

Abstract
Aging greatly increases the risk for intervertebral disc degeneration (IDD) as a result of proteoglycan loss due to reduced synthesis and enhanced degradation of the disc matrix proteoglycan (PG). How disc matrix PG homeostasis becomes perturbed with age is not known. The goal of this study is to determine whether cellular senescence is a source of this perturbation.
We demonstrated that disc cellular senescence is dramatically increased in the DNA repair-deficient Ercc1−/Δ mouse model of human progeria. In these accelerated aging mice, increased disc cellular senescence is closely associated with the rapid loss of disc PG. We also directly examine PG homeostasis in oxidative damage-induced senescent human cells using an in vitro cell culture model system. Senescence of human disc cells treated with hydrogen peroxide was confirmed by growth arrest, senescence-associated β-galactosidase activity, γH2AX foci, and acquisition of senescence-associated secretory phenotype. Senescent human disc cells also exhibited perturbed matrix PG homeostasis as evidenced by their decreased capacity to synthesize new matrix PG and enhanced degradation of aggrecan, a major matrix PG. of the disc. Our in vivo and in vitro findings altogether suggest that disc cellular senescence is an important driver of PG matrix homeostatic perturbation and PG loss.
Keywords: Aging, DNA damage, Cellular senescence, Matrix proteoglycan, Intervertebral disc
INTRODUCTION
Aging is the leading risk factor for intervertebral disc degeneration (IDD), a major contributor to low back pain and other spine-related pathologies that can result in tremendous societal health and economic burden1. With aging, invariably, there is progressive depletion of disc matrix PG. Loss of disc matrix PG, a universal hallmark of IDD, is a consequence of perturbed matrix homeostasis mediated primarily by dysregulated cellular activities2. Matrix homeostasis can be perturbed by a combination of decreased cellular capacity for matrix PG synthesis and increased PG proteolysis from increased production of catabolic enzymes. However, it is still unclear how as disc tissue ages, cells acquire the phenotype that directly perturbs disc matrix homeostasis.
Cellular senescence was first described as an irreversible state of growth arrest3. Early studies identified a type of cellular senescence known as replicative cellular senescence by their growth arrest phenotype due to the critical shortening of telomere length after successive replicative cell cycles. Replicative senescence is thought to have been evolutionarily selected as a tumor suppressor mechanism to prevent the proliferation of cells prone to genomic instability4. This early view of cellular senescence turns out to be oversimplified as subsequent studies began to reveal a much more complex and multifaceted biological role of cellular senescence. For example, there are now other classes of cellular senescence, including stress-induced premature senescence (SIPS)5, that is caused primarily by the accumulation of persistent unrepaired DNA damage, and oncogene induced senescence (OIS). SIPS, in addition to undergoing cell cycle growth arrest, also secrete numerous inflammatory cytokines and matrix proteinases, a feature termed senescence associated secretory phenotype (SASP). SASP can have profound catabolic effects on neighboring cells and extracellular matrix resulting in disruption of tissue structure and function6. These results indicate that cellular senescence is not simply a protective tumor suppressor but also as a driver of diseases. In fact, recent growing discoveries continue to redefine cellular senescence as a trigger of tissue remodeling that acts during normal embryonic development and tissue damage associated with pathologies4. Hence, cellular senescence appears to play a vital role both in normal physiology as well as diseases, the mechanisms behind which are being actively investigated.
Cellular senescence is a complex is Growing evidence support cellular senescence as a major driver of aging and aging-related degenerative diseases. Earlier on Age-dependent accumulation of senescent cells has been shown in various tissues, and SASP has been implicated in the disruption of tissue structure and function leading to various age-associated pathologies in a broad spectrum of tissues, including eye, artery, skeletal muscle, fat, heart, and brain7, 8. p16Ink4a, also known as cyclin-dependent kinase inhibitor 2A, is marker of cellular senescence. Clearance of p16Ink4a-positive senescent cells delays aging-associated disorders in several tissues, including adipose, skeletal muscle and eye, in the BubR1-deficient mouse model of accelerated aging8, 9. In natural aging mice, the clearance of p16Ink4a-positive cells delays tumorigenesis and ameliorates age-related degeneration of kidney, heart and fat8, 9. In addition, treating naturally aged or progeroid mice with drugs known as senolytics that specifically target killing of senescent cells mitigate functional decline in cardiac and skeletal tissues in naturally aged mice and extend healthspan in the Ercc1−/Δ mouse model of accelerated aging10.
Cellular senescence also has been linked to intervertebral disc aging and degeneration. Increased number of senescent cells in aged and degenerative discs of humans and animal models has been reported11, 12. An increased percentage of cells positive for p16INK4a, a marker of senescence, was documented in discs of Ercc1−/Δ mice with reduced DNA repair capacity13. These findings indicate that DNA damage is a key driver behind these processes, consistent with the notion that DNA damage drives cellular senescence14. This idea was further supported by studies of adult mice chronically exposed to different types of genotoxins, including ionizing radiation and tobacco smoke, which showed dramatic up regulation of p16Ink4 in the disc and accelerated loss of disc PG15, 16. These observations established a correlative association between cellular senescence and disc degenerative changes. However it still remains unknown whether senescent cells or cells with other altered phenotype in aged discs are directly responsible for driving disc PG homeostatic imbalance and PG loss.
The goal of this study is to test the hypothesis that, with aging, there is a population of senescent disc cells expressing SASP that severely perturbs PG homeostasis and drives age-dependent disc PG loss. Here we tested this hypothesis by characterizing the phenotype of senescent human disc cells as it directly relates to the modulation of PG matrix homeostasis using a cell culture model system. We also carefully measured the levels of disc cellular senescence in the Ercc1−/Δ mouse model of accelerated aging in order to confirm the robust association between disc cellular senescence and disc PG homeostatic perturbation and loss in vivo.
