


Abstract
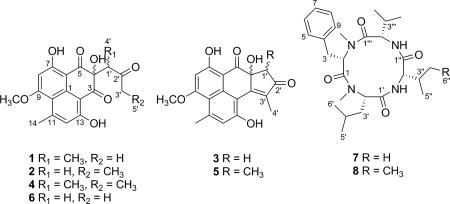
Auxarthrones A–E (1–5), five new phenalenones, and two new naturally occurring cyclic tetrapeptides, auxarthrides A (7) and B (8), were obtained from three different solvent extracts of cultures of the coprophilous fungus Auxarthron pseudauxarthron. Auxarthrones C (3) and E (5) possess an unusual 7a,8-dihydrocyclopenta[a]phenalene-7,9-dione ring system that has not been previously observed in natural products. Formation of 1–5 was found to be dependent on the solvent used for culture extraction. The structures of these new compounds were elucidated primarily by analysis of NMR and MS data. Auxarthrone A (1) was obtained as a mixture of chromatographically inseparable racemic diastereomers (1a and 1b) that cocrystallized, enabling confirmation of their structures by X-ray crystallography. The absolute configurations of 7 and 8 were assigned by analysis of their acid hydrolysates using Marfey’s method. Compound 1 displayed moderate antifungal activity against Cryptococcus neoformans and Candida albicans, but did not affect human cancer cell lines.
Graphical abstract
Cryptococcus species are the world’s most common cause of invasive fungal infections and affect about a million people each year, resulting in over 600 000 deaths worldwide.1 Approximately a third of all HIV/AIDS-associated fatalities are due to cryptococcal disease, exceeding the number of tuberculosis-attributed deaths. Options for treatment of Cryptococcus infections are limited to three classes of antifungal agents, the polyenes, the azoles, and the pyrimidine antimetabolite flucytosine, and are often unsatisfactory. Pharmaceutical high-throughput screening efforts primarily have been devoted to finding cidal agents toward Candida albicans and other Candida species for decades, while Cryptococcus and other emerging invasive pathogens have received only limited attention. Thus, invasive fungal infections are a growing health concern.2
We have continued to build our collection of metabolite-enriched fermentation extracts from unstudied coprophilous ascomycete fungi using methods proven to promote expression of secondary metabolism and have tested these extracts in a growth-inhibition assay against the model pathogen Cryptococcus neoformans strain H99 with the objective of identifying metabolites that selectively inhibit its growth in vitro.3 We observed that a methyl ethyl ketone extract of cultures of an isolate of Auxarthron pseudauxarthron (Eurotiomycetes, Onygenales) obtained from rabbit dung inhibited growth of C. neoformans and C. albicans, but did not affect the growth of Staphylococcus aureus (ATCC 4330). Only a few compounds have been reported from members of this genus, including amauromine, auxarthonoside, methylpenicinoline, dechloroisorumbrin, rumbrin, and auxarconjugatins,4–10 and there are no prior reports of metabolites from this species.
Bioassay-guided fractionation of this extract led to identification of three new phenalenone derivatives designated as auxarthrones A–C (1–3), the previously described, related compound FR-901235 (6),11 and two new cyclic tetrapeptides, auxarthrides A (7) and B (8). After considering features of the structures of new compounds 1–3, as well as known compound 6, it was hypothesized that their formation might be artifactual in nature. Further experiments supported this hypothesis and also led to the generation of two additional new compounds (4 and 5), which we called auxarthrones D and E. Even though phenalenones 1–6 proved to be solvent-induced artifacts, products 1–5 and peptides 7 and 8 are new compounds, some of which showed antifungal activity. Herein, we report on the identification of the producing organism, its fermentation, solvent extraction experiments, and the separation, structure elucidation, and characterization of 1–8, as well as the results of antifungal and cytotoxicity assays for these compounds.
RESULTS AND DISCUSSION
A methyl ethyl ketone extract from strain TTI-0363 grown in an agitated liquid medium (MOF) inhibited growth of both C. neoformans H99 and C. albicans ATCC 10231 in an agar diffusion assay with moderately sized, clear inhibition zones, while growth of S. aureus ATCC 43330 was unaffected at the same test level. On the basis of morphological features on the natural substratum of rabbit dung, features in culture, and sequences of rDNA, strain TTI-0363 was identified as Auxarthron pseudoauxarthon. Fungi of the genus Auxarthron are typically associated with dung, soils, and keratinaceous parts of animals, e.g., hair, skin, and hooves, and occasionally have been isolated from humans.12–15 Their sexual spores are formed within a spiny, cage-like hyphal structure called the reticuloperidium, which is thought to cling to arthropods and other animals to facilitate spore dispersal.14,16 The fermentation was subsequently scaled up to enable separation and identification of the components responsible for the antifungal activity.
NMR data for auxarthrone A (1) consistently showed two sets of signals. Exhaustive efforts to separate this presumed mixture employing column chromatography and HPLC using various stationary and mobile phases were unsuccessful in even partially resolving the components. Ultimately, the sample was determined to be a mixture of racemic diastereomers in a 7:3 ratio, which was confirmed by integration of the best-resolved 1H NMR resonances and the absence of optical rotation. Each diastereomer is present in racemic form, and the ratio of the diastereomeric forms is 7:3, so that all four different stereoisomeric forms are present. Therefore, 2D NMR data were collected on the mixture, and the data for the major and minor diastereomeric forms (1a and 1b, respectively) were considered separately for structure elucidation purposes. The molecular formula was determined as C19H18O7 (11 degrees of unsaturation) on the basis of HRESIMS and NMR data (Table 1). Analysis of the 1H, 13C, and HMQC NMR data for the major diastereomeric form (1a) revealed the presence of three exchangeable protons, including two hydrogen-bonded OH protons (δH 12.27 and 12.79), four methyl groups (including one O-methyl), one sp3 methine, 10 olefinic/aromatic carbons (two of which are protonated and three of which were attached to oxygen), one oxygenated, nonprotonated sp3 carbon, and three ketone carbons (δC 197.8, 198.8, and 208.6). These data accounted for all of the 1H and 13C NMR resonances and suggested that 1a possesses a tricyclic phenalene-dione skeleton. A structure search on the phenalene-dione core identified a known compound, FR-901235 (6),11 a sample of which was also isolated from this extract. Comparison of the NMR spectroscopic data of 1a (Table 1) with those of 6 suggested that they possess the same 4,7,13-trihydroxy-9-methoxy-14-methylphenalene-3,5-dione moiety, but that the side-chain was different. In the HMBC data for 1a, correlations from H3-4′ to C-1′ and C-2′ and from H3-3′ to C-1′ and C-2′ indicated the presence of a 2-butanone moiety (C-1′–C-4′), whereas a key HMBC correlation from H3-4′ to C-4 indicated that the 3-methyl-2-butanone moiety must be attached to C-4 of the phenalene-dione unit. Analysis of the 1D and 2D NMR data (Table 1) for the minor diastereomeric form (1b) established that it has the same planar structure as the major isomer. On the basis of these data, the gross structure of auxarthrone A was established as shown in 1. Crystals were later obtained from the sample, enabling analysis by X-ray crystallography. Both diastereomeric forms were observed in the crystal, and perspective ORTEP plots of the major and minor forms found to be present are shown in Figure 2. Assuming that the major form in the crystal corresponds to the major form observed in solution, the relative configurations of 1a and 1b could be assigned as 4S*,1′R* and 4S*,1′S*, respectively.
