

Abstract
DnaK and other members of the 70-kilodalton heat-shock protein (hsp70) family promote protein folding, interaction, and translocation, both constitutively and in response to stress, by binding to unfoided polypeptide segments. These proteins have two functional units: a substrate-binding portion binds the polypeptide, and an adenosine triphosphatase portion facilitates substrate exchange. The crystal structure of a peptide complex with the substrate-binding unit of DnaK has now been determined at 2.0 Å resolution. The structure consists of a β-sandwich subdomain followed by α-helical segments. The peptide is bound to DnaK in an extended conformation through a channel defined by loops from the β sandwich. An α-helical domain stabilizes the complex, but does not contact the peptide directly. This domain is rotated in the molecules of a second crystal lattice, which suggests a model of conformation-dependent substrate binding that features a latch mechanism for maintaining long lifetime complexes.
Molecular chaperones of the 70-kD heat shock protein (hsp70) family are present in the cells of all organisms, and in eukaryotes they occur in all major cellular compartments. They are named for their selective expression in response to metabolic stress, but they are also expressed normally and participate in various cellular processes, including the folding of nascent polypeptides, assembly and disassembly of multimeric protein structures, membrane translocation of secreted proteins, and protein degradation (1). Current understanding of hsp70 function follows from the initial suggestion by Pelham (2) that they bind to aggregation-prone exposed hydrophobic surfaces that may be induced by stress and from Rothman’s extension (3) that both the response to stress and other functions such as translocation and nascent chain folding involve the antifolding activity of polypeptide binding to prevent aggregation. DnaK, originally identified for its DNA replication by bacteriophage λ in Escherichia coli (4) is the bacterial hsp70 chaperone with both constitutive and stress-induced functions (5). It shares about 40 to 50 percent sequence identity with its eukaryotic homologs.
Polypeptide substrate binding and release by hsp70s is facilitated by adenosine triphosphate (ATP) binding. DnaK and other hsp70s have relatively strong binding affinity for ATP but are slow adenosine triphosphatase (ATPases) (6). Binding of ATP to DnaK leads to the release of substrates and possible rebinding of others (7–12). With the hydrolysis of ATP, DnaK is switched back into the ADP-bound form, which exchanges substrates slowly. The ATPase and substrate-binding activities of DnaK and other hsp70s are divided into two separable functional units: the NH2-terminal half (~44 kD) has ATPase activity but no peptide affinity, and the COOH-terminal half (~27 kD) binds polypeptide substrates (13–15). Genetic data and in vitro studies suggest that DnaK acts together with two other heat-shock proteins, DnaJ and GrpE (5, 10). GrpE is a nucleotide-exchange factor that binds sub-stoichiometrically to the ATPase unit (16, 17), and DnaJ binds both to denatured and to certain native proteins and probably to the substrate-binding unit (18, 19).
The crystal structure of the ATPase unit of bovine hsc70, the constitutively present cellular hsp70 of mammals, has been determined (20); this unit has a four-domain structure with similarities to those of actin and hexokinase. Nuclear magnetic resonance (NMR) analysis has provided secondary structure assignments for a major portion of the substrate-binding unit of hsc70 (21). Biochemical and biophysical experiments directed toward characterizing the specificities of the hsp70 substrate-binding unit with unfolded substrates or synthetic peptides (15, 17, 22–25) indicate that exposed hydrophobic residues are generally required for substrate binding to hsp70. Furthermore, the substrate peptide is shown to bind to hsp70 in an extended conformation (26).
We have crystallized the substrate-binding unit of DnaK in the presence of a synthetic peptide and have determined the structure by multiwavelength anomalous diffraction (MAD) phasing (27) applied to the selenomethionyl analog (28). The analysis reveals that this is a two-domain unit, and the structure in a second crystal lattice shows a relative movement between the two domains. The structure reveals characteristics of the substrate-binding site. Analysis of conformational variations with respect to biochemical data on hsp70s provides us with mechanistic insights into the function of these proteins.
Structure determination
Crystals of peptide complexes with DnaK from E. coli were obtained from recombinant fragments defined by proteolysis (29). The expression, purification, and crystallization procedures are summarized in Table 1. Initial crystals grown from the full-length COOH-terminal fragment 384–638, complexed with the NRLLLTG peptide (15, 30), proved to be derived from proteolytic truncation at residue 609. These orthorhombic type 1 crystals and related type 2 crystals from the fragment at 384 to 609 both grew slowly and not very reproducibly; typically they were small and could not be grown from the selenomethionyl analog (28). Our searches for heavy-atom derivatives failed. Further analysis defined a smaller fragment, 389 to 607. Type 1 crystals grew readily from this fragment and from the corresponding selenomethionyl protein. The NRLLLTG peptide bound to these fragments with the same high affinity as that shown for the complete COOH-terminal fragment and for DnaK itself (15). The type 1 crystals are in space group 1222 (a = 94.5 Å, b = 116.6 Å, and c = 37.2 Å) with one complex in the asymmetric unit and 43 percent solvent content. The type 2 crystals are in space group P21212 with two complexes per asymmetric unit.
Table 1.
Statistics for MAD data and phase determination from a selenomethionyl crystal. The substrate-binding unit of DnaK (389 to 607) was expressed in E. coli (29). A hexahistidine tag followed by a factor Xa cleavage site was introduced at the NH2-terminus of the protein for the purpose of purification. The protein was chromatographed on a Ni-NTA-agarose column and digested with factor Xa. The cleaved protein was then chromatographed on Superdex-200. Crystals were grown from 2.4 M ammonium sulfate and phosphate buffer at pH 7.0 by either the hanging drop or the dialysis method. Most of the crystals are body-centered orthorhombic Se K edge (see Table 2). All data collection was done at the X4A beamline at Brookhaven National Laboratory, using image plates. The crystals were frozen at 100 K with xylitol as a cryoprotectant. The MAD data were collected at four different wavelengths (which were before the Se K edge, at the edge, at the peak, and after the peak, respectively) with 1° oscillation range without overlap. The selenomethionyl crystal was oriented such that the a axis of the crystal was parallel to the oscillation axis. The data were processed with DENZO (53) and SCALEPACK (53). The MADSYS program package (27) was used to evaluate phase differences and amplitudes for the selenium scattering component of the structure factors. Five out of the six selenium atoms were located with the program HASSP (53), and the last site was found by difference Fourier analysis.
| Wavelength (λ) (Å) | Reflections
|
Complete (%) | (l/σ) | Rsym* (%) | ||||
|---|---|---|---|---|---|---|---|---|
| Total | Unique | |||||||
| Data collection (20 to 2.0 Å) | ||||||||
| 0.9873 (remote 1) | 59,575 | 24,415 | 94.9 | 19.5 | 5.2 | |||
| 0.9793 (edge) | 59,962 | 24,662 | 95.6 | 18.6 | 5.2 | |||
| 0.9792 (peak) | 58,583 | 24,560 | 93.9 | 18.6 | 5.1 | |||
| 0.9641 (remote 2) | 60,800 | 25,014 | 96.7 | 17.0 | 5.3 | |||
| Anomalous diffraction ratios† | ||||||||
|
20.0 < d <3.0 Å
|
3.0 < d <2.3 Å
|
|||||||
| X1 | λ2 | X3 | λ4 | λ1 | λ2 | λ3 | λ4 | |
|
|
||||||||
| X1 | .033 (.033) | .051 | .041 | .029 | .059 (.051) | .067 | .058 | .050 |
| X2 | .063 (.033) | .026 | .057 | .090 (.057) | .048 | .074 | ||
| X3 | .084 (.037) | .045 | .109 (.056) | .063 | ||||
| X4 | .058 (.031) | .082 (.053) | ||||||
| MAD phasing statistics‡ | ||||||||
| R(°|Ft|) =0.051 | R(°|FA|) = 0.356 | <Δ(Δϕ)> = 36.5° | <σ(Δϕ)> = 17.2° | |||||
Rsym = 100 × Σhkl Σi |li − <l>|/Σhkl Σili, where Ii is the ith measurement and <l> is the weighted mean of all measurements of l. Unique reflections distinguish Bijvöet mates.
Anomalous diffraction ratios = <Δ|F|2>1/2/<|F]2>1/2. where Δ/F| is the absolute value of the Bijvöet (diagonal elements) or dispersive difference (off-diagonal elements), respectively. Values in parentheses are for centric data.
