

Summary
The importance of extracellular traps (ETs) in innate immunity is well established, but the molecular mechanisms responsible for their formation remain unclear and in scientific dispute. ETs have been defined as extracellular DNA scaffolds associated with the granule proteins of eosinophils or neutrophils. They are capable of killing bacteria extracellularly. Based mainly on results with phosphoinositide 3‐kinase (PI3K) inhibitors such as 3‐methyladenine (3‐MA) and wortmannin, which are commonly used to inhibit autophagy, several groups have reported that autophagy is required for neutrophil extracellular trap (NET) formation. We decided to investigate this apparent dependence on autophagy for ET release and generated genetically modified mice that lack, specifically in eosinophils or neutrophils, autophagy‐related 5 (Atg5), a gene encoding a protein essential for autophagosome formation. Interestingly, neither eosinophils nor neutrophils from Atg5‐deficient mice exhibited abnormalities in ET formation upon physiological activation or exposure to low concentrations of PMA, although we could confirm that human and mouse eosinophils and neutrophils, after pre‐treatment with inhibitors of class III PI3K, show a block both in reactive oxygen species (ROS) production and in ET formation. The so‐called late autophagy inhibitors bafilomycin A1 and chloroquine, on the other hand, were without effect. These data indicate that ET formation occurs independently of autophagy and that the inhibition of ROS production and ET formation in the presence of 3‐MA and wortmannin is probably owing to their additional ability to block the class I PI3Ks, which are involved in signalling cascades initiated by triggers of ET formation.
Keywords: autophagy, eosinophils, extracellular traps, neutrophil extracellular traps, neutrophils
Abbreviations
- 3‐MA
3‐methyladenine
- ATG
autophagy‐related
- Baf A1
bafilomycin A1
- C5a
complement factor 5a
- CQ
chloroquine
- DHR
dihydrorhodamine
- EET
eosinophil extracellular trap
- EPX
eosinophil peroxidase
- ET
extracellular trap
- GM‐CSF
granulocyte/macrophage colony‐stimulating factor
- IL
interleukin
- LPS
lipopolysaccharide
- NET
neutrophil extracellular trap
- PI3K
phosphoinositide 3‐kinase
- PMA
phorbol 12‐myristate 13‐acetate
- ROS
reactive oxygen species
Introduction
Extracellular DNA traps can be generated by several different leucocytes, including neutrophils, eosinophils, basophils and monocytes, as well as mast cells;1 neutrophil extracellular traps (NETs) have been most frequently studied. Extracellular DNA traps can bind and kill microorganisms in the extracellular space,2 and most investigators agree that NETs contribute significantly to innate immunity and are seen in vivo with many infectious, allergic and autoimmune diseases. The molecular mechanisms responsible for their formation remain unclear and in dispute.3 Several groups have reported a requirement for autophagy in the formation of extracellular traps (ETs). The majority of these reports have used pharmacological approaches to demonstrate that inhibition of the phosphatidylinositol 3‐kinase (PI3K) type III signalling, which is required for autophagosome formation, prevents NET formation, implying a role for autophagy in NET formation.4, 5, 6 Moreover, controversial findings have been reported regarding the effect of mammalian target of rapamycin on the kinetics of NET formation; both inhibitory and potentiating effects have been observed.7, 8 Very recently it has been suggested that ageing‐related ATG5 defects might impair NET formation.9
Autophagy is a cellular mechanism responsible for the turnover of macromolecules and organelles via the lysosomal degradative pathway. Cells usually respond with the induction of autophagy following stress.10 Using wortmannin as an inhibitor of class III PI3K to block PMA‐induced NET formation, Remijsen et al. reported that autophagy has a key role in NET formation.5 It is important to note that wortmannin can also completely inhibit reactive oxygen species (ROS) production in human neutrophils following zymosan, hydrocarbon, tumour necrosis factor‐α, or adhesion receptor stimulation.11, 12, 13, 14, 15 Furthermore, 3‐methyladenine (3‐MA), another inhibitor of class III PI3K, has been shown to reduce ROS production in activated neutrophils just as efficiently as diphenylene iodonium, a potent inhibitor of ROS production.13, 16 As ROS have been shown to be essential for NET formation2, 17 and to be a trigger for autophagy,18 we addressed the question whether autophagy has a direct role in ET formation by silencing Atg5 in both eosinophils and neutrophils. Its product, ATG5, is required for autophagy, playing an early role in phagophore development and especially in autophagosome elongation.10
To this end, we generated mice in which Atg5 can be conditionally deleted in eosinophils (hereafter designated Atg5 eoΔ mice) or in neutrophils (hereafter designated Atg5 NΔ mice19). Data obtained in ATG5‐knockout granulocytes were compared with data from experiments in which pharmacological inhibitors of autophagy were employed in normal mouse and human eosinophils and neutrophils. We report here that ET formation is an autophagy‐independent process in both eosinophils and neutrophils.
Methods
Reagents
Human granulocyte–macrophage colony‐stimulating factor (GM‐CSF) was purchased from Novartis Pharma GmbH (Nürnberg, Germany). Mouse GM‐CSF and human interleukin‐5 (IL‐5) were purchased from R&D Systems (Abingdon, UK). Human and mouse complement factor 5a (C5a) were from Hycult Biotech (Uden, the Netherlands). German glass coverslips (#1 thickness, 12 mm diameter) were obtained from Karl Hecht GmbH & Co. KG “Assistent” (Sondheim/Rhön, Germany). Black, glass‐bottom 96‐well plates were from Greiner Bio‐One GmbH (Frickenhausen, Germany). Lipopolysaccharide (LPS, 055:B5), dihydrorhodamine‐123 (DHR123) and saponin were from Sigma‐Aldrich (Buchs, Switzerland). PMA was from Merck Millipore (Zug, Switzerland). DNase I was purchased from Worthington Biochemical Corporation (Lakewood, NJ). The Quant‐iT™PicoGreen®dsDNA Assay Kit, MitoSOX Red, propidium iodide, Prolong Gold mounting media and Hoechst 33342 were from ThermoFisher Scientific (distributed by LuBioScience GmbH, Lucerne, Switzerland). X‐VIVO™ 15 medium and Medium 199, containing l‐glutamine, HEPES and 1·4 g/l NaHCO3, were from Lonza (Walkersville, MD). The protease inhibitor cocktail was from Roche Diagnostics (Rotkreuz, Switzerland). Polyvalent human IgG was a kind gift from CSL Behring (Bern, Switzerland). Normal goat serum was from Santa Cruz Biotechnology, Inc. (Heidelberg, Germany). ChromPure human IgG was obtained from Milan Analytica AG (Rheinfelden, Switzerland).