Table 1.
NMR Data (500 MHz) of 1 in CDCl3
| 1a (major) | 1b (minor) | ||||
|---|---|---|---|---|---|
|
|
|
||||
| position | δC | δH mult., (J in Hz) | HMBCa | δC | δH mult., (J in Hz) |
| 1 | 136.8 | 136.8 | |||
| 2 | 105.6 | 106.2 | |||
| 3 | 197.8b | 196.9c | |||
| 4 | 83.4 | 83.3 | |||
| 5 | 198.8b | 199.8c | |||
| 6 | 102.6 | 102.1 | |||
| 7 | 168.0 | 168.0 | |||
| 8 | 96.6 | 6.41, s | 6, 7, 10 | 96.3 | 6.39, s |
| 9 | 167.1 | 167.1 | |||
| 10 | 113.6 | 113.6 | |||
| 11 | 149.9 | 149.7 | |||
| 12 | 119.1 | 6.79, s | 2, 10, 13, 14 | 119.4 | 6.81, s |
| 13 | 164.9 | 164.8 | |||
| 14 | 26.3 | 2.80, s | 10, 11, 12 | 26.3 | 2.80, s |
| 1′ | 55.4 | 2.92, q (7.4) | 4, 2′, 4′ | 55.7 | 2.92, q (7.4) |
| 2′ | 208.6 | 208.6 | |||
| 3′ | 29.8 | 2.18, s | 1′, 2′ | 29.7 | 2.17, s |
| 4′ | 11.2 | 1.17, d (7.4) | 4, 1′, 2′ | 11.2 | 1.17, d (7.4) |
| 9-OCH3 | 56.1 | 4.01, s | 9 | 56.1 | 4.01, s |
| 4-OH | 4.24, s | 3, 4, 5 | 4.24, s | ||
| 7-OH | 12.79, s | 6, 7, 8 | 12.81, s | ||
| 13-OH | 12.27, s | 2, 12, 13 | 12.25, s | ||
HMBC correlations are from proton(s) to the indicated carbon.
These assignments are interchangeable.
Figure 2.

Thermal ellipsoid representations of 1a and 1b.
Auxarthrone B (2) was assigned the same molecular formula as 1 by HRESIMS analysis. However, in this instance, only a single set of NMR signals was observed. Interpretation of its NMR data (Table 2) again revealed the presence of a phenalene-dione tricyclic ring system with the same substitution pattern found in 1, except that the side-chain (C-1′–C-4′) was different. 1H–1H COSY correlations observed between H2-3′ and H3-5′, as well as HMBC correlations from H-3′ to C-2′ and from H2-1′ to C-3, C-4, C-5, and C-2′, indicated the substitution pattern shown for this unit in 2. A near-zero optical rotation value for the sample of 2 suggested that it was also obtained as a racemic mixture. CD data acquired for samples of both 1 and 2 supported the conclusion that both are racemic, as the data showed only noise-level features and did not show Cotton effects reported for similar compounds obtained in optically active form.17 Notably, the sample of the known related compound FR-901235 (6) obtained in this study was also found to be optically inactive, as in the original report.11
Table 2.
NMR Data (500 MHz) of 2 in CDCl3 and 3 in DMSO-d6
| 2 | 3 | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| position | δC | δH mult. (J in Hz) | HMBCa | δC | δH mult. (J in Hz) | HMBCa |
| 1 | 137.2 | 136.0 | ||||
| 2 | 105.6 | 108.2 | ||||
| 3 | 197.4 b | 138.3 | ||||
| 4 | 77.5 | 76.3 | ||||
| 5 | 199.3 b | 198.5 | ||||
| 6 | 102.1 | 103.0 | ||||
| 7 | 168.9 | 168.1 | ||||
| 8 | 96.4 | 6.40, s | 6, 7, 10 | 95.9 | 6.47, s | 6, 7, 10 |
| 9 | 167.2 | 167.1 | ||||
| 10 | 113.6 | 113.4 | ||||
| 11 | 149.8 | 140.7 | ||||
| 12 | 119.3 | 6.80, s | 2, 10, 13, 14 | 119.1 | 6.90, s | 2, 10, 13, 14 |
| 13 | 165.7 | 156.9 | ||||
| 14 | 26.4 | 2.80, s | 10, 11, 12 | 25.7 | 2.73, s | 10, 11, 12 |
| 1′ | 50.8 | 3.26, s | 3, 4, 5, 2′ | 42.0 | 2.44, d (18.7) | 4, 5, 2′, 3′ |
| 3.14, d (18.7) | 3, 4, 2′, 3′ | |||||
| 2′ | 208.3 | 205.0 | ||||
| 3′ | 37.1 | 2.49, q (7.2) | 2′, 5′ | 161.4 | ||
| 4′ | 11.5 | 1.73, s | 3, 2′, 3′ | |||
| 5′ | 7.3 | 0.99, t (7.2) | 2′, 3′ | |||
| 9-OCH3 | 56.1 | 4.01, s | 9 | 56.9 | 4.00, s | 9 |
| 4-OH | 3.73, s | |||||
| 7-OH | 13.28, s | 6, 7, 8 | 13.48, s | 7 | ||
| 13-OH | 12.71, s | 2, 12, 13 | 11.00, brs | |||
HMBC correlations are from proton(s) to the indicated carbon.
