

Abstract
Proteogenomics, i.e. comprehensive integration of genomics and proteomics data, is a powerful approach identifying novel protein biomarkers. This is especially the case for proteins that differ structurally between disease and control conditions. As tumor development is associated with aberrant splicing, we focus on this rich source of cancer specific biomarkers. To this end, we developed a proteogenomic pipeline, Splicify, which can detect differentially expressed protein isoforms. Splicify is based on integrating RNA massive parallel sequencing data and tandem mass spectrometry proteomics data to identify protein isoforms resulting from differential splicing between two conditions. Proof of concept was obtained by applying Splicify to RNA sequencing and mass spectrometry data obtained from colorectal cancer cell line SW480, before and after siRNA-mediated downmodulation of the splicing factors SF3B1 and SRSF1. These analyses revealed 2172 and 149 differentially expressed isoforms, respectively, with peptide confirmation upon knock-down of SF3B1 and SRSF1 compared with their controls. Splice variants identified included RAC1, OSBPL3, MKI67, and SYK. One additional sample was analyzed by PacBio Iso-Seq full-length transcript sequencing after SF3B1 downmodulation. This analysis verified the alternative splicing identified by Splicify and in addition identified novel splicing events that were not represented in the human reference genome annotation. Therefore, Splicify offers a validated proteogenomic data analysis pipeline for identification of disease specific protein biomarkers resulting from mRNA alternative splicing. Splicify is publicly available on GitHub (https://github.com/NKI-TGO/SPLICIFY) and suitable to address basic research questions using pre-clinical model systems as well as translational research questions using patient-derived samples, e.g. allowing to identify clinically relevant biomarkers.
Approximately 95% of multiexon transcripts undergo alternative splicing, making the human transcriptome far more complex than the protein-coding genome (1). Because of alternative splicing, a single gene can be transcribed into a variety of isoforms which, when translated into proteins, will differ in structure, location, and function. Abnormally spliced RNA can cause or contribute to disease. Aberrant splicing is associated with tumor progression and metastasis, and has been shown to affect each of the biological processes commonly referred to as the hallmarks of cancer (2). Therefore, studying aberrant splicing may reveal additional insights into tumor biology and phenotype. For instance, usage of an alternative 5′ splice site of BCL2L1 causes a switch from a pro- to an antiapoptotic isoform in cancer and contributes to resisting cell death (3). Usage of an alternative 3′ splice site of VEGFA leads to a shift from an anti- to a proangiogenic isoform in cancer and induces angiogenesis (4). As aberrant splicing accompanies tumor progression, splice variants provide a promising source of clinically relevant biomarkers.
Splicing factors play a direct role in splicing regulation and isoform expression. Splicing factors can develop oncogenic activity, e.g. because of aberrant expression or somatic mutations, and through aberrant splicing lead to carcinogenesis (2). SF3B1 is a splicing factor required for the early spliceosome assembly and is also one of the most commonly mutated splicing factors in cancer (5). Recurrent mutations affecting this gene were found in leukemia, melanoma and in pancreatic, breast, and bladder cancer. Even though the specific effects of these alterations on splicing are still to be explored, their features often suggest proto-oncogenic activity (6). In chronic lymphocytic leukemia, mutations in this splicing factor contribute to tumor progression, poor patient survival, and poor chemotherapy response (7, 8). Overexpression of another splicing factor, SRSF1, was observed in different tumor types including breast (9), colon, thyroid, small intestine, kidney, lung, liver, and pancreas (10) and was proven to lead to oncogenic activity (2, 11–13). Transcription of SRSF1 is directly regulated by MYC, a well-known oncogenic transcription factor. Through activation of SRSF1, MYC can affect alternative splicing of a subset of SRSF1 target genes and contribute to tumor development (14). For instance, in breast cancer upregulation of SRSF1 promotes transformation of mammary cells through abnormal splicing of BCL2L11 and BIN1 (15). In colorectal cancer (CRC),1 SRSF1 causes inclusion of exon 4 in RAC1, generating a Rac1b isoform that contributes to cell survival (16, 17).
RNA-seq allows studying the complexity of transcriptomes. Although there is a lot of evidence for alternative splicing on the RNA level, for many of the isoforms it is still not known whether they are translated into proteins. This knowledge is crucial to understanding the biological consequences of alternative splicing, and toward identifying protein biomarkers that result from the translation of splice variants. Protein isoforms have significant potential as biomarkers to increase the accuracy of diagnosis, prognosis, or therapy prediction of the disease (18). Identification of disease-specific protein isoforms enables the discovery of biomarkers with better sensitivity and/or specificity.
Protein isoforms can be studied on the proteome level with the use of in-depth tandem mass spectrometry. Proteogenomics is a dynamic field integrating genomic and proteomic data (19). One of the focus areas in the field is to increase the knowledge of the human proteome and identify novel variant proteins resulting from single nucleotide variants or aberrant splicing (20, 21). The number of bioinformatics tools for performing proteogenomic analysis is rapidly increasing, including tools for proteogenomic database construction (22–27) or visualization of the peptides on the genome (28, 29). However, a number of these tools lack an automated, user-friendly downstream analysis after MS/MS identification to extract interesting outcomes. Moreover, the tools are often designed for single sample or single cohort analysis without the flexibility to perform a differential comparison between case and control groups on both RNA and protein level. To identify disease specific biomarkers resulting from aberrant splicing there is a need for a tool that will perform a differential group analysis.
Here we present a method to identify tumor-specific protein isoforms based on RNA-seq and mass spectrometry (LC-MS/MS) data. In this approach, RNA-seq analysis is used to perform quantitative isoform analysis and identify differential splice variants, and LC-MS/MS confirms translation of these variants into proteins. The method was applied to the CRC cell line SW480 upon downmodulation of the splicing machinery factors SF3B1 and SRSF1. In this way, a controlled setting was created that allowed to monitor changes in alternative splicing and consequently, to design a pipeline for proteogenomic analysis of spliced isoforms. The methodological novelty of this approach lies in differential analysis of alternative splicing between two groups in two molecular domains and could be applied in any comparative setting such as gene knock-down versus control or cancer versus healthy control.
EXPERIMENTAL PROCEDURES
Cell Culture, Gene Knock-down and Cell Viability Assay
SW480 cells cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Bleiswijk, The Netherlands) containing 10% fetal bovine serum (FBS; Perbio Science, Etten-Leur, The Netherlands) were maintained in a humidified 5% CO2 atmosphere at 37 °C. Twenty-four hours after seeding, cells were transfected in duplo with small interfering RNA (siRNA) pools against SF3B1 (siGENOME SF3B1 SMARTpool, M-020061-02; Thermo Fisher Scientific, Waltham, MA) and SRSF1 (siGENOME SRSF1 SMARTpool, M-018672-01), according to manufacturer's recommendations. A final siRNA concentration of 30 nm was obtained using DharmaFECT3 reagent (1:1000 dilution; T-2003-02, Thermo Fisher Scientific). A nontargeting siRNA pool (siGENOME Non-Targeting pool #2, d-001206-14) was used as negative control. Cell viability was determined after transfection using the MTT (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide; ICN Biomedicals, Solon, OH) assay, as described previously (30).