R = Σhkl Σi ||Fi| − <F> |/Σ|F|.
FT is the structure factor due to normal scattering from all the atoms; °FA is the structure factor due to normal scattering from the anomalous scatterers only, and Δϕ is the phase difference between °FT and °FA. Δ(Δϕ) is the difference between two independent determinations of Δϕ.
An exceptionally large type 1 selenomethionyl crystal (~0.4 by 0.06 by 0.06 mm) grown by the sitting-drop dialysis method (31) was frozen and used to measure MAD data at beamline X4A of the National Synchrotron Light Source (Brookhaven National Laboratory). These data were analyzed with the MADSYS implementation of the MAD phasing method (27). The signals were strong (Table 1) since all six of the expected selenium sites proved to be well ordered. The resulting electron density map at 2.3 Å resolution, a portion of which is shown in Fig. 1, was readily interpretable. We used the graphics program O (32) to build an atomic model into this experimental map without ambiguity in the polypeptide chain trace. Diffraction data were also measured from native methionyl type 1 and type 2 crystals (Table 2), and the structure was initially refined against the somewhat superior native type 1 data. After 10 iterative rounds of model building and refinement, both the native and selenomethionyl structures were refined to 2.0 Å resolution with crystallographic R values of 19.3 and 20.6 percent, respectively (Table 2). The type 2 crystal structure was determined by molecular replacement from a partially refined type 1 model. This structure was refined at 2.8 Å resolution to an R value of 22.5 percent (Table 2). Residues 389 to 607 of DnaK and the entire peptide have been included in both type 1 models. Only residues 389 to 599 from one and 389 to 591 from the other DnaK molecule plus residues 2 to 5 from each peptide have been located in the type 2 crystal structure.
Fig. 1.
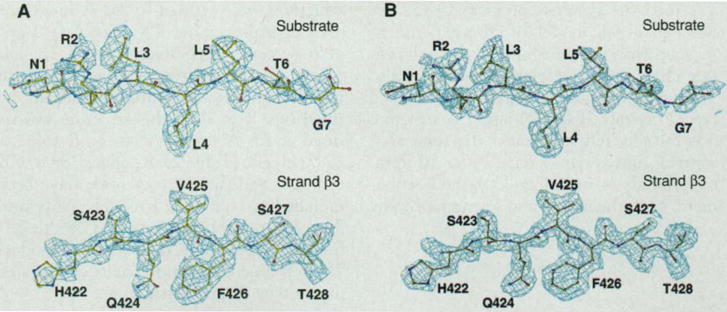
Electron density map. (A) The experimental MAD-phased electron density map at 2.3 Å resolution in the separate regions of the substrate peptide and β strand 3 in the protein, contoured at 1σ. Superposed is the refined atomic model. Only the electron density within 5 Å sphere range of the model is shown [with the use of the “mapcover” feature in program O (32)]. The peptide is shown in full length and residues are numbered 1 to 7. Residues 422 to 428 of DnaK in β strand 3 are also labeled. (B) The corresponding 2Fo−Fc electron density map at 2.0 Å resolution, contoured at 1σ, produced by program O (32).
Table 2.
Lattice and refinement statistics. Fitting of the polypeptide chain into the 2.3 Å MAD-phased electron density map was done with the graphics program O (32). After iterative model building and refinement, the model was refined against both the native and selenomethionyl protein crystal data in space group I222 (crystal form type I). A second crystal form (type 2) was also obtained using the same crystallization conditions. Although the cell constants appeared similar to those of the first crystal form, this crystal form belongs to the space group P21212 without body-centered symmetry. The typical diffraction limit of these crystals was about 2.8 Å. The structure was solved by molecular replacement with AMORE (53) with the protein model determined from the type 1 crystals and was refined to 2.8 Å.
| Type 1 Native | Type 1 Selenomethionine (λ1) | Type 2 Native | |
|---|---|---|---|
| Lattice | |||
| Space group | I222 | I222 | P21212 |
| Cell constants | a=94.4 Å | a=94.5 Å | a=96.4 Å |
| b = 116.7 | b = 116.6 | b = 117.0 | |
| c=37.3 | c=37.2 | c=36.2 | |
| Za* | 1 | 1 | 2 |
| Diffraction data | |||
| dmin | 2.0 Å | 2.0 Å | 2.8 Å |
| RSym1 †(RSym2) (%)† | 5.6 (14.3) | 5.2 (18.5) | 4.3% (11.6) |
| Completeness‡ | 91.5 (60.6) | 94.9 (84.6) | 85.2% (61.6) |
| Total reflections (N) | 57,907 | 59,575 | 23,005 |
| Unique reflections (N) | 13,127 | 13,579 | 9,117 |
| R value (|F|>2σ) (%)§ | 19.3 | 20.6 | 22.5 |
| Rfree(%) | 27.4 | 28.8 | 30.9 |
| Model parameter | |||
| Total non-H atoms | 1629 | 1703 | 2971 |
| Total solvent molecules | 224 | 225 | 28 |
| rms bond length (Å), angle (°) | 0.008, 1.26° | 0.008, 1.30° | 0.010, 2.1° |
| Average B factor (Å) | 22.5 | 23.3 | 30.3 |
| Main-chain rms B (bond, angle) | 1.3, 1.5 | 1.4, 1.6 | 2.5, 3.0 |
| Side-chain rms B (bond, angle) | 2.6, 3.0 | 2.8, 3.2 | 4.3, 5.1 |
Za: number of molecules in the asymmetric unit.
Rsym1 and Rsym2, Rsym for the overall data and last resolution shell, respectively.
Completeness: completeness for overall data and last resolution shell (parentheses, which is 2.1 to 2.0 Å for type 1 crystals and 3.0 to 2.8 Å for type 2 crystals, respectively).
R-value = Σhkl||Fo| − |Fc||/Σhkl|Fo|.
||A subset of the data (5%) was excluded from the refinement and used for the free R-value calculation.
Structure description
The substrate-binding unit of DnaK, which we describe primarily from the better ordered type 1 structure, has two parts. The first half is folded into a compact β sandwich, which is followed by an extended structure of α helices (Fig. 2, A through C). The elements of secondary structure are defined in the topology diagram (Fig. 2D), and the extent of each segment is shown in alignment with the sequence (Fig. 3). From the skeleton (Fig. 2, A through C), the β portion appears to be separable from the α portion. This proves to be somewhat illusory when we consider the disposition of buried side chains (Fig. 2E). Then helix αA and the first half of the long second helix (αB) are integrated with the first domain, whereas the remaining helical segments clearly are folded around a separate hydrophobic core. This division is consistent with sites of proteolytic or genetic truncation that have produced stable fragments of hsc70 (13, 14, 16, 33). Helix αB then spans both of the domains as defined at the hydrophobic-core level of description, and this may have functional implications (see below). The substrate peptide is bound to DnaK in an extended conformation in the β sandwich. Helical elements are not directly involved in binding to the peptide. Instead, the helical domain appears to serve as a lid that encapsulates the substrate through latchlike interactions between the COOH-terminal half of helix αB and the outer loops of the β domain (outer is relative to the domain termini).
Fig. 2.
