

Abstract
Outer membrane lipids in pathogenic mycobacteria are important for virulence and survival. While biosynthesis of these lipids has been extensively studied, mechanisms responsible for their assembly in the outer membrane are not understood. In the study of Gram-negative outer membrane assembly, protein-protein interactions define transport mechanisms, but analogous interactions have not been explored in mycobacteria. Here we identified interactions with the lipid transport protein LprG. Using site-specific photocrosslinking in live mycobacteria, we mapped three major interaction interfaces within LprG. We went on to identify proteins that crosslink at the entrance to the lipid binding pocket, an area likely relevant to LprG transport function. We verified LprG site-specific interactions with two hits, the conserved lipoproteins LppK and LppI. We further showed that LprG interacts physically and functionally with the mycolyltransferase Ag85A, as loss of either protein leads to similar defects in cell growth and mycolylation. Overall, our results support a model in which protein interactions coordinate multiple pathways in outer membrane biogenesis and connect lipid biosynthesis to transport.
Keywords: mycobacteria, outer membrane, cell wall, lipid transport, photocrosslinking, protein interactions, mycolic acid, lipoprotein
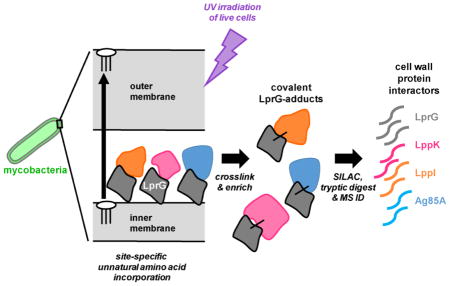
Graphical Abstract
INTRODUCTION
Mycobacteria have a complex cell envelope architecture that includes an outer membrane (OM). The mycobacterial OM is structurally and chemically distinct from canonical bacterial membranes, including the Gram-negative outer membrane. The mycobacterial OM comprises a layer of ultra long-chain mycolic acids esterified onto the underlying arabinogalactan and diverse non-covalently associated lipids. Many of these “free” or “surface” lipids are associated with the virulence of pathogenic mycobacteria such as Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis1. Prominent examples include acyltrehaloses, phthiocerol dimycocerosates (PDIM), trehalose dimycolate (TDM), and lipoarabinomannan (LAM).
The biosynthesis of these virulence-associated lipids has been extensively studied and the deletion of lipid biosynthesis genes reduces Mtb virulence in infection models2–3. Intriguingly, the disruption of OM lipid localization also leads to attenuation. For example, Mtb defective for the predicted transporter MmpL7 or the lipoprotein LppX are less virulent in the mouse4–5. In both mutant strains, PDIM is still made but is absent from the outermost cell layers. Other members of the MmpL transporter family have also been implicated in OM lipid transport. The most extreme example is MmpL3, which is required for Mtb viability6. MmpL3 transports trehalose monomycolate (TMM), which serves as a mycolate donor in the formation of the arabinogalactan-linked mycolates (AGM) and the free lipid TDM7–9. Homologues of LppX, which make up the lipid-binding lipoprotein or Llp family10, have been shown to bind various lipids in vitro, which suggests analogous roles in lipid localization11–12. Indeed, the homologue LprG is required for the surface display of LAM, a triacylated lipoglycan13–14. Loss of function in LprG and the co-transcribed transporter Rv1410c also leads to the intracellular accumulation of triacylglycerides and highly attenuated growth and virulence in the mouse model of infection15. In vitro LprG can transfer triacylglyceride between lipid membranes. Overall the data support a role for LprG and Rv1410c in the export of triacylated glycerolipids.
These studies provide strong evidence for the importance OM lipid transport in mycobacterial pathogenesis, but little is understood about the mechanisms of lipid transport, or indeed of mycobacterial cell envelope assembly in general. The mycobacterial envelope is highly dissimilar to that of canonical Gram-positive bacteria, which lack an outer membrane, so no clues arise from comparison to characterized pathways in better studied Gram positives. Mycobacteria more closely resemble Gram-negative bacteria in their membrane structures, but are genetically distant, which precludes the use of sequence homology to identify functionally relevant pathways. Notably, protein-protein interactions are mechanistically important in all OM transport pathways that have been studied. For example, the transport of lipopolysaccharide and lipoproteins to the Gram-negative OM requires specific interactions between proteins in the Lpt and Lol pathways, respectively16–17.
There is also precedent in mycobacteria for protein interactions that contribute to cell envelope assembly. Elongation complexes and divisomes are complexes of peptidoglycan-modifying enzymes and scaffolding proteins that are required for normal cell growth and division18. Accordingly, components of these complexes such as PonA1 and Wag31 localize to the cell poles and septa19–22. The implied co-localization of these various proteins suggests the coordinated transport and assembly of cell envelope components. Overall these observations suggested to us that methods that report on protein-protein interactions could reveal functionally related components of envelope assembly pathways in mycobacteria.
Protein photocrosslinking approaches have been used to dissect mechanism in the Gram-negative lipoprotein and lipopolysaccharide transport systems16–17. The success of these approaches in Escherichia coli led us to pursue an analogous strategy in mycobacteria. The site-specific metabolic incorporation of a photocrosslinking amino acid allows crosslinking in live cells and can thus capture physiological interactions in the native environment. Our aim was to uncover novel protein interactions with LprG. In addition to be required for virulence in Mtb, LprG is found in all mycobacteria, indicating a conserved role in mycobacterial physiology.
In this study we show that nonsense suppression can be used to site-specifically incorporate a photocrosslinking amino acid into LprG in Mycobacterium smegmatis (Msm). Photo-induced adducts to LprG depend on native cell wall localization and the location of crosslinker incorporation. Using scanning mutagenesis, we identified three major protein interaction interfaces within LprG: the entrance to the lipid-binding pocket, the C-terminus, and the N-terminus. Stable isotope-labeled quantitative proteomics identified several known and predicted cell wall proteins that interact with LprG near the entrance to the binding pocket. One of these was the mycolyltransferase Ag85A, which has been reported as primarily catalyzing the formation of TDM in the cell wall23. Msm strains lacking Ag85A or LprG function displayed similar growth attenuation and reduced incorporation of a TMM analogue, indicating a defect in mycolylation. These results support a physical and functional link between LprG and Ag85A and provide important evidence that protein interactions coordinate multiple cell wall assembly pathways in mycobacteria.
RESULTS
Validation of compartment-specific crosslinking to LprG
LprG is a lipoprotein that is targeted via an N-terminal signal sequence for secretion and via a conserved lipobox sequence for subsequent cleavage and N-terminal lipidation by the Lgt-LspA-Lnt enzymes. The consequent predicted cell-wall localization of LprG has been experimentally confirmed24. We thus hypothesized that all protein interactions with LprG are limited to the cell wall and that an LprG variant expressed in the cytosol would not have access to its native partners. To determine whether interactions are compartment-specific for LprG, we used the bifunctional amine-reactive crosslinker dithiobis(succinimidylpropionate) (DSP) and compared the formation of LprG-containing adducts following DSP treatment of live Msm expressing either full-length LprG or the previously characterized non-acylated mutant NA-LprG, which lacks the secretion signal and lipobox.
DSP has been used to identify protein interactions in the cell wall and in the cytosol, underscoring its utility in multiple cellular contexts25–27. DSP-dependent adducts containing LprG were detected by immunoblot against a C-terminal 6xHis tag and confirmed by loss of higher-molecular weight bands following cleavage of the DSP disulfide by dithiothreitol (Figure 1). Adduct formation was specific to full-length LprG, as no DSP-dependent adducts to NA-LprG were observed. These results indicate not only that the LprG interactions detected by crosslinking are specific to the cell wall, but also that these interactions are likely physiological rather than an artifact of LprG overexpression in this system.
Figure 1. Crosslinker-dependent higher molecular-weight adducts form only in the when LprG is expressed in the cell wall.

Msm expressing C-terminally His-tagged LprG or the soluble truncated form NA-LprG were cultured and split into two samples, one for DSP treatment and the other for DMSO-vehicle control. A portion of the LprG sample was also treated with the reducing agent dithiothreitol (DTT) to cleave the internal disulfide of DSP.
Validation of site-specific incorporation of the photocrosslinking unnatural amino acid pBpa via nonsense suppression in Msm
We first confirmed site-specific unnatural amino acid incorporation (UAAI) in Msm using a GFP reporter and the amino-acyl-tRNA synthetase (aaRS) and Tyr-tRNA from Methanocaldococcus jannaschii (Mj) as previously reported28. The plasmid pSMT-MjtRNA-pHsp60-GFP47TAG encodes a GFP mutant in which codon 47 is mutated to the amber stop codon TAG (originally reported as GFP151TAG; see Supporting Information). In this system GFP fluorescence encoded by GFP47TAG was observed only after addition of the unnatural amino acid pBpa to the culture medium and only when the Mj aaRS/tRNA pair was expressed, confirming the orthogonality of this pair and the specificity of nonsense suppression in Msm (Figure S1). Furthermore, fluorescence from GFP47TAG was comparable to that from GFP(WT), indicating efficient incorporation of pBpa under these conditions.