Atg5NΔ mice
All animal studies were approved by the Cantonal Veterinary Office of Bern, Switzerland. Atg5 flox/flox mice20 were kindly provided by Dr Christian Münz (University of Zurich, Switzerland). Lyz2 Cre/Cre mice (B6.129P2‐Lyz2tm1(cre)Ifo) were purchased from Jackson Laboratories (Bar Harbor, ME) at backcross generation N6 and were further backcrossed to N10 with C57BL/6J mice. Atg5 flox/flox were crossed with Lyz2 Cre/Cre mice to obtain Atg5 flox/flox‐Lyz2 Cre/Cre (Atg5 NΔ) mice. Mice were genotyped as described previously.19
Atg5eoΔ mice
The Epx‐Cre (eoCRE) strain (kindly provided by Dr J.J. Lee, Mayo Clinic, Scottsdale, AZ) generated by homologous recombination in embryonic stem cells was used to create a knock‐in strain of mice (eoCRE) Epx tm1.1(cre)Jlee /Epx + Gt(ROSA)26Sor tm1(DTA)Lky /Gt(ROSA)26Sor + with a chimeric locus that encodes a mammalianized Cre recombinase from the endogenous AUG start codon of the Epx open reading frame on a C57BL/6J background. The eoCRE strain in conjunction with strains of mice carrying the floxed genes of interest permits eosinophil lineage‐specific targeting of gene expression. It expresses Cre from the open reading frame of the endogenous gene encoding eosinophil peroxidase (Epx) and so expresses Cre only after commitment to the eosinophil lineage and is exceedingly specific for eosinophils rather than cells of other haematopoietic lineages.21 Enzymatic and ELISA data derived from hemizygous or homozygous eoCRE mice showed that the internal ribosome entry site (IRES) element was insufficient to rescue eosinophil peroxidase (EPX) expression from the knock‐in locus. Hence, all subsequent studies described here were performed with eoCRE hemizygous mice that displayed EPX expression levels within a factor of two of wild‐type C57BL/6J animals. Hemizygous experimental eoCRE mice were generated by crossing of homozygous or hemizygous eoCRE mice with wild‐type C57BL/6J animals. Haematological examinations (i.e. cell counts and differentials) of eoCRE mice demonstrated that the baseline levels of circulating leucocytes in these knock‐in animals were unaffected relative to wild‐type.21 Atg5 flox/flox mice were crossed with eoCRE mice to generate Atg5 eoΔ mice.
IL5tg_Atg5eoΔ mice
To increase the number of eosinophils in functional in vitro assays, we generated IL5 tg _Atg5 eoΔ mice by crossing IL‐5 tg (C57BL/6J‐Tg(Il5)1638Jlee) (kindly provided by Dr J.J. Lee) and Atg5 eoΔ mice. Control mice (IL5 tg _Atg5 flox/flox) were generated by crossing IL‐5 tg mice with Atg5 flox/flox mice.
Purification of mouse neutrophils and eosinophils
Lyz2 Cre/Cre and Atg5 NΔ mice were analysed between 8 and 12 weeks of age. In additional experiments, mice were between 9 and 10 months old. Primary bone marrow mouse neutrophils were isolated from control and Atg5 NΔ mice19 under animal license BE54/15, by a negative selection technique using an EasySep Mouse Neutrophil Enrichment Kit (Stemcell Technologies, Grenoble, France). Briefly, bone marrow cells were collected by flushing the femur with 5 ml of isolation medium (PBS plus 2% fetal calf serum, no EDTA added) using a 26‐gauge needle and filtering through a sterile 70‐μm nylon cell strainer. Bone marrow single‐cell suspensions were then washed with medium and the cells were counted with a haemocytometer using the Türk's staining solution (Merck Millipore). Primary bone marrow neutrophils were then isolated by a negative selection technique using the EasySep Mouse Neutrophil Enrichment Kit (Stemcell Technologies) and the isolation medium described above. Mouse neutrophil purity was > 93% as assessed by staining with the Hematocolor Set (Merck Millipore) followed by light microscopic analysis.19
Mouse eosinophils were isolated from IL5 tg _Atg5 flox/flox and IL5 tg _Atg5 eoΔ mice, aged between 8 and 12 weeks. For eosinophil isolation, bone marrow cells were collected by flushing the femurs and tibia with medium (2% fetal calf serum in PBS), using a 26‐gauge needle, and filtering through a sterile 70‐μm nylon cell strainer. Erythrocytes were lysed for 5 min by adding 3 ml of lysis buffer (155 mm NH4Cl, 10 mm KHCO3 and 0·1 mm EDTA, pH 7·3). T cells, B cells, macrophages and neutrophils were depleted with antibodies against CD8α, CD19, CD90.2 and Ly‐6G (Miltenyi Biotec, Bergisch Gladbach, Germany), using an EasySep Mouse PE Positive Selection Kit (Stemcell Technologies). Mouse eosinophil purity was > 95% as assessed by staining with the Hematocolor Set (Merck Millipore) followed by light microscopic analysis.22
Purification of human neutrophils and eosinophils
Written, informed consent was obtained from all blood donors. The Ethics Committee of the Canton of Bern approved this study. Human blood neutrophils were purified from healthy individuals by Ficoll‐Hypaque centrifugation as previously described.23 Briefly, peripheral blood mononuclear cells were separated by centrifugation on Pancoll Human from PAN™ BioTech (PAN‐Biotech GmbH, Aidenbach, Germany). The lower layer, consisting mainly of granulocytes and erythrocytes, was treated with erythrocyte lysis solution (155 mm NH4Cl, 10 mm KHCO3 and 0·1 mm EDTA, pH 7·3). The resulting cell populations contained > 95% neutrophils as assessed by staining with the Hematocolor Set followed by light microscopic analysis.