RNA Isolation and Quantitative Reverse Transcription PCR
Total RNA was isolated from viable cells, 48 h after siRNA transfection with siSF3B1 and the siNon-Targeting (siNT) control, and 72 h after transfection with siSRSF1 and its siNT control using Trizol reagent (15596; Invitrogen, Breda, The Netherlands) and the miRNeasy Mini Kit (217004; Qiagen, Venlo, the Netherlands), following the manufacturer's protocol. Concentrations and purities were measured on a Nanodrop ND-1000 spectrophotometer (Isogen, Ijsselstein, The Netherlands). cDNA was synthesized using the Iscript cDNA synthesis kit (170-8891; Bio-Rad Laboratories, Hercules, CA). Quantitative reverse transcription PCR (RT-qPCR) was performed using SYBR Green (4309155, Thermo Fisher Scientific), to monitor SF3B1 and SRSF1 knock-down efficiencies and to evaluate efficiency of alternative splicing for ADD3, CTNND1, RAC1, SYK, MKI67, and OSBPL3. Beta-2-Microglobulin (B2M) was used as a housekeeping reference gene. In brief, gene expression was measured using 2 μl of 10 ng/μl cDNA in a 25 μl SYBR Green reaction (see supplemental Table S1 for primers and conditions), as described previously (30).
cDNA Library Preparation and Illumina RNA Sequencing
cDNA libraries were prepared with the TruSeq Stranded mRNA LT sample Prep kit (RS-122-2101, Illumina, San Diego, CA) according to the TruSeq Stranded mRNA sample preparation guide (Part# 15031047, Revision E, October 2013). cDNA library quality control was performed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Sample libraries were diluted and pooled to obtain a final concentration of 10 nm. Sequencing was performed on an Illumina HiSeq V4 2500, using a 125 bases paired end run with an input of 16 pm cDNA. Quality assessment of RNA-seq data was performed with FastQC version 0.11.4 (31) with default settings and visualized with MultiQC version 0.9 (32) with default parameters.
Protein Isolation and Separation
Proteins were isolated at the same time points as RNA extraction. After thorough washing with PBS, cells were lysed in reducing sample buffer (NuPAGE LDS sample buffer, NP0008, Thermo Fisher Scientific; 65% Milli-Q, 25% 4*LDS, 10% 1 m DTT) to obtain an approximate protein concentration of 1 μg/μl. Cells were scraped and transferred to eppendorf tubes. After heating for 5–10 min at 99 °C and centrifugation for 1 min at 14,000 rpm aliquots of the samples were stored at −80 °C until further use. Approximately 35 μg protein from the supernatant was loaded on a NuPAGE Novex 4–12% Bis-Tris Protein Gel, 1.5 mm, 10-well (NP0335BOX; Thermo Fisher Scientific). Proteins were resolved at 150V for 1 h in 200 ml NuPAGE MES SDS Running buffer (NP0002; Thermo Fisher Scientific) supplemented with 0.5 ml NuPAGE antioxidant (NP0005; Thermo Fisher Scientific). The gel was placed in a container with fixing solution (50% ethanol, 46.5% Milli-Q and 3.5% phosphoric acid) for 15 min and stained with colloïdal Coomassie (48.4% Milli-Q, 34% methanol, 15% ammonium sulfate, 2.5% phosphoric acid, 0.1% Coomassie Brilliant Blue G-250 (20279; Thermo Fisher Scientific)) overnight and destained with multiple changes of Milli-Q water. Each gel lane was sliced in 10 slices.
Whole Gel In-gel Digestion
The in-gel digestion procedure was done as described previously (33) with the following changes: gel pieces were dried in a centrifugal evaporator (SpeedVac) for ∼30 min and peptides were extracted with 100 μl 1% formic acid and two times 150 μl 5% formic acid/50% acetonitrile. Concentrated extracts were transferred to Millipore filters (Millex-HV Syringe driven filter unit, 0.45 μm, SLHVR04NL, Millipore), placed on autosampler vials and centrifuged for 5 min at room temperature in the centrifugal evaporator without vacuum.
LC-MS/MS
Peptides were separated by an Ultimate 3000 nanoLC-MS/MS system (Dionex LC-Packings, Amsterdam, The Netherlands) equipped with a 40 cm × 75 μm ID fused silica column custom packed with 1.9 μm 120 Å ReproSil Pur C18 aqua (Dr Maisch GMBH, Ammerbuch-Entringen, Germany). After injection, peptides were trapped at 10 μl/min on a 10 mm × 100 μm ID trap column packed with 5 μm 120 Å ReproSil Pur C18 aqua at 2% buffer B (buffer A: 0.5% acetic acid in MQ; buffer B: 80% ACN + 0.5% acetic acid in MQ) and separated at 300 nl/min in a 10–40% buffer B gradient in 60 min (90 min inject-to-inject). The nanoLC column was maintained at 50 °C using a column heater (Phoenix S&T, Chester, PA). Eluting peptides were ionized at a potential of +2 kVa into a Q Exactive mass spectrometer (Thermo Fisher Scientific). Intact masses were measured at resolution 70.000 (at m/z 200) in the orbitrap using an AGC target value of 3 × 106 charges. The top 10 peptide signals (charge-states 2+ and higher) were submitted to MS/MS in the HCD (higher-energy collision) cell (1.6 m/z isolation width, 25% normalized collision energy). MS/MS spectra were acquired at resolution 17.500 (at m/z 200) in the orbitrap using an AGC target value of 1 × 106 charges and an underfill ratio of 0.5%. Dynamic exclusion was applied with a repeat count of 1 and an exclusion time of 30 s.
Full Length Isoform Sequencing - Iso-Seq
RNA isolated from siSF3B1- and siNT-treated SW480 cells was subjected to full-length RNA single molecule real time (SMRT) sequencing called Iso-Seq (34). Briefly, RNA (RIN score of ∼9.0 assessed by Agilent Bioanalysis) was amplified using the Clontech Switching Mechanism at 5′ end of RNA Template (SMART) technology which incorporates known sequence at both ends of the cDNA product in the first strand synthesis process without the need for conventional adapter ligation strategies. Four hundred eight nanograms of siSF3B1 and 352 ng siNT cDNA were used as input to the SMART cDNA amplification process to capture full-length, intact isoforms to be reverse transcribed and amplified into full-length cDNA representing the full transcriptome where the known sequences are used to complete SMRTbell library preparation using the cDNA products.
Once ample double stranded cDNA was synthesized, cDNA Iso-Seq sequencing libraries were prepared using the SMRTbell library preparation procedure resulting in a library containing molecular inserts that represent a single isoform per library molecule. These libraries were then size-selected to enrich for isoforms of interest by targeting a population of full-length transcripts to enhance coverage by loading individual size fractions on single SMRTcells. More specifically, the SageELF electrophoretic lateral fractionator instrument was used to separate independent fractions of library where isoforms that are 0–1 kbp, 1 kbp–2 kbp, 2 kbp–3 kbp, and 3 kbp–50 kbp were split into independent SMRTbell libraries for sequencing so that larger isoforms were not detrimentally dominated by smaller isoform library molecules during the sequencing process.
Finally, samples were sequenced using 6-hr movie collection on the PacBio RSII sequencer with two SMRTcells per cDNA size fraction. The RSII data yielded 523k to 750k subreads for each size fraction of the siNT sample, resulting in 66.8k to 98.3k CCS reads with up to 43k full length cDNA reads per size fraction. For siSF3B1, the RSII yield was 321k to 981k subreads for each size fraction, resulting in 47.5k to 97.3k CCS reads with up to 51.7k full-length cDNA reads per size fraction, using default Iso-Seq pipeline settings. Raw sequencing data was processed using Iso-Seq on PacBio SMRTportal (smrtanalysis v2.3.0) and ICE software (35) to predict low and high-quality isoforms and generate high resolution transcriptome references.