MATERIALS AND METHODS
Sample Collection and Cell Isolation
Human nucleus pulposus samples were obtained from surgical specimens from patients with mean age of 47.7 ± 11.4 years (mean ± SD) with mean degeneration grade of 2.41 ± 0.5 on the Thompson grading scale (IRB #: PRO12100603). Cells were isolated from digested tissues in 0.2% pronase (EMD Chemicals 53702) for 60 minutes followed by overnight digestion in 0.02% collagenase P (Roche Applied Science 11213873001) as described17. Lumbar nucleus pulposus were collected under a dissecting microscope and pooled from three 20-week-old wild-type or three 20-week-old Ercc1−/Δ mutant mice and digested for disc cell isolation with 0.02% pronase for 10 minutes and 0.002% collagenase P for 30 minutes. Both human and mouse primary disc cells were cultured under hypoxic conditions (37°C, 5% CO2, and 5% O2 with a bicarbonate buffer to maintain pH 7.2) unless noted otherwise. Ercc1−/Δ mice were bred and genotyped as previously described13, 18. Deficient in the DNA repair protein ERCC1, Ercc1−/Δ mice are asymptomatic until 5 weeks of age, beyond which they age progressively and die prematurely at about 24 weeks of age. 20-week-old Ercc1−/Δ mutant mice were used because at this age they exhibit pronounced progeroid syndrome.
Senescence Induction
It is expected that a fraction of the cells isolated from the disc surgical specimens are senescent since the patients were mostly older individuals (mean age of 47.7 years). However, due to their non-proliferative nature, the native senescent disc cells isolated from the disc tissue were lost during the culture expansion performed to obtain sufficient cell quantity to characterize their phenotype. This is the reason why we decided to induce senescence using hydrogen peroxide. Hydrogen peroxide was used to induce cellular senescence (Fig. 1) as previously described, using an established method19. Primary human nucleus pulposus (hNP) cells were treated with 500 μM hydrogen peroxide in F-12 10% FBS 1% PS (Life Technologies 11765-062, Atlanta Biologicals S12450, Life Technologies 15140-16), for two hours. Culture media was then replaced with fresh media F-12 10% FBS 1% PS without hydrogen peroxide before being plated on new plates and incubated overnight at 37°C at 21% O2. Cells were treated once more with hydrogen peroxide under the same conditions to ensure complete induction of senescence. The cells were then incubated under atmospheric oxygen (F-12 10% FBS 1% PS) for five days without hydrogen peroxide to allow establishment of senescence before being evaluated by the assays below.
Figure 1. Enhanced disc cellular senescence in old DNA repair-deficient Ercc1−/Δ mouse model of accelerated aging.
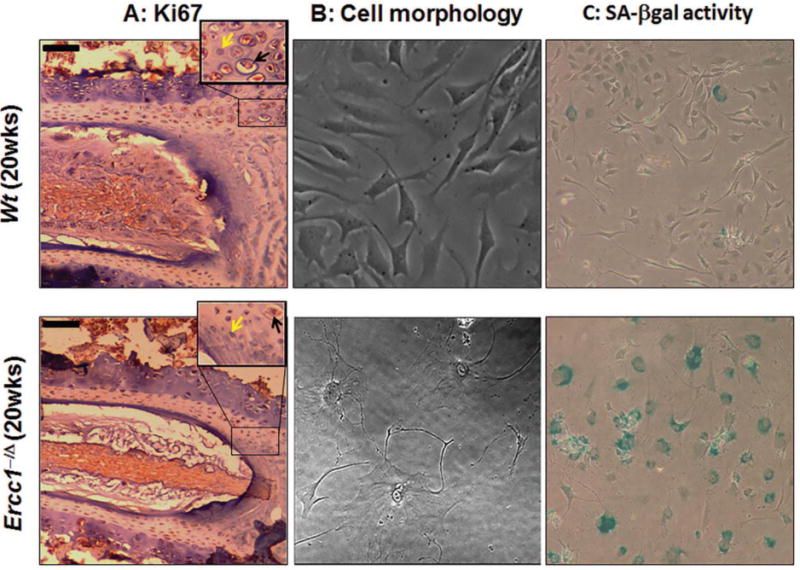
Disc tissue sections and disc cells isolated from the 20 wk old progeroid Ercc1−/Δ mice and their WT littermates were analyzed. (A) Reduced staining (brown) of the cell proliferative marker Ki67 in disc tissue section of 20 wk old progeroid Ercc1−/Δ mice compared to WT littermates (black arrow = Ki67 positive cell; yellow arrow = Ki67 negative cell). Moreover, there is sparser matrix in the nucleus pulposus and overall fewer disc cells in Ercc1−/Δ mice than WT mice. (Black bars = 50μm). Compared to WT mice, most of the disc cells isolated from Ercc1−/Δ mice acquired enlarged flattened morphology (B) and were stained positive for SA-β-gal activity and (C).
Senescence-associated β-galactosidase staining
Senescence-associated β-galactosidase (SAβ-gal) staining was performed as previously described20. Images were taken using brightfield microscopy at 40× magnification.
Proteoglycan Synthesis
35S-sulfate incorporation was performed, as previously described, to measure PG synthesis21. Briefly 35S-sulfate (10 μCi/mL, American Radiolabeled Chemical ARS-105) was added to cell culture media in duplicate wells per condition in a 24 well plate format. Cells were radiolabeled for two days and extracted by addition of homogenization buffer containing 200 mM sodium chloride, 50 mM sodium acetate, 0.1% Triton X-100 (Sigma-Aldrich X-100), 10 mM EDTA, 50 μM DTT (Sigma-Aldrich D9779), and 1x Protease Inhibitor (Sigma-Aldrich P8340) and shaking at 4°C for 1 hr in a separate 1.5 mL microcentrifuge tube. Proteoglycans in cell lysate were extracted with shaking in a guanidine hydrochloride solution (8 M guanidine hydrochloride (Sigma-Aldrich G3272) containing 50 mM sodium acetate, 10 mM EDTA, and 1x protease inhibitor at 4°C for 4 hours22. Extracted samples were mixed with alcian blue solution containing 0.02% alcian blue (Sigma-Aldrich A9186), 50 mM sodium acetate, and 85 mM magnesium chloride for an hour at room temperature then loaded onto nitrocellulose membranes (Millipore HAWP 025 00). The membranes were washed with a buffer containing 100 mM sodium acetate (Sigma-Aldrich S2889), 50 mM magnesium chloride, and 50 mM sodium sulfate (Sigma-Aldrich 239313) to eliminate unincorporated 35S-sulfate. The membranes were dissolved in scintillation fluid (National Diagnostics LS-201) and counted in a scintillation counter (Packard Tri-Carb 2100TR). Counts per minute (CPM) were converted to number of pmoles of sulfate, using the specific activity of 35S-sulfate measured in the conditioned media, and then normalized to the amount of DNA per sample as determined by Picogreen assay (Life Technologies P7589).