For eosinophil isolation, white blood cells were isolated from the lithium‐heparinized blood of healthy donors by Ficoll (Biocoll, Biochrom AG) density gradient centrifugation and magnetic beads as previously described.22 Blood was diluted with PBS, layered onto Ficoll and the peripheral blood mononuclear cells were separated from granulocytes/erythrocytes by centrifugation at 800 g. Erythrocytes were lysed with lysis solution (155 mm NH4Cl, 10 mm KHCO3 and 0·1 mm EDTA, pH 7·3) to obtain granulocytes. Eosinophils were isolated with a kit from Stemcell Technologies. Purity of eosinophils was assessed by Diff‐Quik staining and light microscopy and was > 97%.
Immunoblotting
Protein expression was analysed by immunoblotting. 1 × 106 to 5 × 106 cells were washed with PBS and lysed in modified RIPA buffer (50 mm Tris–HCl pH 8·0, 150 mm NaCl, 1% Triton‐X‐100, 0·5% sodium deoxycholate, 0·1% SDS, 2 mm EDTA supplemented with 0·5 mg/ml pepstatin A and complete protease inhibitor cocktail). Protein concentrations were determined by BCA protein assay (ThermoFisher Scientific) and samples were denatured in reducing Laemmli buffer before separation by SDS–PAGE. Proteins were blotted on a PVDF transfer membrane (Merck Millipore, Billerica, MA) and subsequently probed with the following primary antibodies: mouse anti‐ATG5 monoclonal antibody clone 7C6 (1 : 1000; nanoTools, Teningen, Germany, catalogue number #0262‐100), rabbit anti‐p62 polyclonal antibody (1 : 1000; Novus Biologicals LLC, Littleton, CO, catalogue number #NBP1‐48320), rabbit anti‐LC3B polyclonal antibody (1 : 1000; Novus Biologicals LLC, catalogue number #NB600‐1384) or mouse anti‐GAPDH monoclonal antibody clone 6C5 (1 : 5000; Merck Millipore, catalogue number #MAB374). Secondary antibodies coupled to horseradish peroxidase were from GE Healthcare Life Sciences (distributed by VWR International GmbH, Dietikon, Switzerland) and signals were detected by enhanced chemiluminescence (ECL Western blotting substrate, ThermoFisher Scientific) on photosensitive film (ECL Hyperfilm; GE Healthcare Life Sciences).
Cell activation and confocal laser scanning microscopy
Isolated neutrophils or eosinophils were resuspended in X‐VIVO™ 15 medium (2·5 × 106 per ml) and pre‐incubated for 30 min in the presence or absence of inhibitors; 3‐MA (5 mm), wortmannin (100 nm), chloroquine (CQ) (10 μm), bafilomycin A1 (Baf A1) (250 nm) or rapamycin (1 μm). Cells were primed with 25 ng/ml GM‐CSF and IL‐5, respectfully, for 20 min on untreated glass coverslips, which had previously been washed with acetone, ethanol, ddH2O, and baked at a high temperature (200°) oven for 1 hr. Cells were subsequently stimulated with 10−8 m C5a or 100 ng/ml LPS. Unprimed neutrophils were also incubated with 25 nm PMA for 15 min. Cells were then fixed with 4% paraformaldehyde for 5 min, subsequently washed three times in PBS (pH 7·4), stained with 1 μg/ml Hoechst 33342 and mounted in ProLong Gold mounting medium. For extracellular DNA detection, staining with cell‐permeable fluorescent dyes, such as MitoSOX Red (5 μm), was performed with live cells before fixation according to the corresponding manufacturers’ instructions.
Neutrophil elastase or EPX co‐localization with extracellular DNA was analysed by indirect immunofluorescence as previously described.23, 24 Briefly, cells were fixed with 4% paraformaldehyde and permeabilized with 0·05% saponin. Monoclonal mouse antibody to human neutrophil elastase (1 : 1000, clone NP57; Dako, Baar, Switzerland, catalogue number #M0752) or mouse anti‐human EPX (1 : 100, clone MM25‐82·2; obtained from Lee Laboratories, Mayo Clinic, Scottsdale, AZ) served as primary antibodies. Alexa Fluor® 488‐conjugated secondary antibody was obtained from ThermoFisher and was used at 1 : 400. In these experiments, cells were stained with 10 μg/ml propidium iodide for extracellular DNA detection. Slides were examined and images were acquired by LSM 700 (Carl Zeiss Micro Imaging, Jena, Germany) using a 63×/1·40 Oil DIC objective and followed by analysis with imaris software (Bitplane AG, Zurich, Switzerland) as previously reported.23
Quantification of released dsDNA in culture supernatants
Released dsDNA was quantified as previously described.23 Briefly, 2 × 106 isolated neutrophils or eosinophils in 500 μl of medium (X‐VIVO™ 15 for neutrophils, and Medium 199 for eosinophils) were stimulated as described above. At the end of the incubation time, a low concentration of DNase I (2·5 U/ml; Worthington) was added for an additional 10 min, and proteinase K (0·2 mg/ml; Roche) was added at the same time to eosinophils only. Reactions were stopped by addition of 2·5 mm EDTA, pH 8·0. Cells were centrifuged at 500 g for 5 min. Then, 100 μl supernatant was transferred to black, glass‐bottom 96‐well plates (Greiner Bio‐One GmbH) and the fluorescence activity of PicoGreen dye bound to dsDNA was excited at 502 nm and the fluorescence emission intensity was measured at 523 nm using a spectrofluorimeter (SpectraMax M2; Molecular Devices, Biberach an der Riß, Germany), according to the instructions described in the Quant‐iT™PicoGreen® Assay Kit.