These assignments are interchangeable.
The molecular formula of auxarthrone C (3) was determined to be C19H16O6 (12 unsaturations) by HRESIMS, which was 18 mass units (H2O) less than that of 2. Analysis of its 1H and 13C NMR data again showed only a single set of signals, and HMBC correlations (Table 2) revealed structural features matching those of 2, except that the signals for a ketone carbon (δC 197.4) at C-3 and a methylene group (δH/δC 2.49/37.1) at C-3′ in 2 were replaced by two olefinic carbons (δC 138.3 and 161.4) in 3, respectively, suggesting that a π-bond was formed between C-3 and C-3′ to satisfy the unsaturation requirement of 3. This conclusion was supported by HMBC cross-peaks from H3-4′ to C-3, C-2′, and C-3′. On the basis of these data, structure 3 was assigned to this compound. Compound 3 was less abundant in the extract than 1 or 2 and appears to arise from an intramolecular aldol-type condensation of 2. Interestingly, this ring system does not appear to have been previously reported as a feature of a natural product.
At this stage, after considering the biosynthetic steps leading to herqueinone in Penicillium herquei reported by the Tang group,18 we hypothesized that new phenalenones 1–3 (and the previously reported 6) are actually artifacts arising from spontaneous addition of methyl ethyl ketone or acetone to an unstable triketone-type product (Scheme 1). Two experiments were conducted in order to test this hypothesis. When cultures were extracted with a non-ketone solvent (EtOAc), the phenalenone peaks were absent (Figure 1). As a further test, and to explore a potential means of generating additional new compounds, we employed a different, symmetrical ketone (3-pentanone) as the initial extracting solvent, ultimately leading to the anticipated new homologous products, which we named auxarthrones D and E (4 and 5).
Scheme 1.

Proposed Pathways Leading to Formation of Compounds 1–6
Figure 1.

(A) LC-UV analysis of the different solvent extracts of cultures of the fungus Auxarthron pseudauxarthron. (B) Bioassay results for the different solvent extracts. E: ethyl acetate extract; M: methyl ethyl ketone extract; A: acetone extract; P: 3-pentanone extract; Am: amphotericin B; C.N.: C. neoformans; C.A.: C. albicans.
By analogy to the case of 1, auxarthrone D (4) was also obtained as a mixture of racemic diastereomers, this time in a ratio of 6:4. Its molecular formula was determined to be C20H20O7 (11 unsaturations) by HRESIMS, which was 14 mass units (CH2) more than that of 1. Analysis of its 1H NMR, 13C NMR, and HMBC data (Table 3) revealed structural features nearly identical to those found in 1, except that the methyl group attached to the ketone carbonyl C-2′ (δC 211.1) was replaced by signals for an ethyl unit in the NMR spectra of 4, and this observation was further supported by relevant 1H–1H COSY correlations, as well as HMBC cross-peaks from H3-5′ to C-2′ and C-3′ and from H2-9′ to C-2′ and C-5′. Therefore, the structure of 4 was assigned as shown.
Table 3.
NMR Data (500 MHz) of 4 in CDCl3
| 4a (minor) | 4b (major) | ||||
|---|---|---|---|---|---|
|
|
|
||||
| position | δC | δH mult. (J in Hz) | δC | δH mult., (J in Hz) | HMBCa |
| 1 | 136.8 | 136.8 | |||
| 2 | 105.8 | 106.2 | |||
| 3 | 198.1b | 196.9c | |||
| 4 | 83.4 | 83.3 | |||
| 5 | 198.6b | 200.1c | |||
| 6 | 102.6 | 102.1 | |||
| 7 | 167.9 | 168.0 | |||
| 8 | 96.5 | 6.40, s | 96.4 | 6.39, s | 6, 7, 10 |
| 9 | 167.1 | 167.1 | |||
| 10 | 113.5 | 113.5 | |||
| 11 | 149.8 | 149.7 | |||
| 12 | 119.2 | 6.79, s | 119.3 | 6.80, s | 2, 10, 13, 14 |
| 13 | 164.9 | 164.8 | |||
| 14 | 26.3 | 2.79, s | 26.3 | 2.79, s | 10, 11, 12 |
| 1′ | 54.3 | 2.97, q (7.4) | 54.6 | 2.97, q (7.4) | 3, 4, 5, 2′, 4′ |
| 2′ | 211.1 | 211.1 | |||
| 3′ | 36.3 | 2.36, m | 36.1 | 2.36, m | 2′, 5′ |
| 2.47, m | 2.47, m | 2′, 5′ | |||
| 4′ | 11.4 | 1.16, d (7.4) | 11.4 | 1.16, d (7.4) | 4, 1′, 2′ |
| 5′ | 7.3 | 0.94, t (7.2) | 7.3 | 0.94, t (7.2) | 2′, 3′ |
| 9-OCH3 | 56.1 | 4.01, s | 56.1 | 4.01, s | 9 |
| 7-OH | 12.85, s | 12.90, s | |||
| 13-OH | 12.36, s | 12.30, s | |||
HMBC correlations are from proton(s) to the indicated carbon.
These assignments are interchangeable.
Unlike 3, the aldol product (auxarthrone E; 5) that would arise from 4 could form as a mixture of diastereomers. LC-UV-MS data for the 3-pentanone extract (Figure 1 and Figure S11) did reveal a minor component peak that afforded UV and MS data that were fully consistent with results expected for the condensation product 5, which would be analogous to 3, but this product was not further characterized due to sample limitations.
In view of the demonstration that both 2 and 4 are formed by reaction of a naturally occurring species with ketone-containing solvents, and the relationship with 3 and 5 as apparent aldol condensation products of 2 and 4, respectively, efforts were undertaken to independently convert 2 to 3 by exposure of 2 to standard basic conditions employed for such reactions and under the acidic conditions employed for HPLC; however, no discernible conversion of 2 to 3 was detected under these conditions.