Gene Expression
RNA isolation was performed using Qiagen RNeasy Plus Micro Kit (Qiagen 74034). Quantitative RT-PCR was performed to measure releative gene expression of anabolic and catabolic genes of interest as previously described21.
ELISA
ELISA was performed using 200 μL conditioned media (concentrated 3–5X with Millipore UFC 900324) and normalized by cell number (Brightfield microscopy) using R&D Total Human IL-6, IL-8, MMP-1, and MMP-3 DuoSets (R&D Systems DY206, DY208, DY901, DY513).
Antibody Arrays
Conditioned media was concentrated 5X using a Millipore Amicon Ultra-15 Centrifugal Filter Unit with Ultracel-3 (EMD Millipore UFC900308) membrane and antibody arrays performed using 1 mL concentrated conditioned media and inflammation antibody array (RayBioTech AAH-INF-3-4). Quantification was performed with densitometry analysis. Each protein of the array was normalized to the positive controls provided on the array membrane while background subtraction was performed using negative controls and blanks on the array membrane within each sample.
Immohistochemistry
Ki67 staining was performed on deparaffinized disc tissue sections from 20 wk-old WT and Ercc1−/Δ mice. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 15 minutes. Tissue sections were subjected to heat induced-epitope retrieval by incubation in sodium citrate buffer (10mM, pH 6.0) for 30 min in a decloaker, followed by 30 min cool-down. The primary antibody for Ki67 (rat anti-mouse Ki67 (TEC-3), M-7249, Dako Cytomation, Carpinteria, CA) was applied for 1 hour at a 1:50 dilution at room temperature. The secondary antibody was applied at a 1:300 dilution for 30 minutes, followed by the label antibody (ABC Elite, Vector Laboratories, Burlingame, CA) for 30 minutes. DAB chromagen (Dako Cytomation, Carpinteria, CA) was applied for 6 minutes, followed by 2 rinse steps in distilled water. Hematoxylin was used as a counterstain, and slides were dehydrated, cleared and coverslipped. Images were collected using an Olympus BX51 microscope.
Immunofluorescence
Detection of ADAMTS4 was performed on disc cells on the chamber slides (Thermo Scientific Nunc 154526) that were washed, blocked, followed by a 60 min incubation with 1:100 dilution of the primary antibody, anti-ADAMTS4 (PA1-33177, ThermoFisher Scientific) and subsequent incubation with the goat anti-Rabbit secondary antibody, Alexa Fluor 488 (LifeTechnologies). Detection of phosphorylated γH2A.X, a marker of DNA double strand breaks, was also performed on disc cells using 1:50 dilution of the primary antibody, anti-phospho-γH2A.X (05-636, MilliporeSigma), and the goat anti-mouse secondary antibody, Cyanine3 (lifeTechnologies). The slides were washed and mounted in Prolong Gold anti-fade reagent with DAPI (P36935, Invitrogen, Carlsbad, CA) to stain for nuclei. Images were collected using fluorescent microscope (fluoview1000) to measure the number and distribution of a variety of fluorescently labeled cells.
Immunoblotting
For detection of aggrecan fragments in cell culture conditioned media, 1:1000 rabbit polyclonal anti-Aggrecan antibody (Abcam ab36861) was used as primary antibody followed with 1:10000 anti-rabbit goat secondary antibody with HRP (Thermo Scientific PI-31460) and chemiluminescent detection (Thermo Scientific 34096 and Bio-Rad ChemiDoc MP). Western was performed using Tris-HEPES 4–20% gradient gel (Thermo Scientific 25204), Tris-HEPES-SDS Running Buffer (Thermo 28398), Tris-Glycine Transfer Buffer with 10% Methanol (Thermo Scientific 28380, Fisher Scientific A452-4), and TBST (Sigma-Aldrich T9039). Quantification was performed with densitometry analysis and local background subtraction.
DMMB assay for total GAG
Colorimetric dimethylmethylene blue (DMMB) assay for total glycosaminoglycan (GAG) was used to quantify total matrix PG content as previously described21. DMMB assay was done on hNP cell cultures collected 10 days after the first treatment with H2O2 to allow sufficient GAG accumulation for detection and quantitation. The standard curve was created using known concentrations of chondroitin sulfate in a serial dilution. Total GAG content was normalized to the quantity of DNA per sample as measured by the Picogreen assay (Life Technologies P7589). Extracellular matrix digestion buffer was added to cell culture plate wells and incubated at 37°C (non-CO2 incubator) for 6 hours before sample collection and DMMB assay.
Statistical Analysis
Statistical analysis was performed using the software R. The statistical tests performed for the results include: Student’s one-sample T-test and Student’s paired T-test. All statistical tests were two-tailed. The assumptions of Student’s T-test (equality of variance between groups and normality of residuals) were checked and confirmed using plots of residuals vs. fitted values. Quantile-Quantile (Q-Q) plots of standardized residuals for the data for each outcome measure were made.