Reactive oxygen species measurements
Measurements of ROS were performed in stimulated mouse and human neutrophils by fluorescent detection of ROS activity using flow cytometry as previously described.23 Briefly, 2 × 106/ml neutrophils or eosinophils were resuspended in medium, and 100 μl of cell suspension was pre‐incubated in the presence and absence of different inhibitors for 30 min, before priming with 25 ng/ml GM‐CSF for 20 min. Cells were then stimulated with 10−8 m C5a or 100 ng/ml LPS. As a control, we also stimulated neutrophils with 25 nm PMA for 15 min without GM‐CSF priming. DHR123 was added to the cells at the final concentration of 1 μm. The reaction was stopped by adding 200 μl of ice‐cold PBS, the ROS activity of the samples was measured immediately using flow cytometry (BD FACSCalibur) and quantified using flowjo software (Treestar, Ashland, OR).
Statistical analysis
Mean values (± SEM) are provided. To compare groups, one‐way analysis of variance was applied. P‐values < 0·05 were considered statistically significant.
Results
Atg5‐knockout mouse eosinophils are able to release extracellular DNA
To investigate whether Atg5‐knockout mouse eosinophils are capable of forming ETs, eosinophils were isolated from IL5 tg _Atg5 eoΔ and control IL5 tg _Atg5 flox/flox mice, primed for 20 min with GM‐CSF and subsequently activated with C5a or LPS, or alternatively, with PMA in the absence of priming, all known triggers of eosinophil ET (EET) formation.24 Atg5‐knockout eosinophils were able to form EETs just as well as eosinophils from control IL5 tg _Atg5 flox/flox mice (Fig. 1a). Eosinophils of IL5 tg _Atg5 eoΔ mice displayed a complete ATG5 knockout at the protein level and exhibited strongly reduced autophagic activity as assessed by p62 accumulation and reduced LC3‐II expression (Fig. 1b).
Figure 1.
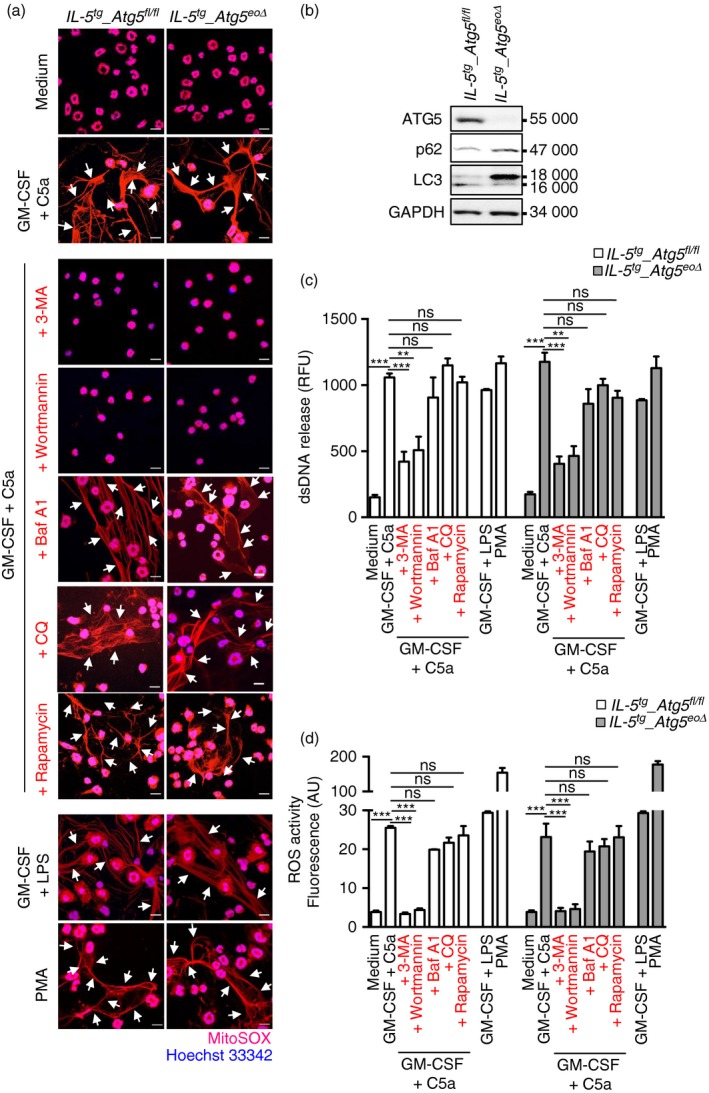
The formation of mouse eosinophil extracellular traps (EETs) is independent of autophagy‐related 5 (ATG5) and autophagy. Mature mouse eosinophils were isolated from the bone marrow of IL5 tg _Atg5 flox/flox and IL5 tg _Atg5 eoΔ mice. (a) Released DNA (MitoSOX Red), indicated by an arrow, and nuclear DNA stained with Hoechst 33342 (blue), were assessed by confocal microscopy. Bars, 10 μm. (b) Eosinophils from control or IL5 tg _Atg5 eoΔ mice were analysed for protein expression of ATG5, p62, LC3 and GAPDH. Results are representative of at least three independent experiments. (c) Quantification of dsDNA in supernatants of non‐activated and activated eosinophils using PicoGreen fluorescent dye. (d) Total reactive oxygen species activity was assessed by DHR123 fluorescence and flow cytometric analysis. All data are presented as means ± SEM of at least three independent experiments. **P < 0·01; ***P < 0·001.