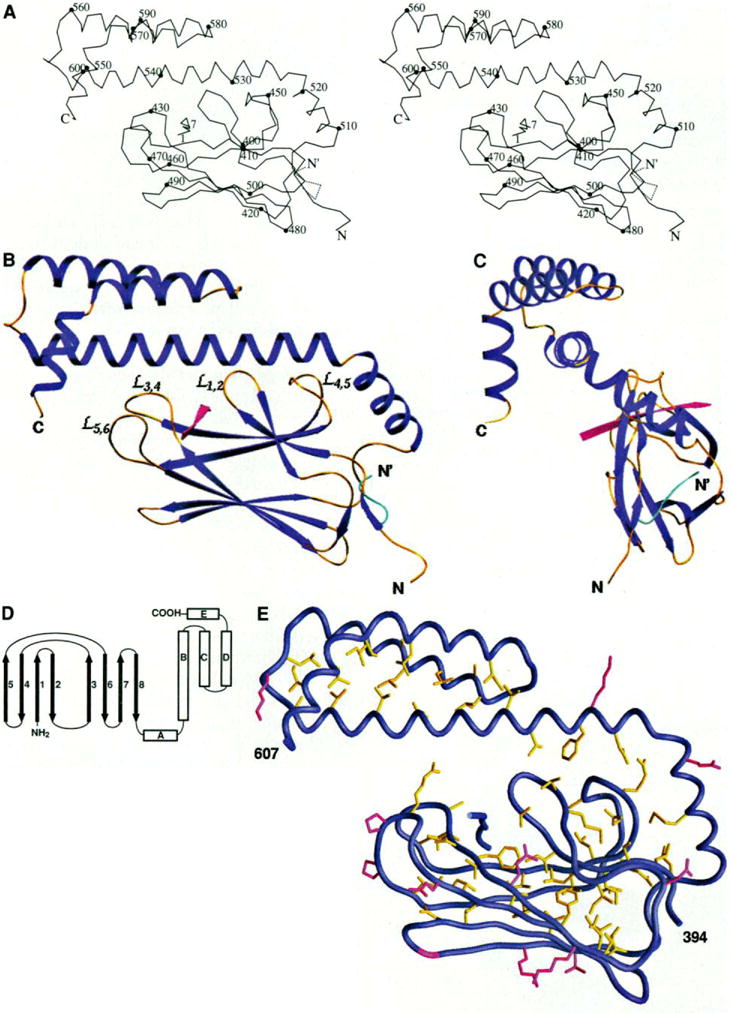
Overall structure, (A) Stereo view of a Cα trace of the structure. Every tenth residue is marked with a filled circle and labeled. The alternative conformation of the NH2-terminus from residues 389 to 394 is shown with the dashed line. N and C are the NH2- and COOH-termini of the polypeptide chain, respectively. (B) Ribbon diagram of the structure in the same orientation as in (A), The α helices and β strands are shown in dark blue, the loops in gold yellow, the substrate peptide in red. The alternative conformation of the NH2-terminus is shown in cyan. The bops between the β strands that interact with the helix αB are labeled. (C) A view of the overall structure rotated 90° around the vertical axis from the view in (B). (D) Schematic representation of the topology of the structure. (E) Buried residues and conserved surface residues in the substrate-binding unit of DnaK. The side chains of those residues with less than 10% solvent accessibility are colored yellow. Also, colored in red are the residues that have more than 40% solvent accessible surface and are, as well, identical in E. coli DnaK, bovine hsc70, and hamster BiP (see Fig. 3). Glycines are colored in red for their backbones. [Part (A) produced by MOLSCRIPT (55), (B) and (C) by SETOR (55), and (E) by GRASP (55)]
Fig. 3.
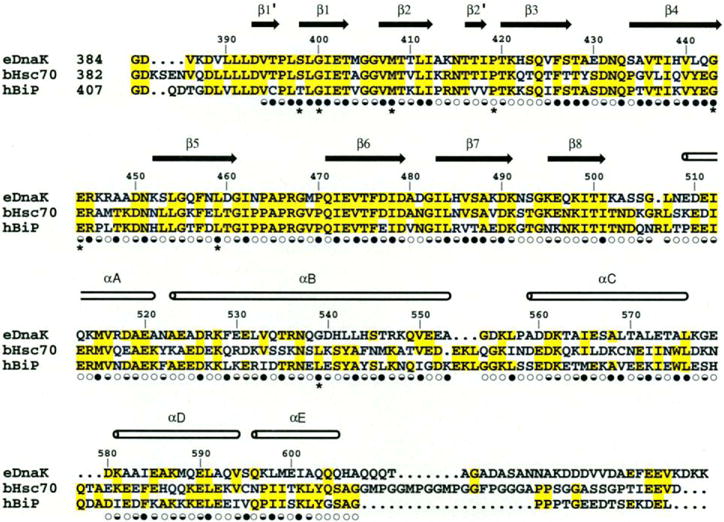
Structure-based sequence alignment of the C-terminal substrate binding unit of E. coli DnaK, bovine hsc70, and hamster BiP. The secondary structure assignments, based on main-chain hydrogen bonding pattern, are shown as arrows and cylinders. Solvent accessibility is indicated for each residue by an open circle if the fractional solvent accessibility is greater than 0.4, a half-closed circle if 0.1 to 0.4, and a closed circle if less than 0.1 (30). Residues changed in binding-defective genetic mutations are identified by asterisks (51).
The overall structure has dimensions of ~60 by 40 by 15 Å. The longest dimension is roughly along the axis of helix αB, and the shortest dimension coincides with the direction of the peptide extension and is perpendicular to the plane of Fig. 2, A and B. This flattened profile (Fig. 2, B and C) corresponds well with the shape deduced from small-angle scattering measurements on a peptide-binding fragment (338 to 554) from bovine hsc70 (34).
The β-sandwich subdomain comprises residues 393 to 502 arranged in two sheets with four antiparallel β strands in each. Both the topology (Fig. 2D) and the extents of strands in these sheets are essentially as found in secondary-structure assignments from the NMR analysis of rat hsc70 (21). The bottom sheet (β3, β6, β7, β8) is a rather regular, twisted β sheet with hairpin β turns connecting the successive strands, whereas the top sheet (β5, β4, β1, β2) is highly irregular (top and bottom are as viewed in Fig. 2). Irregularity in the top sheet is associated with distinctive loops that emanate from the sandwich proper to form the substrate-binding site. Only short segments of strands β1 and β2 are in the sandwich itself; the hairpin end diverges away from the sandwich to form the first loop and, after breaks in hydrogen bonding, the other end is turned to seal off what otherwise would be an open edge of the sandwich. Strands in one sheet are rotated by about 20° clockwise relative to the other when viewed from outside the sandwich (Fig. 4A). We are not aware of other structures with this β-sandwich topology or three-dimensional architecture.
Fig. 4.
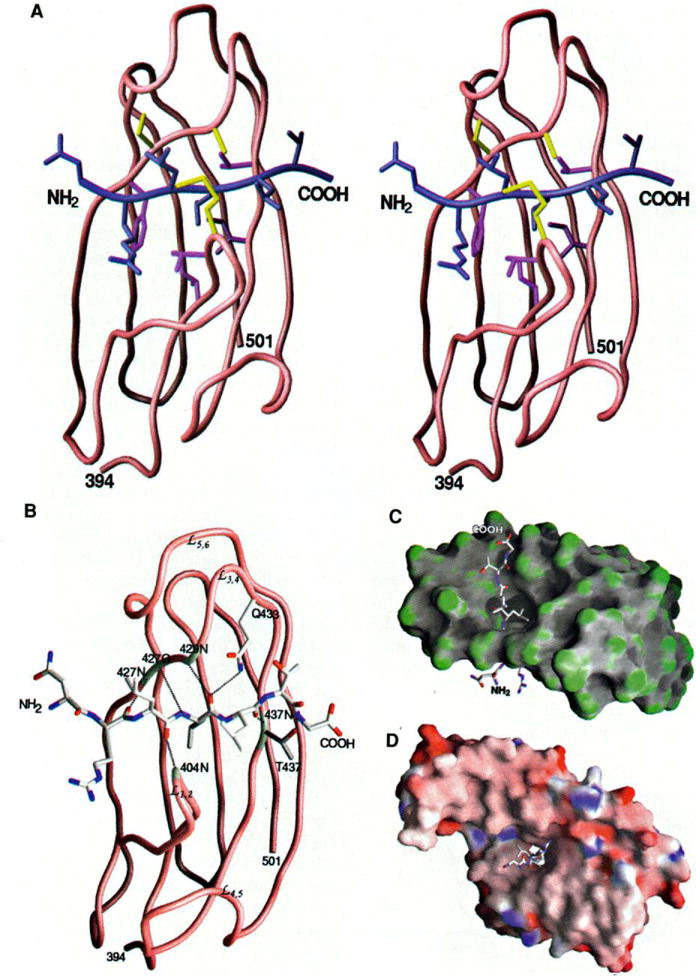
Peptide binding to DnaK (A) Stereo view of the interactions of the three leucines in the peptide with the β subdomain. The protein and the peptide backbones are shown in brown and blue, respectively. The DnaK residues interacting with the side chain of Leu4 of the peptide are colored purple and those interacting with Leu3 and Leu5 of the peptide are colored yellow. (B) Hydrogen bonds formed between the peptide and the β-sandwich subdomain of DnaK. The hydrogen bonds are represented by black dashed lines. The residues of DnaK involved in forming the hydrogen bonds are highlighted white. The backbone amide and carbonyl groups interacting with the peptide are labeled. (C) Surface curvature of the p subdomain (residues 394 to 501) bound to the substrate peptide. The most convex parts of the molecular surface are coded green while the most concave and planar are coded gray and white, respectively. This view is as in (A) and (B) but rotated by 90° so that the NH2-terminal end of the peptide is below. (D) Surface electrostatic potential of the substrate-binding fragment of DnaK complex with the peptide. The orientation of the structure is the same as in Fig. 2B; thus, this side is at the NH2-terminal end of the peptide. The surface is colored according to the local electrostatic potential, ranging from dark blue (most positive region) to deep red (most negative). [Parts (A) through (D) produced by GRASP (55)]
It is the loops off the upper β sheet that particularly distinguish this β structure. Two inner loops, and , are at one side of the substrate-binding channel (inner with respect to the connections to other domains), and two outer loops, and , are at the other side (Fig. 2B). Loop (402 to 409) between β1 and β2 includes hairpin hydrogen bonding and a tight turn. Loop (444 to 451) connects strands β4 and β5 and is wider (more open). These two inner loops are related in part by a quasi-diad axis of symmetry (Fig. 4A) that superimposes defining β bulges in β2 and β5, which in turn project the loops out almost perpendicular to the top β sheet. There are three crossover connections between the sheets. Two of these form the outer loops (428 to 433) and (462 to 470) that take part in substrate binding. The third crossover, between β2 and β3, is effected by the only cis peptide bond in the structure at Pro419. The loops and form the channel for substrate binding directly. The integrity of the substrate-binding channel, however, appears to depend on the buttressing of loop by loop through two main-chain hydrogen bonds and by hydrophobic interactions between the loops. Similarly, loop is stabilized by through both main-chain and side-chain hydrogen bonds.