RESULTS
Ercc1−/Δ mice with accelerated aging exhibit enhanced intervertebral disc cellular senescence
Accumulation of DNA damage is a well-established driver of cellular senescence. The DNA repair-deficient Ercc1−/Δ mice have previously been reported to have increased staining for disc p16INK4a, a marker of senescence. However, emerging evidence suggest that it is not sufficient to characterize cellular senescence by any single marker such as p16INK4a. Thus, here we used multiple senescence markers to fully confirm if there are indeed more senescent cells in disc tissue Ercc1−/Δ mice compared to their WT counterparts. Ercc1−/Δ mouse disc cells were observed to be mostly non-proliferative in cell culture, a feature that is further supported by decreased staining of the cell proliferative marker Ki67 in disc sections of Ercc1−/Δ mice compared to WT mice (Fig. 1A and supplemental Fig. 1SA). Moreover, most of the disc cells isolated from the 20-week-old progeroid Ercc1−/Δ mice showed an enlarged flattened morphology (Fig. 1B) and stained positive for senescence-associated β-galactosidase (Fig. 1C and supplemental Fig. 1SB). Through the use of multiple senescence markers we demonstrated clearly that accelerated aging Ercc1−/Δ mice acquire enhanced intervertebral disc cellular senescence.
We have previously documented accelerated age-associated IDD in the Ercc1−/Δ progeroid mice13. Increased disc cellular senescence is thus associated with disc degenerative changes in these Ercc1−/Δ progeroid mice as well as humans11–13. However, such a correlative link between disc cellular senescence and IDD does not prove that senescent cells, as opposed to other cell phenotypes found in aged or degenerated discs, are directly responsible for driving disc PG homeostatic imbalance and PG loss leading to disc aging and degeneration. It is therefore important to demonstrate that senescent disc cells acquire an altered phenotype that directly negatively impacts PG homeostasis. This could be most effectively achieved using an in vitro cell culture model system to directly demonstrate the phenotype of perturbed PG homeostasis in senescent disc cells. Due to the limited amount of mouse disc tissue, it was not feasible to isolate enough senescent disc cells from the Ercc1−/Δ progeroid mice to characterize their phenotype. Thus, for reasons of physiologic relevance and tissue availability, we chose to characterize human disc cells isolated from surgical specimens that have been induced to undergo senescence by oxidative damage.
Establishment of stress-induced senescent disc cell culture model system
Accumulation of DNA damage, including oxidative DNA damage, is a well-established driver of cellular senescence23, 24. Here, we induced senescence of human nucleus pulposus (hNP) cells by exposure to hydrogen peroxide (H2O2), a strong oxidant commonly used to induce DNA damage and cellular senescence25–27. hNP cells were initially treated with H2O2 on the first day to cause oxidative damage, followed by incubation in culture media without H2O2 for an additional five days to allow the cells to establish oxidative DNA damage-induced senescence (Fig. 2A). This treatment regimen is specifically designed to ensure that any observable phenotypic changes five days post H2O2 treatment are due to senescence of the cells and not acute cellular response to H2O2 exposure.
Figure 2. Establishment of oxidative damage-induced senescent disc cell culture model system.
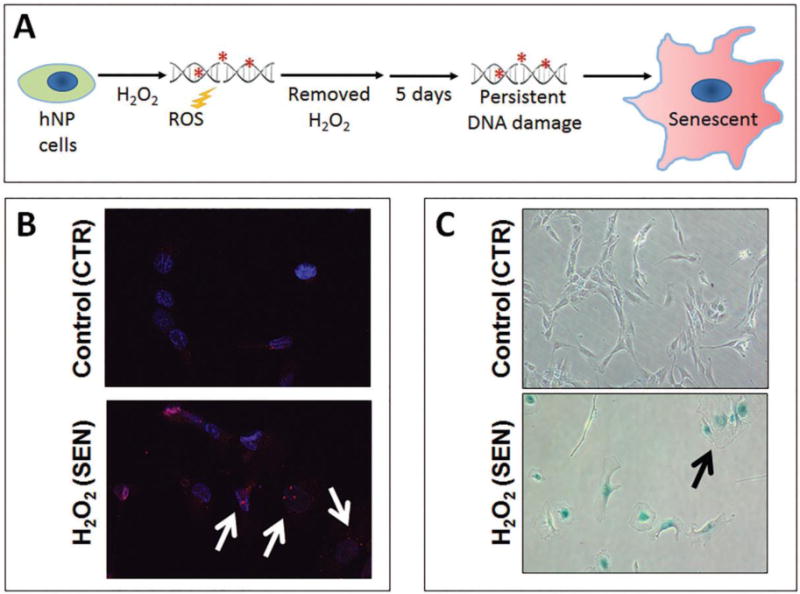
(A). Schematic of senescence induction of hNP cells by H2O2. After the initial H2O2 treatment, cells were incubated for five days without H2O2 to allow establishment of senescence. Most treated hNP cells showed increased nuclear foci of phospho-γH2A.X (white arrows), a marker of DNA damage (B), and were stained (bluish) positive for SA-β-gal activity and exhibit enlarged flattened morphology (black arrow) (C).
At five days post-H2O2 treatment, most hNP cells stopped proliferating as assessed by CCK8 cell proliferation assay (Supplemental Fig. S2) and showed increased nuclear foci of phospho-γH2A.X (Fig. 2B), a modified protein marker of DNA damage-induced cellular senescence. In addition, over 90% of the H2O2-treated human disc cells were stained positive for SA-β-gal activity and exhibit enlarged flattened morphology (Fig. 2C). Detection these key markers of cellular senescence demonstrated that complete senescence of disc cells can be reproducibly induced by oxidative damage using this H2O2 treatment regimen. Successful establishment of this oxidative damage-induced senescent disc cell culture model is vital for characterizing the metabolic phenotype of senescent disc cells as it is related to PG homeostasis.
Senescence-associated secretory phenotype (SASP) in senescent human NP cells
A key feature of SIPS is the acquisition of SASP, whereby the senescent cells overexpress and secrete many different pro-inflammatory cytokines and MMPs. To assess the SASP secretome profile of senescent disc cells, inflammation and MMP antibodies arrays were performed to detect various inflammatory and catabolic factors in the conditioned media of senescent and non-senescent hNP cell cultures. As shown in Fig. 3 and Fig. S3, elevated levels of many pro-inflammatory cytokines (IL-6, IL-8, PDGF-BB, GCSF), chemokines (EOTAXIN-2, IP-10, RANTES), and MMPs (MMP-3, MMP-10, TIMP-2) were observed in culture media of senescent hNP cells compared to those found in nonsenescent hNP cell culture media. Many of these SASP factors have also been reported in senescent fibroblasts and epithelial cells6, 23.
