

Abstract
Cardiac hypertrophy and associated heart fibrosis remain a major cause of death worldwide. Phytochemicals have gained attention as alternative therapeutics for managing cardiovascular diseases. These include the extract from the plant Terminalia arjuna, which is a popular cardioprotectant and may prevent or slow progression of pathological hypertrophy to heart failure. Here, we investigated the mode of action of a principal bioactive T. arjuna compound, arjunolic acid (AA), in ameliorating hemodynamic load-induced cardiac fibrosis and identified its intracellular target. Our data revealed that AA significantly represses collagen expression and improves cardiac function during hypertrophy. We found that AA binds to and stabilizes the ligand-binding domain of peroxisome proliferator-activated receptor α (PPARα) and increases its expression during cardiac hypertrophy. PPARα knockdown during AA treatment in hypertrophy samples, including angiotensin II-treated adult cardiac fibroblasts and renal artery-ligated rat heart, suggests that AA-driven cardioprotection primarily arises from PPARα agonism. Moreover, AA-induced PPARα up-regulation leads to repression of TGF-β signaling, specifically by inhibiting TGF-β-activated kinase1 (TAK1) phosphorylation. We observed that PPARα directly interacts with TAK1, predominantly via PPARα N-terminal transactivation domain (AF-1) thereby masking the TAK1 kinase domain. The AA-induced PPARα-bound TAK1 level thereby shows inverse correlation with the phosphorylation level of TAK1 and subsequent reduction in p38 MAPK and NF-κBp65 activation, ultimately culminating in amelioration of excess collagen synthesis in cardiac hypertrophy. In conclusion, our findings unravel the mechanism of AA action in regressing hypertrophy-associated cardiac fibrosis by assigning a role of AA as a PPARα agonist that inactivates non-canonical TGF-β signaling.
Keywords: angiotensin II, cardiac hypertrophy, collagen, fibroblast, fibrosis, heart failure, transforming growth factor-β (TGF-B), TGF-β-activated kinase 1 (TAK1), arjunolic acid, peroxisome proliferator-activated receptor α (PPARα)
Introduction
Cardiac hypertrophy is accompanied by excess deposition of collagen and other extracellular matrix (ECM)4 proteins in the heart, and this leads to cardiac stiffness and eventual heart failure (1, 2).
Increased hemodynamic load in heart leads to activation of the renin–angiotensin system resulting in heightened local concentration of angiotensinII (AngII), which has been established as a principal causal factor for cardiac hypertrophy and associated fibrosis (3–5). Therefore, ameliorating AngII-induced cardiac fibrosis might be one of the possible measures for prevention of such disease progression with improved efficacy of cardiac performance.
Phytomedicines have been greatly appreciated in recent times as promising candidates toward alternative therapeutic regime in several fibrotic diseases (6, 7). Terminalia arjuna (arjuna), considered as one of the most accepted beneficial plants from ancient times, has also widely been known to have cardiotonic functions (8). Preclinical studies with arjuna extracts as well as clinical trials in the form of combination therapy with standard drugs have shown to be protective in several models of cardiac ailments (9, 10).
Arjunolic acid (AA), a chiral triterpenoid, is one of the principal bioactive components of arjuna extracts. Aqueous extract of arjuna bark containing AA significantly inhibited isoprenaline-induced increase in oxidative stress and also prevented fibrosis without regression of hypertrophy or improvement of cardiac function (11). Purified AA resulted in anti-oxidant, anti-platelet, anti-coagulant, anti-necrotic, anti-apoptotic, free radical-scavenging, anti-inflammatory, hypolipidemic, or hypotensive effects in various cardiac disease models (9, 10, 12, 13). However, the precise molecular mechanism of cardioprotection by AA has still remained elusive. Furthermore, pleiotropic effects of AA make it even harder to annotate specific molecular targets within the cellular milieu.
Thus, the objective of our study was to look into the effect of AA, which is purified and crystallized from ethyl acetate and methanol extracts of the corewood of arjuna plant (14) on cardiac hypertrophy-associated fibrosis, and to seek out its plausible mechanism of action on fibrotic signaling through identification of its specific molecular target.
Our study for the first time establishes PPARα as a molecular target of AA. It is a ligand-dependent transcription factor, and the most abundant PPAR isoform in heart (15). PPARα primarily acts as a modulator of energy metabolism but also acts as an anti-inflammatory agent (16, 17). Growing evidence has shown that PPARα activators are involved in integrating inflammatory and hypertrophic pathways thereby governing the pathological outcome of hypertrophy-associated fibrosis (18–20) in hypertensive rats in vivo. Moreover, PPARα double knock-out mice subjected to transverse aortic constriction (TAC) showed heightened fibrotic and inflammatory gene expressions along with severely aggravated hemodynamic function compared with wild-type mice that has undergone TAC (21).
