was reported that human keratinocytes express both Nox1 and Nox2 (ref. 43). The involvement of Nox2 in TNF-α-mediated cellular H2O2 induction was demonstrated using primary keratinocytes from Nox2 knockout (Nox2 −/−) mice; Nox1 knockdown had little effect (Supplementary Fig. 5c). We found that the TNF-α-induced increase in intracellular H2O2 level was suppressed in Nox2 −/− primary keratinocytes compared with WT cells, while the uptake of exogenous H2O2 was not affected by Nox2 deficiency (Fig. 5f). These results indicated that extracellular H2O2 produced by TNF-α stimulation is dependent on Nox2 in mouse keratinocytes.
On the basis of the rapid increase in intracellular H2O2 in response to TNF-α stimulation, we speculated that AQP3 may associate with Nox2 on the plasma membrane. Immunoprecipitation revealed an association between AQP3 and Nox2 in mouse primary keratinocytes (Fig. 5g).
Together, these results support a model in which TNF-α stimulation induces extracellular H2O2 production by Nox2 activation, which is rapidly transported intracellularly in keratinocytes through AQP3.
AQP3-mediated cellular H2O2 regulates NF-κB activation
Next, we examined whether the intracellular H2O2 level modulates TNF-α-induced NF-κB/p65 activation. Consistent with the immunoblot study in Fig. 4a, immunofluorescence showed that TNF-α stimulation translocated p65 into the nucleus in WT cells following NF-κB activation, with less translocation of p65 in AQP3 −/− cells (Fig. 6a). Treatment of WT cells with DPI or catalase suppressed TNF-α-induced p65 translocation into the nucleus, suggesting that the intracellular H2O2 level modulates TNF-α-induced NF-κB/p65 activation (Fig. 6a).
Figure 6. AQP3-dependent H2O2 accumulation in keratinocytes regulates NF-κB activation.

(a) Left: representative immunofluorescence of p65 in keratinocytes from WT and AQP3 −/− mice. Cells were stimulated with TNF-α (100 ng ml −1) for 1 h. Some cells were incubated with catalase (2,000 U ml−1) or DPI (5 μM) for 30 min before TNF-α stimulation. Scale bar, 20 μm. Right: numbers of p65 positive cells in the nucleus (s.e., over 100 cells from four different fields, *P<0.01 by t-test). (b,c) AQP3 −/− primary keratinocytes were incubated with TNF-α (100 ng ml −1) and/or H2O2 (300 μM). (b) Cellular H2O2 level (1 min, s.e., n = 7, *P<0.01 by t-test). (c) Representative immunoblot with phospho-IKKα/β, IKKβ, phospho-p65, p65 and β-actin stimulated for indicated times. (d–f) AQP3 −/− primary keratinocytes were transfected with mouse AQP3 cDNA or empty vector (pCMV6). (d) Left: mRNA analysed by quantitative RT–PCR. Right: immunoblotting with AQP3 antibody. (e) Cellular H2O2 after stimulation with TNF-α (100 ng ml−1) or H2O2 (100 μM; 1 min, s.e., n = 6, *P<0.05, **P<0.01 by t-test). (f) Immunoblot with phospho-IKKα/β or IKKβ stimulated with TNF-α (100 ng ml−1) for indicated times. Experiments in c and f were performed in two independent sets of experiments with similar results.
Further studies were done to verify that AQP3-mediated H2O2 transport regulates NF-κB activation. Because the addition of a high concentration of H2O2 (over 300 μM) led to increase an intracellular H2O2 level even in AQP3 −/− cells (shown in Fig. 5b), AQP3 −/− cells were stimulated with TNF-α in the presence of 300 μM H2O2. TNF-α stimulation together with the addition of H2O2 increased intracellular H2O2 in AQP3−/− cells, in which H2O2 uptake probably occurs by passive diffusion across the plasma membrane (Fig. 6b). In this setting, both IKKβ and p65 were activated in AQP3 −/− keratinocytes, while addition of 300 μM H2O2 alone had less effect (Fig. 6c, Supplementary Fig. 7). We speculate that AQP3-mediated H2O2 accumulation may increase TNF-α-induced NF-κB activation.
Next, we determined whether forced expression of AQP3 by transfection with mouse AQP3 cDNA in AQP3 −/− cells could rescue TNF-α-induced NF-κB activation by increasing intracellular H2O2. AQP3 expression was seen in AQP3−/− cells following AQP3 transfection (Fig. 6d, Supplementary Fig. 7). Both H2O2 transport and the TNF-α-induced H2O2 level were restored in AQP3 −/− cells by forced expression of AQP3 (Fig. 6e). In this setting, TNF-α-induced IKKβ phosphorylation was increased compared with control (empty vector transfected) AQP3 −/− cells (Fig. 6f, Supplementary Figs 6b and 7).
Taken together, these results suggest that TNF-α-induced NF-κB cell signalling require a threshold concentration of intracellular H2O2 involving AQP3-facilitated membrane transport.
H2O2 inactivates PP2A and regulates NF-κB activation
We next sought to identify the targets of AQP3-mediated H2O2 within the NF-κB pathway. Signalling with H2O2 is believed to be mediated by oxidation of the critical cysteine residues of target proteins, including protein tyrosine phosphatases and many other H2O2 effector molecules17,44. It was hypothesized that AQP3-mediated H2O2 modulates protein phosphatase 2A (PP2A) to regulate NF-κB activation, as PP2A has been shown to be inactivated by H2O2 in redox-sensitive cysteine residues, to interact with the IKK complex and p65, and to regulate NF-κB activation45–51.