Figure 3. Oxidative stress-induced senescent human NP cells secrete catabolic and pro-inflammatory factors.
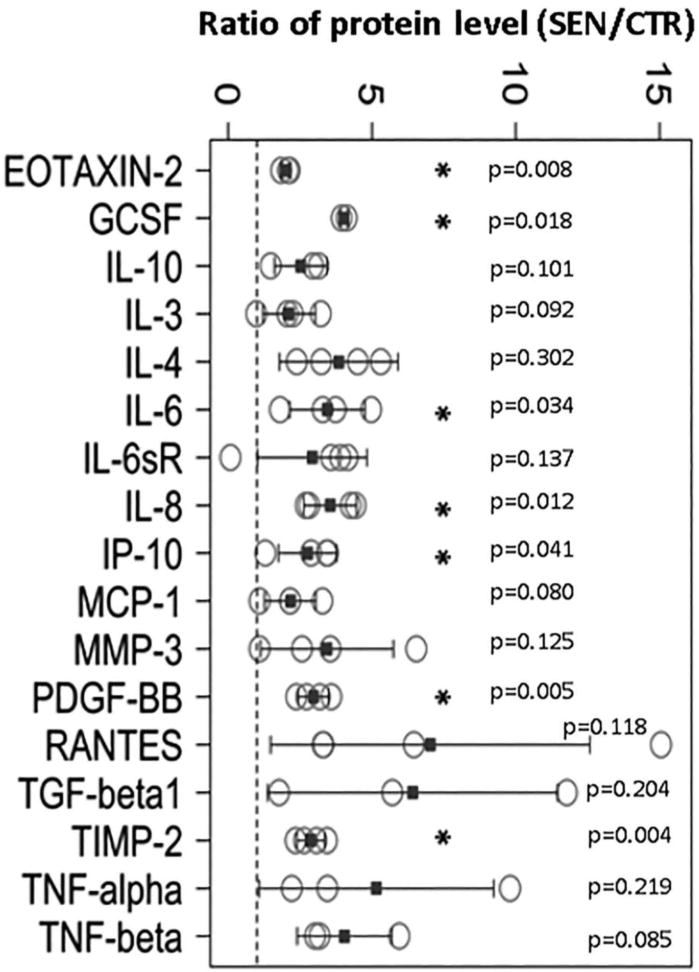
Compared to non-senescent hNP cells, conditioned culture media of senescent hNP cells contained increased levels of numerous catabolic and inflammatory cytokines. Proteins in conditioned media were immunodetected using the inflammation antibody arrays. The dotted line represents a ratio of 1, indicating that the secreted protein level by senescent hNP cells is the same as that by nonsenescent hNP cells. Error bars represent the SEM (n = 4). * denotes p < 0.05.
To further confirm the results of our assays using the inflammation and MMP antibodies arrays, we performed ELISA on conditioned media from senescent hNP cell and non-senescent cultures to detect IL-6, IL-8, MMP-1 and MMP-3, the key common SASP factors produced by many senescent cells6. Compared to non-senescent hNP cells where the levels of MMP-1 and MMP-3 (~0.001 pg/mL) were virtually undetectable, senescent hNP conditioned media contained much higher levels of MMP-1 (0.024 pg/mL) and MMP-3 (0.8 pg/mL per cell) when normalized to cell number (Supplemental Fig S3B). Likewise, when compared to non-senescent hNP cell culture media (IL-6: 0.04 pg/mL and IL-8: 0.025 pg/mL), senescent hNP cell conditioned media also contained significantly higher levels of IL-6 (0.14 pg/mL) and IL-8 (0.14 pg/mL) when normalized to cell number (Supplemental Fig S3B). Upregulation of these four SASP factors appear to occur, at least in part, at the transcription level (Supplemental Fig S4).
Enhanced aggrecan proteolysis in human senescent disc cells
SASP of senescent disc cells suggest a catabolic phenotype that is conducive to matrix degradation. To test this prediction, we assessed the integrity of aggrecan, a major proteoglycan of disc extracellular matrix, by performing immunodetection of its proteolytic products in conditioned media. Western blot analysis revealed a large increase in the level of ADAMTS-mediated and a modest increase in MMP-mediated proteolytic cleavage of the aggrecan interglobular domain (IGD) in the conditioned culture media of senescent hNP cells compared to non-senescent hNP cells (Fig. 4A). Immunofluorescence assay demonstrated an induction of expression of ADAMTS4 protein in senescent hNP cell culture (Fig. 4B). No change in ADAMTS5 expression in the conditioned media of senescent hNP cells relative to non-senescent hNP cells was observed (data not shown). Thus, the enhanced aggrecanolysis in senescent hNP cell culture is likely due to increased expression and enzymatic activity of ADAMTS4.
Figure 4.