Using the same system, we pursued the site-specific incorporation of pBpa into LprG. Sites for pBpa incorporation were chosen based on a homology model of MsmLprG generated by the Protein Homology/Analogy Recognition Engine (Phyre version 2.0) and built on the structure of MtbLprG (PDB: 3MHA)11, 29. To survey the protein surface we chose representative residues across the structure, including positions near the entrance to (F96, Q98, N130, Y132) and within (L70, L75, L93, V213) the lipid-binding pocket; on the α-helical surface (L37, D109, A136, A149, K181, K187); on the β-sheet surface (L60, D102, E160). We also mutated position near the N- (S29, E31, T32, D34) and C-termini (K227, T232, A236), which are largely unresolved in the MtbLprG structure (Figure 2A). For all LprG mutants, the codons at the chosen positions were substituted with the amber stop codon and are hereafter referred to as LprG(pBpa) mutants.
Figure 2. Nonsense suppression mediates site-specific incorporation of pBpa into the mycobacterial protein LprG in Msm.

(A) The amber stop codon was substituted at indicated positions (orange), which were distributed across the surface and within the hydrophobic pocket of Msm LprG. Also highlighted are the positions of tyrosines (underlined; magenta) subject to misincorporation of pBpa (Figure 3). Interaction interfaces at the entrance to the pocket (“mouth”; boxes), the C-terminus (ovals), and the N-terminus (lozenges) were deduced from site-dependent UV-crosslinking patterns (Figure 5). The structure shown is a homology model generated by Phyre2 and based on Mtb LprG (PDB ID: 3MHA). The N- and C-terminii were not resolved in the crystal structure and are shown as dotted lines. The N-terminal lipidation site is at C27 in this numbering scheme and A236 is the C-terminal residue. (B) Msm strains encoding FLAG-tagged LprG(WT) or the indicated LprG(pBpa) mutant were grown with or without 2 mM pBpa. Total lysates were probed for full-length LprG (expected M.W. 24.4 kDa) by SDS-PAGE and immunoblot. GroEL is used here as a loading control. Data are representative of at least 2 biological replicates.
We confirmed pBpa-dependent expression of full-length LprG by immunoblot against a C-terminal FLAG epitope tag. Addition of pBpa to the culture medium did not significantly alter the expression level of LprG(WT) (Figure 2B). In contrast, for representative LprG(pBpa) mutants (L70, Q98, A136, E160, T232), full-length LprG was not detected or was present at significantly lower levels in the absence of pBpa, indicating a low level of read-through at the amber stop codon and robust nonsense suppression. Overall, LprG(pBpa) mutants expressed at lower levels than LprG(WT), consistent with the previously reported ~10% incorporation of p-iodophenylalanine into GFP using the same system28. These data show that nonsense suppression can be successfully applied to the site-specific incorporation of pBpa into a mycobacterial protein in Msm.
Incorporation of pBpa into Tyr codons occurs in the absence of stop codon mutation
In an initial experiment, pBpa treatment and UV irradiation of whole cells expressing LprG(WT) resulted in higher-molecular weight adducts, although no TAG codon was present in the LprG coding sequence (Figure 3). This result was unexpected and implied incorporation of pBpa into wild-type LprG even in the absence of nonsense suppression. We hypothesized that misincorporation of pBpa occurs at positions encoding Tyr when LprG(WT) is expressed at high levels and leads to detectable yields of photocrosslinking to LprG(WT). To test this hypothesis, we mutated the three native Tyr (Y115, Y125, Y132) in LprG to His. Single His mutants (Y115H, Y125H, Y132H) yielded similar crosslinking patterns to that of LprG(WT) and indicated that crosslinking is likely due to pBpa incorporation at more than one position. Correspondingly, the double mutant Y115/125H (Y2xH) showed significantly less crosslinking and the triple mutant Y115/125/132H (Y3xH) showed little to no crosslinking (Figure 3). Thus, photo-induced crosslinking to LprG(WT) when highly overexpressed in the presence of pBpa is due to misincorporation at all three native Tyr positions.
Figure 3. pBpa is incorporated into native Tyr codons in LprG.

Msm expressing Tyr to His mutants of FLAG-tagged LprG were grown with 2 mM pBpa and UV-irradiated for 0 or 30 minutes. Arrowheads indicate examples of prominent UV-dependent higher molecular-weight adducts. Total lysates were probed for LprG by α-FLAG immunoblot. Sample loads were normalized to LprG (expected M.W. 24.4 kDa).
Optimization of conditions for site-specific crosslinking and application to the mapping of LprG interaction interfaces
To optimize UV irradiation time for maximal crosslinking yield, Msm expressing LprG(98pBpa) or LprG(Y3xH) were subjected to 0, 5, 10, 20 or 30 min of UV exposure. The intensity of UV-dependent higher-molecular weight bands increased with irradiation time (Figure 4). The majority of high-yield, exposure time-dependent adducts were unique to LprG(98pBpa), consistent with the site-specific incorporation of pBpa via nonsense suppression. We note that a reproducible UV-dependent band at ~50 kDa was observed in crosslinking experiments with LprG WT, Y3xH and all pBpa mutants, although not in all replicates (Figure 3, 4, 5). This signal appeared in the LprG(Y3xH) mutant despite the mutation of all Tyr residues and suggested pBpa-independent crosslinking or incorporation at non-Tyr residues. The origin of this band was therefore not readily apparent, but because it was ubiquitous to all forms of LprG it did not affect the comparison of crosslinking patterns between the wild type and pBpa mutants that formed the basis of our interpretations below. At time points beyond 10 min, the higher-molecular weight bands intensified, but additional adducts were not detected, suggesting that UV exposure up to 30 min increased the yield of site-specific crosslinks, but did not introduce non-specific crosslinks unrelated to pBpa reactivity (Figure 4).
Figure 4. Optimization of irradiation conditions for site-specific crosslinking.

Msm expressing LprG(Y3xH) or LprG(98pBpa) were grown with 2 mM pBpa and live cells were UV-irradiated for 0, 5, 10, 20 or 30 min. Total lysates were probed for FLAG-tagged LprG by SDS-PAGE and immunoblot.
Figure 5.

Photocrosslinking of mutants containing pBpa identifies multiple distinct LprG interaction interfaces by immunoblot analysis. Msm harboring the indicated LprG(pBpa) mutants of FLAG-tagged LprG were grown with pBpa, and UV-irradiated for 0 or 30 min. Total lysates were probed for LprG by α-FLAG immunoblot. Arrowheads indicate examples of prominent UV-dependent higher molecular-weight adducts. Mutants are grouped by their relative location within Msm LprG. Data are representative of at least 2 biological replicates.
Using an optimized crosslinking time of 30 min, Msm expressing LprG(pBpa) mutants was assayed qualitatively by comparing total lysates from cells grown in pBpa and subjected to either 0 or 30 min of UV irradiation (Figure 5). As expected from their buried positions, pBpa incorporation at sites within the lipid-binding pocket did not lead to significant protein crosslinking, as indicated by the absence of higher molecular-weight bands. In contrast, mutations at the entrance to the lipid-binding pocket or “mouth” yielded comparable patterns of numerous higher molecular-weight adducts. Most mutations on the α-helical and β-sheet surfaces of LprG did not yield significant reproducible adducts with LprG. Of the nine locations that were interrogated on these two surfaces, only pBpa substitution at L37 and K181 consistently yielded significant and reproducible adducts. The proximity of K181 to the pocket entrance (Figure 2A) and the similarity in crosslinking patterns across all mutants in this area suggests that the pocket entrance forms a single interaction interface within LprG. Similarly, the crosslinking patterns resulting from mutations near the N-terminus (D34, L37) and C-terminus (T232, A236) were similar within each group and distinct from each other, suggesting that these regions proximal to the LprG termini constitute additional interaction interfaces. Marked depletion of LprG due to crosslinking was not observed for all mutants that yielded higher molecular-weight adducts, possibly due to sample-to-sample variation in LprG(pBpa) expression. Variation in the intensity of the crosslinked adducts was also observed between replicates, but, importantly, the pattern of crosslinking for each mutant was consistent (data not shown). In summary, the majority of LprG, including the interior of the binding pocket and α-helical and β-sheet surfaces, does not engage in protein interactions captured by pBpa crosslinking. Rather, the patterns of site-specific crosslinking at a limited number of clustered sites indicate at least three unique interaction interfaces that are located near each LprG terminus and around the pocket entrance (Figure 2A).
Quantitative proteomic identification of diverse proteins that interact with LprG
In the Gram-negative lipoprotein transport system, LolA and LolB engage in functionally important interactions between their “mouth” regions, which are proximal to their putative ligand binding pockets. By analogy, we hypothesized that interactions between the “mouth” of LprG and other cell wall proteins modulate their functions. LprG(98pBpa) was chosen for proteomic analysis of protein interactors because this mutant had a consistently high yield of higher molecular-weight adducts. LprG(136pBpa), which did not lead to reproducible crosslinked adducts upon pBpa incorporation, was analyzed as a negative control.