PP2A activity, quantified by PP2A inhibitor okadaic acid (OA)-sensitive phosphatase activity, was measured in primary culture keratinocytes after TNF-α or H2O2 addition. Figure 7a shows reduced PP2A activity in response to H2O2 or TNF-α addition in WT cells, while neither H2O2 nor TNF-α affected PP2A activity in AQP3 −/− cells. Pretreatment of WT cells with catalase attenuated the decrease in PP2A activity in response to TNF-α or H2O2 (Fig. 7a). These data indicate that the TNF-α-mediated increase in H2O2 inactivates PP2A in WT but not AQP3 −/− cells.
Figure 7. TNF-α-mediated H2O2 inactivates PP2A and regulates NF-κB activation.

(a) PP2A activity in WT and AQP3−/− keratinocytes stimulated with TNF-α (100 ng ml −1) or H2O2 (300 μM) for 10 min. Some cells were incubated with catalase (2,000 U ml−1) for 30 min before stimulation. PP2A activity was calculated from the rate of dephosphorylation of a radioactive substrate per minute per microgram protein in the presence and absence of okadaic acid (OA, 10 μM), an inhibitor of PP2A (s.e., n =5, *P<0.01 by t-test). (b) WT keratinocytes were incubated with OA (10 μM, 30 min) and stimulated with TNF-α (100 ng ml−1, 15 min). Immunoblot with anti-phospho-IKKα/β, IKKβ, phospho-p65, p65 and β-actin. (c–e) Ppp2ca knockdown in WT and AQP3 −/− keratinocytes by siRNA transfection. (c) PP2A expression by quantitative RT–PCR and immunoblotting (s.e., n =4, *P<0.01 by t-test). (d) Cells were stimulated with TNF-α (100 ng ml −1). Representative immunoblot with phospho-IKKα/β, IKKβ, phospho-p65 and p65. (e) WT cells with control- or Ppp2ca-siRNA transfection were incubated with catalase (2,000 U ml −1, 30 min) before TNF-α stimulation. (f,g) WT keratinocytes were transfected with empty vector (pCMV6) or plasmid-expressing mouse Ppp2ca. (f) PP2A overexpression was analysed by quantitative RT–PCR and immunoblotting (s.e., n =4, *P<0.01 by t-test). (g) Representative immunoblot of phospho-IKKα/β, IKKβ, phospho-p65 and p65 with TNF-α (100 ng ml −1) stimulation. (h) Mice were injected IL-23 into the ear as described in Fig. 1 and PP2A activity was measured in epidermal homogenates. Some WT mice were injected DPI (2 μg g −1 weight) intravenously 1 h before IL-23 injection (s.e., n =3–5, *P<0.01 by t-test). Experiments in d, e and g were performed in two additional sets of independent experiments with similar results.
We next determined the effect of PP2A inhibition on TNF-α-induced NF-κB activation in WT keratinocytes. Figure 7b shows that OA treatment before TNF-α stimulation greatly enhanced phosphorylation of IKKβ and p65 even in the absence of TNF-α stimulation (Supplementary Fig. 7). To further investigate the role of PP2A in TNF-α-induced NF-κB activation, we knocked down Ppp2ca, the catalytic subunit of PP2A, in WT and AQP3 −/− keratinocytes (Fig. 7c, Supplementary Fig. 7). Ppp2ca knockdown of AQP3 −/− cells rescued the reduced TNF-α-induced NF-κB activation, including phosphorylation of IKKβ and p65 (Fig. 7d, Supplementary Figs 6c and 7). Moreover, pretreatment with catalase suppressed the TNF-α-induced phosphorylation of IKKβ and p65 in WT cells, while Ppp2ca knockdown ameliorated the effect of catalase on WT cells (Fig. 7e, Supplementary Fig. 7). As further evidence for the involvement of PP2A in TNF-α-NF-κB cell signalling, Ppp2ca was overexpressed in WT keratinocytes (Fig. 7f, Supplementary Fig. 7) and NF-κB activation in response to TNF-α was analysed. Figure 7g shows that Ppp2ca over-expression suppressed TNF-α-induced phosphorylation of IKKβ and p65 (Supplementary Figs 6d and 7).
Last, we determined the effect of IL-23 application on PP2A activity in vivo. PP2A activity was significantly decreased in WT epidermal homogenates with IL-23 application compared with control, while PP2A activity was unaffected by IL-23 treatment in AQP3 −/− epidermis (Fig. 7h). The administration of low-dose DPI has been used to inhibit Nox in vivo52. DPI administration before IL-23 injection suppressed the decrease in PP2A activity, implying an effect of Nox-derived H2O2 on PP2A activity in vivo.
Collectively, these data suggest that PP2A is a target of AQP3-mediated H2O2 elevation involved in NF-κB activation.