RNA-seq and LC-MS/MS Data Analysis Within the Proteogenomic Pipeline Splicify
The schematic overview of the proteogenomic pipeline, Splicify, is presented in Fig. 1A. Low quality reads and adapter sequences were trimmed by Trimmomatic (36) version 3 to average quality score for a 4-base wide sliding window of 20, both at the beginning and at the end of the sequences (ILLUMINACLIP:TruSeq3-PE.fa:2:30:10, LEADING:20, TRAILING:20, AVGQUAL:20, SLIDINGWINDOW:4:20). Because of the requirements of the further analysis (rMATS (37)) reads were processed to match length of 120 bp, shorter reads were discarded and longer reads were trimmed (CROP:120, MINLEN:120). Mapping was performed with the use of STAR aligner (38) version 2.4.2a to the human genome (USCS RefSeq hg19 annotation, as STAR option genomeDir) with the following parameters; outSAMtype: BAM SortedByCoordinate, readFilesCommand: zcat, runThreadN: 28, outSAMattributes: All. Differential splice variants were identified with rMATS version 3.2.5 using UCSC RefSeq hg19 GTF file as annotation in the unpaired analysis type (parameters; len: 120, t: paired, analysis: U). Significant events were extracted (FDR≤0.05). Both inclusion- and exclusion-isoforms of spliced genomic fragments were taken into account for further analysis. Nucleotide acid sequences of splicing regions (upstream and downstream exons with and without spliced fragment) were obtained and translated in forward frame to amino acid sequences. In this way, a database was obtained with protein sequences of potential splice variants that were all added to the human reference proteome database (Uniprot, release January 2014, no fragments, canonical and isoform, 42104 entries (39)) forming an enriched human protein database. Peptide identification was performed by MaxQuant 1.5.3.8 (40) with the use of the enriched human protein database. Enzyme specificity was set to trypsin and up to two missed cleavages were allowed. Cysteine carboxamidomethylation was treated as fixed modification and methionine oxidation and N-terminal acetylation as variable modifications. Peptide precursor ions were searched with a maximum mass deviation of 4.5 ppm and fragment ions with a maximum mass deviation of 20 ppm. Peptide and protein identifications were filtered at an FDR of 1% using the decoy database strategy. Common contaminants were included in the MS/MS search. Evidence and peptides files were taken along for further analysis. Peptides specific for splice variants were extracted. Additionally, human database of canonical proteins (Swissprot, canonical, 20197 entries) was used to detect which of the splice variants represented non-canonical isoforms. Peptide intensities were normalized to the average of the samples' medians and log 10 transformed. Imputation was performed on the normalized and transformed matrix, where missing values were imputed from the normal distribution of mean equal to minimal intensity observed and standard deviation equal to mean of standard deviations calculated for each peptide. Differential peptide expression analysis was performed with a Bioconductor package limma (41) and log10 fold changes and p values were obtained. Splicify is available at (https://github.com/NKI-TGO/SPLICIFY).
Fig. 1.

Splicify, the proteogenomic pipeline for identification of differential splice variants. A, The schematic overview of the Splicify data analysis. Within Splicify RNA-seq data analysis is performed by combining exemplar open-source RNA-seq analysis software, including quality and adapter trimming with Trimmomatic (36), reads mapping with STAR (38), differential splicing analysis with rMATS (37), where differential splice variants on RNA level are identified. These splice variants undergo 3-frame translation into potential protein isoform sequence database (FASTA). This database together with the human protein database from Uniprot (39) can be further used with MaxQuant (40), a search engine to identify MS/MS spectra originating from the same samples as RNA-seq reads. Downstream analysis of MaxQuant output is performed with the use of the results from RNA-seq analysis. Isoform-specific peptides are extracted and quantified and based on these peptides differential protein isoforms are identified. Splicify produces a final table with both RNA and protein isoform information. B, Example of peptides supporting translation of splicing events for skipped exon and retained intron. Split peptides map to both sides of an exon-exon junction, spanning peptides span exon-intron junctions (specific for inclusion variants for retained intron, alternative 3′ and 5′ splice sites) and peptides on target map to a spliced fragment.
Isoform Identification with the Use of Full Length Transcripts
Redundant transcripts were removed by first aligning them to the human genome (hg19) with GMAP (42) and collapsing highly similar transcripts predicted across FASTA files from various size fractions with the software cupcake ToFU (v1.3). In these steps both BAM and GTF files were produced for each sample. Samples were chained, to standardize transcript IDs and merge the transcripts from both experiments. Details of the workflow can be found here (43). The merged file was used as input to rMATS instead of human reference annotation GTF file. In this way, the program can use the exon-exon and exon-intron junctions introduced by Iso-seq. Splice variants identified by rMATS were annotated by changing Iso-Seq transcript IDs into gene names based on genomic location, with the use of biomaRt Bioconductor package version 2.26.1(44). In case the Iso-Seq transcript was on the opposite strand than the gene, “otherstrand” was added to the gene symbol. In case there was no gene matching the coordinates of the transcript, “intergenic” was used as a gene symbol. The annotated output of rMATS was further processed as described in the RNA-seq and LC-MS/MS Data Analysis Within the Proteogenomic Pipeline section, with the exclusion of the quantification step.
RESULTS
Experimental Model System To Test the Proteogenomic Pipeline Design
The schematic overview of Splicify, the proteogenomic data analysis pipeline for identification of differential splice variants, is presented in Fig. 1A. To test the design of the proteogenomic pipeline, a model system needed to be established in which modulation of isoform changes could be controlled experimentally. For this purpose, the splicing factors SF3B1 and SRSF1, which play a key role in the splicing machinery, were downmodulated in the CRC cell line SW480, followed by RNA-seq-based transcriptomics and mass spectrometry-based proteomics analyses. A general overview of the experimental design is presented in Fig. 2.
Fig. 2.

General overview of the experimental design and data analysis. Downmodulation of splicing factors SF3B1 (48 h) and SRSF1 (72 h) was performed in CRC cell line SW480 three times. RT-qPCR of known splicing events obtained from literature (skipped exon in ADD3 and in CTNND1) were used as positive controls of alternative splicing to functionally verify that downmodulation of the splicing machinery caused differential splicing. The knock-downs and the paired non-targeting (NT) controls were subjected to RNA-sequencing and LC-MS/MS tandem mass spectrometry, followed by data analysis using the proteogenomic pipeline Splicify (see Fig. 1). Differential mRNA splice variants were identified and several candidates were validated with RT-qPCR. Isoform specific peptides were identified and differential expression of these peptides was performed. Downmodulation of SF3B1 was repeated in a separate experiment, including PacBio Iso-Seq sequencing of full length transcripts while excluding isoform-specific peptide quantitative analysis because of the lack of replicates.
The efficiency of siRNA-mediated downmodulation of SF3B1 and SRSF1 in SW480 CRC cells was determined by RT-qPCR, and reached on average up to a 50 and 40% reduction of mRNA expression for SF3B1 and SRSF1, respectively (supplemental Fig. S1). Cell viability was reduced by 10–30% by downmodulation of SF3B1 at 48 h after transfection, whereas no changes in cell viability were observed after the knockdown of SRSF1 at 72 h after transfection (data not shown). To assure that downmodulation of SF3B1 and SRSF1 resulted functionally in changes in expression of certain isoforms, monitoring of positive controls was included in the experiment. Skipped exons in ADD3 and CTNND1 were identified by literature search as positive controls for alternative splicing in colorectal cancer tissue compared with normal colon tissue (35). Indeed, RT-qPCR analysis for ADD3 exon 14 and CTNND1 exon 20 indicated that exclusion of these exons served as functional splicing controls for knock-down of SF3B1 and SRSF1, respectively (supplemental Fig. S2). These data demonstrate that a model system was established in which isoform switches can be modulated in a CRC cell line, suited to test the design of the proteogenomic pipeline.