To identify proteins crosslinked to LprG at these positions, we pursued quantitative proteomics by stable isotope labeling of amino acids in culture (SILAC). This approach increases both accuracy and sensitivity by removing instrument run-to-run variation and allowing quantitative analysis of spectral counts, but has not to our knowledge been applied to mycobacteria before. Because mycobacteria synthesize nitrogen-containing amino acids such as lysine and arginine and auxotrophs were not readily available, we took a non-specific approach and cultured Msm in Roisin medium in which isotopically labeled ammonium chloride is the sole nitrogen source and is incorporated into all amino acids (see Methods).
Hits were defined as proteins detected in the irradiated vs. non-irradiated control with a spectral count ratio >1 and at least 2 peptides per protein (Table S4). Using these initial cutoff criteria, 48 proteins were detected, 23 of which were found in at least 2 biological replicates (Table 1). Abundant cytosolic species such as ribosomal proteins were notably absent, underscoring the power of the quantitative stable isotope labeling approach to discriminate against non-specific contaminants and provide accurate hit identification. In general, the detection of known or predicted cell-wall and membrane proteins supported the identification of native interactions with LprG. For example, 10 annotated periplasmic binding proteins (2 with homologues in Mtb), which are typically associated with ABC transporters as part of polar solute import systems, were found (Table S4). Among the membrane protein hits were six related to respiration, including components of the electron transport chain (QcrA, QcrB, CtaC) and ATP synthase (AtpA, AtpD, AtpFH).
Table 1.
Quantitative proteomics identifies LprG protein interactions at the entrance to the ligand-binding pocket (position 98). Proteins listed in this table were detected with an isotope ratio ≥2, were identified in at least one technical replicate of both biological replicates and with the number of peptides ≥2.
| Msm mc2155 Locus | Mtb H37Rv Locus | Gene Name | Functional Category | Description |
|---|---|---|---|---|
| MSMEG_0643 | Cell Wall & Cell Processes | Putative ABC transporter, extracellular solute-binding protein | ||
| MSMEG_0835 | Rv0432 | sodC | Virulence, Detoxification, Adaptation | Periplasmic superoxide dismutase [Cu-Zn] |
| MSMEG_0913 | Rv0469 | umaA | Lipid Metabolism | Methoxy mycolic acid synthase |
| MSMEG_0965 | mspA | Cell Wall & Cell Processes | Porin | |
| MSMEG_2410 | Rv2969c | Cell Wall & Cell Processes | Putative conserved membrane or secreted serine-threonine protein kinase | |
| MSMEG_2727 | Cell Wall & Cell Processes | Periplasmic glutamate-binding periplasmic protein | ||
| MSMEG_3058 | Cell Wall & Cell Processes | Lipoprotein | ||
| MSMEG_3235 | Cell Wall & Cell Processes | Secreted component of ABC-type amino acid transport system | ||
| MSMEG_3618 | Rv1860 | apa | Cell Wall & Cell Processes | Alanine and proline-rich secreted protein, fibronectin-attachment family protein |
| MSMEG_3851 | Rv2046 | lppI | Cell Wall & Cell Processes | Probable lipoprotein |
| MSMEG_3903 | Rv2376c | cfp2 | Cell Wall & Cell Processes | Low molecular weight antigen |
| MSMEG_3904 | Rv2116 | lppK | Cell Wall & Cell Processes | Conserved lipoprotein |
| MSMEG_4217 | Rv2145c | wag31 | Cell Wall & Cell Processes | DivIVA family protein, cell wall synthesis protein |
| MSMEG_4262 | Rv2195 | qcrA | Intermediary Respiration & Metabolism | Ubiquinol-cytochrome c reductase iron-sulfur subunit |
| MSMEG_4326 | Rv2244 | acpM | Lipid Metabolism | Meromycolate extension acyl carrier protein |
| MSMEG_4533 | Rv2400c | subI | Cell Wall & Cell Processes | Probable sulfate-binding lipoprotein |
| MSMEG_4561 | Cell Wall & Cell Processes | ABC Fe3+-siderophores transporter; periplasmic binding protein | ||
| MSMEG_4692 | Rv2468c | Conserved hypotheticals | Conserved protein | |
| MSMEG_6049 | Secreted protein | |||
| MSMEG_6394 | Rv3802c | Cell Wall & Cell Processes | Probable conserved membrane cutinase | |
| MSMEG_6398 | Rv3804c | ag85A | Lipid Metabolism | Secreted antigen 85-a, mycolyl transferase 85A, fibronectin-binding protein A |
| MSMEG_6595 | Cell Wall & Cell Processes | Glutamine ABC transporter, glutamine-binding periplasmic protein | ||
| MSMEG_6759 | glpK | Glycerol 3-phosphotransferase |
We predicted that the integral membrane transporter Rv1410c interacts with LprG because lprG and rv1410c are co-transcribed and functionally related15. While membrane proteins are often difficult to identify by mass spectrometry due to low abundance, limited solubility and restricted access to proteolytic enzymes, Rv1410c was detected as a hit in one technical replicate (Table S4). Interestingly, five proteins with known or probable roles in cell wall biosynthesis, specifically of mycolates (Ag85A, Rv3802c, UmaA, Wag31, AcpM); three predicted lipoproteins (including LppI and LppK); and the Msm outer membrane porin MspA were detected in both biological replicates with a ratio >2 (Table 1). Crucially, no hits were identified crosslinking to the negative control LprG(136pBpa), which reinforces the specificity and thus the probable physiological relevance of the crosslinking interactions identified with LprG(98pBpa).
LprG interacts site-specifically with itself, LppI and LppK
The transport of both lipopolysaccharide and lipoproteins to the OM involves protein self-association and/or the interaction of structurally homologous proteins16–17. By analogy, we hypothesized that LprG interacts with itself. Because LprG was used as bait in immunoprecipitation prior to proteomics, LprG was the dominant protein in these samples and the resulting data did not report reliably on the abundance of LprG in one isotopically labeled sample condition compared to another. Therefore, we pursued co-expression and affinity enrichment as an independent means of confirming LprG interactions.
To detect the association of LprG with individual proteins in Msm, we co-expressed LprG(pBpa) mutants with His-tagged putative interactors, starting with LprG(WT). Following pBpa treatment and UV irradiation of live cells as above, total lysates were subjected to nickel-affinity enrichment for His-tagged LprG(WT) and probed for FLAG-tagged LprG(pBpa). Immunoblot analysis showed that a UV-dependent band consistent with an LprG dimer at ~50 kDa was detected for the LprG(98pBpa) mutant, but not for LprG(232pBpa) or LprG(136pBpa), in which the crosslinker is near the C-terminus or within the α-helical face (Figure 6). These results validated the site-specificity of LprG self-association.
Figure 6. LprG interacts site-specifically with itself, LppI and LppK.

Msm co-expressing FLAG-tagged LprG(pBpa) mutants and His-tagged putative interactors LprG, LppI or LppK were grown with 2 mM pBpa and subjected to UV irradiation. Ni-NTA enriched cell wall fractions were probed by SDS-PAGE and immunoblot. Arrowheads indicate examples of prominent UV-dependent higher molecular-weight adducts. Each His-tagged interactor was used as a corresponding loading control. Expected M.W. for His-tagged proteins: 25.3 kDa, 22.7 kDa and 20.2 kDa for LprG, LppI and LppK respectively. Data are representative of at least 2 biological replicates.
We used the same co-expression system further to verify two proteins identified as LprG interactors by proteomics. Of the putative interactors that met our cutoff criteria (Table 1), we prioritized the predicted lipoproteins LppK and LppI as the two most robust hits that are also conserved in Mtb (Table S4). To determine the site-specificity of these interactions, we assayed for crosslinking to LprG at positions 98, 232, and 136 as described above. LprG interaction with LppI was confirmed by an LprG-associated band at ~50 kDa in the α-FLAG immunoblot, consistent with a covalent adduct between these two proteins (Figure 6). (We note that the migrations of LppI and LppK are higher than the predicted M.W. for each, perhaps due to their acyl modifications.) This adduct was detected when LprG(98pBpa) but not LprG(232pBpa) or LprG(136pBpa) was co-expressed with LppI. Using the identical approach, we confirmed that LppK also interacts with LprG. In contrast with LppI, however, a band consistent with the predicted size of the LprG-LppK adduct was detected with both LprG(98pBpa) and LprG(232pBpa), but not LprG(136pBpa). In addition, for LprG(232pBpa), an additional ~75 kDa adduct was observed that is consistent with crosslinking to an additional LprG(pBpa) monomer. In summary, while LppI interacts with LprG specifically near the entrance to the ligand-binding pocket, LppK either has multiple modes of binding to LprG, or has a larger interaction interface that encompasses not only the mouth, but also the C-terminal tail of LprG.