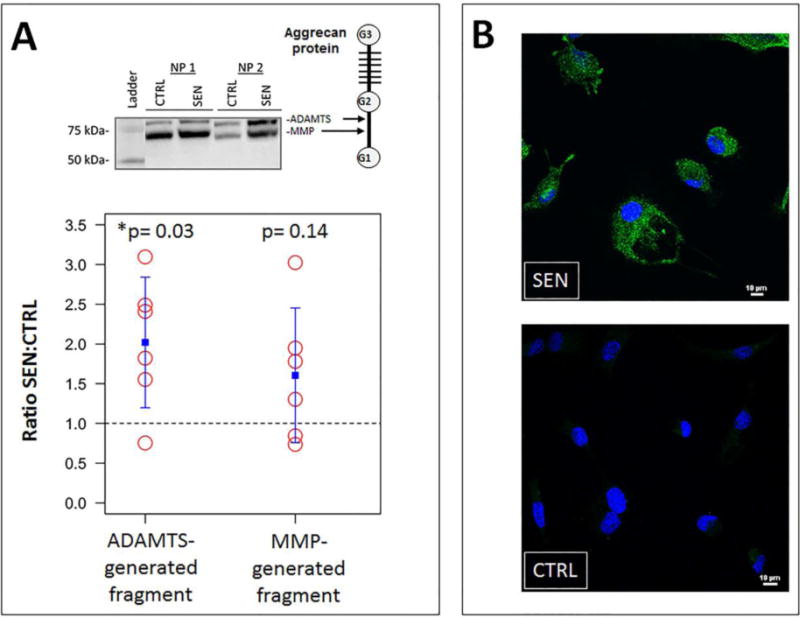
Increased aggrecanolysis and ADAMTS4 expression in oxidative stress-induced senescent hNP cell culture. (A) Western analysis of aggrecan in hNP senescent and control conditioned media. MMP- and ADAMTS-mediate cleavage sites within the interglobular domain of aggrecan (top right). Bottom graphs showed quantification of aggrecan fragments using densitometry. The dotted line represents a ratio of 1, indicating that the amount of aggrecan fragments generated by senescent hNP cells is that same as that by nonsenescent hNP cells. The blue square represents the mean. The error bars represent the SEM (n = 6). (B) Immunofluorescence revealed upregulated expression of ADAMTS4 protein (green) in senescent hNP cells.
Reduced PG matrix production in human senescent disc cells
Having demonstrated enhanced catabolic features in senescent disc cells, we next assessed whether the capacity for matrix anabolism of these cells were also affected. We performed 35S-sulfate incorporation assay for hNP senescent and control cells. Senescent hNP cells showed a significant decrease (2x) in new PG synthesis compared to non-senescent cells (Fig. 5). There was also a significant decrease (2x) in total GAG content in senescent hNP cell cultures compared to the non-senescent cells (Fig. 5). The net reduction in total GAG content in senescent hNP cells is consistent with the reduced ability of these cells to synthesize PG and their enhanced catabolic phenotype.
Figure 5. Oxidative stress-induced hNP cellular senescence resulted in reduced PG synthesis and decreased total GAG content.
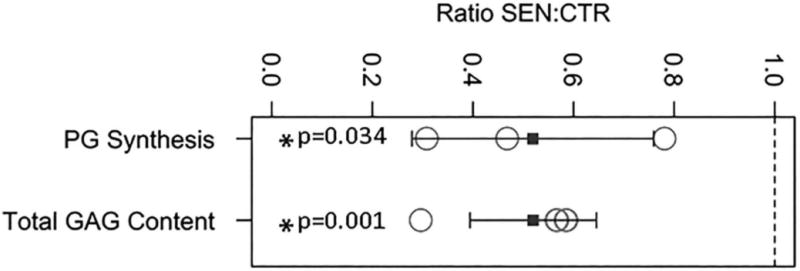
Cell culture PG synthesis was measured by 35S-sulfate incorporation and the GAG content by the DMMB colorimetric assay. Both PG synthesis and GAG were normalized to DNA. Y axis: ratio value of senescent hNP to control non-senescent hNP cells. The dashed line represents a ratio of 1 which indicates no change in PG synthesis or GAG content between senescent hNP and control hNP cells. The blue square represents the mean. The error bars represent the SEM (n = 3).
DISCUSSION
This study initiated and established a basic framework of how aging affects disc health through the action of senescent disc cells on matrix metabolism. Here we confirmed using multiple markers of cellular senescence that there is more disc cellular senescence in the DNA repair-deficient Ercc1−/Δ mouse model of accelerated aging than the WT littermate. In fact, in a separate concurrent study we demonstrated the presence of other common senescence markers, including increased p21Cip1 and cytoplasmic HMGB1 proteins, in disc tissue of Ercc1−/Δ mice when compared to WT mice28. We previously reported that Ercc1−/Δ have progressive, accelerated disc degeneration, similar to natural aging. We also demonstrated recently that treatment of Ercc1−/Δ mice with the senolytic drug combination dasatinib and quercetin, small molecules that selectively induce death of senescent cells, slowed down disc degeneration10. Senescent disc cells thus appear to contribute to driving age-dependent disc degeneration, but until now how they do so remained unclear. In the current study, we examined the effects of senescence on matrix PG homeostasis that could explain how disc senescent cells contribute to the progressive loss of disc PG in disc aging and degeneration. Our findings demonstrate that oxidative damage-induced senescence greatly perturbs matrix homeostasis in human disc cells. This is evident by a decreased capacity for PG synthesis (Fig. 5), enhanced aggrecanolysis (Fig. 4), as well as increased expression and secretion of numerous pro-inflammatory cytokines, chemokines, and matrix proteinases (Fig. 3). Reduced PG synthesis capability, enhanced PG degradation, and an overall reduction of PG GAG content in senescent disc cells provide strong supporting evidence that these cells acquire a perturbed matrix homeostatic phenotype. It should be noted that a recent study also reported a catabolic phenotype in hNP cells treated with H2O229. However, it is not clear from this previous study if the observed changes were due to senescence of the cells or acute cellular response to H2O2 stress since the authors performed the analyses shortly after H2O2 treatment without allowing the cells time to establish senescence and express SASP.
A hallmark feature of stress-induced premature senescence is SASP, which is thought to disrupt tissue structure and function, and promote aging7, 30. This study showed that oxidative damage-induced senescence of human disc cells resulted in induction of SASP (Fig. 3), which could have catabolic effects on neighboring disc cells and the extracellular matrix. Senescent hNP cells secreted IL-6, IL-6sR, IL-8, MMP-1, MMP-3, MMP-10 proteins as well as other cytokines and growth factors such as TNF-α, RANTES, EOTAXIN-2, and GCSF. In particular, IL-6 and IL-8 are well known SASP factors that can promote chronic inflammation, drive aging and increase the risk of age-related pathologies7. Serum levels of IL-6 in low back pain patients are higher than those measured in control patients31. Similarly, the levels of matrix remodeling factors MMP-1, MMP-3, and MMP-10 are elevated in degenerative disc tissue32, 33. RANTES (CCL5) is produced by human disc cells in response to inflammatory stimuli in vitro and is implicated in immune signaling34. Both IL-1β and TNF-α play a key role in driving disc degeneration by increasing both pro-inflammatory and catabolic processes35. The chemokine EOTAXIN-2 (CCL24) is a chemoattractant for eosinophils and the cytokine GCSF stimulates granulocytes and stem cell production and release into bloodstream. Therefore, our findings of increased levels of SASP factors in senescent cells provide support for a contribution to degenerative matrix changes.