Identification of Differentially Expressed RNA and Protein Isoforms by Applying the Proteogenomic Pipeline
To investigate alternative splicing in both the RNA and protein molecular domains, the transcriptome and the proteome of each sample were analyzed with RNA-seq and tandem mass spectrometry. Quality assessment of RNA-seq and LC-MS/MS data is available in supplemental Figs. S3–S5. Within the RNA-seq data analysis, isoforms were identified with the use of reads spanning exon-exon and exon-intron junctions. These splice-variant specific reads, together with reads mapping to the spliced fragment, were further quantified to distinguish differential events between two conditions. In the proteomics data analysis, exon-exon and exon-intron junction-spanning peptides and peptides mapping on the spliced fragment were used to confirm translation of the isoforms detected on the RNA level into proteins (Fig. 1B). The intensities of these peptides were used for quantification to identify differentially expressed protein isoforms. For details, see Fig. 1A.
Differential mRNA Isoforms Induced by Downmodulation of SF3B1 and SRSF1
Transcriptome analysis revealed a number of significantly differentially spliced events for siSF3B1 and siSRSF1 in comparison to their controls (Fig. 3A; see supplemental Tables S3–S12 for details of all the events), proving that manipulation of the splicing machinery resulted in differential splicing. Alternative splicing was more affected upon manipulation of SF3B1 compared with SRSF1, as the number of alternatively spliced events was larger for this splicing factor, for the events like skipped exon and mutually exclusive exons (Fig. 3A). This might be because of the different roles that these splicing factors play in the spliceosome complex. The significantly skipped exon events included the positive controls of alternative splicing, higher exclusion levels of ADD3 exon 14 upon downmodulation of SF3B1 and higher exclusion levels of CTNND1 exon 20 upon downmodulation of SRSF1 (supplemental Fig. S6). These data show that the intermediate mRNA results of the proteogenomic pipeline reproduced the expected outcome, and yielded information about hundreds (for SRSF1) to thousands (for SF3B1) of additional alternative splicing events.
Fig. 3.

The number of splicing events identified on RNA and protein level upon knock-down of SF3B1 and SRSF1. A, Number of significant alternatively spliced events on RNA level upon downmodulation of SF3B1 and SRSF1 versus their controls. B, The number of alternative splicing events for which at least one variant (inclusion/exclusion) was confirmed by identification of isoform-specific peptides. S.E. - skipped exon; MXE - mutually exclusive exons; A5SS - alternative 5′ splice site; A3SS - alternative 3′ splice site; RI - retained intron.
To further validate our approach, four skipped exon splicing events were selected for confirmation by RT-qPCR, comprising SYK exon 7, RAC1 exon 4, OSBPL3 exon 9, and MKI67 exon 7 (Fig. 4, supplemental Table S2). These isoforms are also known as SYK(S) and SYK(L), Rac1b and MKI67 long and short isoforms. According to the RNA-seq analysis, all the events were differentially spliced upon downmodulation of SRSF1 whereas OSBPL3 and MKI67 were affected by downmodulation of SF3B1. The differences in the expression of inclusion and exclusion variants between downmodulation and controls were validated with RT-qPCR (supplemental Fig. S7–S9).
Fig. 4.
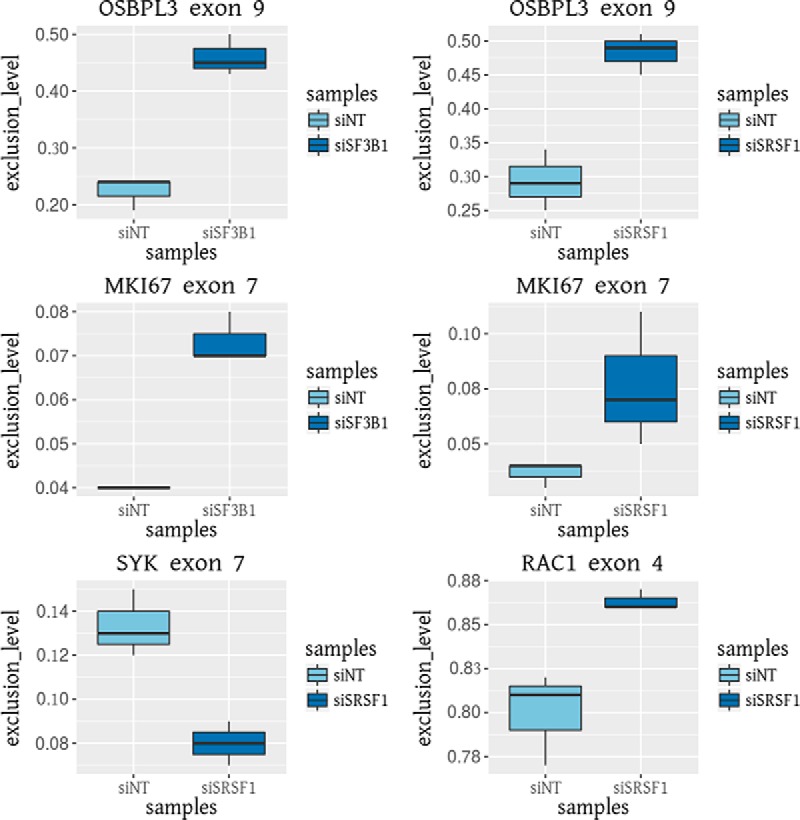
RT-qPCR validation of differential splicing events identified by RNA-seq data analysis with the proteogenomic pipeline, Splicify. The exclusion isoforms of OSBPL3 exon 9 and MKI67 exon 7 are higher expressed upon downmodulation of SF3B1 and SRSF1. The inclusion isoform of SYK exon 7 and the exclusion isoform of RAC1 exon 4 are higher expressed upon downmodulation of SRSF1. Exclusion levels were calculated by dividing exclusion spanning reads by the sum of inclusion and exclusion spanning reads.
Differential Protein Isoforms Induced by Downmodulation of SF3B1 and SRSF1
All significant events identified on RNA level, comprising both exclusion and inclusion variants, were taken along for database construction for mass spectra identification. To prove that these splicing events are translated into proteins we searched for the peptides specific for the splice isoforms (Fig. 1B). Over 5070 and 370 isoform-specific peptides were identified for differential isoforms upon downmodulation of SF3B1 and SRSF1, respectively (Table I, see supplemental Fig. S10 for quality control of isoform-specific peptides). The differences in these numbers correspond to the sizes of the splice variant databases of the two experiments. Overall around 60% of the isoform-specific peptides turned out to map on target, peptides spanning exon-exon junction comprised around 40% and exon-intron junctions were identified far less frequently (Table II).
Table I. Overview of isoform-specific peptides identified upon knock-down of SF3B1 and SRSF1. The numbers of peptides specific for inclusion and exclusion isoforms are listed. Some peptides map to multiple isoforms, being inclusion-specific for one isoform and exclusion-specific for the other.
| Experiment | Isoform-specific peptides | Inclusion-specific peptides | Exclusion-specific peptides |
|---|---|---|---|
| siSF3B1 vs siNT | 5079 | 4525 | 833 |
| siSRSF1 vs siNT | 374 | 309 | 87 |
Table II. Overview of categories of isoform-specific peptides identified upon knock-down of SF3B1 and SRSF1. Peptides on target map fully on the spliced fragment, spanning peptides span exon-intron junctions and split peptides span exon-exon junctions (see also Fig. 1B).
| Experiment | On target | Spanning peptide | Split peptide |
|---|---|---|---|
| siSF3B1 vs siNT | 3278 | 9 | 1794 |
| siSRSF1 vs siNT | 217 | 3 | 154 |
Based on all the isoform-specific peptides, 2172 and 149 isoforms on protein level were identified for siSF3B1 and siSRSF1, respectively (Table III). On average for ∼15% of the splicing events peptide confirmation was observed for both inclusion and exclusion variants of the same event. Most of these isoforms are considered canonical proteins based on the Swissprot canonical sequence database. Approximately 5 and 25% of the identified isoforms were classified as noncanonical for siSF3B1 and siSRSF1, respectively. A subset of peptides mapped to two or more isoforms, usually because of the overlapping exons between the different isoforms. More confirmation for inclusion variants was obtained than for exclusion variants, because of the longer sequences of the inclusion variants. Among the identified isoforms all categories of alternatively spliced events were represented, with most peptides supporting the skipped exon splicing category because of the predominance of this class already at the RNA level. Relatively, looking at the ratios of number of splicing events on RNA and protein level, mutually exclusive exons are more frequently detected (Fig. 3B). This is mainly because mutually exclusive exons do not have an exclusion variant as both isoforms include an additional exon, thereby increasing the overall fragment length and consequently the probability of peptide identification within the spliced region. Even though for the splicing controls ADD3 and CTNND1 no variant-specific peptides were detected, other events such as alternatively skipped exon in SYK, RAC1, OSBPL3, and MKI67 were confirmed on peptide level (supplemental Tables S13–S14).