To quantify the extracellular DNA scaffolds of EETs formed by activated mouse eosinophils, we collected culture supernatants and measured the amount of released dsDNA using PicoGreen fluorescent dye.23 As expected, there were large statistically significant differences in dsDNA release between unstimulated and activated eosinophils whether derived from control or from IL5 tg _Atg5 eoΔ mice (Fig. 1c). However, we observed no differences in the dsDNA release between activated Atg5‐knockout and control mouse eosinophils (Fig. 1c).
Pharmacological inhibitors of autophagy, namely CQ and Baf A1, exhibited no effect on EET formation, whereas other inhibitors, such as 3‐MA and wortmannin, blocking both class I and class III PI3Ks, prevented EET formation and significantly reduced the amount of dsDNA release. Rapamycin, known to stimulate autophagy, had no effect on EET formation in either control or Atg5‐knockout mouse eosinophils (Fig. 1a and 1c). In control experiments, we observed an increased autophagic flux following exposure to rapamycin and a reduced autophagic flux as a consequence of 3‐MA or wortmannin treatment. Moreover, CQ and Baf A1 treatment resulted in an accumulation of LC3‐II (data not shown).
Atg5‐knockout and control eosinophils were able to generate the same amounts of ROS (Fig. 1d), which is known to be required for EET formation.24 Taken together, these data suggest that ROS production and DNA release by mouse eosinophils are independent of ATG5 expression and autophagy, but, as expected, do require class I PI3K activity.
Atg5‐knockout mouse neutrophils are able to release extracellular DNA
To investigate whether the formation of NETs is also independent of ATG5, mouse neutrophils were isolated from Atg5 NΔ and control Lyz2 Cre/Cre mice and primed for 20 min with GM‐CSF and subsequently activated with C5a or LPS, or alternatively, with PMA in the absence of priming, all known triggers of NET formation.17 Atg5‐knockout neutrophils were able to form NETs just as well as neutrophils isolated from control Lyz2 flox/flox mice (Fig. 2a). Neutrophils of Atg5 NΔ mice exhibited no detectable ATG5 expression as well as strongly reduced autophagic activity as assessed by p62 accumulation and LC3‐II expression (Fig. 2b).19
Figure 2.
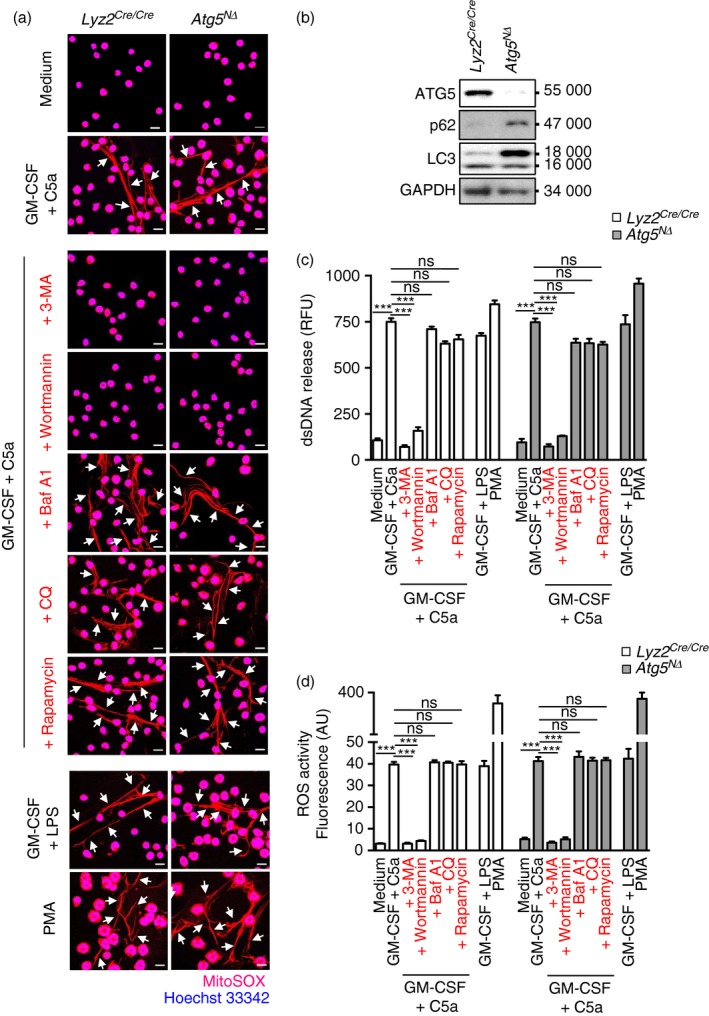
The formation of mouse neutrophil extracellular traps (NETs) is independent of autophagy‐related 5 (ATG5) and autophagy. Mature mouse neutrophils were isolated from bone marrow of Lyz2 Cre/Cre mice as control and Atg5 NΔ mice. (a) Released DNA (MitoSOX Red), indicated by an arrow, and nuclear DNA stained with Hoechst 33342 (blue), were assessed by confocal microscopy. Bars, 10 μm. (b) Neutrophils from control or Atg5 NΔ mice were analysed for protein expression of ATG5, p62, LC3 and GAPDH. Results are representative of at least three independent experiments. (c) Quantification of dsDNA in supernatants of non‐activated and activated neutrophils using PicoGreen fluorescent dye. (d) Total reactive oxygne species activity was assessed by DHR123 fluorescence and flow cytometric analysis. All data are presented as means ± SEM of at least three independent experiments. ***P < 0·001.