The NH2-terminal residues 389 to 393, which begin in the hydrophobic sequence Val-Leu-Leu-Leu, have two alternative conformations. One (40 percent occupancy) is folded back into a hydrophobic pocket near the COOH terminus of the β domain (the “in” conformation), and the other (60 percent) is out into a lattice contact where it occupies the equivalent pocket in another molecule (the “out” conformation). The linker that connects β8 to αA (503 to 508) starts with aβ turn and then adopts a notch-like conformation from which the NH2-terminus emerges for the “in” conformation.
The second half of the substrate-binding fragment of DnaK consists of five a helices, which are improbably elongated if taken out of the context of the entire structure. Helices αA and αB are joined by a kink of 72° at Asn522, and together they span 58 Å from residues 509 to 553. The segment in rat hsc70 corresponding to residues 507 to 530 in DnaK was designated as a single helix from the NMR analysis (21), but the possibility of a kink around the position corresponding to residue 521 in DnaK was noted from chemical shift and hydrogen exchange data. Thus, a similar tertiary structure for the two molecules seems probable. The long (31 residues) helix αB interacts through its COOH-terminal half with αC, αD, and αE. The successive interhelical angles of 151°, 169°, and 69° produce an antiparallel three-helical bundle from αB–αD, whereas αE lies across one end of the bundle at 68° from αB. This helical domain (538 to 607) appears to be a relatively stable structural unit with a well-defined hydrophobic core (Fig. 2E).
The five-helix unit in DnaK is similar in topology and general structure to a portion of the helical domain found in the α subunits of G proteins (35). Helices αB–αE in DnaK correspond to αA–αD in transducin, and αA in DnaK occupies a place similar to linker 1 in transducin (35). The respective helices and joining segments are similar in length, and the inter-helix angles are similar (24° average difference). The helical units in DnaK and Gα subunits are disposed in a similar manner with respect to their associated domains and, just as this results in encapsulation of the substrate peptide in DnaK, it entraps the guanine nucleotide in Gα. There are 31 COOH-terminal residues (608 to 638) missing from this DnaK fragment; coincidentally, the segment in Gα proteins between αD and the rejoining to a Ras-like domain is similar in length.
The α segment interacts with the β domain through αA and through its long helix αB. Helix αA makes hydrophobic contacts with loop and αB is in contact with all four loops ( , , , and ) that form the substrate-binding site. A total area of 1713 Å2 is buried in this interface. Of this total, only 390 Å2 is from interactions with the outer loops ( and ) with the COOH-terminal half of αB (538 to 607). The interactions of αB with the inner loops contribute 742 Å2, including a small hydrophobic patch of three residues between the domains. The relatively small buried surface area between αB and the outer loops of the β subdomain is compatible with the suggestion (see mechanistic implications below) that the helical domain acts as a lid that must be displaced as a unit for substrate binding and release.
Substrate binding to DnaK
The substrate peptide, which has the sequence NR-LLLTG (15, 30), was identified in a phage display screen for high affinity binding to DnaK (15). It also conforms well to the profile of preferences found for heptapeptides binding to other hsp70s (22–24). This peptide is bound in an extended conformation (Fig. 1), as observed by Landry et al. (26), in a channel that cuts through the thin dimension of the substrate-binding unit (Fig. 4). The binding channel has a limiting cross-section of 5 by 7 Å, and it is formed primarily by loops and from the β-sandwich domain. There are no contacts of the peptide with helical segments. Binding interactions between the peptide and DnaK center on Leu4, which is completely buried in a deep pocket. In anticipation of generality in substrate binding, we designate this as “hsp70 site 0” and number the sites occupied by other peptide residues plus and minus relative to site 0 (Table 3).
Table 3.
Peptide-substrate interactions in the DnaK binding site. DnaK site, numbered relative to site 0, the hydrophobic pocket into which Leu4 is buried; φ and ψ, the torsion angles of the peptide residue; <B>, average main-chain temperature factors of the peptide residue; S, fraction of the peptide residue accessible to solvent; φO, the total number of hydrophobic atoms (carbon and sulfur) in DnaK making van der Waals contacts with the peptide side chains (first number) and the number of these from DnaK side-chain atoms (second number). φi, the total number of hydrophilic atoms (nitrogen and oxygen) of DnaK in contact with the peptide side chain (first number) and the number of these from DnaK side-chain atoms (second number); hydrogen bond, residue number and atom of DnaK contributing to the bond is cited first followed by peptide residue number and atom; residues in contact, each residue making van der Waals contact with the peptide side chains is listed. The designation of van der Waals contacts was done as described in (54), except that the upper limit was set at rmin + 2(rmin − rO) where rmin is the radius at potential energy minimum and rO is the crossing radius for zero energy.
| DnaK site | Peptide residue | φ, ψ (°) | <B> (Å2) | S | van der Waals contacts
|
Hydrogen bonds | Residues in van der Waals contacts | |
|---|---|---|---|---|---|---|---|---|
| φo | φi | |||||||
| −3 | Asn1 | –, 109 | 41.0 | 0.84 | – | – | – | – |
| −2 | Arg2 | 119, 133 | 30.8 | 0.30 | 6,6 | 3,0 | 427N:20 | Thr403, Phe426, Thr409, Val407 |
| −1 | Leu3 | 106, 155 | 23.2 | 0.07 | 17,10 | 6,0 | 404N:3O | Met404, Ala429, Ser427 |
| 0 | Leu4 | 122, 153 | 25.2 | 0.00 | 17,16 | 2,0 | 429N:40 433Nε2:40 4270:4N |
Phe426, Val436, Ile401, Thr403, Ile438 |
| 1 | Leu5 | −96, 182 | 29.7 | 0.42 | 3,3 | 0,0 | 437N:50 | Met404, Ala429 |
| 2 | Thr6 | 105, 177 | 44.3 | 0.42 | 7,5 | 0,0 | – | Gln433, Ala435 |
| 3 | Gly7 | 106, – | 52.2 | 0.72 | – | – | 4370γ:7N | |
The peptide interacts with DnaK through numerous van der Waals contacts from its side chains (Fig. 4A) and seven main-chain hydrogen bonds (Fig. 4B), as well as additional main-chain van der Waals interactions. Side-chain contacts with DnaK are dominantly hydrophobic; overall, 75 percent of the van der Waals contacts are to carbon or sulfur atoms and this rises to 95 percent when contacts to the main chain are excluded. The binding interactions are concentrated near site 0 on DnaK and the center of the peptide, but contacts occur over the length of the peptide except for residue 1 at site −3. There is strong correlation among the number of contacts, the degree of solvent exposure, and atomic mobility (average B factor) in the peptide (Table 3). Leu3 and Leu4 are relatively well ordered and highly buried, and they also contribute most of the contacts. Arg2, Leu5, and Thr6 in sites −2, 1, and 2 are moderately buried, and they contribute the rest of the side-chain contacts. Five of the seven hydrogen bonds with the peptide are contributed by protein backbone groups. The hydrogen bond between Gln433 Nε2 and Leu4 O in site 0 is critical, but that between Gly7 N and Thr437 Oγ in site 3 is somewhat questionable because of the poor ordering of Gly7. Thus, the binding is almost completely determined by the interactions of a five-residue substrate core with hsp70 sites −2 to +2. Perhaps relevant to substrate release, all but one of the hydrogen bonds are made to groups on the outer, side of the binding channel.