Msm lacking LprG or Ag85A function is attenuated for growth under carbon restriction and has mycolylation defects
We hypothesized that protein interactions with LprG have functional consequences. We tested this hypothesis by targeting the LprG interactor Ag85A, a mycolyltransferase in the cell wall. Ag85A is one of three enzymes in the Antigen 85ABC complex, which is responsible for transferring mycolate from TMM donor to either arabinogalactan or another molecule of TMM to form trehalose dimycolate30. We first verified the interaction between LprG(98pBpa) and Ag85A co-expression, crosslinking and affinity enrichment as performed above for LppI and LppK (Figure 7A). A band with the appropriate size for the LprG-Ag85A adduct (predicted M.W. 58.6 kDa) was detected. Interestingly, there was an additional band between 75 and 100 kDa that is consistent with an LprG-Ag85A heterotrimer containing two LprG(pBpa) molecules (predicted M.W. 83.0 kDa).
Figure 7. LprG interacts physically and functionally with the mycolyltransferase Ag85A.

a) Msm co-expressing FLAG-tagged LprG(98pBpa) and His-tagged Ag85A were grown with 2 mM pBpa and subjected to UV irradiation. Cell envelope-enriched fractions were subjected to nickel-affinity enrichment and probed for full-length LprG by SDS-PAGE and immunoblot. Arrowheads indicate UV-dependent higher molecular-weight adducts. Expected M.W. for Ag85A is 34.2 kDa. Data are representative of two independent experiments. b) Wild-type and mutant Msm were cultured on either glycerol or propionate as a primary carbon source and growth was monitored by OD595. Data shown are the mean ± S.D. for 3 independent replicates per group. c) Msm lipid extracts were analyzed by thin-layer chromatography for TDM and TMM. Lane 1) wild type, 2) ΔlprG, 3) ΔlprG::lprG, 4) ΔlprG-rv1410c::rv1410c(Mtb), 5) ΔlprG-rv1410c::lprG-rv1410c(Mtb), 6) ag85A::Tn; 7) TDM standard. TDM and TMM as a percent of the total lipid and the TMM/TDM ratio are shown for each lane. Mobile phase was 80:20:2 chloroform:methanol:water. d) Mean fluorescence intensity (MFI) by flow cytometry for Msm treated with alkynyl-TMM analogues and subsequently chemically coupled to carboxyrhodamine 110 azide. For the mutant strains, “null” and “comp” designate the corresponding gene-disrupted and complemented strains. For lprG, strains used were based on ΔlprG-rv1410c, as described in c). Data shown are the mean ± S.D. for 3 independent replicates per group. * p < 0.005 vs. wild type by multiple t-tests.
We further predicted that loss of function in Ag85A would phenocopy the growth defect of ΔlprG under carbon restriction, as reported previously for a null mutant of lprG in Mtb15. Indeed, both the deletion strain ΔlprG and the transposon mutant Msm ag85A::Tn23 were attenuated for log-phase growth in defined medium containing propionate as the primary carbon source (Figure 7B). There was also less severe but reproducible attenuation with glycerol. As deletion of lprG can disrupt transcription of its operonic partner rv141031, we verified that ΔlprG-rv1410c mutant complemented with Mtb rv1410c retains the growth phenotype (Figure S2)15, 32. Growth on either carbon source was restored by in both strains by complementing with a single copy of lprG (Figure S2).
To determine whether loss of LprG function affects the mycolyltransferase activity of Ag85A, total lipid extracts were analyzed by thin-layer chromatography for TDM and TMM content. As expected, TMM accumulated in the ag85A::Tn mutant and the TMM/TDM ratio was elevated compared to the wild type (Figure 7C). In contrast to previous reports23, 33, however, TDM as a percent of the total lipid was not reduced. No significant difference in the TMM/TDM ratio or the TDM or TMM content was observed for ΔlprG or ΔlprG-rv1410c::rv1410c(Mtb). These results were not specific to growth on defined medium, as similar results were obtained in 7H9 medium (Figure S3).
To further investigate mycolate biosynthesis, we assayed directly for mycolate incorporation using TMM analogues. N-AlkTMM and O-AlkTMM are incorporated into TDM or primarily cell wall mycolates, respectively, by the activity of the Ag85 complex34. We treated Msm with either N-AlkTMM or O-AlkTMM and chemically coupled the resulting alkyne-bearing bacteria to a fluorescent dye for flow cytometry analysis. Surprisingly, significantly elevated N-AlkTMM incorporation was detected in ag85A::Tn and trended higher in the LprG null mutant compared to the wild type (Figure 7D). On the other hand, a significant reduction in O-AlkTMM incorporation was observed for both mutant strains. Both the N-AlkTMM and O-AlkTMM phenotypes could be complemented. These results suggest that the two null strains are similarly perturbed for the incorporation of cell wall mycolates, but that in LprG null mutants, this occurs without the accumulation of TMM. Overall, the comparable changes in growth and AlkTMM incorporation strongly support a role for LprG in modulating Ag85A activity.
DISCUSSION
We defined several distinct protein interaction interfaces with LprG at the mouth and proximal to the N- and C-termini. After identifying proteins that crosslink near the mouth, we showed that LprG interacts with itself and validated several hits for the site-specificity of their interactions. Overall, the dependence of crosslinking on native cell wall localization and the position of the pBpa crosslinker strongly supports the detection of physiological interactions by this approach. The identification of many known or predicted cell envelope proteins, with very few abundant cytosolic proteins, also supports the robustness and accuracy of hit identification afforded by our quantitative proteomics methods. We identified dozens of proteins that crosslink with LprG(98pBpa), which suggests that numerous interactors compete at the mouth of LprG. Indeed, the involvement of LprG in TAG and LAM localization and our new results on LprG regulation of mycolylation suggest that LprG is part of diverse protein complexes that function in different pathways. How the formation of these complexes is regulated remains to be determined.
As previously noted28, the incidence of TAG stop codons is significantly higher in Mtb (and also in Msm) than in E. coli35. Wang et al. maintained that suppression by the MjtRNA/aminoacyl-tRNA synthetase pair is poor and most translated proteins would be terminated normally. Indeed, they reported normal growth of Mtb under nonsense suppression by para-iodophenylalanine. In contrast we observed significant growth attenuation in Msm expressing the MjtRNA/aaRS pair in the presence of pBpa (doubling time of ~6 vs. ~4h in Sauton medium). This might suggest that the detected protein interactions are induced by nonsense suppression. However, our results on the influence of LprG on Ag85A function indicate that the interaction is not specific to the conditions of pBpa incorporation.
Among high-confidence hits are proteins known or likely to be involved in cell wall biosynthesis, including Ag85A23, 30, the meromycolate acyl carrier protein AcpM36, the cell wall synthesis scaffolding protein Wag3121–22, the putative methoxy mycolic acid synthase UmaA37, and the putative lipase Rv3802c38. While Ag85A is a known cell wall protein, UmaA, Rv3802c, AcpM and Wag31 all lack predicted secretion signals and have therefore been assumed to be confined to the cytosol. However, previous proteomics studies39–43 on mycobacterial cell wall or membrane fractions have identified numerous proteins without secretion signals, including the four found crosslinked to LprG in our study. Given the known localization of LprG to the cell wall24, our results provide additional strong support for the export of these proteins to this compartment, especially since our methods do not involve fractionation or detergent extraction approaches that are subject to contamination by cytosolic proteins.
As noted earlier, Ag85A has been implicated as primarily a TMM acyltransferase based on a reduction in TDM and no change in AGM in ag85A-disrupted strains33, 44. However, we observed the opposite phenotype for ag85A::Tn: Although TMM accumulated, TDM levels were not reduced, but TMM incorporation into AGM was compromised. The discrepancy in phenotypes could be due to kinetic effects. Nguyen et al. measured AGM as the total mycolic acid methyl esters (MAMEs) at steady-state, whereas we detected O-AlkTMM incorporation after 4 hours. Kinetics may also underlie the difference in TDM phenotypes. Nguyen et al. and Ojha et al. labeled TDM with 14C-acetate or 14C-glucose for 2–6 h, similar to our labeling with N-AlkTMM for 4 hours. However, N-AlkTMM is incorporated directly into TDM in the cell wall, whereas acetate and glucose may take longer to be transformed into TDM. Our TLC results support the idea that while TDM synthesis may be slower in the absence of Ag85A, TDM at steady-state is present at a similar level as it is in the wild type. Overall, the relative TDM vs. AGM mycolyltransferase activities of Ag85A clearly remain to be resolved. Regardless, our data show that loss of either LprG or Ag85A disrupts mycolate formation compared to the wild type and supports a role for LprG in Ag85A function. In one possible model, LprG binds to the highly hydrophobic mycolate of TMM and thereby facilitates Ag85A access to its substrate. While the cavity of LprG is too small to accommodate the C60–C90 mycolate chain, two LprG monomers may be sufficient. Our detection of LprG dimerization and an adduct consistent with an LprG2Ag85A heterotrimer provide preliminary support for this mechanism.