As in the eosinophil system, we also quantified the dsDNA release between Atg5‐knockout and control mouse neutrophils. There were large statistically significant differences in dsDNA release between unstimulated and activated neutrophils whether derived from control or from Atg5 NΔ mice (Fig. 2c). However, we observed no differences in the dsDNA release between activated Atg5‐knockout and control mouse neutrophils (Fig. 2c). The two pharmacological inhibitors of autophagy CQ and Baf A1 exhibited no effect on NET formation, whereas 3‐MA and wortmannin prevented NET formation and significantly reduced the amount of dsDNA release. Furthermore, rapamycin, which stimulates autophagy, had no effect on NET formation (Fig. 2a and 2c).
Moreover, Atg5‐knockout neutrophils were able to generate the same amounts of ROS as controls (Fig. 2d), known to be required for NET formation.17 It should be noted that neutrophils from older wild‐type mice (9–10 months old) also exhibited no defect in ROS production or NET formation (data not shown). Taken together, silencing the Atg5 gene in mouse eosinophils and neutrophils resulted in autophagy‐deficient cells. However, both Atg5‐knockouts were able to produce the same amounts of ROS and ETs as the corresponding wild‐type granulocytes.
Pharmacological inhibitors of autophagy do not prevent EET formation by human eosinophils
As mouse and human eosinophils differ in some functional responses, we also performed experiments with human eosinophils. To trigger EET formation, we stimulated human eosinophils in the same two ways as mouse eosinophils. We defined the formation of EETs by demonstrating the presence of extracellular DNA fibres associated with granule proteins.24 3‐MA and wortmannin completely abrogated EET formation upon physiological activation with IL‐5 and C5a or after exposure to low concentrations of PMA (Fig. 3a). In contrast, Baf A1, CQ, and rapamycin had no effect as could be confirmed by quantitative assessment of the released DNA (Fig. 3b). These results indicate that the PI3K inhibitors 3‐MA and wortmannin affect EET formation as a consequence of diminished ROS production and not because of blocked autophagy. Indeed, 3‐MA and wortmannin abrogated the ability of human eosinophils to produce ROS upon activation, whereas ROS production was not affected by CQ, Baf A1 or rapamycin (Fig. 3c).
Figure 3.
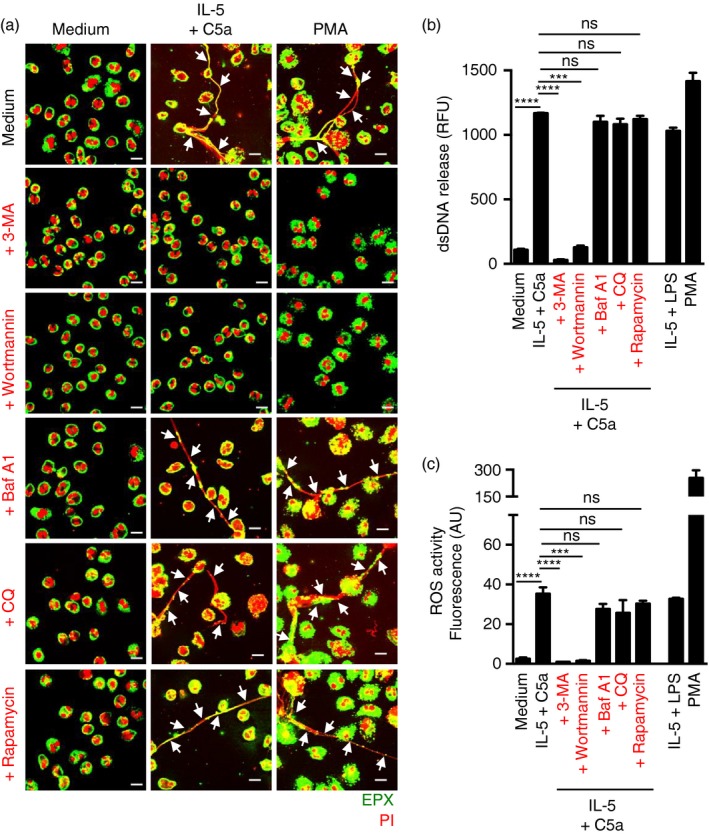
The formation of human eosinophil extracellular traps (EETs) is autophagy‐independent. Human blood eosinophils were isolated from healthy donors by negative selection using antibody‐conjugated magnetic beads. (a) EET formation, indicated by an arrow, following short‐term stimulation (total 45 min) of human eosinophils with the indicated triggers in the presence and absence of chloroquine (CQ) (10 μm), bafilomycin A1 (Baf A1) (250 nm), 3‐methyladenine (3‐MA) (5 mm), wortmannin (100 nm) or rapamycin (1 μm), was assessed by confocal microscopy. Extracellular eosinophil peroxidase (green) together with released DNA (propidium iodide, red) is shown outside the cells. Bars, 10 μm. (b) Quantification of dsDNA in supernatants of activated eosinophils using PicoGreen fluorescent dye. (c) Total reactive oxygen species activity was assessed by DHR123 fluorescence and flow cytometric analysis. All data are presented as means ± SEM of at least three independent experiments. ***P < 0·001; ****P < 0·0001.
Pharmacological inhibitors of autophagy do not prevent NET formation by human neutrophils
We also performed experiments in the human neutrophil system and stimulated NET formation in the same way as in mouse neutrophils. We defined the formation of NETs by demonstrating the presence of extracellular DNA fibres associated with released elastase.17 The results were similar to those seen in the eosinophil system: 3‐MA and wortmannin completely blocked NET formation upon physiological activation of neutrophils with GM‐CSF and C5a or after exposure to low concentrations of PMA. In contrast, CQ, Baf A1 and rapamycin had no effect on NET formation (Fig. 4a). These data were confirmed by quantification of dsDNA release (Fig. 4b). Also, as with eosinophils, the ROS data showed that CQ, Baf A1 and rapamycin had no effect, while the PI3K inhibitors 3‐MA and wortmannin completely blocked ROS production (Fig. 4c).
