

Abstract
Marine sponges (phylum Porifera) are a diverse, phylogenetically deep-branching clade known for forming intimate partnerships with complex communities of microorganisms. To date, 16S rRNA gene sequencing studies have largely utilised different extraction and amplification methodologies to target the microbial communities of a limited number of sponge species, severely limiting comparative analyses of sponge microbial diversity and structure. Here, we provide an extensive and standardised dataset that will facilitate sponge microbiome comparisons across large spatial, temporal, and environmental scales. Samples from marine sponges (n = 3569 specimens), seawater (n = 370), marine sediments (n = 65) and other environments (n = 29) were collected from different locations across the globe. This dataset incorporates at least 268 different sponge species, including several yet unidentified taxa. The V4 region of the 16S rRNA gene was amplified and sequenced from extracted DNA using standardised procedures. Raw sequences (total of 1.1 billion sequences) were processed and clustered with (i) a standard protocol using QIIME closed-reference picking resulting in 39 543 operational taxonomic units (OTU) at 97% sequence identity, (ii) a de novo clustering using Mothur resulting in 518 246 OTUs, and (iii) a new high-resolution Deblur protocol resulting in 83 908 unique bacterial sequences. Abundance tables, representative sequences, taxonomic classifications, and metadata are provided. This dataset represents a comprehensive resource of sponge-associated microbial communities based on 16S rRNA gene sequences that can be used to address overarching hypotheses regarding host-associated prokaryotes, including host specificity, convergent evolution, environmental drivers of microbiome structure, and the sponge-associated rare biosphere.
Keywords: marine sponges, archaea, bacteria, symbiosis, microbiome, 16S rRNA gene, microbial diversity
Data Description
Purpose of data acquisition
Sponges (phylum Porifera) are an ancient metazoan clade [1], with more than 8500 formally described species [2]. Sponges are benthic organisms that have important ecological functions in aquatic habitats [3, 4]. Marine sponges are often found in symbiotic association with microorganisms, and these microbial communities can be very diverse and complex [5, 6]. Sponge symbionts perform a wide range of functional roles, including vitamin synthesis, production of bioactive compounds, and biochemical transformations of nutrients or waste products [7–9]. The diversity of microorganisms associated with sponges has been the subject of intense study (the search of “sponge microbial diversity” returned 348 publications in the Scopus database) [10]. Most of these studies were performed on individual species from restricted geographic regions [e.g., 11, 12]. A comparative assessment of these studies is often hindered by differences in sample processing and 16S rRNA gene sequencing. However, 2 recent studies incorporating a large number of sponge microbiomes (>30) [5, 13] revealed the potential of large-scale, standardised, high-throughput sequencing for gaining insights into the diversity and structure of sponge-associated microbial communities. The purpose of this global dataset is to provide a comprehensive 16S rRNA gene-based resource for investigating and comparing microbiomes more generally across the phylum Porifera.
Sample collection, processing, and 16S rRNA gene sequencing
Sample collection and processing, species identification, and DNA extractions were conducted as previously described [13]. A total of 3569 sponge specimens were collected, representing at least 268 species, including several yet unidentified taxa (hereafter collectively referred to as species) (Supplementary Table S1). Of all species, 213 were represented by at least 3 specimens. Carteriospongia foliascens had the highest replication, comprising 150 individuals. Seawater (n = 370), sediment (n = 65), algae (n = 1), and echinoderm (n = 1) samples as well as biofilm swabs (n = 21) of rock surfaces were collected in close proximity to the sponges for comparative community analysis. Six negative control samples (sterile water) were processed to identify any potential contaminations. Of the samples included in this current dataset, 973 samples had been analysed previously [13]. Samples were collected from a wide range of geographical locations (Fig. 1; Supplementary Table S1). Total DNA was extracted as previously described [13] and used as templates to amplify and sequence the V4 region of the 16S rRNA gene using the standard procedures of the Earth Microbiome Project (EMP) [14 15].
Figure 1:

Global sample collection sites. Bubbles indicate collection sites of (A) marine sponges, (B) seawater, and (C) marine sediment samples. Bubble sizes are proportional to number of samples as indicated.