TNF-α-induced NF-κB activation depends on AQP3 in NHEK
The involvement of AQP3 in TNF-dependent NF-κB activation was studied using AQP3 knockdown in human primary keratinocytes (NHEK). Transfection of short interfering RNA (siRNA)-AQP3 into NHEK successfully reduced AQP3 mRNA and protein expression (Fig. 8a, Supplementary Fig. 7). H2O2 transport was decreased in AQP3 knockdown cells compared with control-siRNA transfected cells (Fig. 8b). The increase in intracellular H2O2 following TNF-α was also reduced by AQP3 knockdown (Fig. 8b), as was TNF-α-induced NF-κB activation as shown by phosphorylation of IKKβ and p65 (Fig. 8c, Supplementary Figs 6e and 7). Quantitative RT–PCR showed that TNF-α stimulation increased IL-17C and IL-6 transcript expression, with lesser increases in AQP3 knockdown keratinocytes (Fig. 8d).
Figure 8. TNF-α induced NF-κB activation depends on AQP3 in human primary keratinocytes.

NHEK were transfected with AQP3 or non-targeting (Ct) siRNA. (a) Left: relative mRNA expression of AQP3/GAPDH (s.e., n =4, *P<0.01 by t-test). Right: immunoblot of membrane fraction with anti-AQP3 and anti-Na+/K +-ATPase. (b) Cellular H2O2 levels determined by CM-H2DCFDA after TNF-α (100 ng ml−1) or H2O2 (10 μM) stimulation (30 s, s.e., n = 5, *P<0.01 by t-test). (c) Immunoblot with phospho-IKKβ, IKKβ, phospho-p65 and p65 after TNF-α stimulation. (d) mRNA expression of IL-17C and IL-6 by real-time RT–PCR. Cells were incubated with TNF-α (50 ng ml −1) for 24 h. Data are expressed as the ratio to GAPDH (s.e., n =5, *P<0.05, **P<0.01 by t-test). (e) Ppp2ca knockdown in NHEK by siRNA transfection (s.e., n =4, *P<0.01 by t-test). (f) Immunoblot with phospho-p65 and p65 after TNF-α stimulation in AQP3 and/or Ppp2ca knockdown cells. Experiments in c and f were performed in two additional sets of independent experiments with similar results. (g) NHEK cell lysates were immunoprecipitated with anti-AQP3 or anti-Nox2 antibodies, showing interaction between AQP3 and Nox2.
Last, we investigated the involvement of PP2A in AQP3-facilitated NF-κB activation in NHEK. Ppp2ca knockdown was done in control or AQP3 knockdown NHEK (Fig. 8e, Supplementary Fig. 7). Ppp2ca knockdown restored the impaired TNF-α-induced p65 activation in AQP3 knockdown cells (Fig. 8f, Supplementary Fig. 7). Again, reciprocal immunoprecipitation analysis showed association of AQP3 with Nox2 in NHEK (Fig. 8g).
These results in human primary keratinocytes support the findings in mouse primary keratinocytes on the involvement of AQP3 in TNF-α-dependent H2O2 induction and NF-κB signalling (Fig. 9).
Figure 9. Model of AQP3-mediated NF-κB activation.
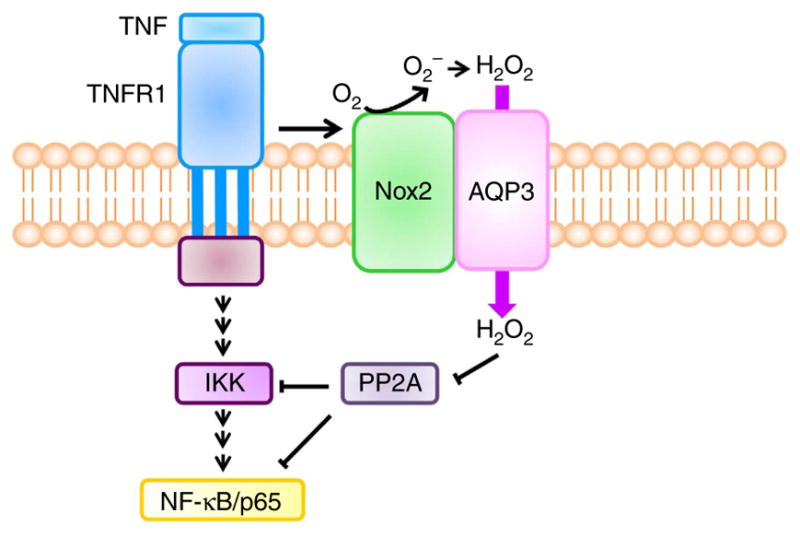
TNF-α binds to TNFR1 in keratinocytes and induces the production of H2O2 by Nox2. Extracellular H2O2 is rapidly transported intracellularly through AQP3. H2O2 modifies PP2A, regulating IKKβ and/or NF-κB/p65 activation.
Discussion
We discovered here a novel role for AQP3 in the pathogenesis of psoriasis, which involved its H2O2 transport function in keratinocytes. The uptake of extracellular H2O2 by keratinocytes generated in response to TNF-α, which was required for NF-κB activation, was dependent on AQP3 as diagrammed in Fig. 9. Keratinocytes made AQP3-deficient by gene knockout or knockdown showed reduced TNF-α-induced increase in intracellular H2O2 and NF-κB activation. The reduced intracellular H2O2 suppressed TNF-α-dependent NF-κB activation, while either exogenous H2O2 supplementation or expression of AQP3 rescued the impaired NF-κB activation with restored intracellular H2O2 levels in AQP3−/− cells. In a mouse model of psoriasis produced by IL-23 application, there was continuous NF-κB activation and a higher H2O2 concentration in keratinocytes of WT psoriatic skin, which was dependent on TNF-α signalling. AQP3 −/− mice showed remarkably impaired development of psoriasis, with reduced NF-κB activation and cellular H2O2. Together, these findings support the involvement of AQP3 in the development of psoriasis by a mechanism involving its H2O2 transport function and downstream TNF-α-dependent NF-κB activation in keratinocytes.