As noted above, the known compound 6 was also isolated as a minor component in this work and was readily identified as FR-901235 by comparison of its NMR and MS data with those reported.11 Experiments rigorously excluding acetone from the process showed no evidence for the presence of 6, while preparation of an extract using acetone as the solvent clearly afforded much more of this product (Figure 1), strongly suggesting that the original report of 6 is also likely to have been associated with artifactual formation, even though the published protocol for its isolation made no mention of acetone.11
Auxarthride A (7) was obtained as a white powder with a molecular formula of C27H42N4O4 (nine degrees of unsaturation), as determined by HRESIMS. Analysis of its 1H and 13C NMR (Table 4) and HSQC data revealed the presence of two amide N-H protons (δH 8.05 and 8.11), eight methyl groups including two N-methyls, two methylenes, seven methines (four of which are heteroatom-bonded), one phenyl group, and four carboxylic carbons (δC 170.1–171.7). These data accounted for all of the NMR resonances of 7 and eight of the nine unsaturations, suggesting that 7 is a monocyclic peptide. Interpretation of 1H–1H COSY (Figure 3) and HSQC data of 7 established two valine (Val) residues, one N-methylphenylalanine (N-MePhe) unit, and one N-methylleucine (N-MeLeu) unit. The amino acid sequence of 7 was proposed by using the HMBC correlations (Figure 3) from relevant N-H or N-CH3 protons to neighboring carboxylic and methine carbons. Specifically, the signals for the N-CH3 of N-MePhe, the N-CH3 of N-MeLeu, the N-H of Val1, and the N-H of Val2 were correlated to the carboxylic carbon signals of the Val2 (δC 171.2), N-MePhe (δC 170.1), N-MeLeu (δC 171.7), and Val1 (δC 171.2) residues, respectively, establishing the sequence N-MePhe–N-MeLeu–Val1–Val2 for 7. Marfey’s method19 was applied to assign the absolute configurations of the amino acid residues resulting from acid hydrolysis of auxarthride A (7). The 1-fluoro-2,4-dinitrophenyl-5-l-alanine amide derivatives of the amino acids present in the acid hydrolysate of 7 were analyzed by HPLC-MS along with those of authentic d- and l-amino acids. All of the amino acid residues were determined to have the l-configuration (Figure S16).
Table 4.
NMR Data (500 MHz) of 7 and 8 in DMSO-d6
| 7 | 8 | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| position | δC | δH mult. (J in Hz) | HMBCa | δC | δH mult. J in Hz) | HMBCa |
| 1 | 170.1 | 170.0 | ||||
| 2 | 62.4 | 4.24, dd (11.6, 3.0) | 1, 3, 4, 2-NCH3 | 62.4 | 4.24, dd (11.6, 3.0) | 1, 3, 4, 2-NCH3 |
| 3 | 34.2 | 2.95, d (15.0) | 2, 4, 5/9 | 34.2 | 2.94, d (15.0) | 2, 4, 5/9 |
| 3.45, dd (15.0, 3.0) | 4, 5/9 | 3.46, dd (15.0, 3.0) | 4, 5/9 | |||
| 4 | 138.4 | 138.4 | ||||
| 5/9 | 128.6 | 7.18, d (8.2) | 3, 7 | 128.6 | 7.18, d (8.2) | 3, 7 |
| 6/8 | 129.0 | 7.32, t (8.5) | 4, 8/6 | 129.0 | 7.32, t (8.5) | 4, 8/6 |
| 7 | 127.0 | 7.22, t (8.5) | 5/9 | 127.0 | 7.22, t (8.5) | 5/9 |
| 2-NCH3 | 30.7 | 2.67, s | 1‴, 2 | 30.7 | 2.66, s | 1‴, 2 |
| 1′ | 171.7 | 171.6 | ||||
| 2′ | 59.0 | 3.93, dd (11.3, 3.2) | 1′, 3′, 4′, 2′-NCH3 | 59.0 | 3.94, dd (11.3, 3.2) | 1′, 3′, 4′, 2′-NCH3 |
| 3′ | 37.5 | 1.70, m | 2′, 4′, 5′ | 37.5 | 1.71, m | 2′, 4′, 5′ |
| 1.82, m | 2′, 4′, 5′ | 1.83, m | 2′, 4′, 5′ | |||
| 4′ | 24.9 | 1.32, m | 24.9 | 1.32, m | ||
| 5′ | 21.3 | 0.88, d (6.0) | 3′, 4′, 6′ | 21.3 | 0.88, d (6.0) | 3′, 4′, 6′ |
| 6′ | 23.6 | 0.92, d (6.0) | 3′, 4′, 5′ | 23.6 | 0.92, d (6.0) | 3′, 4′, 5′ |
| 2′-NCH3 | 30.5 | 2.64, s | 1, 2′ | 30.5 | 2.64, s | 1, 2′ |
| 1″ | 171.2 | 171.3 | ||||
| 2″ | 55.4 | 4.37, dd (9.0, 6.5) | 1″, 3″, 4″, 5″ | 54.9 | 4.43, dd (9.0, 6.5) | 1″, 3″, 4″, 5″ |
| 3″ | 29.6 | 2.16, m | 2″, 4″, 5″ | 36.4 | 1.90, m | 2″, 4″, 5″ |
| 4″ | 18.6 | 0.89, d (6.8) | 2″, 3″, 5″ | 24.8 | 0.99, m | 3″, 5″, 6″ |
| 1.60, m | 2″, 3″, 5″, 6″ | |||||
| 5″ | 21.1 | 0.93, d (6.8) | 2″, 3″, 4″ | 17.2 | 0.93, d (6.8) | 2″, 3″, 4″ |
| 6″ | 12.3 | 0.88, t (6.8) | 3″, 4″ | |||
| 2″-NH | 8.11, d (9.0) | 1′, 2″ | 8.11, d (9.0) | 1′, 2″ | ||
| 1‴ | 171.2 | 171.2 | ||||
| 2‴ | 55.0 | 4.04, t (8.3) | 1‴, 3‴, 4‴, 5‴ | 55.0 | 4.04, t (8.3) | 1‴, 3‴, 4‴, 5‴ |
| 3‴ | 29.2 | 1.92, m | 2‴, 4‴, 5‴ | 29.2 | 1.92, m | 2‴, 4‴, 5‴ |
| 4‴ | 18.6 | 0.74, d (6.8) | 2‴, 3‴, 5‴ | 18.6 | 0.74, d (6.8) | 2‴, 3‴, 5‴ |
| 5‴ | 20.8 | 0.64, d (6.8) | 2‴, 3‴, 4‴ | 20.8 | 0.64, d (6.8) | 2‴, 3‴, 4‴ |
| 2‴-NH | 8.05, d (8.3) | 1″, 2‴ | 8.02, d (8.3) | 1″, 2‴ | ||
HMBC correlations are from proton(s) to the indicated carbon.