Table III. Overview of mRNA splicing events confirmed by proteomics upon knock-down of SF3B1 and SRSF1. RNA isoforms were considered to be translated if there was at least one splice-specific peptide identified. For a subset of alternatively spliced events both inclusion and exclusion variants were confirmed by identification of splice-specific peptides. Based on the database of canonical proteins a small number of non-canonical proteins was identified.
| Experiment | Alternatively spliced events | Inclusion isoforms | Exclusion isoforms | Events with both isoforms | Non-canonical isoforms |
|---|---|---|---|---|---|
| siSF3B1 vs siNT | 2172 | 2006 | 400 | 234 | 93 |
| siSRSF1 vs siNT | 149 | 128 | 47 | 26 | 36 |
Differential peptide expression analysis was performed for all of the splice-specific peptides and revealed that a subset of these peptides did significantly differ between splice factor knock-downs and controls, indicating concordant events between mRNA genomic and proteomic results (Table IV, supplemental Tables S13–S14). For both experiments around 65% of the significantly differentially expressed isoform-specific peptides showed concordant expression differences as observed on the RNA level. For instance, upon downmodulation of SF3B1 three split peptides spanning inclusion of exon 9 in OSBPL3 and one split peptide supporting the exclusion of this exon were identified. Two of the inclusion specific peptides show significantly lower expression upon downmodulation of SF3B1 whereas the exclusion specific peptide indicates higher expression in comparison to the control (Fig. 5; supplemental Table S13). Another example is lower expression of the Rac1b isoform, resulting from the inclusion of exon 4 in RAC1 gene, upon downmodulation of SRSF1, which is in line with the current knowledge of the SRSF1 effect on alternative splicing of RAC1 in colorectal cancer (16). This result was detected in the proteogenomic pipeline at RNA level, both by RNA-seq and by RT-qPCR (Fig. 4; supplemental Fig. S9B). On protein level only inclusion specific peptides were identified. Even though the differences in peptide intensities between siSRSF1 downmodulation and the control were not significant, log10 fold changes suggest a similar effect as on RNA level (supplemental Fig. S11; supplemental Table S14).
Table IV. The number of isoform specific-peptides showing consistent or opposite expression changes as detected on RNA level. Isoform-specific peptides were filtered based on p value ≤0.1 and absolute value of log10 transformed fold change ≥0.5, to extract only the peptides differentially expressed between the two conditions; siRNA mediated down-modulation of a splicing factor and the non-targeting control. For inclusion-specific peptides, a peptide was labelled as “consistent” if the log10 fold change of the peptide expression showed the same direction of change as the Inclusion Level Difference for the RNA splice variant. For exclusion-specific peptides, a peptide was labelled as “consistent” if the direction of change was the opposite of the RNA-derived Inclusion Level Difference. As a subset of peptides maps to multiple isoforms, the percentages might exceed 100%.
| Experiment | Number (and percentage) of the isoform-specific peptides |
|
|---|---|---|
| Consistent | Opposite | |
| siSF3B1 vs siNT | 267 (65%) | 157 (38%) |
| siSRSF1 vs siNT | 16 (64%) | 9 (36%) |
Fig. 5.

Splicing isoforms of OSBPL3 presented in two molecular domains. A, Screenshot from IGV of the spliced region of OSBPL3 exon 9; in blue -RefSeq genes, in black - inclusion and exclusion variants identified with RNA-seq, in pink - inclusion and exclusion specific peptides identified in mass spectrometry. B, Peptide intensities upon downmodulation of SF3B1 and its control for two inclusion specific peptides and one exclusion specific peptide for exon 9 in OSBPL3. Peptide number on the x axis corresponds to the peptide sequence in the table. Intensities of the overlapping peptides TYSAPAINAIQGGCFESPK and TYSAPAINAIQGGCFESPKK were manually summed and annotated as TYSAPAINAIQGGCFESPK[K] in the table and as peptide number 2 on the figure. Differential peptide expression analysis was performed with limma with no imputation for all the isoform-specific peptides including the merged peptide TYSAPAINAIQGGCFESPK[K] instead of the two. Even though not all of the peptides are significantly up or downregulated, the signal is concordant with RT-qPCR and RNA-seq results, with higher exclusion and lower inclusion of the exon upon downmodulation of SF3B1.
Full-length Transcripts Validation
To examine if sequencing of full length transcripts can validate the isoforms identified within Splicify and enrich these results with novel transcripts, Iso-Seq was performed in SW480 cells upon downmodulation of SF3B1 and its siNT control (see Fig. 2). As Iso-Seq provides qualitative information, transcripts detected by this technique were used as the source of transcriptome variation instead of the human reference annotation, which could be further quantified upon mapping back the shorter, but higher density Illumina reads. On RNA level, within each alternative splicing category, the number of significantly differential isoforms identified with the use of Iso-Seq data exceeded the results compared with the approach making use of the reference annotation (Fig. 6A; see supplemental Tables S15–S24 for details). There was a large overlap between detection of alternatively spliced events by Illumina-sequencing using the human reference annotation and the analysis that used Illumina reads with the Iso-Seq full length transcripts, thereby validating detection of alternatively spliced events by Splicify (Fig. 6B). Additionally, full length isoform sequencing revealed several novel events that were not detected with the standard Splicify approach before because of absence of these events in the reference genome annotation. The largest effect is noticed for detection of retained intron events, where rMATS uses a database of annotated retained introns instead of all the introns in the genome. On the protein level, the majority of the isoform-specific peptides were identified with both approaches (Fig. 6C). However, the protein database composed of the Iso-seq based findings increased the number of identified isoform-specific peptides compared with the use of the human reference annotation (supplemental Tables S25–S26). For example, three peptides supporting intron retention in FXR1 were identified by sequencing of full-length transcripts that included this intron, which therefore was included in the annotation file. Illumina short reads supported this event and provided quantitative proof that it is higher expressed upon downmodulation of SF3B1 compared with its control (Fig. 6D). These data indicate that to unravel differential splicing events more comprehensively, one should provide annotation files enriched with novel transcripts from e.g. transcriptome assembly tools or full-length transcript sequencing.
Fig. 6.