Processing of sequencing data
Clustering using the EMP standard protocols in QIIME
Raw sequences were demultiplexed and quality controlled following the recommendations of Bokulich et al. [16]. Quality-filtered, demultiplexed fastq files were processed using the default closed-reference pipeline from QIIME v. 1.9.1 (QIIME, RRID:SCR_008249). Briefly, sequences were matched against the GreenGenes reference database (v. 13_8 clustered at 97% similarity). Sequences that failed to align (e.g., chimeras) were discarded, which resulted in a final number of 300 140 110 sequences. Taxonomy assignments and the phylogenetic tree information were taken from the centroids of the reference sequence clusters contained in the GreenGenes reference database (Greengenes, RRID:SCR_002830). This closed-reference analysis allows for cross-dataset comparisons and direct comparison with the tens of thousands of other samples processed in the EMP and available via the Qiita database [17].
Clustering using Mothur
Quality-filtered, demultiplexed fastq files were also processed using Mothur v. 1.37.6 (Mothur, RRID:SCR_011947) [18] and Python v. 2.7 (Python Programming Language, RRID:SCR_008394) [19] custom scripts with modifications from previously established protocols [13]. Detailed descriptions and command outputs are available at the project notebook (see Availability of supporting data). Briefly, sequences were quality-trimmed to a maximum length of 100 bp. To minimize computational effort, the dataset was reduced to unique sequences, retaining total sequence counts. Sequences were aligned to the V4 region of the 16S rRNA gene sequences from the SILVA v. 123 database (SILVA, RRID:SCR_006423) [20]. Sequences that aligned at the expected positions were kept, and this dataset was again reduced to unique sequences. Further, singletons were removed from the dataset, and the remaining sequences were preclustered if they differed by 1 nucleotide position. Sequences classified as eukaryote, chloroplast, mitochondria, or unknown according to the Greengenes (v. 13_8 clustered at 99% similarity) [21] and SILVA taxonomies [22] were removed. Chimeras were identified with UCHIME (UCHIME, RRID:SCR_008057) [23] and removed. Finally, sequences were de novo clustered into operational taxonomic units (OTUs) using the furthest neighbour method at 97% similarity. Representative sequences of OTUs were retrieved based on the mean distance among the clustered sequences. Consensus taxonomies based on the SILVA, Greengenes, and RDP (v. 14_03 2015; Ribosomal Database Project, RRID:SCR_006633) [24] databases were obtained based on the classification of sequences clustered within each OTU. The inclusion of these taxonomies is helpful considering that they have substantial differences, as recently discussed [25]. For example, Greengenes and RDP have the taxon Poribacteria, a prominent sponge-enriched phylum [26], which did not exist in the SILVA version used.
De-noising using Deblur
Recently, sub-OTU methods that allow views of the data at single-nucleotide resolution have become available. One such method is Deblur [27], which is a de-noising algorithm for identification of the actual bacterial sequences present in a sample. Using an upper bound on the polymerase chain reaction and read-error rates, Deblur processes each sample independently and outputs the list of sequences and their frequencies in each sample, enabling single nucleotide resolution. For creating the deblurred biom table, quality-filtered, demultiplexed fasta files were used as input to Deblur using a trim length of 100 and min-reads of 25 (removing sOTUs with <25 reads total in all samples combined). Taxonomy was added to the resulting biom table using QIIME [28], RDP classifier [29], and Greengenes v. 13.8 [21].
Database metadata category enrichment
For enrichment analysis of metadata terms in a set of sequences, each unique metadata value is tested using both a binomial test and a ranksum test. All analysis is performed on a randomly subsampled (5000 reads/sample) table.
The binomial (presence/absence) P-value for enrichment calculated as follows
For a bacterial sequence s and metadata value v, denote N the total number of samples, O(s) the number of samples where s is present, Kv(s) the number of samples with value v where s is present, and T(v) the total number of samples with value v.
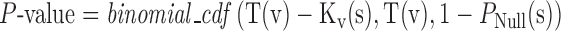 |
where PNull(s) = O(s)/N
The ranksum (frequency aware) P-value is calculated using the Kruskal-Wallis test (implemented in scipy 0.19) as follows.