Figure 3.

Selected 2D NMR correlations for 7 and 8.
The molecular formula of auxarthride B (8) was determined to be C28H44N4O4 (nine degrees of unsaturation) by HRESIMS, indicating the presence of one CH2 unit more than that of 7. Analysis of its 1H and 13C NMR spectroscopic data (Table 4) revealed structural features nearly identical to those found in 7, except that the Val1 residue was replaced by an Ile unit, which was confirmed by relevant 1H–1H COSY and HMBC correlations (Figure 3). The absolute configurations of all of the amino acid residues in 8 were again assigned to have l-configurations (Figure S17) by using Marfey’s method, although notably, as documented in the literature,20 this method does not enable l-Ile to be definitively differentiated from the l-allo-isomer.
All of these compounds were tested for antifungal activity against C. neoformans H99 and C. albicans ATCC 10231 (Table 5). Phenalenone derivative 1 (i.e., the mixture of 1a and 1b) showed a moderate inhibitory effect against both pathogens, with MIC values of 3.2 µg/mL in each case, while the positive control amphotericin B showed an MIC value of 0.8 µg/mL. The other phenalenones (2–4 and 6) showed weaker anti-Cryptococcus and anti-Candida activity, with MIC values ranging from 6.4 to 51.2 µg/mL. These compounds were also evaluated for cytotoxicity against two human cancer cell lines, but none showed significant cytotoxic effects against MDA-MB-231 or MDA-MB-451 breast cancer cell lines (IC50 > 20 µM in each case).
Table 5.
Antifungal Activities of 1–4 and 6
| MIC (µg/mL) | ||
|---|---|---|
|
|
||
| compound | C. neoformans H99 | C. albicans ATCC 10231 |
| 1 | 3.2 | 3.2 |
| 2 | 12.8 | 25.6 |
| 3 | 25.6 | 51.2 |
| 4 | 6.4 | 6.4 |
| 6 | 51.2 | 51.2 |
| amphotericin B | 0.8 | 0.8 |
Phenalenones comprise a unique class of polyketides.21 Members of this group of natural products have been reported from both higher plants and microbial sources and have shown significant biological activities, including antimicrobial and cytotoxic effects.21 To date, more than 80 fungal-derived phenalenones have been characterized, some of which are homodimers and heterodimers, reflecting substantial chemical diversity.18,21–25 Auxarthrones A–E (1–5) are new variants in this class. Structurally, they are most closely related to the co-isolated compound FR-901235 (6),11 a compound previously identified from extracts of Paecilomyces carneus cultures. As discussed above, 1–6 are not natural products, but are derived from a natural precursor upon exposure to ketone-containing solvents. However, auxarthrones C (3) and E (5) possess a novel 7a,8-dihydrocyclopenta[a]phenalene-7,9-dione ring system not previously observed in natural products, and synthetic compounds with this ring system have only very rarely been reported.26,27 Compounds 1–6 share the same putative biosynthetic precursor heptaketide and intermediate triketone, subsequently undergoing acetone, 2-butanone, or 3-pentanone addition at C-6 to generate 1, 2, 4, and 6, while 3 and 5 are further aldol condensation products of 2 and 4, respectively.18,21,22 The production of phenalenone oxidative cleavage artifacts leading to the formation of naphthalic anhydrides has been discussed previously, as has the possibility that certain metabolites of this type might be formed as artifacts.21 However, potential for reactions of fermentation products from phenalenone biosynthesis with ketone-containing solvents (or other reagents) seems to have been neither fully verified nor explored in detail. Ultimately, even though the new phenalenones isolated in this work proved to be artifactual in nature, some of them showed antifungal activity.
Cyclic peptides are among the most frequently encountered fungal secondary metabolites. Over 40 fungal cyclic tetrapeptides are known, although interestingly, aside from auxarthrides A (7) and B (8), hirsutide28 and tentoxins29 appear to be the only previously reported fungal cyclic tetrapeptides that contain N-methylated amino acid units.
EXPERIMENTAL SECTION
General Experimental Procedures
Optical rotations were acquired using a Rudolph Research AUTOPOL III automatic polarimeter. UV data were obtained for solutions in MeOH with a Varian Cary III UV/vis spectrophotometer. Electronic circular dichroism data were recorded with an Olis Cary-17 spectrophotometer (1 cm cell) using MeOH as solvent. All 1H, 13C, and 2D NMR (COSY, HSQC, and HMBC) data were collected using a Bruker AVANCE-500 MHz spectrometer with a 5 mm triple resonance cryoprobe at 298 K using solvent signals (CDCl3: δH/δC, 7.26/77.2; DMSO-d6: δH/δC, 2.50/39.5) as references. The HSQC and HMBC experiments were optimized for 145 and 8 Hz, respectively. 13C NMR multiplicities were established by APT experiments and were consistent with the position assignments in Tables 1–4. ESIMS data were recorded on an Agilent 6120 single quadrupole LC-MS using positive ion electrospray ionization. High-resolution mass spectra were acquired with an Orbitrap Elite (Thermo Scientific) instrument. HPLC separations were performed on an Agilent 1260 instrument (Agilent, USA) equipped with a variable-wavelength UV detector.
Fungal Strain and Identification
Strain TTI-0363 was isolated from ascomata formed on rabbit dung (Figure S1), likely from desert cottontail (Sylvilagus audubonii), collected in October 2015 along U.S. Route 385, between Marathon and the north entrance of Big Bend National Park, Brewster County, Texas. Dung samples were rehydrated with deionized water and incubated at room temperature in deep-dish Petri plates lined with moist filter paper until ascomata formed on the dung surface. Ascospores and asci were dissected from mature ascomata and transferred to cornmeal-dextrose agar supplemented with 50 µg/mL chlortetracycline and streptomycin sulfate. Agar plates with dissected ascospores and asci were sealed and incubated overnight at 45 °C to promote ascospore germination. Germinated ascospores were then transferred to YMA medium (1% maltose extract, 0.2% yeast extract, and 2% agar) to establish axenic cultures. The isolate was assigned the accession number TTI-0363 in Texas Therapeutics Institute’s culture collection at the Brown Foundation Institute of Molecular Medicine, University of Texas Health Science Center at Houston, Houston, TX, USA.