Comparison of the standard Splicify approach with the reference annotation and Splicify analysis with Iso-seq full length transcripts used as annotation. A, Number of significant alternatively spliced events on RNA level for downmodulation of SF3B1 versus the non-targeting controls with the use of Iso-Seq full-length transcripts or Reference Annotation, S.E. - skipped exon, MXE - mutually exclusive exons, A5SS - alternative 5′ splice site, A3SS - alternative 3′ splice site, RI - retained intron. Illumina reads were quantified on alternatively spliced events originating from reference annotation or Iso-Seq full-length transcripts. B, Overlap analysis between alternatively spliced events upon downmodulation of SF3B1 and its control, identified with reference annotation or Iso-Seq used as annotation. Overlap was defined by chromosome number and coordinates of the spliced fragment. In case of skipped exon, retained intron and alternatively spliced sites it was one fragment, in case of mutually exclusive exons, coordinates of both exons were taken into the overlap. C, Overlap analysis of splice-specific peptides identified with the databases based on the approach including reference annotation or Iso-Seq data. Differential splicing events were translated in 3-frame into potential proteins. These databases were used for mass spectra identification with MaxQuant. Splice-specific peptides were extracted from the MaxQuant output. Overlap analysis was performed based on unique peptide sequences. D, IGV screenshot of retained intron in FXR1 gene. Blue and red coverage plots represent Illumina reads for samples siSF3B1-4 and siNT-4, respectively. Below in dark blue - reference annotation, in green - Iso-Seq transcripts obtained from the same samples, in black - retained intron event identified with Iso-Seq and quantified by Illumina reads, in pink - 3 peptides spanning the exon-intron junction and supporting intron retention on protein level.
DISCUSSION
Splicify was designed to identify differentially expressed splice variants on RNA and protein level. Splicify was applied on CRC cell line SW480 upon downmodulation of splicing machinery and nontargeting controls. We showed that this method can successfully identify condition-specific aberrant splicing events on protein level, by performing comparative splice variant analysis on both RNA and protein level. A subset of the RNA-seq based results of Splicify was validated by RT-qPCR. This proved that the pipeline yielded real splice variants on RNA level. Additionally, applying Splicify using PacBio Iso-Seq full-length transcript sequencing confirmed the existence of the identified isoforms and increased the transcriptomic space to detect novel events. These were especially prevalent in the retained intron and alternative 3′ and 5′ splice site splicing events, where the overlap between Splicify with reference annotation and Splicify with Iso-Seq full length transcripts was smaller than for skipped exon and mutually exclusive exons splicing events. This shows that the reference annotation is still lacking several alternatively spliced isoforms which include whole or a part of the intronic sequence. A number of the novel events were also detected on protein level. This indicates that Splicify, next to the standard approach with the use of the human reference annotation, can also be applied using an alternative transcriptome annotation file that extends isoform identification with novel splicing events. On protein level, we identified several noncanonical isoforms, which is a valuable finding as it indicates their translation into proteins that may play a different functional role in comparison to their canonical counterparts. This is known for the Rac1b isoform, which has been shown to have a different functional role than the canonical RAC1 protein enhancing cell survival (45). Splicing of RAC1 is known to be dependent on SRSF1 activity, which was confirmed with the Splicify pipeline applied to the SW480 CRC cell line upon downmodulation of SRSF1. These data indicate that the results on protein level are in line with current literature. Other interesting findings include the detection of differential splice variants of OSBPL3. These isoforms have been shown to be differentially expressed on RNA level in various tissues, indicating that the OSBPL3 splice variants might have different functionality (46). Translation of these splice-variants into proteins and differential protein isoform expression was now shown by Splicify. These results demonstrate that Splicify successfully identifies differentially expressed mRNA and protein isoforms.
Our findings include identification of several other biologically interesting isoforms that might be linked to SF3B1 and SRSF1 activity. For instance, SYK splice variants, SYK(S) and SYK(L), have been shown to play a role in breast, liver and colorectal cancers (47). Alternative splicing of SYK has been demonstrated to regulate colorectal cancer progression and sensitivity of CRC cells to chemotherapy (48). Here, identification of differential splicing of SYK upon downregulation of SRSF1 might indicate possible impact of SRSF1 on alternative splicing of SYK and subsequently on colorectal cancer progression and chemotherapy resistance. Another interesting finding is identification of differential expression of MKI67 long and short isoforms upon modulation of SF3B1 as well as SRSF1 expression. It is speculated that MKI67 long isoform plays a role in cell differentiation by causing the cell to exit the cell cycle, whereas the short isoform leads to permanent cell cycle (49). Based on our findings, one could hypothesize that SF3B1 and SRSF1 might regulate cell proliferation through alternative splicing of MKI67. However, further studies are needed to support these statements. In addition to these examples, Splicify provided many other differentially spliced isoforms. Studies that aim to investigate gene function or biomarker utility could focus on splice events with peptide evidence, as these events confirm RNA translation that implies functional consequences. Moreover, different filtering approaches can be applied, e.g. based on fold change in RNA and protein expression, false discovery rates or the number of split peptides required.
The small number of protein isoforms that were detected compared with the results obtained based on analyses of RNA-seq data demonstrated the current struggles in the field of proteogenomics. There might be various reasons why many mRNA splice variants were not identified on protein level, including biological and technical ones. First, not all of the aberrant isoforms are translated into proteins. For instance, if there is a stop codon on the fragment that is alternatively spliced in, it will lead to degradation of the shorter transcript via nonsense-mediated decay. There are also splicing events called detained introns that may not exit the nucleus and therefore do not undergo translation (50). Another reason might be the kinetics of transcription and translation, concerning the siRNA mediated downmodulation. It is possible that although transcripts are already present on the RNA level, they might not be translated into proteins yet at the time of RNA and protein isolation. Also, low protein isoform count can be a result of post-translational modifications of the spliced regions, for instance phosphorylation, which requires alternative sample processing preceding mass spectrometry to obtain high resolution of phosphopeptide identifications. There are also technical issues that limit the identification of splice-specific peptides, especially for the exclusion variants. If one would exclude the peptides with missed cleavages, there can only be one split peptide spanning an exclusion variant. This peptide needs to have a suitable distribution of lysine and arginine so that it spans the junction, while also having the required length and physicochemical features to be identified by a mass spectrometer. Inclusion isoforms are identified more frequently because of their longer sequence and therefore higher probability to contain a suitable tryptic peptide within the fragment of interest.
All these issues explain the current advantage of RNA-seq over mass spectrometry in terms of performing quantitative analyses of splice fragments. The aberrant isoforms are often lower expressed than canonical proteins, which further complicates differential isoform expression analysis on protein level (5, 51). The 65% consistency of splice variant expression differences on RNA and protein level was expected in the context of multiple studies reporting modest correlation between RNA and protein expression (21, 52, 53). However, the qualitative information provided by mass spectrometry is highly valuable and crucial to determine what isoforms are translated into proteins. Detection of protein isoforms gives more confidence in the functional relevance of splice variants identified on RNA level, and enables to prioritize candidate biomarkers for further studies when identified in both molecular domains. In terms of biomarkers studies, Splicify can be applied in a clinically relevant setting, e.g. to compare a large series of cancer samples to healthy control tissues, and reveal differentially expressed isoforms. As the proteogenomic approach within Splicify is an unbiased first discovery step, these candidate biomarkers should be further quantified by e.g. multiple reaction monitoring or data independent acquisition, preferably both in human tissues and in relevant human body fluids for which a biomarker test is being developed (54–57). Ultimately, a highly robust approach of detecting these isoforms is necessary that could be applied in a clinical setting. For instance, antibodies targeting the spliced region could be incorporated into an immunoassay for testing large cohorts of human samples (56, 57). In conclusion, the output of proteogenomic analysis within Splicify provides answers to basic research and translational research questions, allowing identifying biologically and clinically relevant isoform-specific biomarkers.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (58) partner repository with the dataset identifier PXD006486.
Supplementary Material
Acknowledgments
We thank Bo Han, Sarah Kingan, and Elizabeth Tseng from PacBio for the support with data analysis and David Cozijnsen for his contribution. The transcriptomics data was obtained at Leiden Genome Technology Center, VUmc Clinical Genetics, and Genome Core Facility at the NKI. We also thank HPC facility at the NKI for making the computational analysis possible.