For a bacterial sequence s and metadata value v, denote by Fv(s) the vector of relative frequencies of bacteria s in all samples with metadata value v, and denote by  the vector of relative frequencies of bacteria s in all samples with metadata other than v. The ranksum P-value is then calculated using the Kruskal-Wallis test for Fv(s) and
the vector of relative frequencies of bacteria s in all samples with metadata other than v. The ranksum P-value is then calculated using the Kruskal-Wallis test for Fv(s) and  and shown only if significantly enriched in samples containing v (i.e., rank difference of Fv(s)—
and shown only if significantly enriched in samples containing v (i.e., rank difference of Fv(s)—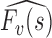 > 0).
> 0).
We have set up a webserver [30] that performs this enrichment analysis for user-defined sequence submissions. The code for the webserver is also available in Github for a local installation.
Data description
The dataset covers 4033 samples with a total of 1 167 226 701 raw sequence reads. These sequence reads clustered into 39 543 OTUs using QIIME’s closed-reference processing, 518 246 OTUs from de novo clustering using Mothur (not filtered for OTU abundances), and 83 908 sOTUs using Deblur (with a filtering of at least 25 reads total per sOTU). We recommend that data users consider the differences in sequencing depths per sample and abundance filtering for certain downstream analyses, such as when calculating diversity estimates [16] and comparing OTU abundances across samples [31]. In terms of taxonomic diversity, most Mothur OTUs were assigned to the phylum Proteobacteria, although more than 60 different microbial phyla were recovered from the marine sponge samples according to SILVA (n = 63) and Greengenes classifications (n = 72) (Fig. 2).
Figure 2:

Microbial taxonomic profile of marine sponge samples processed with Mothur. (A) SILVA, (B) Greengenes, and (C) RDP taxonomies are shown. OTU sequence counts were grouped according to phylum and class. Taxa with relative abundances ≤0.5% were grouped as “others.” Classes with relative abundances >1% are shown in the legend (phylum “;” class). Relative abundances are represented on the x-axes.
Potential uses
This dataset can be utilised to assess a broad range of ecological questions pertaining to host-associated microbial communities generally or to sponge microbiology specifically. These include: (i) the degree of host specificity, (ii) the existence of biogeographic or environmental patterns, (iii) the relation of microbiomes to host phylogeny, (iv) the variability of microbiomes within or between host species, (v) symbiont co-occurrence patterns, and (vi) assessing the existence of a core sponge microbiome. An example of this type of analysis is shown in Fig. 3, where samples were clustered using unweighted UniFrac data [10] with a Principal Coordinates Analysis and visualization in Emperor [15] based on their origins from sponges, seawater, or kelps [17].
Figure 3:

Unweighted UniFrac Principal Coordinates Analysis (PCA) of samples from sponges (“animal-associated habitat”), kelp forest, and ocean water. Samples were rarefying to 10 000 sequences per sample. A movie showing the PCA plot in 3D is provided in the supporting information.
Availability and requirements
Project name: The Sponge Microbiome Project
Project home page: www.spongeemp.com; https://github.com/amnona/SpongeEMP
Operating system(s): Unix
Programming language: Python and R
Other requirements: Python v. 2.7, Biopython v. 1.65, Python 3.5, R v. 3.2.2, Mothur v. 1.37.6, QIIME v. 1.9.1, Deblur
License: MIT
Any restrictions to use by non-academics: none
Availability of supporting data
Raw sequence data were deposited in the European Nucleotide Archive (accession number: ERP020690). Quality-filtered, demultiplexed fastq files, QIIME resulting OTU tables are available at the Qiita database (Study ID: 10 793) [17]. The additional datasets that support the results of this article are available in the GigaScience repository, GigaDB [32] and include an OTU abundance matrix (the output “.shared” file from Mothur, which is tab delimited), an OTU taxonomic classification table (tab delimited text file), an OTU representative sequence FASTA file, a table of samples’ metadata, the biom files from QIIME and Deblur analyses, and the QIIME-generated tree file. The project workflow, Mothur commands, and additional scripts are available as HTML in GigaDB [32].