H2O2 is emerging as a second messenger in cell signalling. Previous papers have proposed that the intracellular concentration of H2O2 should rapidly exceed a certain threshold, probably 10–100 μM, to serve as a signalling molecule30,53,54. Extracellular H2O2 produced by membrane Nox activation in response to receptor activation by ligands, such as growth factors, hormones and cytokines, has classically been thought to freely diffuse across biological membranes17,55. However, because H2O2 is rapidly degraded in the cells by various enzymes, including catalase, glutathione peroxidases and peroxiredoxins53,56, continuous influx of H2O2 may be necessary to elevate intracellular H2O2 levels needed for cell signalling. Here, we found that TNF-α-induced production of extracellular H2O2 was mainly dependent on Nox2 in keratinocytes, and that a membrane-associated AQP3–Nox2 complex may allow constitution of a localized signalling mechanism following TNF-α stimulation. AQP3 may thus be required to achieve a sufficiently high local concentration of H2O2 near the membrane to act as a second messenger.
H2O2 has been implicated in the regulation of NF-κB; however, there is still no consensus on its role in NF-κB activation30,57. A recent study showed that dual oxidase 1-derived H2O2 is involved in NF-κB signalling in skin in a zebrafish model39. Nevertheless, the molecular mechanism of NF-κB regulation by H2O2 has been shown to be cell-type specific and to involve quite different mechanisms. There is evidence for cysteine residues being the most likely targets of Nox-mediated H2O2 signalling17,58. Exposure to excess H2O2 causes irreversible oxidation of the cysteine residues at the active sites of specific phosphatases such as PTEN and SHP1/2, resulting in their inhibition, which suppresses cell signalling17. The Ser/Thr phosphatases, including PP2A and PP1a, are also sensitive to H2O2 modification, having H2O2-sensitive cysteine residues59. PP2A was reported to be inactivated by H2O2 exposure or TNF-α stimulation60,61. Previous studies also indicated that PP2A-regulated NF-κB cell signalling involves modulation of IKK45–48,51,62. Consistent with these findings, a large-scale RNAi screen revealed that a PP2A catalytic or regulatory subunit associated IKK complex, p65, and/or Traf2, and led to the regulation of NF-κB49. Here, we showed that PP2A inactivation by H2O2 exposure or TNF-α stimulation was dependent on AQP3 expression in WT primary keratinocytes. Ppp2ca knockdown in AQP3 −/− keratinocytes rescued the reduced IKKβ and p65 activation, while Ppp2ca knockdown in WT cells abrogated the effect of catalase on TNF-α-induced NF-κB activation. In contrast, Ppp2ca overexpression in WT keratinocytes suppressed IKKβ and p65 activation. Taken together, our data suggest that PP2A is one of the target proteins of AQP3-mediated H2O2 uptake during TNF-α-induced NF-κB cell signalling, in which the increased H2O2 may oxidize and inactivate PP2A through the cysteine residue and sequentially activate IKK and p65. Further studies are needed to establish the precise mechanism(s) linking PP2A with the regulation of TNF-α-induced NF-κB activation in keratinocytes.
Regarding the redox state in psoriasis, there are increasing studies that showed higher levels of oxidative stress markers in psoriatic subjects, such as nitric oxide63, superoxide dismutase, catalase64, superoxide anions65 and peroxiredoxin 2 (refs 66,67). A recent report also showed significantly higher H2O2 and Nox activity in white blood cells of psoriatic patients compared with controls68. Although the activated redox state was proposed to cause DNA modification or inflammation, the underlying mechanisms involving redox state in the pathogenesis of psoriasis are not clear. Our findings suggest that keratinocyte-generated H2O2 triggers psoriasis via modulation of NF-κB cell signalling, which clarifies the role of H2O2- and redox-mediated cell signalling in psoriasis. Previously, we reported impaired tumorigenesis in AQP3 −/− mice in a DMBA/PMA-induction model, but the underlying mechanism was unclear13. A number of studies support the involvement of ROS in cell growth, survival, metastasis and inflammation during cancer development and progression69. AQP3-mediated H2O2 transport may provide an additional or alternative mechanism for the impaired skin tumorigenesis in AQP3 −/− mice.
There is currently a controversy regarding the primary pathogenic functions of keratinocytes versus T cells in psoriasis1–3. The psoriasis model in this study uses IL-23, which may induce downstream signal molecules including TNF-α and IL-22 (refs 23,24). Our results suggested the requirement of AQP3 expression in both keratinocytes and T cells for the development of psoriasis induced by IL-23 application. We here identified a link between AQP3 expression and NF-κB cell signalling in keratinocytes that was associated with the development of psoriasis. Regarding the function of AQP3 in T cells, the current study together with our previous findings16 suggest the involvement of AQP3 in T cell chemotaxis into the dermal and epidermal layer, a critical step for initiation of the immune response in the development of psoriasis. Further studies will determine the role of AQP3 in the crosstalk between keratinocytes and T cells in the pathogenesis of psoriasis.
In summary, our data implicate a novel role of AQP3 in the pathogenesis of psoriasis in which AQP3-mediated intracellular H2O2 uptake is required for TNF-α-induced NF-κB activation in epidermal keratinocytes. Our findings support AQP3 as a novel determinant in psoriasis and hence a potential new therapeutic target.