The strain was identified as a species of Auxarthron based on the following combination of characteristics (Figure S1). The ascomata were buff- to cinnamon-colored, spherical, and consisting of a loosely interwoven reticuloperidium. The peridial hyphae were yellowish brown to rusty brown, finely to coarsely asperate, with short spines (10–100 µm long) forming on the angled joints of the peridial hyphae. The asci formed in the center of the reticuloperidium were globose to subgloblose and dissolved quickly. The ascospores were globose and pale yellow, with the wall ornamented by a punctate reticulum, and had an average diameter of 3.3 ± 0.3 µm (n = 20). The short peridial spines, small ascospores, and lack of a conidial state distinguished the strain as A. pseudauxarthron. A. pseudauxthron was first described from rabbit dung in Utah and has been reported from Texas and Argentina.30,31
To corroborate the morphological identification, we estimated its approximate phylogenetic relationships based on the internal transcribed spacer (ITS) and large subunit (LSU) rDNA sequences. DNA extraction from cultured mycelium and DNA cloning and sequencing protocols were the same as previously described.32 The DNA sequence was accessioned in GenBank (KY014424, TTI-0363, ITS + LSU). Several recent phylogenetic studies of the Onygenales and the genus Auxarthron have focused on defining the generic boundaries and placement of new species within the existing phylogenetic framework.12,13,15 The sequence of the ITS region of TTI-0363 was intercalated within previous alignments of ITS sequences from other Auxarthron species. A maximum likelihood analysis of this alignment was executed in Mega 6.033 and resulted in a phylogenetic tree with topology similar to those previously reported (Figure S2). On the basis of alignment of its ITS sequence, TTI-0363 fell among a group of strains that have been loosely referred to as the A. kuehnii complex12,13,15 with the nearest match being a strain from soil in Utah identified as A. kuehnii (RV 22810, Figure S2). TTI-0363 fell outside of a monophyletic branch of the tree with the type strain of A. kuehnii and among a loosely organized branch that included the extype strain of A. pseudauxarthron and other strains that have been previously referred to as A. kuehnii. The phylogenetic heterogeneity of strains identified as A. kuehnii has prompted investigators to consider this species as a complex that includes A. pseudauxthron, A. pseudoreticulum, and other possibly unrecognized species. Therefore, we refer to TTI-0363 as A. pseudauxarthron based on its morphological features and its phylogenetic proximity to the ex-type strain.
Fermentation
Agar plugs from YMA cultures were inoculated into 250 mL Erlenmeyer flasks containing 50 mL of SMYA seed medium (1% Bacto neopeptone, 4% maltose, 1% yeast extract, and 0.4% agar). Seed media were incubated for 5 days with agitation at 220 rpm at 23 °C. A scaled-up fermentation was then carried out in 10 Erlenmeyer flasks (500 mL) each containing 100 mL of MOF medium34 consisting of mannitol 75 g; oat flour 15 g; yeast extract 5 g; l-glutamic acid 4 g; and 2-(N-morpholino)ethanesulfonic acid 16.2 g in 1 L of deionized H2O. A 5 mL amount of seed medium was inoculated into each flask and cultivated at 25 °C with agitation at 220 rpm for 14 days.
Extraction and Isolation
The fermented cultures of TTI-0363 were combined and extracted repeatedly with methyl ethyl ketone (3 × 1.0 L), and the organic solvent was evaporated to dryness under vacuum to afford a crude extract (3.0 g), which was fractionated by silica gel VLC using n-hexane–EtOAc gradient elution. The fraction (300 mg) eluted with 40% to 60% EtOAc were combined and further separated by Sephadex LH-20 column chromatography using 1:1 CH2Cl2–MeOH as the eluent, and resulting subfractions were combined and further purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 µm; 9.4 × 250 mm; gradient elution 55–70% MeCN in H2O with 0.1% formic acid over 35 min; 2 mL/min) to afford 3 (2.5 mg, tR 10.1 min), 6 (1.5 mg, tR 13.4 min), 1 (12.0 mg, tR 15.5 min), 2 (14.5 mg, tR 17.4 min), 7 (8.5 mg, tR 23.6 min), and 8 (10.5 mg, tR 30.2 min).
Similarly, 100 mL of fermented cultures of TTI-0363 were extracted repeatedly with 3-pentanone (3 × 100 mL), and the solvent was evaporated to dryness under vacuum to afford a crude extract (300 mg), which was fractionated on a Grace RP C18 column (10–100% MeOH in water over 35 min; flow rate was 20 mL/min) coupled to a Grace Reveleris X2 flash chromatography system. Fractions with desired compounds were combined and further purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 µm; 9.4 × 250 mm; gradient elution 55–64% MeCN in H2O with 0.1% formic acid over 21 min; 2 mL/min) to afford compound 4 (5.3 mg, tR 19.75 min). A metabolite with LCMS and LCUV spectra consistent with structure 5 was detected, but was not present in sufficient quantities to enable its isolation.
Auxarthrone A (1): yellow needles; mp 166–168 °C; UV (MeOH) λmax (log ε) 248 (4.58), 330 (4.51) nm; 1H and 13C NMR and HMBC data see Table 1; HRESIMS m/z 381.0944 [M + Na]+ (calcd for C19H18O7Na, 381.0945).
X-ray Crystallographic Analysis of 1.35
Upon crystallization from CHCl3–H2O (30:1) using the vapor diffusion method, colorless crystals were obtained for 1. A crystal (0.020 × 0.110 × 0.230 mm) was separated from the sample and mounted on a glass fiber, and data were collected using a Bruker-Nonius Kappa ApexII diffractometer with graphite-monochromated Mo Kα radiation, λ = 0.710 73 Å at 150(2) K. Crystal data: C19H18O7·H2O, M = 376.35, space group orthorhombic, P2(1)2(1)2(1); unit cell dimensions a = 10.1479(10) Å, b = 10.1826(10) Å, c = 10.2083(10) Å, V = 857.44(15) Å3, Z = 2, Dcalcd = 1.458 mg/m3, μ = 0.115 mm−1, F(000) = 396. A total of 1461 frames were collected. The total exposure time was 20.29 h. The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm. All non-hydrogen atoms were refined with anisotropic displacement parameters, and all hydrogen atoms were placed in idealized positions and refined as riding atoms with the relative isotropic parameters. Data were corrected for absorption effects using the multiscan method (SADABS). The ratio of minimum to maximum apparent transmission was 0.968. The calculated minimum and maximum transmission coefficients (based on crystal size) are 0.9740 and 0.9980. The 24 943 measurements yielded 3382 independent reflections after equivalent data were averaged, and Lorentz and polarization corrections were applied. The final refinement gave R1 = 0.0529 and wR2 = 0.1504 [I > 2σ(I)]. The crystal structure was found to contain a mixture of the (4R′,1′S′) diastereomer (Occ. = 0.772(3)) and the (4R′,1′R′) diastereomer (Occ. = 0.228(3)).