In summary, we applied the site-specific incorporation of a UV-crosslinking unnatural amino acid to the mapping of protein interaction interfaces in LprG. This approach also enabled us to identify the interacting proteins. We expect UAAI and the quantitative proteomics methods to be broadly useful and important tools in the study of mycobacteria. As for the implications for our understanding of LprG function and cell wall biogenesis, we provide evidence that LprG affects mycolylation by the mycolyltransferase Ag85A. Cell growth and lipid biosynthesis have been previously linked via the physical association of Wag31 with fatty acid and lipid biosynthetic enzymes (FASII, AccA3, AccD5), and the inner membrane mycolate transporter MmpL320, 22, 45. Also, enzymes in the phosphatidyl inositol mannoside lipid and phosphatidic acid biosynthetic pathways co-localize at the cell poles, pointing to analogous coordination of inner membrane synthesis46. Our results with LprG support these models and provide novel insights into the composition of protein complexes that we suggest are integral to the regulation of OM maintenance. The coordination of multiple OM assembly pathways is an exciting area for future exploration inspired by these results.
METHODS
Bacterial strains, growth media, growth conditions, vector construction and oligonucleotide primers
M. smegmatis str. mc2155 (ATCC 700084) (Msm) was grown in Middlebrook 7H9 (liquid), Middlebrook 7H11 (solid), Sauton medium47 or Roisin medium48. In addition, modified Sauton medium15 containing 0.2% (v/v) glycerol or 10 mM sodium propionate was used for some assays as specified below. Middlebrook 7H9 was supplemented with 0.2% casamino acids (BD), 0.5% (v/v) glycerol, 0.05% (w/v) glucose and 0.05% (v/v) Tween80. For selective media, antibiotic concentrations were as follows: 25 μg/mL hygromycin and 12.5 μg/mL kanamycin for strains harboring pMV361- and pSMT-derived plasmids or 25 μg/mL hygromycin for strains containing only pSMT-derived plasmids. Note that Msm harboring both plasmids has a ~6-hour doubling time in Sauton medium. The Msm ΔlprG null mutant was constructed by recombineering (see Supporting Information)49. Msm ag85A::Tn (also known as Tn:fbpA) was a kind gift of Charles Thompson44. Msm ΔlprG-rv1410c::rv1410c(Mtb) and ΔlprG-rv1410c::rv1410c(Mtb) were the kind gift of Eric Rubin15, 32. See Tables S1–S3 for vector constructs, oligonucleotide primer sequences and cloning strategies used in this study. All cultures were incubated at 37 °C with shaking at 225 rpm.
Protein expression and chemical crosslinking
Msm strains expressing C-terminally 6xHis-tagged LprG or NA-LprG (see Table S5) were grown in 100 mL per experimental condition to an OD600 ~1. Cells were pelleted at 3000g for 10 min and washed twice with an equal volume of PBS with 0.05% Tween 80 (PBS-Tw80). The final cell pellet was resuspended in 10 mL PBS-Tw80. Dithiobis-(succinimidylpropionate) (DSP; Thermo Fisher) was added to a 1 mM final concentration from a 100 mM stock in DMSO. An equal volume of DMSO was added to negative control samples. Cell suspensions were incubated at 22 °C for 30 min with gentle rotation. Each sample was quenched with 0.5 mL 1 M Tris pH 7.5 and incubated for 15 min. Cells were harvested by centrifugation as above and the decanted cell pellet stored at −20 °C until further processing. Thawed cell pellets were resuspended in 1 mL PBS. Cells were lysed at 4 °C in 2 mL screwcap tubes containing 0.1 mm silica beads (Spectrum Chemical Manufacturing) at 5.0 m/s with 1 min on/off for 2 min total processing time (FastPrep-24 beat beater; MP Biomedical). Beads were allowed to settle and the lysate was removed. Beads were washed twice with 1 mL PBS and the washes combined with the initial lysate. SDS was added to a 1% final concentration from a 10% stock and the lysate incubated with gentle rotation at 22 °C for 30 min. Cell debris was pelleted at 5000g for 15 min. The supernatant was removed and incubated with 150 μL Ni-NTA agarose (Qiagen) with gentle rotation at 22 °C for 30 min. Resin was pelleted at 3000g for 2 min and the supernatant removed. The resin was washed 5 times with 1 mL PBS each. Enriched proteins were eluted 3 times, each with 100 μL PBS with 250 mM imidazole. A 95-uL aliquot was removed incubated with 5 μL 1 M DTT to reduce disulfide bonds. All samples were combined with non-reducing loading dye prior to analysis as described in “Immunoblot Analysis.”
Protein expression and photocrosslinking
A 10-mL culture of Msm harboring pSMT-pHsp60-MjtRNA-FLAG and pMV361-pHsp60-MjTyrRS was cultured in selective Sauton medium for 15 hours overnight to an optical density at 600 nm (OD600) ~1.5. Cells were pelleted and resuspended in Sauton medium supplemented with 25 μg/mL hygromycin only and used to inoculate 30 mL of the same medium at an OD600 ~0.05. Cultures were incubated for a further 9 hours. Filter-sterilized 100 mM pBpa dissolved in 1:5 1 N NaOH:water was added to the cells at OD600 0.2 to a final concentration of 2 mM pBpa and incubated in the dark for another 15 hours. Control cells were treated with an equivalent volume of vehicle. At OD600 1.0 – 1.5, cells were harvested at 5,000 × g for 25 min and resuspended in PBS at 1/10th the original culture volume. For crosslinking, cells were irradiated with 365-nm UV light for 30 min on ice with stirring every 5 min. Negative control cells were not exposed to UV irradiation, shielded from ambient light, and incubated on ice throughout. All cell suspensions were then pelleted at 4,000 × g for 10 min and stored at 20 °C until further processing.
Protein expression, photocrosslinking, cell fractionation and immunoprecipitation for proteomic analysis
The protocol was the same as described above in the “Protein expression and photocrosslinking” section except culture volumes were 100 mL for the initial culture and 2 L for the subculture. For stable isotope labeling in culture Msm was grown in Roisin medium containing either 14NH4Cl or 15NH4Cl (Cambridge Isotope Laboratories) as the sole nitrogen source. To control for isotope-dependent effects on protein levels, the isotopic labeling was reversed in a second biological replicate. Live cells were UV-irradiated for 0 or 30 min and the labeled and unlabeled cells were mixed prior to pelleting and storage as described above.
Cell pellets were thawed on ice and resuspended in PBS at 1/40th the original culture volume. Protease inhibitor was added at 1X from a 100X stock [17 g/L phenylmethanesulfonylfluoride (PMSF, Sigma-Aldrich), 33 g/L benzamidine hydrochloride (Sigma-Aldrich), 0.137 g/L pepstatin A (MP Biomedicals), 0.03 g/L leupeptin (Roche), 0.2 g/L chymostatin (Sigma-Aldrich) in ethanol]. Cells were lysed at 4 °C in 2-mL screw cap tubes containing 0.1 mm silica beads (Spectrum Chemical Manufacturing) at 5.0 m/s with 1 min on/off for 8 min total processing time (FastPrep-24 beat beater; MP Biomedical). Cell debris was pelleted by centrifuging at 5,000 × g for 10 min and the cleared lysate was transferred to 4 mL ultracentrifuge tubes (Fisher). The cytosol-enriched fraction was separated from the cell envelope (CE)-enriched fraction by ultracentrifugation at 100,000 × g for 1 hour. The CE-enriched fraction was solubilized in 8 mL 1% N-dodecyl-β-D-maltoside (DDM; Anatrace) in PBS at 4 °C for 15 hours overnight with gentle agitation. The detergent-solubilized CE was subjected to ultracentrifugation at 100,000 × g for 1 hour to remove insoluble debris. The supernatant was transferred to a 15-mL conical tube containing 0.5 mL equilibrated in buffer A (300 mM NaCl, 1x protease inhibitor, 0.02% NaN3, 1% DDM in PBS, pH 7.4) anti-FLAG M2 Affinity Gel (Sigma-Aldrich) resin and incubated on a rotator at 25 °C for 2 hours. The resin was pelleted at 1,000 × g for 5 min and the supernatant was removed. The resin was washed with 60 column volumes (6 × 5 mL buffer A) by pelleting the resin at 1,000 × g for 5 min between wash steps. The resin was resuspended in a final volume of 1 mL buffer A and transferred to a fresh 5-mL tube. Residual resin was recovered with 3 × 0.5 mL buffer A and pooled. The resin was pelleted at 1,000 × g for 5 min, supernatant removed, and 500 μL of 300 μg/mL FLAG peptide (Sigma-Aldrich) in buffer A was added to elute the α-FLAG-enriched species from the resin. The elution slurry was incubated for 1 hour at 25 °C on a rotator before the resin was pelleted at 1,000 × g for 5 min and the eluent removed. A second 500 μL elution fraction was collected in the same manner, but without the 1 hour incubation step.