Enhanced aggrecanolysis was observed in cultures of senescent hNP cells (Fig. 4). This is not surprising given the SASP feature of these cells. Western blot analysis revealed a substantial increase in ADAMTS-generated aggrecan fragments with ADAMTS-4 apparently being the predominant aggrecanase responsible for ADAMTS-mediated aggrecanolysis. The increase in MMP-generated aggrecan fragments is surprisingly modest given the large increase in MMP mRNA and protein production (MMP-1, MMP-3, MMP-10) by senescent hNP cells (Fig. 3 and supplemental Fig. S3A, S3B). It is possible that in our disc cell cultures there may be post-translational regulation or some suppression of enzymatic activities of the MMPs36, 37, e.g., protein turnover or TIMP inhibitory action on MMPs. Indeed, MMP enzymatic activity in conditioned culture media, quantified using the MMP fluorogenic substrate, showed similar levels in both control and senescent hNP cells (supplemental Fig. S5). TIMP-2 could be a candidate since its expression is induced in senescent hNP cells (Fig. 3). Further investigation is needed to determine the specific ADAMTS(s) and MMP(s) in senescent disc cells primarily responsible for aggrecanolysis.
It is important to note that the complete SASP profile of senescent hNP cells still needs to be defined since the inflammatory and MMP antibody arrays used here cover only a limited number of targets6, 38, 39. Additional studies are required to provide a comprehensive picture of what other protein factors are affected senescent disc cells. For instance, while our study looked at only the major matrix PG, aggrecan, there are likely other PGs and collagens that may change in abundance or integrity as disc cells senesce2, 40 which may contribute to the compositional and structural changes in the disc extracellular matrix. More importantly, in vivo studies are needed to investigate whether clearance of senescent cells in naturally aging mice affects disc tissue structure and function8. This is needed to establish causal relationship between cellular senescence and age-related IDD.
In summary, our findings support the hypothesis that senescent disc cells acquire imbalanced matrix metabolic homeostasis, i.e., decreased anabolism and increased catabolism. An important question arising from this study is how disc cellular senescence is established during the natural course of aging. Since DNA damage drives cellular senescence, it is important to explore the potential sources of DNA damage in disc, including oxidative stress, inflammation, replication, and limited nutrition12, 41. Understanding disc cellular senescence is critical to understand the biology of disc aging. This is useful in guiding the development of effective therapeutic interventions to treat age-related IDD disorders, which are expected to increase rapidly with the growing aging population.
Supplementary Material
Table 1.
Primers used for qRT-PCR for matrix anabolism and matrix catabolism
| Gene [Homo sapiens] |
Forward (5′ to 3′) | Reverse (5′ to 3′) |
|---|---|---|
| ADAMTS-4 | TCACTGACTTCCTGGACAATGG | ACTGGCGGTCAGCATCATAGT |
| ADAMTS-5 | CTGACCTACCACGAAAGCAGATC | ATGCCGGACACACGGAGTA |
| GAPDH | ACCCACTCCTCCACCTTTGAC | TCCACCACCCTGTTGCTGTAG |
| IL-6 | AGCCACTCACCTCTTCAGAACG | TGCCTCTTTGCTTTCACAC |
| IL-8 | GGCCGTGGCTCTCTTGGCAG | TGTGTTGGCGCAGTGTGGTCC |
| MMP-1 | GAGCTCAACTTCCGGGTAGA | CCCAAAAGCGTGTGACAGTA |
| MMP-3 | CAAGGAGGCAGGCAAGACAGC | GCCACGCACAGCAACAGTAGG |
| TIMP-1 | TGGCTTCTGGCATCCTGTTGTTG | CGCTGGTATAAGGTGGTCTGGTTG |
| TIMP-3 | AGGACGCCTTCTGCAACTC | GTACTGCACATGGGGCATCT |
Highlights.
Aging enhances loss of disc matrix proteoglycan that drives intervertebral disc degeneration
Senescent human disc cells exhibit enhanced catabolic activities and proteoglycan loss
Cellular senescence is closely associated with proteoglycan loss in intervertebral discs of a mouse model of accelerated aging
Cellular senescence is a likely driver of disc proteoglycan loss resulting in intervertebral disc aging and degeneration
Acknowledgments
We would like to thank the Ferguson laboratory administrative and research staff for their support. The work was supported by the National Institute of Health (AG044376 to NV and AG043376 to PDR and LJN). We would like to acknowledge the NIH supported microscopy resources in the Center for Biologic Imaging. Specifically the confocal microscope supported by grant number 1S10OD019973-01”.