The central site 0 is a crucial determinant of peptide binding. It contributes three hydrogen bonds to the substrate backbone, and it provides a relatively large hydrophobic pocket that is occupied here by Leu4. This is a pocket of ~5 Å wide and 3 Å deep on the floor of the binding channel (Fig. 4C). The side chain of Leu4 makes contact with Phe426 from β3; Val436 and Ile438 from β4; and Ile401 and Thr403 from β1 and Moreover, Ile472 from β6 is also slightly accessible to a probe of solvent accessibility. The peptide conformation is such that with Leu4 down into this pocket, the adjacent side chains of Leu3 and Leu5 project up away from the β sandwich. The long hydro-phobic side chain of Met404 at the tip of loop wraps over the peptide at site 0 to make contact via both Cγ and Sδ with Ala429 from loop (Fig. 4A). This generates a hydrophobic arch over the top of the binding channel that contributes on either side to sites −1 and +1 for Leu3 and Leu5 (Fig. 4C). These sites are relatively unrestricted otherwise. Sites outside this central triad are even more open, and the electrostatic potential of this surface is largely negative. This is evident in Fig. 4D for the NH2-terminal side of the substrate binding channel, but it is even more pronounced at the opposite, COOH-terminal side.
Given the presumed spectrum of substrates for DnaK, one wonders what other peptides might be accommodated in the binding channel. Clearly, selectivity will be greatest at site 0. A simple contact analysis of this hydrophobic pocket indicates that leucine is ideal, and that methionine or isoleucine could fit into the site in favorable conformations if slightly close contacts are tolerated. Smaller side chains could also fit, but presumably with an energetic penalty for the residual cavity (36). For threonine or serine, however, a hydrogen bond from Oγ to 437 O would be possible. Adjustments would be needed in the protein or the peptide backbone to accept phenylalanine, and the price to fit tyrosine or tryptophan seems unacceptable. The hydrophobic character of this site makes the binding of charged or polar side chains less likely, and proline seems to be excluded by the hydrogen-bonding approach of O 427 to N 4. In general, the observed peptide φ angles (Table 3) are disallowed for proline. Apart from this, the geometric constraints on residues other than the anchor residue at site 0 appear to be relatively unrestrictive. There is, however, a general hydrophobic character at sites +1 and −1, and a somewhat negative electrostatic potential at more remote sites, which would be expected to bias substrate affinities. These findings are remarkably compatible with data on hsp70 selections from random sequences. In particular, large aliphatic residues are strongly preferred in the central region, negative charges tend to be excluded throughout, and positive charges are favored at the ends of heptamers (15, 24). An observed prevalence of aromatics and leucine in alternating positions (23) is difficult to reconcile with the structure, however. The dominating influence of site 0 in substrate registration makes it difficult to align peptide sequences. Finally, whereas the polarity of the peptide in this structure is unambiguous, we cannot exclude by modeling the possibility of reverse-direction binding for other sequences, as happens for some SH3 domains (37).
There are a number of other proteins that, like hsp70, bind to polypeptide segments in proteins or to free peptides and do so with incomplete restriction on the sequences of ligands that can be accommodated. These include the class I and class II major histocompatibility complex (MHC) antigen-presenting molecules, SH2 and SH3 domains, protein kinases and phosphatase, proteases, calmodulin, antibodies, the bacterial chaperone protein PapD, and the bacterial periplasmic transport protein OppA. Three-dimensional structures are known for peptide complexes with several such molecules (38). Apart from calmodulin, which binds to helical conformations, all of these are like DnaK in binding to peptide segments in extended conformations. Typically, the binding to extended segments involves both generic hydrogen bonding to the backbone and a few key specificity determining pockets. The binding interaction in DnaK is like this except that there is only one strongly determining pocket.
Peptide binding to MHC molecules, to which the hsp70 interaction with peptides has been likened (39) (somewhat inappropriately), is typical in having the peptide in an open groove. This is, of course, essential for the role of antigen presentation, but it is unnecessary or perhaps even undesirable for the role of hsp70s in preventing unwanted aggregation. Where MHC and hsp70 are similar is in their slow kinetics of release and binding. The half-lives of class II MHC-peptide complexes are measured in days (40) and hsp70-peptide complexes are also long-lived in the absence of ATP (9, 11, 12, 41). The functional reasons for such kinetic behavior differ in the two cases, but the structural basis for the phenomenon probably lies in the requisite for high activation energy conformational changes in association with complex formation and dissociation.
Comparison with other hsp70 proteins
We have selected bovine hsc70 and hamster BiP as well-studied representatives of major hsp70 classes and have prepared a structure-based sequence alignment with DnaK for the substrate-binding units of these proteins (Fig. 3). As has been appreciated (42), members of the hsp70 family are highly similar proteins. Overall, for the substrate-binding fragment that we studied (394 to 607) the pairwise identities range from 46 to 62 percent for the sequences aligned in Fig. 3, which is slightly less than the corresponding identities in the ATPase unit (51 to 70 percent). What may be less well appreciated is the asymmetric distribution of this similarity. The β-sandwich sub-domain (394 to 502) is exceptionally conserved with a range of 65 to 76 percent pairwise identity and 60 percent identity among all three. By contrast, the compact α-helical domain (538 to 607), which serves as a lid on the substrate complex, is much less well conserved with pairwise identities of 18 to 49 percent. The connecting helical portions (503 to 537) are relatively more similar at 31 to 49 percent pairwise identity, which is in keeping with their more intimate association with the β sandwich. The COOH-terminal segment (608 to 638) for which we have no structural information has very little commonality in sequence or length among hsp70 proteins.
The high degree of conservation in the β subdomain suggests that the backbone conformation in this domain would be virtually identical in all the members of the hsp70 family. One then expects that the substrate binding channel in other hsp70 proteins would be like that in DnaK. In particular, the major binding interactions of DnaK with the substrate through backbone hydrogen bonds should be preserved. Furthermore, Gln433 of DnaK, which makes a hydrogen bond to the peptide through its side chain, is absolutely conserved for the 30 hsp70 proteins that we checked. Residues that contact side chains of the peptide in this complex with DnaK differ from site to site in degree of variability in the hsp70 family. The hydrophobic pocket at site 0 is identically the same among the three proteins in Fig. 3, although conservative changes of these residues are observed in other members of the hsp70 family members.
There is greater variation among hsp70 homologs outside the central binding site. First, the Met404 to Ala429 arch, which provides important hydrophobic contacts both at site −1 and at site +1, is highly variable. In bovine hsc70, the residues corresponding to Met404 and Ala429 are an alanine and a tyrosine, respectively. These can be seen as complementary changes that would preserve the hydrophobic arch. In hamster BiP they are a valine and an alanine. Other observed combinations at the 404 to 429 arch include valine-alanine (as in mammalian BiP), leucine-alanine, threonine-ala-nine, and glutamine-cysteine. All of these could maintain the hydrophobic character of these sites. Second, there are distinctions in the electrostatic potential surfaces surrounding the substrate-binding channel. Whereas DnaK is electronegative on both sides, but especially so on the COOH-terminal side of the peptide, an homology-built model for hsc70 has both positive and negative patches near the COOH-terminal side and the model for BiP has slight positivity close to the NH2-terminal side. In summary, subtle differences in sequence preferences might be expected as has been observed in peptide binding studies (43, 44).
A few of the identically conserved residues (Fig. 3) are exposed on the surface (Fig. 2E). Some of these exposed residues, such as Pro464 and Pro466, which help to form the substrate-binding channel, appear to have a role in stabilizing the structure. Others, such as Lys421, Lys498, and Thr500, which are clustered on the bottom surface of the β sandwich and Glu520 and Lys528 near the αA to αB kink, having no other obvious role, become candidates for interaction with the ATPase unit.