Methods
Mice
AQP3 −/− mice (C57BL/6 genetic background) were generated by targeted gene disruption70. Nox2 knockout mice (Nox2 −/−, gp91 phox−, C57BL/6 genetic background) were purchased from The Jackson Laboratory (Bar Harbor, MA). Eight- to 10-week-old female mice were used. All animal experiments were approved by the Committee on Animal Research of Kyoto University.
Psoriasis-like mouse models
IL-23 (500 ng in 40 μl PBS, Miltenyi Biotec) or vehicle control (PBS, 40 μl) was intradermally injected daily into the ear skin of WT and AQP3 −/− mice for 4 days23,24. Mice were killed at 24 h after the final IL-23 injection and skin samples were excised.
Human subjects
A total of 18 patients with psoriasis and five healthy volunteers were enrolled in this study. Informed consent was obtained from all the subjects. The study was approved by the Ethics Committee of the Kyoto University.
Primary culture of mouse and human keratinocytes
Full-thickness skin samples from 1-day-old mice were incubated in dispase II (5 U ml −1; Boehringer Mannheim) overnight at 4 °C. The epidermis was separated from the dermis, cut into fragments and incubated in 0.25% trypsin-0.1% EDTA for 10 min. Cells were seeded on collagen type I plates (BD Biosciences) and cultured in keratinocyte growth medium (Lonza) at 37 °C under 5% CO2.
Neonatal human keratinocytes (NHEK, KK-4009, Kurabo, Japan) were grown in keratinocyte growth medium (Lonza) at 37 °C under 5% CO2.
Cellular H2O2 analysis
Cellular H2O2 was assayed using the CM-H2DCFDA reagent (Invitrogen) according to the manufacturer’s instructions using flow cytometry (FACS Fortessa system, Becton Dickinson, San Diego, CA) or a microplate reader (Flex station, Molecular Devices; or Envision, PerkinElmer Life Sciences). To monitor cellular H2O2 in the skin, mice were injected with H2DCFDA intravenously (10 nmol g−1, Invitrogen) 1 h before being killed. Cellular H2O2 was observed in frozen section by fluorescence microscopy. To quantify cellular H2O2 in mouse skin by FACS analysis, the excised skin was trypsinized and single epidermal cells were incubated with H2DCFDA.
Immunoblotting and immunoprecipitation
Cells were lysed with RIPA buffer (Cell Signaling Technology) for cell signalling analysis or with membrane extraction buffer (HEPES pH 7.4, 250 mM sucrose, 1 mM EDTA, 1 mM EGTA, 1% protease inhibitor cocktail) for membrane protein analysis. The supernatant (10,000g, 10 min, 4 °C) was used for immunoblotting with antibodies against phospho-p65, p65, phospho-IKKβ, IKKβ, phosphor-Iκβα, Iκβα (Cell Signaling, 1:1,000), phospho- IKKα/β (Abcam, 1:500), AQP3 (Millipore, 1:1,000), Nox2 (Santa Cruz, 1:200 or Millipore, 1:1,000), PP2A or β-actin (Sigma, 1:2,000). For immunoprecipitation, protein lysate in RIPA was incubated with anti-TNF receptor I (R&D system, 1 μg antibody per 200 μg total protein), anti-AQP3 (Santa Cruz, 2 μg antibody per 400 μg total protein) or anti-Nox2 (Santa Cruz, 2 μg antibody per 400 μg total protein), and protein A or G agarose beads (GE Healthcare) at 4 °C overnight. Immunoblots were performed with anti-Traf2, RIP1 (Cell Signaling, 1:1,000), TNFR1 (R&D system, 1:1,000), AQP3 (Millipore, 1:1,000), Nox2 (Santa Cruz, 1:200 or Millipore, 1:1,000) or Nox1 (Millipore, 1:1,000). The horseradish peroxidase-conjugated secondary anti-rabbit or anti-mouse IgG antibody (Cell Signaling Technology, 1:1,000) were used and visualized by chemiluminescence (GE Healthcare). The optical density of the bands was quantified using Image J (National Institutes of Health, Supplementary Fig. 6).
PP2A assay
PP2A activity was assessed in primary keratinocytes using ProFluor Ser/Thr PPase assay kit (Promega) according to the manufacturer’s instructions.
RNAi and plasmid DNAs
Cells were transfected using Lipofectamine 2000 (Invitrogen) with Ppp2ca-, AQP3- or non-targeting-siRNA (ON-TARGET plus SMART pool, Thermo Scientific). The cDNA plasmids for AQP3, Ppp2ca (pCMV6 vector, Origine) or Hyper (pHyPer-cyto vector, Evrogen) was also transfected with Lipofectamine 2000. Using these methods, we generally found that 65–80% cells become positive after transfection.
Immunofluorescence and immunohistochemistry
For p65 immunofluorescence, cells were fixed with 4% formalin and permeabilized with 0.1% Triton X-100 and immunostained with p65 (Cell Signaling, 1:100) and anti-rabbit secondary antibody (FITC, Sigma, 1:200). Paraffin-embedded sections were stained with haematoxylin and eosin, anti-phospho-p65 antibody (Cell Signaling, 1:200) or Ki67 (Cell Signaling, 1:100).