Abbreviations
- PG
Proteoglycan
- IDD
Intervertebral disc degeneration
- SASP
Senescence-associated secretory phenotype
- SAβ-gal
Senescence-associated β-galactosidase
- MMP
Matrix metalloproteinase
- ADAMTS
A Disintegrin and Metalloproteinase with Thrombospondin motifs
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luo X, Pietrobon R, Sun SX, Liu GG, Hey L. Estimates and patterns of direct health care expenditures among individuals with back pain in the United States. Spine. 2004;29:79–86. doi: 10.1097/01.BRS.0000105527.13866.0F. [DOI] [PubMed] [Google Scholar]
- 2.Roughley PJ. Biology of intervertebral disc aging and degeneration: involvement of the extracellular matrix. Spine (Phila Pa 1976) 2004;29:2691–9. doi: 10.1097/01.brs.0000146101.53784.b1. [DOI] [PubMed] [Google Scholar]
- 3.Hayflick L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res. 1965;37:614–36. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 4.Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–96. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 5.Toussaint O, Royer V, Salmon M, Remacle J. Stress-induced premature senescence and tissue ageing. Biochem Pharmacol. 2002;64:1007–9. doi: 10.1016/s0006-2952(02)01170-x. [DOI] [PubMed] [Google Scholar]
- 6.Coppe JP, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–56. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker DJ, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–6. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker DJ, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184–9. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu Y, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14:644–58. doi: 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gruber HE, Ingram JA, Norton HJ, Hanley EN., Jr Senescence in cells of the aging and degenerating intervertebral disc: immunolocalization of senescence-associated beta-galactosidase in human and sand rat discs. Spine. 2007;32:321–7. doi: 10.1097/01.brs.0000253960.57051.de. [DOI] [PubMed] [Google Scholar]
- 12.Le Maitre CL, Freemont AJ, Hoyland JA. Accelerated cellular senescence in degenerate intervertebral discs: a possible role in the pathogenesis of intervertebral disc degeneration. Arthritis Res Ther. 2007;9:R45. doi: 10.1186/ar2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vo N, et al. Accelerated aging of intervertebral discs in a mouse model of progeria. J Orthop Res. 2010;28:1600–7. doi: 10.1002/jor.21153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Q, et al. DNA damage drives accelerated bone aging via an NF-kappaB-dependent mechanism. J Bone Miner Res. 2013;28:1214–28. doi: 10.1002/jbmr.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang D, et al. Spine degeneration in a murine model of chronic human tobacco smokers. Osteoarthritis Cartilage. 2012;20(8):896–905. doi: 10.1016/j.joca.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–21. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 17.Vo NV, et al. Expression and regulation of metalloproteinases and their inhibitors in intervertebral disc aging and degeneration. Spine J. 2013;13:331–41. doi: 10.1016/j.spinee.2012.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niedernhofer LJ, et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–43. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- 19.Chen JH, O S, Hales CN. Methods of cellular senescence induction using oxidative stress. Methods Mol Biol. 2007:178–89. doi: 10.1007/978-1-59745-361-5_14. [DOI] [PubMed] [Google Scholar]
- 20.Dimri GP, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nasto LA, et al. Mitochondrial-derived reactive oxygen species (ROS) play a causal role in aging-related intervertebral disc degeneration. J Orthop Res. 2013;31:1150–7. doi: 10.1002/jor.22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang D, et al. Bupivacaine decreases cell viability and matrix protein synthesis in an intervertebral disc organ model system. Spine J. 2011;11:139–46. doi: 10.1016/j.spinee.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodier F, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–9. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodier F, et al. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci. 2011;124:68–81. doi: 10.1242/jcs.071340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen JH, Ozanne SE, Hales CN. Methods of cellular senescence induction using oxidative stress. Methods Mol Biol. 2007;371:179–89. doi: 10.1007/978-1-59745-361-5_14. [DOI] [PubMed] [Google Scholar]
- 26.Deyo RA, Weinstein JN. Low back pain. N Engl J Med. 2001;344:363–70. doi: 10.1056/NEJM200102013440508. [DOI] [PubMed] [Google Scholar]
- 27.Nakajima EC, Van Houten B. Metabolic symbiosis in cancer: refocusing the Warburg lens. Mol Carcinog. 2013;52:329–37. doi: 10.1002/mc.21863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yingchao Han JT, Hartman Robert A, Dong Qing, Li Hongshuai, McGowan Sara J, Zhao Jing, Sowa Gwendolyn A, Kang James D, Niedernhofer Laura J, Robbins Paul D, Nam NVo. Attenuation of ATM signaling mitigates age-associated spine degeneration. Journal of Annals of the Rheumatic Diseases. 2017 Submitted. [Google Scholar]
- 29.Ross JM, et al. High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc Natl Acad Sci U S A. 2010;107:20087–92. doi: 10.1073/pnas.1008189107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 31.Weber KT, et al. Serum levels of the proinflammatory cytokine interleukin-6 vary based on diagnoses in individuals with lumbar intervertebral disc diseases. Arthritis Res Ther. 2016;18:3. doi: 10.1186/s13075-015-0887-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bachmeier BE, et al. Matrix metalloproteinase expression levels suggest distinct enzyme roles during lumbar disc herniation and degeneration. Eur Spine J. 2009;18:1573–86. doi: 10.1007/s00586-009-1031-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richardson SM, Doyle P, Minogue BM, Gnanalingham K, Hoyland JA. Increased expression of matrix metalloproteinase-10, nerve growth factor and substance P in the painful degenerate intervertebral disc. Arthritis Res Ther. 2009;11:R126. doi: 10.1186/ar2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gruber HE, et al. Production and expression of RANTES (CCL5) by human disc cells and modulation by IL-1-beta and TNF-alpha in 3D culture. Exp Mol Pathol. 2014;96:133–8. doi: 10.1016/j.yexmp.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 35.Johnson ZI, Schoepflin ZR, Choi H, Shapiro IM, Risbud MV. Disc in flames: Roles of TNF-alpha and IL-1beta in intervertebral disc degeneration. Eur Cell Mater. 2015;30:104–16. doi: 10.22203/ecm.v030a08. discussion 116–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang LW, Leonhard-Melief C, Haltiwanger RS, Apte SS. Post-translational modification of thrombospondin type-1 repeats in ADAMTS-like 1/punctin-1 by C-mannosylation of tryptophan. J Biol Chem. 2009;284:30004–15. doi: 10.1074/jbc.M109.038059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stanton H, Melrose J, Little CB, Fosang AJ. Proteoglycan degradation by the ADAMTS family of proteinases. Biochim Biophys Acta. 2011;1812:1616–29. doi: 10.1016/j.bbadis.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 38.Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sivan SS, Wachtel E, Roughley P. Structure, function, aging and turnover of aggrecan in the intervertebral disc. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbagen.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 41.Miyazaki S, et al. Recombinant human SIRT1 protects against nutrient deprivation-induced mitochondrial apoptosis through autophagy induction in human intervertebral disc nucleus pulposus cells. Arthritis Res Ther. 2015;17:253. doi: 10.1186/s13075-015-0763-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.