The deblurred dataset has also been uploaded to an online server [19] that supplies both html and REST-API access for querying bacterial sequences and obtaining the observed prevalence and enriched metadata categories where the sequence is observed (Figure 4). This allows an interactive view of which sequences are associated with which specific parameters, such as depth or salinity.
Figure 4:

Output of the enrichment analysis through the online server www.spongeemp.com. Top line shows taxonomic assignment for the user-submitted sequence in the second line. Pie charts below show the total number of samples (right) and the number of samples where the submitted sequence is present (left) based on the scientific names of the host, followed by the significantly enriched host names containing the submitted sequence (using either presence/absence binomial test or relative frequency–based ranksum test). At the bottom, fields can be opened to show results of the enrichment analyses for other metadata types (e.g., country).
Additional file
sample.metadata
Abbreviations
EMP: Earth microbiome project; bp: base pairs; OTU: operational taxonomic unit; rRNA: ribosomal RNA.
Funding
T.T. and N.S.W were funded by Australian Research Council Future Fellowships FT140100197 and FT120100480, respectively. T.T. received funds from the Gordon and Betty Moore Foundation. This work was also supported in part by the W.M. Keck Foundation and the John Templeton Foundation. R.K. received funding as a Howard Hughes Medical Institute Early Career Scientist.
Competing interests
The authors declare that they have no competing interests.
Author contributions
L.M.-S., N.S.W., and T.T. designed the study. C.A.G., D.S., F.L., G.S., G.K., G.McC., G.-F. F, J.J.B., J.V., J.R.B., J.M.M., J.R., L.S., M.C.P, M.V.M., M.W.T., N.S.W., P.P., P.M.E., P.J.S., R.L.S, R.W.T., R.C., R.T.H., S.L-L., T.D., T.R., U.H., and Z-Y. L. collected samples. C.A.G., D.S., J.V., J.R.B., L.S., M.C.P., M.W.T., N.S.W., P.M.E., R.L.S, R.W.T., S.L-L., and U.H. extracted DNA. G.L.A. and R.K. sequenced DNA. L.M.-S., S.N., A.A., A.G., G.L.A., and T.T. performed data processing and analysis. L.M.-S., N.S.W., and T.T. wrote the manuscript. All authors contributed to the writing of the manuscript.
Supplementary Material
References
- 1. Li CW, Chen JY, Hua TE. Precambrian sponges with cellular structures. Science 1998;279(5352):879–82. [DOI] [PubMed] [Google Scholar]
- 2. Van Soest RWM, Boury-Esnault N, Vacelet J et al. Global diversity of sponges (Porifera). PLoS One 2012;7(4):e35105. doi: 10.1371/journal.pone.0035105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bell JJ. The functional roles of marine sponges. Estuar Coast Shelf Sci 2008;79(3):341–53. [Google Scholar]
- 4. De Goeij JM, Van Oevelen D, Vermeij MJA et al. Surviving in a marine desert: the sponge loop retains resources within coral reefs. Science 2013;342(6154):108–10. [DOI] [PubMed] [Google Scholar]
- 5. Schmitt S, Tsai P, Bell J et al. Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J 2012;6(3):564–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Webster NS, Taylor MW, Behnam F et al. Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ Microbiol 2010;12(8):2070–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Siegl A, Kamke J, Hochmuth T et al. Single-cell genomics reveals the lifestyle of Poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J 2011;5(1):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taylor MW, Radax R, Steger D et al. Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol Mol Biol Rev 2007;71(2):295–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilson MC, Mori T, Ruckert C et al. An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature 2014;506(7486):58–62. [DOI] [PubMed] [Google Scholar]
- 10. Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 2005;71(12):8228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moitinho-Silva L, Bayer K, Cannistraci CV et al. Specificity and transcriptional activity of microbiota associated with low and high microbial abundance sponges from the Red Sea. Mol Ecol 2014;23(6):1348–63. [DOI] [PubMed] [Google Scholar]
- 12. Montalvo NF, Hill RT. Sponge-associated bacteria are strictly maintained in two closely related but geographically distant sponge hosts. Appl Environ Microbiol 2011;77(20):7207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thomas T, Moitinho-Silva L, Lurgi M et al. Diversity, structure and convergent evolution of the global sponge microbiome. Nat Commun 2016;7:11870. doi: 10.1038/ncomms11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gilbert JA, Jansson JK, Knight R. The Earth Microbiome project: successes and aspirations. BMC Biol 2014;121:69. doi: 10.1186/s12915-014-0069-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vazquez-Baeza Y, Pirrung M, Gonzalez A et al. EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2013;2(1):16. doi: 10.1186/2047-217X-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bokulich NA, Subramanian S, Faith JJ et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 2013;10(1):57–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marzinelli EM, Campbell AH, Zozaya Valdes E et al. Continental-scale variation in seaweed host-associated bacterial communities is a function of host condition, not geography. Environ Microbiol 2015;17(10):4078–88. [DOI] [PubMed] [Google Scholar]
- 18. Schloss PD, Westcott SL, Ryabin T et al. Introducing Mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 2009;75(23):7537–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sponge microbiome project deblurred dataset online server. http://www.spongeemp.com. Accessed 31 March2017.