Auxarthrone B (2): yellow, amorphous solid; UV (MeOH) λmax (log ε) 248 (4.58), 330 (4.51) nm; 1H and 13C NMR and HMBC data see Table 2; HRESIMS m/z 359.1126 [M + H]+ (calcd for C19H19O7, 359.1125).
Auxarthrone C (3): yellow, amorphous solid; UV (MeOH) λmax (log ε) 219 (4.20), 258 (4.40), 334 (4.01) nm; 1H and 13C NMR and HMBC data see Table 2; HRESIMS m/z 341.1012 [M + H]+ (calcd for C19H17O6, 341.1020).
Auxarthrone D (4): green, amorphous solid; UV (MeOH) λmax (log ε) 217 (4.23), 255 (4.14), 331 (3.76) nm; 1H and 13C NMR and HMBC data see Table 3; HRESIMS m/z 373.1290 [M + H]+ (calcd for C20H21O7, 373.1287).
FR-901235 (6): 1H and 13C NMR and MS data were consistent with literature values.11
Auxarthride A (7): white powder; [α]20D −149 (c 0.07, MeOH); UV (MeOH) λmax (log ε) 206 (4.14) nm; 1H and 13C NMR and HMBC data see Table 4; HRESIMS m/z 487.3273 [M + H]+ (calcd for C27H43N4O4, 487.3279).
Auxarthride B (8): white powder; [α]20D −165 (c 0.07, MeOH); UV (MeOH) λmax (log ε) 206 (4.19) nm; 1H and 13C NMR and HMBC data see Table 4; HRESIMS m/z 501.3429 [M + H]+ (calcd for C28H45N4O4, 501.3435).
Marfey’s Analysis of Auxarthride A (7) and Auxarthride B (8).12
A sample (ca. 1.0 mg) of the target compound (7 or 8) was hydrolyzed by heating in 6 N HCl (1 mL) at 110 °C for 24 h. Upon removal of excess HCl under vacuum, the hydrolysate was placed in a 2 mL reaction vial and treated with a 1% solution of 1-fluoro-2,4-dinitrophenyl-5-l-alanine amide (FDAA; 150 µL) in acetone, followed by 1.0 M NaHCO3 (40 µL). The reaction mixture was heated at 45 °C for 1.5 h, cooled to room temperature, and acidified with 2.0 N HCl (20 µL). Similarly, the standard l- and d-amino acids were also derivatized with FDAA following the same procedure. The derivatives of the hydrolysates and the standard amino acids were subjected to HPLC-ESIMS analysis (Agilent Eclipse Plus C18 column; 5 µm, 4.6 × 150 mm; 1.0 mL/min) at 35 °C using the following gradient program: solvent A, H2O (0.1% formic acid); solvent B, MeCN (0.1% formic acid); linear gradient, 15–45% of B in A over 60 min with UV detection at 340 nm. The retention times and ESIMS data for FDAA derivatives of the hydrolysates and the standard amino acids are summarized in Figures S16 and S17.
Antifungal Assays
In vitro antifungal activity was measured according to National Committee for Clinical Laboratory Standards (NCCLS) recommendations.36 The minimum inhibitory concentration (MIC) was determined by means of the serial dilution method in 96-well plates with YM (1% maltose extract, 0.2% yeast extract, and 0.4% agar) as the test medium. Amphotericin B was used as the positive control. Test compounds were dissolved in DMSO and serially diluted in growth medium. Visual end point and optical density readings of microplate wells were measured relative to positive and negative controls. The strains were incubated at 25 °C, and the MICs were determined at 24 h for C. albicans ATCC 10231 and at 48 h for C. neoformans H99. Viability was determined with the aid of a plate reader using PrestoBlue resazurin dye (Life Technologies) as the viability indicator. The spectrophotometric MIC value was defined as the lowest concentration of a test compound that resulted in a culture with a density consistent with 100% inhibition when compared to the growth of the untreated control.
Breast Cancer Cell Viability Assay
Cell viability of the breast cancer cell lines MDA-MB-231 and MDA-MB-451 was determined using the Cell Titer Glo assay kit (Promega). The Cell Titer Glo buffer was added to the media of each well at a 1:1 ratio as recommended by the manufacturer. The plates were wrapped in foil to minimize light, and the samples were mixed on a shaker for 2 min at room temperature. After a 2 min pause, the plates were shaken for a further 2 min. Luminescence intensity was determined using a plate reader (PHERAstar FSX).
Supplementary Material
Acknowledgments
This work was supported by University of Texas Health Science Center at Houston new faculty start-up funds, the Kay and Ben Fortson Endowment (to G.B.), and a grant from the NIH (R01 GM121458).
Footnotes
ASSOCIATED CONTENT
The authors declare no competing financial interest.