Footnotes
Author contributions: M.A.K., G.A.M., C.R.J., and R.J.F. designed research; M.A.K., A.C.H., S.R.P., A.S.B., T.S., P.M.D.-v.D., M.T., and R.P.S. performed research; M.A.K., T.P., R.P.S., and M.A. contributed new reagents/analytic tools; M.A.K., G.A.M., C.R.J., and R.J.F. analyzed data; M.A.K., A.C.H., S.R.P., A.S.B., R.P.S., G.A.M., C.R.J., and R.J.F. wrote the paper.
* This work was supported by KWF Kankerbestrijding, project number 2013-6025 and PacBio AACR SMRT Grant Award. This work was carried out on the Dutch national e-infrastructure with the support of SURF Foundation.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- CRC
- colorectal cancer
- CCS
- circular consensus sequencing
- A3SS
- alternative 3′ splice site
- A5SS
- alternative 5′ splice site
- MXE
- mutually exclusive exons
- RI
- retained intron
- RNA-seq
- RNA sequencing
- RT-qPCR
- quantitative reverse transcription PCR
- SE
- skipped exon
- siNT
- siNon-Targeting
- siSF3B1
- siRNA mediated downmodulation of SF3B1
- siSRSF1
- siRNA mediated downmodulation of SRSF1
- SMART
- Switching Mechanism at 5′ End of RNA Template
- SMRT
- single molecule real time.
REFERENCES
- 1. Pan Q., Shai O., Lee L. J., Frey B. J., and Blencowe B. J. (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Gen. 40, 1413–1415 [DOI] [PubMed] [Google Scholar]
- 2. Oltean S., and Bates D. O. (2014) Hallmarks of alternative splicing in cancer. Oncogene 33, 5311–5318 [DOI] [PubMed] [Google Scholar]
- 3. Boise L. H., Gonzalez-Garcia M., Postema C. E., Ding L., Lindsten T., Turka L. A., Mao X., Nunez G., and Thompson C. B. (1993) bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell 74, 597–608 [DOI] [PubMed] [Google Scholar]
- 4. Ladomery M. R., Harper S. J., and Bates D. O. (2007) Alternative splicing in angiogenesis: the vascular endothelial growth factor paradigm. Cancer Lett. 249, 133–142 [DOI] [PubMed] [Google Scholar]
- 5. Sveen A., Kilpinen S., Ruusulehto A., Lothe R. A., and Skotheim R. I. (2016) Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 35, 2413–2427 [DOI] [PubMed] [Google Scholar]
- 6. Dvinge H., Kim E., Abdel-Wahab O., and Bradley R. K. (2016) RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 16, 413–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Quesada V., Conde L., Villamor N., Ordonez G. R., Jares P., Bassaganyas L., Ramsay A. J., Bea S., Pinyol M., Martinez-Trillos A., Lopez-Guerra M., Colomer D., Navarro A., Baumann T., Aymerich M., Rozman M., Delgado J., Gine E., Hernandez J. M., Gonzalez-Diaz M., Puente D. A., Velasco G., Freije J. M., Tubio J. M., Royo R., Gelpi J. L., Orozco M., Pisano D. G., Zamora J., Vazquez M., Valencia A., Himmelbauer H., Bayes M., Heath S., Gut M., Gut I., Estivill X., Lopez-Guillermo A., Puente X. S., Campo E., and Lopez-Otin C. (2011) Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Gen. 44, 47–52 [DOI] [PubMed] [Google Scholar]
- 8. Oscier D. G., Rose-Zerilli M. J., Winkelmann N., Gonzalez de Castro D., Gomez B., Forster J., Parker H., Parker A., Gardiner A., Collins A., Else M., Cross N. C., Catovsky D., and Strefford J. C. (2013) The clinical significance of NOTCH1 and SF3B1 mutations in the UK LRF CLL4 trial. Blood 121, 468–475 [DOI] [PubMed] [Google Scholar]
- 9. Anczuków O., Akerman M., Cléry A., Wu J., Shen C., Shirole N. H., Raimer A., Sun S., Jensen M. A., Hua Y., Allain F. H. T., and Krainer A. R. (2015) SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol. Cell 60, 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Karni R., de Stanchina E., Lowe S. W., Sinha R., Mu D., and Krainer A. R. (2007) The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Structural Mol. Biol. 14, 185–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. David C. J., and Manley J. L. (2010) Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Develop. 24, 2343–2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ladomery M. (2013) Aberrant alternative splicing is another hallmark of cancer. Int. J. Cell Biol. 2013, 463786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moore M. J., Wang Q., Kennedy C. J., and Silver P. A. (2010) An Alternative Splicing Network Links Cell Cycle Control to Apoptosis. Cell 142, 625–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Das S., Anczukow O., Akerman M., and Krainer A. R. (2012) Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Reports 1, 110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Anczukow O., Rosenberg A. Z., Akerman M., Das S., Zhan L., Karni R., Muthuswamy S. K., and Krainer A. R. (2012) The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Structural Mol. Biol. 19, 220–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goncalves V., Henriques A. F., Pereira J. F., Neves Costa A., Moyer M. P., Moita L. F., Gama-Carvalho M., Matos P., and Jordan P. (2014) Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor-related Rac1b in colorectal cells. RNA 20, 474–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matos P., and Jordan P. (2008) Increased Rac1b expression sustains colorectal tumor cell survival. Mol. Cancer Res. 6, 1178–1184 [DOI] [PubMed] [Google Scholar]
- 18. Mischak H., Apweiler R., Banks R. E., Conaway M., Coon J., Dominiczak A., Ehrich J. H., Fliser D., Girolami M., Hermjakob H., Hochstrasser D., Jankowski J., Julian B. A., Kolch W., Massy Z. A., Neusuess C., Novak J., Peter K., Rossing K., Schanstra J., Semmes O. J., Theodorescu D., Thongboonkerd V., Weissinger E. M., Van Eyk J. E., and Yamamoto T. (2007) Clinical proteomics: A need to define the field and to begin to set adequate standards. Proteomics. Clin. Appl. 1, 148–156 [DOI] [PubMed] [Google Scholar]
- 19. Ruggles K. V., Krug K., Wang X., Clauser K. R., Wang J., Payne S. H., Fenyo D., Zhang B., and Mani D. R. (2017) Methods, tools and current perspectives in proteogenomics. Mol. Cell. Proteomics 16, 959–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu S., Im H., Bairoch A., Cristofanilli M., Chen R., Deutsch E. W., Dalton S., Fenyo D., Fanayan S., Gates C., Gaudet P., Hincapie M., Hanash S., Kim H., Jeong S. K., Lundberg E., Mias G., Menon R., Mu Z., Nice E., Paik Y. K., Uhlen M., Wells L., Wu S. L., Yan F., Zhang F., Zhang Y., Snyder M., Omenn G. S., Beavis R. C., and Hancock W. S. (2013) A chromosome-centric human proteome project (C-HPP) to characterize the sets of proteins encoded in chromosome 17. J. Proteome Res. 12, 45–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang B., Wang J., Wang X., Zhu J., Liu Q., Shi Z., Chambers M. C., Zimmerman L. J., Shaddox K. F., Kim S., Davies S. R., Wang S., Wang P., Kinsinger C. R., Rivers R. C., Rodriguez H., Townsend R. R., Ellis M. J., Carr S. A., Tabb D. L., Coffey R. J., Slebos R. J., and Liebler D. C. (2014) Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang X., and Zhang B. (2013) customProDB: an R package to generate customized protein databases from RNA-Seq data for proteomics search. Bioinformatics 29, 3235–3237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li Y., Wang X., Cho J. H., Shaw T., Wu Z., Bai B., Wang H., Zhou S., Beach T. G., Wu G., Zhang J., and Peng J. (2016) JUMPg: an Integrative Proteogenomics Pipeline Identifying Unannotated Proteins in Human Brain and Cancer Cells. J. Proteome Res. 15, 2309–2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wen B., Xu S., Sheynkman