Figure 4.
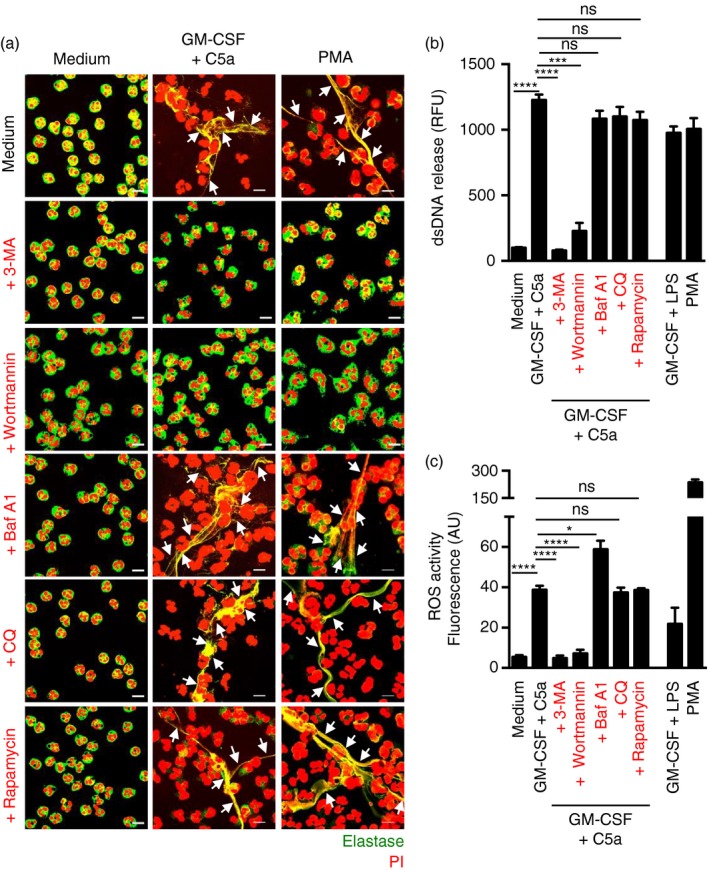
The formation of human neutrophil extracellular traps (NETs) is autophagy‐independent. Human blood neutrophils were isolated from healthy donors by Ficoll‐Hypaque centrifugation. (a) NET formation, indicated by an arrow, following short‐term stimulation (total 45 min) of human neutrophils with the indicated triggers in the presence and absence of chloroquine (CQ) (10 μm), bafilomycin A1 (Baf A1) (250 nm), 3‐methyladenine (3‐MA) (5 mm), wortmannin (100 nm) or rapamycin (1 μm), was assessed by confocal microscopy. Extracellular neutrophil elastase (green) together with released DNA (propidium iodide, red) is shown outside the cells. Bars, 10 μm. (b) Quantification of dsDNA in supernatants of activated neutrophils using PicoGreen fluorescent dye. (c) Total reactive oxygen species activity was assessed by DHR123 fluorescence and flow cytometric analysis. All data are presented as means ± SEM of at least three independent experiments. *P < 0·05; ***P < 0·001; ****P < 0·0001.
Discussion
Previously published work pointed to the possibility that NET formation required autophagy.4, 5, 9 It should be noted that these studies used pharmacological inhibitors to block class III PI3K in order to stop the initiation of autophagosome formation. However, it is probable that these inhibitors have additional targets, resulting in an indirect inhibition of NET formation. Moreover, rapamycin, a drug that can induce autophagy, has revealed contrasting results in different studies regarding its influence on NET formation.7, 8 In a recent study, it was suggested that neutrophils from older mice exhibit decreased expression levels of ATG5 and reduced levels of autophagy. Such neutrophils also had a reduced capacity to generate NETs and the authors implied that autophagy could regulate NET formation.9
In this study, in order to verify such previous findings, we silenced the Atg5 gene in both mouse eosinophils and neutrophils. We have described the generation of Atg5 NΔ mice previously,19 but this is the first report in which Atg5 was specifically deleted in eosinophils. Clearly, the Atg5 eoΔ mice will be valuable tools to dissect the role of autophagy in eosinophils in future studies. Silencing of Atg5 was followed by the loss of ATG5 protein and reduced levels of autophagy in both granulocyte types. In contrast to the previously published work, however, our data suggest that lack of ATG5 does not influence the formation of ETs in either mouse eosinophils or neutrophils.
Both EET and NET formation depend on the activation of the NADPH oxidase,17, 24 which requires type I PI3K activity.14 GM‐CSF treatment followed by C5a stimulation activates type I PI3K and NADPH activities.13, 25, 26 Consequently, pharmacological inhibitors targeting type I PI3K will block ROS production and ET formation in both eosinophils and neutrophils. As wortmannin and 3‐MA not only block type III, but also type I PI3K activity, the conclusion that autophagy is required for the formation of ETs is unwarranted. In fact, our study supports the assumption that these agents block EET and NET formation by blocking type I PI3K activity. This view is further supported by the lack of any effect after treatment with pharmacological inhibitors (CQ and Baf A1) and activators (rapamycin) of autophagy.
Taken together, this report indicates that neither mature eosinophils nor neutrophils have a requirement for autophagy for EET and NET formation. The strength of our study is that we have obtained the same data in both mouse and human cells and that we have used both genetic and pharmacological approaches. Future work will be required to determine which granulocyte functions depend on autophagy.
Disclosures
The authors declare no financial or commercial conflict of interest.