Bone marrow transplantation
For bone marrow (BM) transplantation, red blood cells from WT and AQP3 −/− mice were subjected to hypotonic cell lysis. WT and AQP3 −/− recipients (8–10 weeks old) were γ-irradiated with two doses of 600 rad, 3 h apart. After irradiation, the mice received 106 BM cells intravenously. This protocol consistently gave >95% reconstitution of the recipient by donor haematopoietic cells, as evaluated by separate transplantation experiments using BM from C57BL/6-CD45.1 congenic mice (Supplementary Fig. 2a). The psoriasis mouse model was performed 2 months later.
RNA extraction and real-time quantitative RT–PCR
Total RNA was extracted using TRIZOL (Invitrogen). The cDNA was reverse transcribed from total RNA using the Prime Script RT reagent kit (Takara Bio, Otsu, Japan). Quantitative RT–PCR was performed using SYBR Green I (Takara Bio) and the Light Cycler real-time PCR apparatus (Roche). Primer sequences are listed in Supplementary Table 1.
Statistical analysis
Statistical analysis was performed using the two-tailed Student’s t-test or analysis of variance.
Supplementary Material
Acknowledgments
We thank Atsuko Shibayama for mouse breeding, Drs Shu Narumiya and Shunsuke Chikuma for helpful discussions. This work was supported by grants from Astellas Pharma Inc. in the Creation of Innovation Centers for Advanced Inter disciplinary Research Areas Program, and the Ministry of Education, Culture, Sports, Science and Technology of Japan (M.H.-C.), and grant DK35124 from the U.S. National Institutes of Health (A.S.V.).
Footnotes
Author contributions
M.H.-C. conceived and designed the experiments. M.H.-C. and S.W. performed the experiments. M.H.-C., S.W., H.S., T.H., T.W., Y.M. and A.S.V. analysed the data. T.H. and Y.M. contributed to collect human subjects. M.H.-C. and A.S.V. wrote the manuscript.
Supplementary Information accompanies this paper at http://www.nature.com/naturecommunications
Competing financial interests: The authors declare no competing financial interests.
References
- 1.Perera GK, Di Meglio P, Nestle FO. Psoriasis. Annu Rev Pathol. 2012;7:385–422. doi: 10.1146/annurev-pathol-011811-132448. [DOI] [PubMed] [Google Scholar]
- 2.Elder JT, et al. Molecular dissection of psoriasis: integrating genetics and biology. J Invest Dermatol. 2010;130:1213–1226. doi: 10.1038/jid.2009.319. [DOI] [PubMed] [Google Scholar]
- 3.Martin DA, et al. The emerging role of IL-17 in the pathogenesis of psoriasis: preclinical and clinical findings. J Invest Dermatol. 2013;133:17–26. doi: 10.1038/jid.2012.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Capon F, Burden AD, Trembath RC, Barker JN. Psoriasis and other complex trait dermatoses: from Loci to functional pathways. J Invest Dermatol. 2012;132:915–922. doi: 10.1038/jid.2011.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oka A, Mabuchi T, Ozawa A, Inoko H. Current understanding of human genetics and genetic analysis of psoriasis. J Dermatol. 2012;39:231–241. doi: 10.1111/j.1346-8138.2012.01504.x. [DOI] [PubMed] [Google Scholar]
- 6.Gudjonsson JE, Johnston A, Ellis CN. Novel systemic drugs under investigation for the treatment of psoriasis. J Am Acad Dermatol. 2012;67:139–147. doi: 10.1016/j.jaad.2011.06.037. [DOI] [PubMed] [Google Scholar]
- 7.Langley RG, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371:326–338. doi: 10.1056/NEJMoa1314258. [DOI] [PubMed] [Google Scholar]
- 8.Leonardi C, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. 2012;366:1190–1199. doi: 10.1056/NEJMoa1109997. [DOI] [PubMed] [Google Scholar]
- 9.Goldminz AM, Au SC, Kim N, Gottlieb AB, Lizzul PF. NF-kappaB: an essential transcription factor in psoriasis. J Dermatol Sci. 2013;69:89–94. doi: 10.1016/j.jdermsci.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 10.Zhou Q, Mrowietz U, Rostami-Yazdi M. Oxidative stress in the pathogenesis of psoriasis. Free Radic Biol Med. 2009;47:891–905. doi: 10.1016/j.freeradbiomed.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 11.Verkman AS, Anderson MO, Papadopoulos MC. Aquaporins: important but elusive drug targets. Nat Rev Drug Discov. 2014;13:259–277. doi: 10.1038/nrd4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakahigashi K, et al. Upregulation of aquaporin-3 is involved in keratinocyte proliferation and epidermal hyperplasia. J Invest Dermatol. 2011;131:865–873. doi: 10.1038/jid.2010.395. [DOI] [PubMed] [Google Scholar]
- 13.Hara-Chikuma M, Verkman AS. Prevention of skin tumorigenesis and impairment of epidermal cell proliferation by targeted aquaporin-3 gene disruption. Mol Cell Biol. 2008;28:326–332. doi: 10.1128/MCB.01482-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hara-Chikuma M, Verkman AS. Aquaporin-3 facilitates epidermal cell migration and proliferation during wound healing. J Mol Med (Berl) 2008;86:221–231. doi: 10.1007/s00109-007-0272-4. [DOI] [PubMed] [Google Scholar]