- 20. Quast C, Pruesse E, Yilmaz P et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 2013;41(D1):D590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Desantis TZ, Hugenholtz P, Larsen N et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006;72(7):5069–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yilmaz P, Parfrey LW, Yarza P et al. The SILVA and All-species Living Tree Project (LTP)–taxonomic frameworks. Nucl Acids Res 2014;42(D1):D643–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Edgar RC, Haas BJ, Clemente JC et al. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011;27(16):2194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cole JR, Wang Q, Fish JA et al. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucl Acids Res 2014;42(D1):D633–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Balvociute M, Huson DH. SILVA, RDP, Greengenes, NCBI and OTT – how do these taxonomies compare? BMC Genomics 2017;18(S2):114. doi: 10.1186/s12864-017-3501-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fieseler L, Horn M, Wagner M et al. Discovery of the novel candidate phylum "Poribacteria" in marine sponges. Appl Environ Microbiol 2004;70(6):3724–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Amir A, Mcdonald D, Navas-Molina JA et al. Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems 2017;2(2). doi: 10.1128/mSystems.00191-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Caporaso JG, Kuczynski J, Stombaugh J et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010;7(5):335–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Q, Garrity GM, Tiedje JM et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 2007;73(16):5261–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. www.spongeemp.com.
- 31. Mcmurdie PJ, Holmes S, Mchardy AC. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol 2014;10(4):e1003531. doi: 10.1371/journal.pcbi.1003531.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moitinho-Silva L, Nielsen S, Amir A et al. Supporting data for “The sponge microbiome project.” GigaScience Database. 2017. 10.5524/100332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. SpongeEMP GitHub. https://github.com/amnona/SpongeEMP. Accessed 31 March2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequence data were deposited in the European Nucleotide Archive (accession number: ERP020690). Quality-filtered, demultiplexed fastq files, QIIME resulting OTU tables are available at the Qiita database (Study ID: 10 793) [17]. The additional datasets that support the results of this article are available in the GigaScience repository, GigaDB [32] and include an OTU abundance matrix (the output “.shared” file from Mothur, which is tab delimited), an OTU taxonomic classification table (tab delimited text file), an OTU representative sequence FASTA file, a table of samples’ metadata, the biom files from QIIME and Deblur analyses, and the QIIME-generated tree file. The project workflow, Mothur commands, and additional scripts are available as HTML in GigaDB [32].
The deblurred dataset has also been uploaded to an online server [19] that supplies both html and REST-API access for querying bacterial sequences and obtaining the observed prevalence and enriched metadata categories where the sequence is observed (Figure 4). This allows an interactive view of which sequences are associated with which specific parameters, such as depth or salinity.
Figure 4:
Output of the enrichment analysis through the online server www.spongeemp.com. Top line shows taxonomic assignment for the user-submitted sequence in the second line. Pie charts below show the total number of samples (right) and the number of samples where the submitted sequence is present (left) based on the scientific names of the host, followed by the significantly enriched host names containing the submitted sequence (using either presence/absence binomial test or relative frequency–based ranksum test). At the bottom, fields can be opened to show results of the enrichment analyses for other metadata types (e.g., country).