Conformational variation
Solution scattering and biochemical studies show that the function of hsp70 proteins involves very substantial conformational change (17, 33, 34, 45–47). The extent of change within the substrate-binding unit, as opposed to segmental changes relative to the ATPase unit (34), has been less clear. From the structure of this complex, however, it now seems obvious that large internal movements must take place to permit substrate binding and release. Information relevant to conformational change comes from conformational differences between the two lattices, the patterns of atomic mobilities (B factors), and conformational heterogeneity within each lattice.
A comparison of the structure of the complex in type 1 and type 2 crystals reveals a conformational difference that may be relevant for substrate exchange (see below). The β-sandwich subdomains superimpose closely on one another [root mean square (rms) deviation of 0.37 Å in Cα positions], but after this superposition the structures diverge in the middle of helix αB (Fig. 5A). With the exception of local changes in the loop between αB and αC, this is a rigid-body transformation fitted by a rotation of 11°. The juncture between the two rigid bodies is between residues 536 and 538, and as a consequence order is lost in the latchlike contact of the α domain with the outer loops. In the type 1 structure, this involves hydrogen bonds from Arg467 to Asp540 and His544 to Asp431 and a salt bridge (4.0 Å) between Lys548 and Asp431 (Fig. 5B), but the side chains of Arg467, His544, and Lys548 are all disordered in the type 2 structure. The substantial interactions between the NH2-terminal half of αB and the inner loops of the β-subdomain remain completely intact in the type 2 crystal. The peptide remains bound but is less well ordered. We view the change from type 1 to type 2 as incipient to the full-scale change required for the peptide exchange.
Fig. 5.
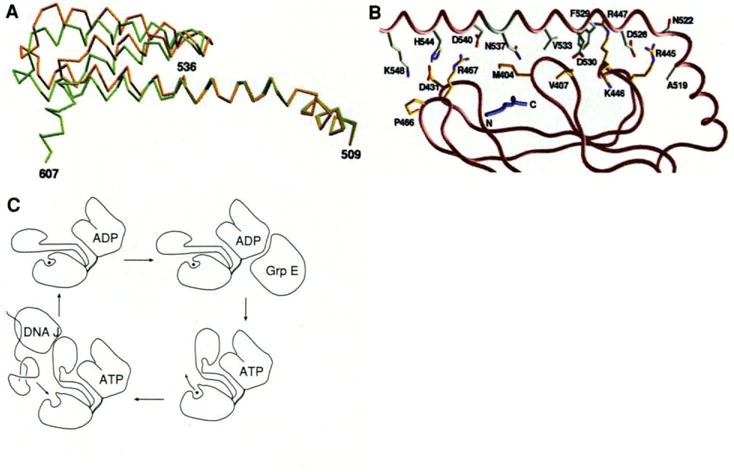
(A) Superposition of the Cα trace of the helices in two different crystal forms, based on the Cα positions in the β-sandwich subdomain (396–501). The type 2 (P21212) structure is shown in brown and the type 1 (I222) is shown in green. (B) Residues at the interface between the β-sandwich subdomain and helix αB. The orientation of the structure is similar to that in Fig, 2B, The peptide is colored blue. The carbon atoms of the residues from αB are colored white and the residues from the β domain are colored yellow. Nitrogen and oxygen atoms are colored blue and red, respectively. (C) Schematic representation of the mechanistic function of DnaK. [Parts (A) and (B) were produced by GRASP (55)]
Atomic mobility factors for the type 2 structure are generally higher than for the type 1 structure, and there is considerable variation in the pattern as well. The α domain (538 to 607) is most affected with a change in average main-chain B factor from 19 Å2 in type 1 to 41 Å2 and 50 Å2 for the two type 2 molecules. The average main-chain B factor in the β subdomain rises from 16 Å2 to 23 Å2 and 24 Å2, respectively. But for the outer loops ( and ) where the latch to the α domain is broken, the rise is much larger than average (from 16 Å2 to 30 Å2), whereas for the inner loops ( and ) it is less than average (from 18 to 23 Å2). This loosening of the outer, hydrogen-bond contributing side of the substrate channel may be a progenitor of peptide release.
The two conformations of the NH2-terminal segment (389 to 393), one extended and the other folded into a hydrophobic pocket, leave the alternative Cα positions of residue 389 22 Å apart. Alternative states for this connecting segment between the ATP and substrate-binding units may be relevant to the ATP-dependent conformational change. The modeling of small-angle scattering data from bovine hsc70 in the presence of ATP versus ADP required a 14 Å movement of the two fixed domains (34), although dimerization might also account for the observations (47).
Mechanistic implications
By its very nature, the image of the peptide embedded in a channel through the substrate-binding unit of DnaK (Fig. 2) raises the question of how the peptide got there and how it might leave. The high degree of sequence conservation and functional similarity among hsp70 proteins make it very likely that the answer to this question in the specific case will also apply generally to this entire family of molecular chaperones. Moreover, evidence on substrate binding to any of the hsp70 proteins can justifiably be brought to bear on the question for this particular complex.
A mechanism by which the substrate threads its way in and out through the channel seems implausible even for a short peptide such as the one in this complex, and it is even more unlikely when an internal loop from a protein is considered. It is clear that the hsc70-mediated uncoating of clathrin-coated vesicles involves the binding of hsc70 to an internal epitope of clathrin light chains that becomes exposed when the calcium level increases (25). The binding of hsp70 proteins to internal segments of other proteins has also been demonstrated (48), and in general hsp70 proteins are thought to bind to diverse segments of unfolded protein. Moreover, in vitro, longer peptides bind as effectively as short ones (22). We believe that substrate binding and release must involve conformational changes both at a grand scale to displace the helical domain from its blockage of the site and at the local level of the binding loops in the β subdomain to open the channel. We know that peptide binding and release occur in COOH-terminal fragments of DnaK and postulate that the required conformational changes in the substrate-binding unit will be like those for the intact molecule. Indeed, it is plausible that the roles of the ATPase domain and DnaJ may be to affect the equilibrium among conformers. We suggest that the ADP state of an hsp70 molecule will have the α-helical lid domain closed as in the type 1 structure reported here, and that the ATP state that accelerates peptide exchange (9, 11, 12) will have the lid displaced.
The differences in conformation and mobility patterns between the type 1 and type 2 crystals provide important clues about conformational changes that might accompany substrate exchange. This demonstrates a juncture of flexibility between 536 and 538. By analogy with calmodulin, in which an exposed interdomain segment of a long helix becomes disordered and adopts a defined loop structure in calmodulin-peptide complexes (49), we suggest that this interdomain segment of αB (536 to 538) may melt out to permit the α-helical domain to move away as an entity. The increased flexibility in the outer loops of the substrate-binding channel would, in turn, be expected to free up the binding site for release and rebinding.
Several pieces of biochemical evidence support this model for conformation-dependent substrate binding. First, DnaK becomes susceptible to tryptic cleavage after Arg467 upon ATP binding, whereas it is protected with ADP or in the absence of nucleotide (33). Arg467 is part of the latch between outer loops and the α-helical domain in type 1 crystals, and it is disordered in type 2 crystals. By contrast, there is no cleavage under any examined circumstances after Arg445 and Arg447, which are at the interface between the inner loops and αB (Fig. 5B). Second, the stability of peptide complexes with the COOH-terminal fragment is identical to the stability of complexes formed with the peptide by the ADP-bound form of DnaK (15). By contrast, a 60-kD chymotryptic fragment of hsc70—which by approximate size may be truncated after a residue corresponding to 539, 544, or 556 in DnaK—was shown to bind clathrin triskelions more efficiently than did intact hsc70 (13). Third, the kinetics of peptide binding by hsp70s are greatly increased in the presence of ATP (9, 11), and they involve large activation energy barriers (50). Even in the presence of ATP, both on and off rates are relatively slow, which is consistent with the barriers intrinsic to the β subdomain. Finally, several mutations in the substrate-binding unit of DnaK that were selected for deficiency in binding function cluster in the inner loops that remain fixed in this model, whereas there are none in the outer loops, which by their flexibility may be more accommodating (51). In addition, one of the mutations is at the proposed intersegment junction at residue 539.