- 15.Miller EW, Dickinson BC, Chang CJ. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc Natl Acad Sci USA. 2010;107:15681–15686. doi: 10.1073/pnas.1005776107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hara-Chikuma M, et al. Chemokine-dependent T cell migration requires aquaporin-3-mediated hydrogen peroxide uptake. J Exp Med. 2012;209:1743–1752. doi: 10.1084/jem.20112398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paulsen CE, Carroll KS. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem Biol. 2010;5:47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 19.Bowcock AM, et al. Insights into psoriasis and other inflammatory diseases from large-scale gene expression studies. Hum Mol Genet. 2001;10:1793–1805. doi: 10.1093/hmg/10.17.1793. [DOI] [PubMed] [Google Scholar]
- 20.Lee Y, et al. Changes in transepidermal water loss and skin hydration according to expression of aquaporin-3 in psoriasis. Ann Dermatol. 2012;24:168–174. doi: 10.5021/ad.2012.24.2.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Voss KE, et al. Abnormal aquaporin-3 protein expression in hyperproliferative skin disorders. Arch Dermatol Res. 2011;303:591–600. doi: 10.1007/s00403-011-1136-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jabbari A, Suarez-Farinas M, Dewell S, Krueger JG. Transcriptional profiling of psoriasis using RNA-seq reveals previously unidentified differentially expressed genes. J Invest Dermatol. 2012;132:246–249. doi: 10.1038/jid.2011.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng Y, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 24.Chan JR, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. 2006;203:2577–2587. doi: 10.1084/jem.20060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cai Y, et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity. 2011;35:596–610. doi: 10.1016/j.immuni.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mabuchi T, Chang TW, Quinter S, Hwang ST. Chemokine receptors in the pathogenesis and therapy of psoriasis. J Dermatol Sci. 2012;65:4–11. doi: 10.1016/j.jdermsci.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 27.Lizzul PF, et al. Differential expression of phosphorylated NF-kappaB/RelA in normal and psoriatic epidermis and downregulation of NF-kappaB in response to treatment with etanercept. J Invest Dermatol. 2005;124:1275–1283. doi: 10.1111/j.0022-202X.2005.23735.x. [DOI] [PubMed] [Google Scholar]
- 28.Doger FK, et al. Nature of cell kinetics in psoriatic epidermis. J Cutan Pathol. 2007;34:257–263. doi: 10.1111/j.1600-0560.2006.00719.x. [DOI] [PubMed] [Google Scholar]
- 29.Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oliveira-Marques V, Marinho HS, Cyrne L, Antunes F. Role of hydrogen peroxide in NF-kappaB activation: from inducer to modulator. Antioxid Redox Signal. 2009;11:2223–2243. doi: 10.1089/ars.2009.2601. [DOI] [PubMed] [Google Scholar]
- 31.Matoba T, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. 2000;106:1521–1530. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Fits L, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. 2009;182:5836–5845. doi: 10.4049/jimmunol.0802999. [DOI] [PubMed] [Google Scholar]
- 33.Chu WM. Tumor necrosis factor. Cancer Lett. 2013;328:222–225. doi: 10.1016/j.canlet.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wajant H, Scheurich P. TNFR1-induced activation of the classical NF-kappaB pathway. FEBS J. 2011;278:862–876. doi: 10.1111/j.1742-4658.2011.08015.x. [DOI] [PubMed] [Google Scholar]
- 35.Li Q, Spencer NY, Oakley FD, Buettner GR, Engelhardt JF. Endosomal Nox2 facilitates redox-dependent induction of NF-kappaB by TNF-alpha. Antioxid Redox Signal. 2009;11:1249–1263. doi: 10.1089/ars.2008.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-kappaB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 37.Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009;129:1339–1350. doi: 10.1038/jid.2009.59. [DOI] [PubMed] [Google Scholar]
- 38.Sano S, Chan KS, DiGiovanni J. Impact of Stat3 activation upon skin biology: a dichotomy of its role between homeostasis and diseases. J Dermatol Sci. 2008;50:1–14. doi: 10.1016/j.jdermsci.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 39.Candel S, et al. Tnfa signaling through tnfr2 protects skin against oxidative stress-induced inflammation. PLoS Biol. 2014;12:e1001855. doi: 10.1371/journal.pbio.1001855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grecco HE, Schmick M, Bastiaens PI. Signaling from the living plasma membrane. Cell. 2011;144:897–909. doi: 10.1016/j.cell.2011.01.029. [DOI] [PubMed] [Google Scholar]
- 41.Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med. 2009;47:1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Belousov VV, et al. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat Methods. 2006;3:281–286. doi: 10.1038/nmeth866. [DOI] [PubMed] [Google Scholar]
- 43.Chamulitrat W, et al. A constitutive NADPH oxidase-like system containing gp91phox homologs in human keratinocytes. J Invest Dermatol. 2004;122:1000–1009. doi: 10.1111/j.0022-202X.2004.22410.x. [DOI] [PubMed] [Google Scholar]