G. M., Feng Q., Lin L., Wang Q., Xu X., Wang J., and Liu S. (2014) sapFinder: an R/Bioconductor package for detection of variant peptides in shotgun proteomics experiments. Bioinformatics 30, 3136–3138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ruggles K. V., Tang Z., Wang X., Grover H., Askenazi M., Teubl J., Cao S., McLellan M. D., Clauser K. R., Tabb D. L., Mertins P., Slebos R., Erdmann-Gilmore P., Li S., Gunawardena H. P., Xie L., Liu T., Zhou J. Y., Sun S., Hoadley K. A., Perou C. M., Chen X., Davies S. R., Maher C. A., Kinsinger C. R., Rodland K. D., Zhang H., Zhang Z., Ding L., Townsend R. R., Rodriguez H., Chan D., Smith R. D., Liebler D. C., Carr S. A., Payne S., Ellis M. J., and Fenyo D. (2016) An Analysis of the Sensitivity of Proteogenomic Mapping of Somatic Mutations and Novel Splicing Events in Cancer. Mol. Cell. Proteomics 15, 1060–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Woo S., Cha S. W., Merrihew G., He Y., Castellana N., Guest C., MacCoss M., and Bafna V. (2014) Proteogenomic database construction driven from large scale RNA-seq data. J. Proteome Res. 13, 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ghali F., Krishna R., Perkins S., Collins A., Xia D., Wastling J., and Jones A. R. (2014) ProteoAnnotator–open source proteogenomics annotation software supporting PSI standards. Proteomics 14, 2731–2741 [DOI] [PubMed] [Google Scholar]
- 28. Wang X., Slebos R. J., Chambers M. C., Tabb D. L., Liebler D. C., and Zhang B. (2016) proBAMsuite, a bioinformatics framework for genome-based representation and analysis of proteomics data. Mol. Cell. Proteomics 15, 1164–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Askenazi M., Ruggles K. V., and Fenyo D. (2016) PGx: putting peptides to BED. J. Proteome Res. 15, 795–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sillars-Hardebol A. H., Carvalho B., Tijssen M., Belien J. A., de Wit M., Delis-van Diemen P. M., Ponten F., van de Wiel M. A., Fijneman R. J., and Meijer G. A. (2012) TPX2 and AURKA promote 20q amplicon-driven colorectal adenoma to carcinoma progression. Gut 61, 1568–1575 [DOI] [PubMed] [Google Scholar]
- 31. Andrews S. (2015) FastQC a quality control tool for high throughput sequence data. [Google Scholar]
- 32. Ewels P., Magnusson M., Lundin S., and Kaller M. (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Piersma S. R., Warmoes M. O., de Wit M., de Reus I., Knol J. C., and Jimenez C. R. (2013) Whole gel processing procedure for GeLC-MS/MS based proteomics. Proteome Sci. 11, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., Peluso P., Rank D., Baybayan P., Bettman B., Bibillo A., Bjornson K., Chaudhuri B., Christians F., Cicero R., Clark S., Dalal R., Dewinter A., Dixon J., Foquet M., Gaertner A., Hardenbol P., Heiner C., Hester K., Holden D., Kearns G., Kong X., Kuse R., Lacroix Y., Lin S., Lundquist P., Ma C., Marks P., Maxham M., Murphy D., Park I., Pham T., Phillips M., Roy J., Sebra R., Shen G., Sorenson J., Tomaney A., Travers K., Trulson M., Vieceli J., Wegener J., Wu D., Yang A., Zaccarin D., Zhao P., Zhong F., Korlach J., and Turner S. (2009) Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138 [DOI] [PubMed] [Google Scholar]
- 35. Gordon S. P., Tseng E., Salamov A., Zhang J., Meng X., Zhao Z., Kang D., Underwood J., Grigoriev I. V., Figueroa M., Schilling J. S., Chen F., and Wang Z. (2015) Widespread Polycistronic Transcripts in Fungi Revealed by Single-Molecule mRNA Sequencing. PloS One 10, e0132628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bolger A. M., Lohse M., and Usadel B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shen S., Park J. W., and Lu Z. X. (2014) rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U.S.A. 111, E5593–E5601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., and Gingeras T. R. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. The Uniprot Consortium. (2017) UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 41. Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., and Smyth G. K. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu T. D., and Watanabe C. K. (2005) GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859–1875 [DOI] [PubMed] [Google Scholar]
- 43. Magdoll (March/14/2017) https://github.com/Magdoll/cDNA_Cupcake/wiki/Cupcake-ToFU%3A-supporting-scripts-for-Iso-Seq-after-clustering-step GitHub
- 44. Durinck S., Spellman P. T., Birney E., and Huber W. (2009) Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protocols 4, 1184–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Singh A., Karnoub A. E., Palmby T. R., Lengyel E., Sondek J., and Der C. J. (2004) Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 23, 9369–9380 [DOI] [PubMed] [Google Scholar]
- 46. Collier F. M., Gregorio-King C. C., Apostolopoulos J., Walder K., and Kirkland M. A. (2003) ORP3 splice variants and their expression in human tissues and hematopoietic cells. DNA Biol. 22, 1–9 [DOI] [PubMed] [Google Scholar]
- 47. Krisenko M. O., and Geahlen R. L. (2015) Calling in SYK: SYK's dual role as a tumor promoter and tumor suppressor in cancer. Biochim. Biophys. Acta 1853, 254–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ni B., Hu J., Chen D., Li L., Chen D., Wang J., and Wang L. (2016) Alternative splicing of spleen tyrosine kinase differentially regulates colorectal cancer progression. Oncol. Lett. 12, 1737–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schmidt M. H., Broll R., Bruch H. P., Finniss S., Bogler O., and Duchrow M. (2004) Proliferation marker pKi-67 occurs in different isoforms with various cellular effects. J. Cell. Biochem. 91, 1280–1292 [DOI] [PubMed] [Google Scholar]
- 50. Boutz P. L., Bhutkar A., and Sharp P. A. (2015) Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Develop. 29, 63–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gonzàlez-Porta M., Frankish A., Rung J., Harrow J., and Brazma A. (2013) Transcriptome analysis of human tissues and cell lines reveals one dominant transcript per gene. Gen. Biol. 14, R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gry M., Rimini R., Strömberg S., Asplund A., Pontén F., Uhlén M., and Nilsson P. (2009) Correlations between RNA and protein expression profiles in 23 human cell lines. BMC Genomics 10, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kosti I., Jain N., Aran D., Butte A. J., and Sirota M. (2016) Cross-tissue analysis of gene and protein expression in normal and cancer tissues. Scientific Reports 6, 24799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gillet L. C., Navarro P., Tate S., Röst H., Selevsek N., Reiter L., Bonner R., and Aebersold R. (2012) Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anderson L., and Hunter C. L. (2006) Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteomics 5, 573–588 [DOI] [PubMed] [Google Scholar]
- 56. Carr S. A., and Anderson L. (2008) Protein Quantitation Through Targeted Mass Spectrometry: the Way Out of Biomarker Purgatory? Clin. Chem. 54, 1749–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rifai N., Gillette M. A., and Carr S. A. (2006) Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat. Biotech. 24, 971–983 [DOI] [PubMed] [Google Scholar]
- 58. Vizcaino J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (58) partner repository with the dataset identifier PXD006486.