The substrate binding activity of DnaK is modulated directly or indirectly by the accessory chaperones DnaJ and GrpE as well as by its own ATPase unit (5, 10). The effects of these components must ultimately be felt at the substrate binding site seen in the fragment studied here. Moreover, substrate binding stimulates ATP hydrolysis (27, 31, 57). Although these structures of a peptide complex with the substrate-binding unit cannot on their own explain how these interactions take place, they do limit the options. The foremost questions concern the nature of interactions of this substrate-binding unit with the ATPase unit in its alternative ADP-bound and ATP-bound states. Our suggested α-domain movements further the emerging model for DnaK action (Fig. 5C).
Changes monitored by tryptophan fluorescence (33, 45, 46), small-angle x-ray scattering (34, 47), and proteolytic susceptibility (17, 33) indicate that the ADP and ATP states are different and that the conformational transition is induced after ATP binding but before hydrolysis (46). It seems likely that large differences in the mode of association between the units, rather than changes within the units, are responsible for the observations. Remarkably, although these ATP-induced effects do not occur in isolated ATPase units, they do occur for molecules truncated at 554 of hsc70 (34) or at 538 of DnaK (33) as well as for intact hsp70s. Although COOH-terminal portions do play a role (19, 52), direct interactions between the ATPase unit and the α domain identified here as moveable, do not appear to be required for the transition. Instead, it appears that there must be a mechanism for transmitting the change through the substrate-binding unit to the flexible juncture at 536 to 538. Possibly the alternative conformations of the NH2-terminus observed in the structure here, which happens to be the inter-unit segment, play a role. The ATPase domain is joined to the β subdomain next to the juncture between β8 and αA, and if the linker were displaced from its hydrophobic pocket at the β8-αA juncture, the precise positioning of αB might be affected. This could then affect the equilibrium between open and closed states of the α-helical lid. Conversely, the stimulatory effect on ATP hydrolysis from the binding of substrate of a DnaJ analog might be caused by a reciprocal effect on conformational equilibria. Contacts between the α-helical domain and the β-sandwich subdomain must be affected by the bound peptide. In addition, DnaJ appears to bind to the COOH-terminal region of DnaK (19), which is shown here to comprise the α-helical domain. Thus, the binding of peptide or DnaJ could stabilize the closed state of the α-helical lid, which by this model would in turn favor the ADP state over the ATP state, hence promoting hydrolysis.
The ATP-driven lid opening transition of DnaK that we describe above could couple with DnaJ to explain the concerted action by DnaK with DnaJ, or their homologs; DnaK and DnaJ can both interact with the same protein and with one another. When the conformational transition occurs in such a ternary complex, it should exert a force on the substrate to pull the DnaK-bound, loose polypeptide out from the DnaJ-bound remainder of the protein. In this way, during heat shock, misfolded aggregates might become disen tangled. In the translocation of proteins across membranes, coupling of the hsp70 conformational transition with the membrane-tethered DnaJ homolog should serve to pull the polypeptides from one compartment to another.
Contributor Information
Xiaotian Zhu, Department of Biochemistry and Molecular Biophysics, College of Physicians and Surgeons, Columbia University, New York, NY 10032, USAHoward Hughes Medical Institute, Columbia University.
Xun Zhao, Department of Biochemistry and Molecular Biophysics, College of Physicians and Surgeons, Columbia University, New York, NY 10032, USA.
William F. Burkholder, Department of Biochemistry and Molecular Biophysics, College of Physicians and Surgeons, Columbia University, New York, NY 10032, USAInstitute of Cancer Research, Columbia University, New York, NY 10032, USA
Alexander Gragerov, Department of Biochemistry and Molecular Biophysics, College of Physicians and Surgeons, Columbia University, New York, NY 10032, USA.
Craig M. Ogata, Howard Hughes Medical Institute, National Synchrotron Light Source, Brookhaven National Laboratory, Stony Brook, New York, USA
Max E. Gottesman, Department of Biochemistry and Molecular Biophysics, College of Physicians and Surgeons, Columbia University, New York, NY 10032, USAInstitute of Cancer Research, Columbia University, New York, NY 10032, USA
Wayne A. Hendrickson, Department of Biochemistry and Molecular Biophysics, College of Physicians and Surgeons, Columbia University, New York, NY 10032, USAHoward Hughes Medical Institute, Columbia University
REFERENCES AND NOTES
- 1.Gething MJ, Sambrook J. Nature. 1992;355:33. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]; Hendrick JP, Hartl FU. Annu Rev Biochem. 1993;62:349. doi: 10.1146/annurev.bi.62.070193.002025. [DOI] [PubMed] [Google Scholar]; Morimoto RI, Tissieres A, Georgopoulos C. In: The Biology of Heat Shock Proteins and Molecular Chaperones. Morimoto RI, Tissieres A, Georgopoulos C, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1994. [Google Scholar]
- 2.Pelham HRB. Cell. 1986;46:959. doi: 10.1016/0092-8674(86)90693-8. [DOI] [PubMed] [Google Scholar]
- 3.Rothman JE. ibid. 1989;59:591. doi: 10.1016/0092-8674(89)90005-6. [DOI] [PubMed] [Google Scholar]
- 4.Saito H, Uchida H. Mol Gen Genet. 1978;164:1. doi: 10.1007/BF00267592. [DOI] [PubMed] [Google Scholar]; Yochem J, Uchida H. ibid. 1978;164:9. doi: 10.1007/BF00267593. [DOI] [PubMed] [Google Scholar]
- 5.Georgopoulos C. In: The Biology of Heat Shock Proteins and Molecular Chaperones. Morimoto RI, Tissieres A, Georgopoulos C, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1994. pp. 209–249. [Google Scholar]
- 6.Welch WJ, Feramisco JR. Mol Cell Biol. 1985;5:1229. doi: 10.1128/mcb.5.6.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]; McKay DBW, et al. In: The Biology of Heat Shock Proteins and Molecular Chaperones. Morimoto RI, Tissieres A, Georgopoulos C, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1994. p. 209. [Google Scholar]
- 7.Palleros DR, Welch WJ, Fink AL. Proc Natl Acad Sci USA. 1991;88:5719. doi: 10.1073/pnas.88.13.5719. [DOI] [PMC free article] [PubMed] [Google Scholar]; Palleros DR, Shi L, Reid KL, Fink AL. J Biol Chem. 1994;269:13107. [PubMed] [Google Scholar]
- 8.Palleros DR, Reid KL, Shi L, Welch WJ, Fink AL. Nature. 1993;365:664. doi: 10.1038/365664a0. [DOI] [PubMed] [Google Scholar]
- 9.Schmid D, Baici A, Gehring H, Christen P. Science. 1994;263:971. doi: 10.1126/science.8310296. [DOI] [PubMed] [Google Scholar]
- 10.Szabo A, Langer T, Schroder H, Flanagan J, Bukau B, Hartl FU. Proc Natl Acad Sci USA. 1994;91:10345. doi: 10.1073/pnas.91.22.10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greene LE, Zinner R, Naficy S, Eisenberg E. J Biol Chem. 1995;270:2967. doi: 10.1074/jbc.270.7.2967. [DOI] [PubMed] [Google Scholar]
- 12.McCarty JS, Buchberger A, Reinstein J, Bukau B. J Mol Biol. 1995;249:126. doi: 10.1006/jmbi.1995.0284. [DOI] [PubMed] [Google Scholar]
- 13.Chappell TG, Konforti BB, Schmid SL, Rothman JE. J Biol Chem. 1987;262:746. [PubMed] [Google Scholar]
- 14.Wang TF, Chang JH, Wang C. ibid. 1993;268:26409. [Google Scholar]
- 15.Gragerov A, Zeng L, Zhao X, Burkholder W, Gottesman ME. J Mol Biol. 1994;235:848. doi: 10.1006/jmbi.1994.1043. [DOI] [PubMed] [Google Scholar]
- 16.Buchberger A, Valencia A, McMacken R, Sander C, Bukau B. EMBO J. 1994;13:1687. doi: 10.1002/j.1460-2075.1994.tb06433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liberek K, Marszalek J, Ang D, Georgopoulos C, Zylicz M. Proc Natl Acad Sci USA. 1991;88:2874. doi: 10.1073/pnas.88.7.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cyr DM, Langer T, Douglas MG. Trends Biochem Sci. 1994;19:176. doi: 10.1016/0968-0004(94)90281-x. [DOI] [PubMed] [Google Scholar]
- 19.Wawrzynow A, Zylicz M. J Biol Chem. 1995;270:19300. doi: 10.1074/jbc.270.33.19300. [DOI] [PubMed] [Google Scholar]
- 20.Flaherty KM, DeLuca-Flaherty C, McKay DB. Nature. 1990;346:623. doi: 10.1038/346623a0. [DOI] [PubMed] [Google Scholar]
- 21.Morshauser RC, Wang H, Flynn GC, Zuderweg RP. Biochemistry. 1995;34:6261. doi: 10.1021/bi00019a001. [DOI] [PubMed] [Google Scholar]
- 22.Flynn GC, Chappell TG, Rothman JE. Science. 1989;245:385. doi: 10.1126/science.2756425. [DOI] [PubMed] [Google Scholar]
- 23.Blond-Elguindi S, Cwirla SE, Dower WJ, Lipshutz RJ, Sprang SR, Sambrook JF, Gething MJH. Cell. 1993;75:717. doi: 10.1016/0092-8674(93)90492-9. [DOI] [PubMed] [Google Scholar]
- 24.Flynn GC, Pohl J, Flocco MT, Rothman JE. Nature. 1991;353:726. doi: 10.1038/353726a0. [DOI] [PubMed] [Google Scholar]
- 25.DeLuca-Flaherty C, McKay DB, Parham P, Hill BL. Cell. 1990;62:875. doi: 10.1016/0092-8674(90)90263-e. [DOI] [PubMed] [Google Scholar]
- 26.Landry SJ, Jordan R, McMacken R, Gierasch LM. Nature. 1992;355:455. doi: 10.1038/355455a0. [DOI] [PubMed] [Google Scholar]
- 27.Hendrickson WA. Science. 1991;254:51. doi: 10.1126/science.1925561. [DOI] [PubMed] [Google Scholar]
- 28.Hendrickson WA, Horton JR, LeMaster DM. EMBO J. 1990;9:1665. doi: 10.1002/j.1460-2075.1990.tb08287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao X. in preparation [Google Scholar]
- 30.Abbreviations for the amino acids are: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile, K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser, T, Thr; V, Val; W, Trp; Y, Tyr.