- 44.Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 45.Shifera AS. Proteins that bind to IKKgamma (NEMO) and down-regulate the activation of NF-kappaB. Biochem Biophys Res Commun. 2010;396:585–589. doi: 10.1016/j.bbrc.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 46.Barisic S, Strozyk E, Peters N, Walczak H, Kulms D. Identification of PP2A as a crucial regulator of the NF-kappaB feedback loop: its inhibition by UVB turns NF-kappaB into a pro-apoptotic factor. Cell Death Differ. 2008;15:1681–1690. doi: 10.1038/cdd.2008.98. [DOI] [PubMed] [Google Scholar]
- 47.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 48.Kray AE, et al. Positive regulation of IkappaB kinase signaling by protein serine/threonine phosphatase 2A. J Biol Chem. 2005;280:35974–35982. doi: 10.1074/jbc.M506093200. [DOI] [PubMed] [Google Scholar]
- 49.Li S, Wang L, Berman MA, Zhang Y, Dorf ME. RNAi screen in mouse astrocytes identifies phosphatases that regulate NF-kappaB signaling. Mol Cell. 2006;24:497–509. doi: 10.1016/j.molcel.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prajapati S, Verma U, Yamamoto Y, Kwak YT, Gaynor RB. Protein phosphatase 2Cbeta association with the IkappaB kinase complex is involved in regulating NF-kappaB activity. J Biol Chem. 2004;279:1739–1746. doi: 10.1074/jbc.M306273200. [DOI] [PubMed] [Google Scholar]
- 51.Witt J, et al. Mechanism of PP2A-mediated IKK beta dephosphorylation: a systems biological approach. BMC Syst Biol. 2009;3:71. doi: 10.1186/1752-0509-3-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jaquet V, Scapozza L, Clark RA, Krause KH, Lambeth JD. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid Redox Signal. 2009;11:2535–2552. doi: 10.1089/ars.2009.2585. [DOI] [PubMed] [Google Scholar]
- 53.Rhee SG, Woo HA. Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger H(2)O(2), and protein chaperones. Antioxid Redox Signal. 2011;15:781–794. doi: 10.1089/ars.2010.3393. [DOI] [PubMed] [Google Scholar]
- 54.Stone JR, Yang S. Hydrogen peroxide: a signaling messenger. Antioxid Redox Signal. 2006;8:243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 55.Schroder E, Eaton P. Hydrogen peroxide as an endogenous mediator and exogenous tool in cardiovascular research: issues and considerations. Curr Opin Pharmacol. 2008;8:153–159. doi: 10.1016/j.coph.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 56.Woo HA, et al. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell. 2010;140:517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 57.Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 58.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 59.Gough DR, Cotter TG. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis. 2011;2:e213. doi: 10.1038/cddis.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rao RK, Clayton LW. Regulation of protein phosphatase 2A by hydrogen peroxide and glutathionylation. Biochem Biophys Res Commun. 2002;293:610–616. doi: 10.1016/S0006-291X(02)00268-1. [DOI] [PubMed] [Google Scholar]
- 61.Guy GR, Philp R, Tan YH. Activation of protein kinases and the inactivation of protein phosphatase 2A in tumour necrosis factor and interleukin-1 signal-transduction pathways. Eur J Biochem. 1995;229:503–511. [PubMed] [Google Scholar]
- 62.Miskolci V, et al. Okadaic acid induces sustained activation of NFkappaB and degradation of the nuclear IkappaBalpha in human neutrophils. Arch Biochem Biophys. 2003;417:44–52. doi: 10.1016/s0003-9861(03)00336-9. [DOI] [PubMed] [Google Scholar]
- 63.Zalewska A, Wyczolkowska J, Narbutt J, Sysa-Jedrzejowska A. Nitric oxide levels in plasma and fibroblast cultures of psoriasis vulgaris patients. J Dermatol Sci. 2007;48:237–240. doi: 10.1016/j.jdermsci.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 64.Therond P, et al. Antioxidant enzymes in psoriatic fibroblasts and erythrocytes. J Invest Dermatol. 1996;106:1325–1328. doi: 10.1111/1523-1747.ep12349055. [DOI] [PubMed] [Google Scholar]
- 65.Guerard S, Allaeys I, Martin G, Pouliot R, Poubelle PE. Psoriatic keratinocytes prime neutrophils for an overproduction of superoxide anions. Arch Dermatol Res. 2013;305:879–889. doi: 10.1007/s00403-013-1404-z. [DOI] [PubMed] [Google Scholar]
- 66.Besgen P, Trommler P, Vollmer S, Prinz JC. Ezrin, maspin, peroxiredoxin 2, and heat shock protein 27: potential targets of a streptococcal-induced autoimmune response in psoriasis. J Immunol. 2010;184:5392–5402. doi: 10.4049/jimmunol.0903520. [DOI] [PubMed] [Google Scholar]
- 67.Ryu J, et al. Proteomic analysis of psoriatic skin tissue for identification of differentially expressed proteins: Up-regulation of GSTP1, SFN and PRDX2 in psoriatic skin. Int J Mol Med. 2011;28:785–792. doi: 10.3892/ijmm.2011.757. [DOI] [PubMed] [Google Scholar]
- 68.Barygina VV, et al. Altered redox status in the blood of psoriatic patients: involvement of NADPH oxidase and role of anti-TNF-alpha therapy. Redox Rep. 2013;18:100–106. doi: 10.1179/1351000213Y.0000000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gius D, Spitz DR. Redox signaling in cancer biology. Antioxid Redox Signal. 2006;8:1249–1252. doi: 10.1089/ars.2006.8.1249. [DOI] [PubMed] [Google Scholar]
- 70.Ma T, et al. Nephrogenic diabetes insipidus in mice lacking aquaporin-3 water channels. Proc Natl Acad Sci USA. 2000;97:4386–4391. doi: 10.1073/pnas.080499597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.