Proteomic sample prep and analysis
For proteomic analysis each sample comprised a 1:1 mixture of α-FLAG-immunoprecipitated LprG from pBpa-treated, UV-irradiated Msm and a corresponding immunoprecipitated, non-irradiated negative control. Samples were reduced with 4 mM DTT for 30 min at 25 °C and alkylated in 8.4 mM iodoacetamide for 30 min at 25 °C in the dark. The solution was diluted to 1.7 M urea with 50 mM (NH4)HCO3 and 1 μg trypsin in 25 mM (NH4)HCO3 (Trypsin Gold, Mass Spectrometry Grade, Promega, USA) was added. The digestion mixture was incubated at 37 °C for approximately 18 hours.
The digests were brought to 2% formic acid (FA) and the volume reduced by vacuum centrifugation to a final concentration of about 4 M urea. This solution was desalted with Supel-Tips C18 Micropipette Tips (Sigma-Aldrich) using FA-containing solutions with varied acetonitrile (ACN), essentially as described in vendor’s bulletin. The eluted peptides were dried by vacuum centrifugation and dissolved in 2% ACN, 0.1% FA (buffer MS1) for analysis by automated microcapillary liquid chromatography-tandem mass spectrometry. Fused-silica capillaries (100 μm inner diameter) were pulled using a P-2000 CO2 laser puller (Sutter Instruments, Novato, CA) to a 5 μm inner diameter tip and packed with 10 cm of 5 μm ProntoSil 120-5-C18H (Bischoff Chromatography, Leonberg, Germany) using a pressure bomb.
This column was then placed in-line with a Dionex Ultimate 3000 equipped with an autosampler. The column was equilibrated in buffer MS1, and the peptide mixture was loaded onto the column using the autosampler at a flow rate of 2 μl/min. The HPLC pump flowed at 100 μL/min and the flow rate to the electrospray tip was reduced to ~ 200–300 nL/min by a split. The HPLC separation was provided by a gradient between Buffer MS1, Buffer MS2 (98% ACN, 0.1% FA) and Buffer MS3 (50% ACN, 0.1% FA). The HPLC gradient was held constant at 100% buffer MS1 for 5 min after peptide loading followed by an 80 min gradient from 100% buffer MS1 to 100% buffer MS3. Then, there was a 1 min transition as the gradient was switched to 51% buffer MS2 and this was followed by a 34 min gradient to 80% buffer MS2 and then held constant for 3 min. Finally, the gradient was changed from 80% buffer MS2 to 100% buffer MS1 over 2 min and then held constant at 100% buffer MS1 for 40 more min. The application of a 1.8 kV distal voltage electrosprayed the eluted peptides directly into a Thermo Fisher Scientific LTQ XL ion trap mass spectrometer equipped with a nanoLC electrospray ionization source (ThermoFinningan, San Jose, CA). Full mass spectra were recorded on the peptides over a 400–2000 m/z range, followed by five tandem mass (MS/MS) events sequentially generated in a data-dependent manner on the first, second, third, fourth and fifth most intense ions selected from the full MS spectrum (at 35% collision energy). Mass spectrometer scan functions and HPLC solvent gradients were controlled by the Xcalibur data system (ThermoFinnigan, San Jose, CA).
MS/MS spectra were extracted from the RAW file with ReAdW.exe (http://sourceforge.net/projects/sashimi). The resulting mzXML data files were searched using The GPM X!Tandem search engine (http://www.thegpm.org/tandem/) against the Uniprot Mycobacterium smegmatis (strain ATCC 700084/mc2155) proteome (dated 12/08/2014) with added sequences for common contaminants. Stable isotope ratios were quantified using in-house CIMAGE software to compare the intensity ratios of the MS1 chromatograms of heavy and light peptides50.
Interactor enrichment for crosslinking validation
After cell culture, crosslinking, and lysis as described above in “Protein expression and photocrosslinking”, the cell envelope (CE)-enriched fraction was pelleted by ultracentrifugation at 100,000 × g for 1 hour. The CE-enriched fraction was solubilized in 250 μL PBS with 1% N-dodecyl-β-D-maltoside (DDM; Anatrace) at 4 °C for 15 hours overnight with gentle agitation. The detergent-solubilized CE was subjected to ultracentrifugation at 100,000 × g for 1 hour to remove insoluble debris. The supernatant was transferred to a 1.5-ml microcentrifuge tube containing 30 μL Ni-NTA resin (Qiagen) pre-equilibrated in PBS with 1 % DDM and incubated at 25 °C with rotation for 30 min. The resin was pelleted at 4,000 × g for 1 min and the supernatant was removed. The resin was washed with 2 × 300 μL 1% DDM PBS by pelleting the resin at 4,000 × g for 1 min between washes. After the second wash, 60 μL of PBS with 1% DMM and 300 mM imidazole (Acros Organics) was added to elute. The resin was pelleted at 1,000 × g for 5 min and the eluent removed. The elution step was repeated and the eluents pooled. Enriched fractions were then probed by immunoblot as described below
Immunoblot analysis
For reducing SDS-PAGE, 30 μg protein/well was loaded for whole-cell lysate samples and 2 μg protein/well for NiNTA-enriched samples. Protein was transferred to nitrocellulose membranes (0.45 μm, Bio-Rad), blocked and probed with mouse α-FLAG (1:1,000; Sigma Aldrich, clone M2, product #F1804); rabbit α-6xHis HRP (1:10,000; Abcam, product #ab1187); or mouse α-GroEL (1:5,000; Abcam, product #ab20519) for 1 hour with gentle agitation at 25 °C. After washing, membranes were probed with secondary antibodies goat α-mouse IgG HRP (1:10,000; Abcam, product # ab6789) or goat α-rabbit IgG HRP (1:10,000; Abcam, product # ab6721). Immunoblots were visualized by chemiluminescence (Immobilon Western Chemiluminescent HRP Substrate; Millipore).
Growth of Msm strains in defined medium
Starter cultures in 2 mL modified Sauton medium with 0.2% (v/v) glycerol were grown to mid-logarithmic phase. The cultures were then diluted to an OD595 of 0.05 in 3 ml modified Sauton medium with 0.2% bglycerol or 10 mM propionate to initiate the growth assay. Growth was monitored by OD595 using a F5 Filtermax plate reader (Molecular Devices).
Lipid extraction and analysis
Msm was inoculated in 50 mL modified Sauton medium with 10 mM sodium propionate at OD600 ~0.04 and grown for 18 h. Cells were centrifuged at 3000 × g for 10 min at 22 °C. The cell pellets were each extracted in 4 mL 1:1 chloroform:methanol for 20 h at 22 °C. Cell debris was removed by centrifugation and the clarified lipid extracts were dried under a stream of nitrogen gas. The final residue was resolubilized at a concentration of 50 μL 1:1 chloroform:methanol per 9 OD600-mL. Samples were analyzed by thin-layer chromatography in 80:20:2 chloroform:methanol:water and visualized by staining with 5% sulfuric acid in ethanol followed by heating at 130 °C for 5 min. TDM standard was obtained from BEI Resources. Lipid quantification was performed using ImageJ.
Labeling of Msm whole cells by AlkTMM analogues
N-(6-heptynoyl)-6-amino-6-deoxy-α,α-D-trehalose (N-AlkTMM) and 6-O-(6-heptynoyl)-α,α-D-trehalose (O-AlkTMM) were the kind give of Dr. Benjamin Swartz. Incorporation of N-AlkTMM and O-AlkTMM into Msm was performed largely as reported34. Briefly, Msm was grown in 3 mL modified Sauton medium with 10 mM propionate to mid-logarithmic phase. Cultures were diluted to OD600 0.2 in 2 ml modified Sauton medium with 10 mM propionate and 100 μm N-AlkTMM, 50 μm O-AlkTMM, or no probe and incubated for an additional 4 hours. Cells were pelleted at 5,000 × g for 10 min at 22 °C and washed three times with an equivalent volume of PBS containing 0.5% bovine serum albumin (PBSB). Cells were fixed in 500 μL 4% paraformaldehyde in PBS for 10 minutes and washed three more times with 500 μL PBSB each with a final resuspension in 552 μl PBS. The cell suspension was subjected to copper(I)-catalyzed alkyne-azide cycloaddition (CuACC) by sequential addition of 12 μL 1 mM carboxyrhodamine 110 azide (Click Chemistry Tools), 12 μL 60 mM sodium ascorbate, 2.5 μL 30 mM TBTA, and 12 μL 50 mM CuSO4 to give a final reaction volume of 600 μL. After thorough mixing, reactions were incubated in the dark at 22 °C for 30 min. Cells were pelleted and washed three times with an equivalent volume of PBS followed by a final resuspension in PBS. Flow cytometry was carried out on a BD FACScan. For each sample 10,000 events were recorded at an event rate of 500–1,000 events/sec and processed using Cyflogic software (CyFlo Ltd). Events were gated to exclude aggregates from the analysis.