- 31.Athappilly FK, Hendrickson WA. Structure. 1995;3:1407. doi: 10.1016/s0969-2126(01)00277-5. [DOI] [PubMed] [Google Scholar]
- 32.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Acta Crystallogr A. 1991;47:110. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 33.Buchberger A, et al. J Biol Chem. 1995;270:16903. doi: 10.1074/jbc.270.28.16903. [DOI] [PubMed] [Google Scholar]
- 34.Wilbanks SM, Chen L, Tsuruta H, Hodgson KO, McKay DB. Biochemistry. 1995;34:12095. doi: 10.1021/bi00038a002. [DOI] [PubMed] [Google Scholar]
- 35.Noel JP, Hamm HE, Sigler PB. Nature. 1993;366:654. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]; Coleman D, Berghuis AM, Lee E, Linder ME, Gilman AG, Sprang SR. Science. 1994;265:1405. doi: 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- 36.Sharp A, Nicholls A, Fine R, Honig B. Science. 1991;252:106. doi: 10.1126/science.2011744. [DOI] [PubMed] [Google Scholar]
- 37.Lim WA, Richards FM, Fox RO. Nature. 1994;372:375. doi: 10.1038/372375a0. [DOI] [PubMed] [Google Scholar]
- 38.Stanfield RL, Wilson IA. Curr Opin Struct Biol. 1995;5:103. doi: 10.1016/0959-440x(95)80015-s. [DOI] [PubMed] [Google Scholar]
- 39.Rippmann F, Taylor WR, Rothbard JB, Green NM. EMBO J. 1991;10:1053. doi: 10.1002/j.1460-2075.1991.tb08044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Germain RN, Margulies DH. Annu Rev Immunol. 1993;11:403. doi: 10.1146/annurev.iy.11.040193.002155. [DOI] [PubMed] [Google Scholar]
- 41.Prasa K, Heuser J, Eisenberg E, Greene L. J Biol Chem. 1994;269:6831. [PubMed] [Google Scholar]
- 42.Boorstein WR, Ziegelhoffer T, Craig EA. J Mol Evol. 1994;38:1. doi: 10.1007/BF00175490. [DOI] [PubMed] [Google Scholar]; Rensing SA, Maier UG. J Mol Evol. 1994;39:80. doi: 10.1007/BF00178252. [DOI] [PubMed] [Google Scholar]
- 43.Fourie AM, Sambrook JF, Gething MJH. J Biol Chem. 1994;269:30470. [PubMed] [Google Scholar]
- 44.Gragerov A, Gottesman ME. J Mol Biol. 1994;241:133. doi: 10.1006/jmbi.1994.1482. [DOI] [PubMed] [Google Scholar]
- 45.Palleros D, Reid KL, McCarty JS, Walker GC, Fink AL. J Biol Chem. 1992;267:5279. [PubMed] [Google Scholar]
- 46.Ha JH, McKay DB. Biochemistry. 1995;34:11635. doi: 10.1021/bi00036a040. [DOI] [PubMed] [Google Scholar]
- 47.Shi L, Kataka M, Fink AL. ibid. 1996;35:3297. doi: 10.1021/bi951984l. [DOI] [PubMed] [Google Scholar]
- 48.Inoue A, et al. J Biol Chem. 1995;270:22571. doi: 10.1074/jbc.270.38.22571. [DOI] [PubMed] [Google Scholar]; Roman E, Moreno C, Young D. Eur J Biochem. 1994;222:65. doi: 10.1111/j.1432-1033.1994.tb18842.x. [DOI] [PubMed] [Google Scholar]; Pratt WB, Welsh MJ. Sem Cell Biol. 1994;5:83. doi: 10.1006/scel.1994.1012. [DOI] [PubMed] [Google Scholar]
- 49.Meador WE, Means AR, Quiocho FA. Science. 1993;262:1718. doi: 10.1126/science.8259515. [DOI] [PubMed] [Google Scholar]; Ikura M, et al. ibid. 1992;256:632. [Google Scholar]; Babu YS, et al. Nature. 1985;315:37. doi: 10.1038/315037a0. [DOI] [PubMed] [Google Scholar]
- 50.Farr CD, Galiano FJ, Witt SN. Biochemistry. 1995;34:15574. doi: 10.1021/bi00047a024. [DOI] [PubMed] [Google Scholar]
- 51.Burkholder WF, et al. in preparation [Google Scholar]
- 52.Freeman BC, Myers MP, Schumacher R, Morimoto RI. EMBO J. 1995;14:2281. doi: 10.1002/j.1460-2075.1995.tb07222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.For DENZO and SCALEPACK, see; Otwinowski Z. In: Data Collection and Processing. Sawyer L, Isaacs N, Bailey S, editors. SERC Daresbury Laboratory; Warrington, UK: 1993. pp. 56–62. [Google Scholar]; for HASSP, see; Twerwiliger T. CCP4: A Suite of Programs for Protein Crystallography. SERC Collaborative Computing Project No. 4, Daresbury Laboratory; Warrington, UK: 1979. [Google Scholar]; for AMORE, see; Navaza J. Acta Crystallogr. 1994;A50:157. [Google Scholar]
- 54.Sheriff S, Hendrickson WA, Smith JL. J Mol Biol. 1987;197:273. doi: 10.1016/0022-2836(87)90124-0. [DOI] [PubMed] [Google Scholar]
- 55.MOLSCRIPT P Kraulis. J Appl Crystallogr. 1991;24:946. [Google Scholar]; SETOR SV Evans. J Mol Graph. 1993;11:134. doi: 10.1016/0263-7855(93)87009-t. [DOI] [PubMed] [Google Scholar]; Nicholls GRASPA, Sharp KA, Honig B. Proteins Struct Funct Genet. 1991;11:281. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
- 56.We thank Y. Liu for help in synchrotron data collection; F. Athappilly for his help in crystallization of selenomethionyl DnaK by dialysis; M. Gawinowitz for amino-acid sequencing; N. Pileggi for synthesis of selenomethionyl peptides; D. King and B. Pramanik for mass spectrometric data; and laboratory colleagues for discussion, especially C. Lima, Y. Liu, L. Shapiro, P. Kwong, H. Wu, D. Fremont, and X. Jiang. Supported in part by NIH grants GM34102 (W.A.H.) and GM37219 (M.E.G.). Beamline X4A at the National Synchrotron Light Source, a Department of Energy facility, is supported by the Howard Hughes Medical Institute. The coordinates have been deposited in PDB (1DKX, 1DKY, and 1DKZ).