References
- 1.Harrison TS. AIDS. 2009;23:531–532. doi: 10.1097/QAD.0b013e328322ffc3. [DOI] [PubMed] [Google Scholar]
- 2.Olsen L, Choffnes ER, Relman DA, Pray L. Fungal Diseases: An Emerging Threat to Human, Animal, and Plant Health: Workshop Summary. Washington DC: 2011. [PubMed] [Google Scholar]
- 3.Li Y, Yue Q, Krausert NM, An Z, Gloer JB, Bills GF. J. Nat. Prod. 2016;79:2357–2363. doi: 10.1021/acs.jnatprod.6b00498. [DOI] [PubMed] [Google Scholar]
- 4.Alvi KA, Rabenstein J. J. Ind. Microbiol. Biotechnol. 2004;31:11–15. doi: 10.1007/s10295-003-0106-5. [DOI] [PubMed] [Google Scholar]
- 5.Nazir M, Harms H, Loef I, Kehraus S, El Maddah F, Arslan I, Rempel V, Muller CE, Konig GM. Planta Med. 2015;81:1141–1145. doi: 10.1055/s-0035-1545979. [DOI] [PubMed] [Google Scholar]
- 6.Oh IJ, Ju WT, Kim YJ, Jung WJ, Kim KY, Park RD. Nematology. 2014;16:427–436. [Google Scholar]
- 7.Elsebai MF, Rempel V, Schnakenburg G, Kehraus S, Muller CE, Konig GM. ACS Med. Chem. Lett. 2011;2:866–869. doi: 10.1021/ml200183z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang Z, Liao PC, Yang YL, Yang FL, Chen YL, Lam Y, Hua KF, Wu SH. J. Med. Chem. 2010;53:7967–7978. doi: 10.1021/jm100619x. [DOI] [PubMed] [Google Scholar]
- 9.Hosoe T, Fukushima K, Takizawa K, Miyaji M, Kawai K. Phytochemistry. 1999;52:459–463. [Google Scholar]
- 10.Yamagishi Y, Shindo K, Kawai H. J. Antibiot. 1993;46:888–891. doi: 10.7164/antibiotics.46.888. [DOI] [PubMed] [Google Scholar]
- 11.Shibata T, Nishikawa M, Tsurumi Y, Takase S, Terano H, Kohsaka M. J. Antibiot. 1989;42:1356–1361. doi: 10.7164/antibiotics.42.1356. [DOI] [PubMed] [Google Scholar]
- 12.Hubka V, Dobiasova S, Lyskova P, Mallatova N, Chlebkova J, Skorepova M, Kubatova A, Dobias R, Chudickova M, Kolarik M. Med. Mycol. 2013;51:614–624. doi: 10.3109/13693786.2013.770608. [DOI] [PubMed] [Google Scholar]
- 13.Sarrocco S, Diquattro S, Baroncelli R, Cimmino A, Evidente A, Vannacci G, Doveri F. Mycol. Prog. 2015;14:112. [Google Scholar]
- 14.Skinner SJ, Tsuneda A, Currah RS. Mycologia. 2006;98:447–454. doi: 10.3852/mycologia.98.3.447. [DOI] [PubMed] [Google Scholar]
- 15.Solé M, Cano J, Guarro J. Mycol. Res. 2002;106:388–396. [Google Scholar]
- 16.Greif MD, Currah RS. Mycol. Res. 2003;107:77–81. doi: 10.1017/s0953756202007104. [DOI] [PubMed] [Google Scholar]
- 17.Komatsu K, Shigemori H, Mikami Y, Kobayashi Ji. J. Nat. Prod. 2000;63:408–409. doi: 10.1021/np990452t. [DOI] [PubMed] [Google Scholar]
- 18.Gao SS, Duan A, Xu W, Yu P, Hang L, Houk KN, Tang Y. J. Am. Chem. Soc. 2016;138:4249–4259. doi: 10.1021/jacs.6b01528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marfey P. Carlsberg Res. Commun. 1984;49:591. [Google Scholar]
- 20.Vijayasarathy S, Prasad P, Fremlin LJ, Ratnayake R, Salim AA, Khalil Z, Capon RJ. J. Nat. Prod. 2016;79:421–427. doi: 10.1021/acs.jnatprod.5b01125. [DOI] [PubMed] [Google Scholar]
- 21.Elsebai MF, Saleem M, Tejesvi MV, Kajula M, Mattila S, Mehiri M, Turpeinen A, Pirttila AM. Nat. Prod. Rep. 2014;31:628–645. doi: 10.1039/c3np70088g. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L-H, Feng B-M, Sun Y, Wu H-H, Li S-G, Liu B, Liu F, Zhang W-Y, Chen G, Bai J, Hua H-M, Wang H-F, Pei Y-H. Tetrahedron Lett. 2016;57:645–649. [Google Scholar]
- 23.Elsebai MF, Ghabbour HA, Mehiri M. Molecules. 2016;21:178. doi: 10.3390/molecules21020178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Intaraudom C, Nitthithanasilp S, Rachtawee P, Boonruangprapa T, Prabpai S, Kongsaeree P, Pittayakhajonwut P. Phytochemistry. 2015;120:19–27. doi: 10.1016/j.phytochem.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 25.Rukachaisirikul V, Rungsaiwattana N, Klaiklay S, Phongpaichit S, Borwornwiriyapan K, Sakayaroj J. J. Nat. Prod. 2014;77:2375–2382. doi: 10.1021/np500324b. [DOI] [PubMed] [Google Scholar]
- 26.Ried W, Mehrotra ML. Angew. Chem., Int. Ed. Engl. 1967;6:172–173. [Google Scholar]
- 27.Ried W, Mehrotra ML. Justus Liebigs Ann. Chem. 1968;718:120–126. [Google Scholar]
- 28.Lang G, Blunt JW, Cummings NJ, Cole ALJ, Munro MHG. J. Nat. Prod. 2005;68:1303–1305. doi: 10.1021/np0501536. [DOI] [PubMed] [Google Scholar]
- 29.Koncewicz M, Mathiaparanam P, Uchytil TF, Sparapano L, Tam J, Rich DU, Durbin RD. Biochem. Biophys. Res. Commun. 1973;53:653–658. doi: 10.1016/0006-291x(73)90711-0. [DOI] [PubMed] [Google Scholar]
- 30.Anonymous. CBS-KNAW Fungal Biodiversity Center Culture Collection. 2016 [Google Scholar]
- 31.Godeas AM. Mycopathologia. 1972;46:189–204. [Google Scholar]
- 32.Jayanetti DR, Yue Q, Bills GF, Gloer JB. J. Nat. Prod. 2015;78:396–401. doi: 10.1021/np5007718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. Mol. Biol. Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mandala SM, Harris GH. Methods in Enzymology. Vol. 311. Academic Press; 2000. Isolation and characterization of novel inhibitors of sphingolipid synthesis: Australifungin, viridiofungins, rustmicin, and khafrefungin; pp. 335–348. [DOI] [PubMed] [Google Scholar]
- 35.Crystallographic data for 1 have been deposited with the Cambridge Crystallographic Data Centre (deposition number CCDC 1518585). Copies of the data can be obtained, free of charge, on application to the director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: + 44 1223 336033 or deposit@ccdc.cam.ac.uk).
- 36.National Committee for Clinical Laboratory Standards. Reference method for broth dilution antifungal susceptibility testing of yeasts. Approved standard M27-A3. 3. Clinical and Laboratory Standards Institute; Wayne, PA: 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.