Supplementary Material
Acknowledgments
We thank David Thanassi, Nadine Henderson and members of the Seeliger Lab for their helpful discussions, Hiren V. Patel for assistance with statistical analysis, and Temitope O. Nathan for synthesizing the TMM analogues. This work was supported by a SUNY Health Network of Excellence Planning Grant and a Stony Wold-Herbert Fund Grant-In Aid (J.C.S.); NIH/NIGMS F32 GM108208 (K.M.B); and NIH/NCRR 1 S10 RR023680-1 (Proteomics Center at Stony Brook University).
ABBREVIATIONS USED
- OM
outer membrane
- Mtb
Mycobacterium tuberculosis
- PDIM
phthiocerol dimycocerosate
- TDM
trehalose dimycolate
- LAM
lipoarabinomannan
- TMM
trehalose monomycolate
- AGM
arabinogalactan-linked mycolate
- DSP
dithiobis(succinimidylpropionate)
- DTT
dithiothreitol
- SILAC
stable isotope labeling of amino acids in culture
- pBpa
p-benzoylphenylalanine
- UAAI
unnatural amino acid incorporation
- Msm
Mycobacterium smegmatis
- aaRS
amino-acyl-tRNA synthetase
- Mj
Methanocaldoccus janaschii
- CuACC
copper(I)-catalyzed alkyne-azide cycloaddition
- N-AlkTMM
N-(6-heptynoyl)-6-amino-6-deoxy-α,α-D-trehalose
- O-AlkTMM
6-O-(6-heptynoyl)-α,α-D-trehalose
Footnotes
Author Contributions
M.H.T., E.R.V.V., L.B., J.K. and J.C.S. designed the experiments. M.H.T., E.R.V.V., performed the biochemical experiments. M.L.P. and E.R.V.V. performed the molecular cloning. J.K. performed the growth assays. E.R.V.V. performed the whole-cell labeling experiments. L.B. performed lipid analysis. K.M.B. established and shared the isotopic labeling conditions for proteomics. D.W.M., A.B.C. and B.F.C. performed the mass spectrometry and quantitative proteomics analysis. M.H.T., E.R.V.V., and J.C.S. analyzed the data and wrote the manuscript.
CONFLICT OF INTEREST
None to declare.
Materials and methods for GFP fluorescence measurements and cloning; validation of UAAI in Msm; complemented growth attenuation of ΔlprG; accumulation of TMM without loss of TDM in ag85A::Tn independent of growth medium; UAAI expression constructs and oligonucleotide primers; constructs and oligonucleotide primers used for the production of ΔlprG and ΔlprG:lprG; LprG(98pBpa) proteomic data. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Neyrolles O, Guilhot C. Recent advances in deciphering the contribution of Mycobacterium tuberculosis lipids to pathogenesis. Tuberculosis (Edinb) 2011;91(3):187–95. doi: 10.1016/j.tube.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 2.Kirksey MA, Tischler AD, Siméone R, Hisert KB, Uplekar S, Guilhot C, McKinney JD. Spontaneous Phthiocerol Dimycocerosate-Deficient Variants of Mycobacterium tuberculosis Are Susceptible to Gamma Interferon-Mediated Immunity. Infect Immun. 2011;79(7):2829–2838. doi: 10.1128/iai.00097-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Day TA, Mittler JE, Nixon MR, Thompson C, Miner MD, Hickey MJ, Liao RP, Pang J, Shayakhmetov D, Sherman DR. Mycobacterium tuberculosis strains lacking the surface lipid phthiocerol dimycocerosate are susceptible to killing by an early innate host response. Infect Immun. 2014 doi: 10.1128/IAI.01340-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox JS, Chen B, McNeil M, Jacobs WR., Jr Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. 1999;402(6757):79–83. doi: 10.1038/47042. [DOI] [PubMed] [Google Scholar]
- 5.Sulzenbacher G, Canaan S, Bordat Y, Neyrolles O, Stadthagen G, Roig-Zamboni V, Rauzier J, Maurin D, Laval F, Daffé M, Cambillau C, Gicquel B, Bourne Y, Jackson M. LppX is a lipoprotein required for the translocation of phthiocerol dimycocerosates to the surface of Mycobacterium tuberculosis. EMBO J. 2006;25(7):1436–1444. doi: 10.1038/sj.emboj.7601048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Domenech P, Reed MB, Barry CE., 3rd Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance. Infect Immun. 2005;73(6):3492–501. doi: 10.1128/IAI.73.6.3492-3501.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tahlan K, Wilson R, Kastrinsky DB, Arora K, Nair V, Fischer E, Barnes SW, Walker JR, Alland D, Barry CE, 3rd, Boshoff HI. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2012;56(4):1797–809. doi: 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Varela C, Rittmann D, Singh A, Krumbach K, Bhatt K, Eggeling L, Besra Gurdyal S, Bhatt A. MmpL Genes Are Associated with Mycolic Acid Metabolism in Mycobacteria and Corynebacteria. Chemistry & Biology. 2012;19(4):498–506. doi: 10.1016/j.chembiol.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grzegorzewicz AE, Pham H, Gundi VA, Scherman MS, North EJ, Hess T, Jones V, Gruppo V, Born SE, Kordulakova J, Chavadi SS, Morisseau C, Lenaerts AJ, Lee RE, McNeil MR, Jackson M. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat Chem Biol. 2012;8(4):334–41. doi: 10.1038/nchembio.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Touchette MH, Seeliger JC. Transport of outer membrane lipids in mycobacteria. Biochim Biophys Acta. 2017 doi: 10.1016/j.bbalip.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Drage MG, Tsai HC, Pecora ND, Cheng TY, Arida AR, Shukla S, Rojas RE, Seshadri C, Moody DB, Boom WH, Sacchettini JC, Harding CV. Mycobacterium tuberculosis lipoprotein LprG (Rv1411c) binds triacylated glycolipid agonists of Toll-like receptor 2. Nat Struct Mol Biol. 2010;17(9):1088–95. doi: 10.1038/nsmb.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brülle JK, Grau T, Tschumi A, Auchli Y, Burri R, Polsfuss S, Keller PM, Hunziker P, Sander P. Cloning, expression and characterization of Mycobacterium tuberculosis lipoprotein LprF. Biochem Biophys Res Commun. 2011;391(1):679–684. doi: 10.1016/j.bbrc.2009.11.120. [DOI] [PubMed] [Google Scholar]
- 13.Shukla S, Richardson ET, Athman JJ, Shi L, Wearsch PA, McDonald D, Banaei N, Boom WH, Jackson M, Harding CV. Mycobacterium tuberculosis Lipoprotein LprG Binds Lipoarabinomannan and Determines Its Cell Envelope Localization to Control Phagolysosomal Fusion. PLoS Pathog. 2014;10(10):e1004471. doi: 10.1371/journal.ppat.1004471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaur RL, Ren K, Blumenthal A, Bhamidi S, Gibbs S, Jackson M, Zare RN, Ehrt S, Ernst JD, Banaei N. LprG-Mediated Surface Expression of Lipoarabinomannan Is Essential for Virulence of Mycobacterium tuberculosis. PLoS Pathog. 2014;10(9):e1004376. doi: 10.1371/journal.ppat.1004376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinot AJ, Farrow M, Bai L, Layre E, Cheng TY, Tsai JH, Iqbal J, Annand JW, Sullivan ZA, Hussain MM, Sacchettini J, Moody DB, Seeliger JC, Rubin EJ. Mycobacterial Metabolic Syndrome: LprG and Rv1410 Regulate Triacylglyceride Levels, Growth Rate and Virulence in Mycobacterium tuberculosis. PLoS Pathog. 2016;12(1):e1005351. doi: 10.1371/journal.ppat.1005351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freinkman E, Okuda S, Ruiz N, Kahne D. Regulated assembly of the transenvelope protein complex required for lipopolysaccharide export. Biochemistry. 2012;51(24):4800–6. doi: 10.1021/bi300592c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okuda S, Tokuda H. Model of mouth-to-mouth transfer of bacterial lipoproteins through inner membrane LolC, periplasmic LolA, and outer membrane LolB. Proc Natl Acad Sci U S A. 2009;106(14):5877–82. doi: 10.1073/pnas.0900896106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kieser KJ, Rubin EJ. How sisters grow apart: mycobacterial growth and division. Nat Rev Microbiol. 2014;12(8):550–62. doi: 10.1038/nrmicro3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hett EC, Chao MC, Rubin EJ. Interaction and modulation of two antagonistic cell wall enzymes of mycobacteria. PLoS Pathog. 2010;6(7):e1001020. doi: 10.1371/journal.ppat.1001020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carel C, Nukdee K, Cantaloube S, Bonne M, Diagne CT, Laval F, Daffe M, Zerbib D. Mycobacterium tuberculosis proteins involved in mycolic acid synthesis and transport localize dynamically to the old growing pole and septum. PLoS One. 2014;9(5):e97148. doi: 10.1371/journal.pone.0097148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang CM, Nyayapathy S, Lee JY, Suh JW, Husson RN. Wag31, a homologue of the cell division protein DivIVA, regulates growth, morphology and polar cell wall synthesis in mycobacteria. Microbiology. 2008;154(Pt 3):725–35. doi: 10.1099/mic.0.2007/014076-0. [DOI] [PubMed] [Google Scholar]