Acknowledgements
This work was supported by the Swiss National Science Foundation to SY (grant number 31003A_173215) and HUS (grant number 310030_166473). N.G. is a PhD student of the Graduate School of Cellular and Biomedical Sciences of the University of Bern. Images were acquired on equipment supported by the Microscopy Imaging Centre of the University of Bern. The authors dedicate this work to the memory of Dr Jamie Lee from whom we received the eoCRE and IL‐5 transgenic mice and who is recently deceased.
References
- 1. Simon D, Simon HU, Yousefi S. Extracellular DNA traps in allergic, infectious, and autoimmune diseases. Allergy 2013; 68:409–16. [DOI] [PubMed] [Google Scholar]
- 2. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS et al Neutrophil extracellular traps kill bacteria. Science 2004; 303:1532–5. [DOI] [PubMed] [Google Scholar]
- 3. Yousefi S, Simon HU. NETosis – does it really represent nature's “suizide bomber”? Front Immunol 2016; 7:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I et al Neutrophil extracellular trap formation is associated with IL‐1β and autophagy‐related signaling in gout. PLoS ONE 2011; 6:e29318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R et al Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res 2011; 21:290–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kenno S, Perito S, Mosci P, Vecchiarelli A, Monari C. Autophagy and reactive oxygen species are involved in neutrophil extracellular traps release induced by C. albicans morphotypes. Front Microbiol 2016; 7:879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McInturff AM, Cody MJ, Elliott EA, Glenn JW, Rowley JW, Rondina MT et al Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia‐inducible factor 1 α . Blood 2012; 120:3118–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Itakura A, McCarty OJT. Pivotal role for the mTOR pathway in the formation of neutrophil extracellular traps via regulation of autophagy. Am J Physiol Cell Physiol 2013; 305:C348–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xu F, Zhang C, Zou Z, Fan EKY, Chen L, Li Y et al Aging related Atg5 defect impairs neutrophil extracellular traps formation. Immunology 2017. https://doi.org/10.1111/imm.12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simon HU, Friis R, Tait SWG, Ryan KM. Retrograde signaling from autophagy modulates stress responses. Sci Signal 2017; 10:eaag2791. [DOI] [PubMed] [Google Scholar]
- 11. Dreiem A, Myhre O, Fonnum F. Involvement of the extracellular signal regulated kinase pathway in hydrocarbon‐induced reactive oxygen species formation in human neutrophil granulocytes. Toxicol Appl Pharmacol 2003; 190:102–10. [DOI] [PubMed] [Google Scholar]
- 12. Makni‐Maalej K, Chiandotto M, Hurtado‐Nedelec M, Bedouhene S, Gougerot‐Pocidalo M, Dang PMC et al Zymosan induces NADPH oxidase activation in human neutrophils by inducing the phosphorylation of p47phox and the activation of Rac 2: involvement of protein tyrosine kinases, PI3Kinase, PKC, ERK1/2 and p38MAPkinase. Biochem Pharmacol 2013; 85:92–100. [DOI] [PubMed] [Google Scholar]
- 13. Mihalache CC, Yousefi S, Conus S, Villiger PM, Schneider M, Simon HU. Inflammation‐associated autophagy‐related programmed necrotic death of human neutrophils characterized by organelle fusion events. J Immunol 2011; 186:6532–42. [DOI] [PubMed] [Google Scholar]
- 14. Geering B, Gurzeler U, Federzoni E, Kaufmann T, Simon HU. A novel TNFR1‐triggered apoptosis pathway mediated by class 1A PI3Ks in neutrophils. Blood 2011; 117:5953–62. [DOI] [PubMed] [Google Scholar]
- 15. Wang X, He Z, Liu H, Yousefi S, Simon HU. Neutrophil necroptosis is triggered by ligation of adhesion molecules following GM‐CSF priming. J Immunol 2016; 197:4090–100. [DOI] [PubMed] [Google Scholar]
- 16. You RN, Ho CC, Dai MS, Hung HM, Chen CS, Chao TY. Autophagy regulation in heme‐induced neutrophil activation is associated with microRNA expression on transfusion‐related acute lung injury. Biomarkers Genomic Med 2014; 6:150–3. [Google Scholar]
- 17. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ 2009; 16:1438–44. [DOI] [PubMed] [Google Scholar]
- 18. Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 2015; 22:377–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rozman S, Yousefi S, Oberson K, Kaufmann T, Benarafa C, Simon HU. The generation of neutrophils in the bone marrow is controlled by autophagy. Cell Death Differ 2015; 22:445–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki‐Migishima R et al Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885–9. [DOI] [PubMed] [Google Scholar]
- 21. Doyle AD, Jacobsen EA, Ochkur SI, Willetts L, Shim K, Neely J et al Homologous recombination into the eosinophil peroxidase locus generates a strain of mice expressing Cre recombinase exclusively in eosinophils. J Leukoc Biol 2013; 94:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stoeckle C, Geering B, Yousefi S, Rozman S, Andina N, Benarafa C et al RhoH is a negative regulator of eosinophilopoiesis. Cell Death Differ 2016; 23:1961–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Amini P, Stojkov D, Wang X, Wicki S, Kaufmann T, Wong WWL et al NET formation can occur independently of RIPK3 and MLKL signaling. Eur J Immunol 2016; 46:178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E et al Catapult‐like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med 2008; 14:949–53. [DOI] [PubMed] [Google Scholar]
- 25. Akasaki T, Koga H, Sumimoto H. Phosphoinositide 3‐kinase‐dependent and ‐independent activation of the small GTPase Rac2 in human neutrophils. J Biol Chem 1999; 274:18055–9. [DOI] [PubMed] [Google Scholar]
- 26. Simon HU. Neutrophil apoptosis pathways and their modifications in inflammation. Immunol Rev 2003; 193:101–10. [DOI] [PubMed] [Google Scholar]