- 22.Meniche X, Otten R, Siegrist MS, Baer CE, Murphy KC, Bertozzi CR, Sassetti CM. Subpolar addition of new cell wall is directed by DivIVA in mycobacteria. Proc Natl Acad Sci U S A. 2014;111(31):E3243–51. doi: 10.1073/pnas.1402158111.;. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen L, Chinnapapagari S, Thompson CJ. FbpA-Dependent biosynthesis of trehalose dimycolate is required for the intrinsic multidrug resistance, cell wall structure, and colonial morphology of Mycobacterium smegmatis. J Bacteriol. 2005;187(19):6603–11. doi: 10.1128/JB.187.19.6603-6611.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bigi F, Espitia C, Alito A, Zumarraga M, Romano MI, Cravero S, Cataldi A. A novel 27 kDa lipoprotein antigen from Mycobacterium bovis. Microbiology. 1997;143(Pt 11):3599–605. doi: 10.1099/00221287-143-11-3599. [DOI] [PubMed] [Google Scholar]
- 25.Huang BX, Kim HY. Effective identification of Akt interacting proteins by two-step chemical crosslinking, co-immunoprecipitation and mass spectrometry. PLoS One. 2013;8(4):e61430. doi: 10.1371/journal.pone.0061430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maguire PB, Briggs FN, Lennon NJ, Ohlendieck K. Oligomerization is an intrinsic property of calsequestrin in normal and transformed skeletal muscle. Biochem Biophys Res Commun. 1997;240(3):721–7. doi: 10.1006/bbrc.1997.7729. [DOI] [PubMed] [Google Scholar]
- 27.de Gunzburg J, Riehl R, Weinberg RA. Identification of a protein associated with p21ras by chemical crosslinking. Proc Natl Acad Sci U S A. 1989;86(11):4007–11. doi: 10.1073/pnas.86.11.4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang F, Robbins S, Guo J, Shen W, Schultz PG. Genetic incorporation of unnatural amino acids into proteins in Mycobacterium tuberculosis. PLoS ONE. 2010;5(2):e9354. doi: 10.1371/journal.pone.0009354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–58. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belisle JT, Vissa VD, Sievert T, Takayama K, Brennan PJ, Besra GS. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science. 1997;276(5317):1420–2. doi: 10.1126/science.276.5317.1420. [DOI] [PubMed] [Google Scholar]
- 31.Bigi F, Gioffre A, Klepp L, Santangelo MP, Alito A, Caimi K, Meikle V, Zumarraga M, Taboga O, Romano MI, Cataldi A. The knockout of the lprG-Rv1410 operon produces strong attenuation of Mycobacterium tuberculosis. Microbes Infect. 2004;6(2):182–7. doi: 10.1016/j.micinf.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Farrow MF, Rubin EJ. Function of a mycobacterial major facilitator superfamily pump requires a membrane-associated lipoprotein. J Bacteriol. 2008;190(5):1783–91. doi: 10.1128/JB.01046-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ojha AK, Trivelli X, Guerardel Y, Kremer L, Hatfull GF. Enzymatic hydrolysis of trehalose dimycolate releases free mycolic acids during mycobacterial growth in biofilms. J Biol Chem. 2010;285(23):17380–9. doi: 10.1074/jbc.M110.112813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foley HN, Stewart JA, Kavunja HW, Rundell SR, Swarts BM. Bioorthogonal Chemical Reporters for Selective In Situ Probing of Mycomembrane Components in Mycobacteria. Angew Chem Int Ed Engl. 2016;55(6):2053–7. doi: 10.1002/anie.201509216. [DOI] [PubMed] [Google Scholar]
- 35.Korkmaz G, Holm M, Wiens T, Sanyal S. Comprehensive analysis of stop codon usage in bacteria and its correlation with release factor abundance. J Biol Chem. 2014;289(44):30334–42. doi: 10.1074/jbc.M114.606632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaeffer ML, Agnihotri G, Kallender H, Brennan PJ, Lonsdale JT. Expression, purification, and characterization of the Mycobacterium tuberculosis acyl carrier protein, AcpM. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2001;1532(1–2):67–78. doi: 10.1016/s1388-1981(01)00116-0. [DOI] [PubMed] [Google Scholar]
- 37.Laval F, Haites R, Movahedzadeh F, Lemassu A, Wong CY, Stoker N, Billman-Jacobe H, Daffe M. Investigating the function of the putative mycolic acid methyltransferase UmaA: divergence between the Mycobacterium smegmatis and Mycobacterium tuberculosis proteins. J Biol Chem. 2008;283(3):1419–27. doi: 10.1074/jbc.M708859200. [DOI] [PubMed] [Google Scholar]
- 38.Parker SK, Barkley RM, Rino JG, Vasil ML. Mycobacterium tuberculosis Rv3802c encodes a phospholipase/thioesterase and is inhibited by the antimycobacterial agent tetrahydrolipstatin. PLoS ONE. 2009;4(1):e4281. doi: 10.1371/journal.pone.0004281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gunawardena HP, Feltcher ME, Wrobel JA, Gu S, Braunstein M, Chen X. Comparison of the membrane proteome of virulent Mycobacterium tuberculosis and the attenuated Mycobacterium bovis BCG vaccine strain by label-free quantitative proteomics. J Proteome Res. 2013;12(12):5463–74. doi: 10.1021/pr400334k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mawuenyega KG, Forst CV, Dobos KM, Belisle JT, Chen J, Bradbury EM, Bradbury AR, Chen X. Mycobacterium tuberculosis functional network analysis by global subcellular protein profiling. Mol Biol Cell. 2005;16(1):396–404. doi: 10.1091/mbc.E04-04-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malen H, De Souza GA, Pathak S, Softeland T, Wiker HG. Comparison of membrane proteins of Mycobacterium tuberculosis H37Rv and H37Ra strains. BMC Microbiol. 2011;11:18. doi: 10.1186/1471-2180-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolfe LM, Mahaffey SB, Kruh NA, Dobos KM. Proteomic definition of the cell wall of Mycobacterium tuberculosis. J Proteome Res. 2010;9(11):5816–26. doi: 10.1021/pr1005873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He Z, De Buck J. Cell wall proteome analysis of Mycobacterium smegmatis strain MC2 155. BMC Microbiol. 2010;10:121. doi: 10.1186/1471-2180-10-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen L, Chinnapapagari S, Thompson CJ. FbpA-Dependent biosynthesis of trehalose dimycolate is required for the intrinsic multidrug resistance, cell wall structure, and colonial morphology of Mycobacterium smegmatis. J Bacteriol. 2005;187(19):6603–11. doi: 10.1128/JB.187.19.6603-6611.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu WX, Zhang L, Mai JT, Peng RC, Yang EZ, Peng C, Wang HH. The Wag31 protein interacts with AccA3 and coordinates cell wall lipid permeability and lipophilic drug resistance in Mycobacterium smegmatis. Biochem Biophys Res Commun. 2014;448(3):255–60. doi: 10.1016/j.bbrc.2014.04.116. [DOI] [PubMed] [Google Scholar]
- 46.Hayashi JM, Luo CY, Mayfield JA, Hsu T, Fukuda T, Walfield AL, Giffen SR, Leszyk JD, Baer CE, Bennion OT, Madduri A, Shaffer SA, Aldridge BB, Sassetti CM, Sandler SJ, Kinoshita T, Moody DB, Morita YS. Spatially distinct and metabolically active membrane domain in mycobacteria. Proc Natl Acad Sci U S A. 2016;113(19):5400–5. doi: 10.1073/pnas.1525165113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allen BW. Mycobacteria. 1998;101:15–30. doi: 10.1385/0-89603-471-2:15. [DOI] [PubMed] [Google Scholar]
- 48.Beste DJ, Peters J, Hooper T, Avignone-Rossa C, Bushell ME, McFadden J. Compiling a molecular inventory for Mycobacterium bovis BCG at two growth rates: evidence for growth rate-mediated regulation of ribosome biosynthesis and lipid metabolism. J Bacteriol. 2005;187(5):1677–84. doi: 10.1128/JB.187.5.1677-1684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murphy KC, Papavinasasundaram K, Sassetti CM. Mycobacterial recombineering. Methods Mol Biol. 2015;1285:177–99. doi: 10.1007/978-1-4939-2450-9_10. [DOI] [PubMed] [Google Scholar]
- 50.Cognetta AB, 3rd, Niphakis MJ, Lee HC, Martini ML, Hulce JJ, Cravatt BF. Selective N-Hydroxyhydantoin Carbamate Inhibitors of Mammalian Serine Hydrolases. Chem Biol. 2015;22(7):928–37. doi: 10.1016/j.chembiol.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.