During pressure overload hypertrophy, autocrine and paracrine actions of locally expressed AngII induce TGF-β signaling in cardiac fibroblasts (2, 3). Upon ligand binding, TGF-β receptor II (TβRII) transphosphorylates TGF-β receptor I (TβRI). In the canonical branch, active TβRI phosphorylates the “suppressor of mothers against decapentaplegic” (SMAD 2/3), which combines with co-SMADs (SMAD 4) for nuclear translocation. Active nuclear SMADs up-regulate transcription of several genes involved in ECM synthesis (3, 22). SMAD3−/− mice have shown reduced cardiac fibrosis after TAC (23). Independent of the SMAD kinase activity, the non-canonical pathway is principally governed by MAPKs, and TAK1 is a principal MAP3K. Phosphorylation-dependent activation of TAK1 requires interaction of its kinase domain with the C terminus of TAK1-binding protein (TAB1) (24, 25). Once activated, TAK1 phosphorylates the inhibitor of κB kinase β (IKKβ) (but not IKKα), MKK4/7, and MKK3/6, which in turn phosphorylate NF-κB, JNK, and p38 MAPK, respectively (25, 26). Ultimately, NF-κB and other transcription factors downstream of JNK and p38 MAPK are activated resulting in transcription of the target genes (27). The importance of the non-canonical branch via TAK1 has also been well-documented in cardiac hypertrophy and fibrosis (28). Dominant-negative TAK1 has been found to inhibit TGF-β-induced hypertrophic events in mouse cardiomyocytes and fibroblasts, including ECM production (29).
The inhibitory effect of PPAR activation upon TGF-β signaling has been extensively documented (30–32). Our work thus aims to study the role of PPARα in AA-driven modulation of the TGF-β axes during cardiac hypertrophy and associated fibrosis. This work identifies the PPARα-TAK1 interaction as a causal factor preventing TAK1 phosphorylation, and it further analyzes the relative strength of different PPARα domains contributing toward inhibition of non-canonical TGF-β axes and regression of subsequent collagen synthesis during cardiac hypertrophy.
Results
Arjunolic acid regresses collagen expression and improves cardiac function during cardiac hypertrophy
AngII treatment in cardiac fibroblasts in vitro (Fig. 1A) and renal artery ligation in vivo (Fig. 1B) confirmed a significant increase in collagen-1 (col-1; 2.29 ± 0.032-fold in vitro and 1.71 ± 0.017-fold in vivo) and collagen-3 (col-3; 1.96 ± 0.035-fold in vitro and 1.76 ± 0.015-fold in vivo) gene expressions compared with respective DMSO-treated control samples as revealed by RT-PCR analyses (Fig. 1, A and B). Hydroxyproline assay also showed significant up-regulation in total secreted collagen content in the fibroblast culture supernatant in vitro (471.99 ± 20.188 ng/ml vis à vis 220.57 ± 10.289 ng/ml culture supernatant; Fig. 1C) as well as significantly increased total left ventricular collagen content in vivo (446.63 ± 12.808 μg/g vis à vis 214.91 ± 18.828 μg/g of wet tissue; Fig. 1D) in hypertrophy samples compared with respective control samples in vitro and in vivo (Fig. 1, C and D).
Figure 1.

AA regresses collagen expression and improves cardiac function during cardiac hypertrophy. A, RT-PCR analyses showing significant increase in collagen-1 (col-1) and collagen-3 (col-3) gene expressions in AngII-treated adult cardiac fibroblasts in vitro compared with control fibroblasts. AA-infused AngII-treated cells showed significant decrease in col-1 and col-3 gene expressions compared with hypertrophied fibroblasts. Rpl-32 was used as internal loading control. Control cells treated with either DMSO or AA yielded similar results. AngII-treated cells were also treated with equivalent amounts of DMSO. n = 10 for each experimental group. All the results were expressed as ±S.E. of three independent experiments. Representative graphs showing relative alterations in collagen gene expressions among different experimental groups. ***, p < 0.001 with respect to DMSO-treated control cells; ###, p < 0.001 with respect to AngII-treated cells. B, RT-PCR analyses showing significant increase in col-1 and col-3 gene expressions in right renal artery-ligated rat heart compared with sham-operated control rat group. AA treatment in ligated animals showed significant decrease in col-1 and col-3 gene expressions compared with ligated animals. Rpl-32 was used as internal loading control. Sham-operated control animals and renal artery-ligated animals were also treated with equivalent amounts of DMSO. n = 7 for each experimental group. All the results were expressed as ±S.E. of three independent experiments. Representative graphs showing relative alterations in collagen gene expressions in different in vivo experimental groups. **, p < 0.01 with respect to sham-operated control rat group; ***, p < 0.001 with respect to sham-operated control rat group; ##, p < 0.01 with respect to renal artery-ligated rat group; ###, p < 0.001 with respect to renal artery-ligated rat group. C, graphical representation of in vitro hydroxyproline assay showing significantly increased collagen content in AngII-treated fibroblast culture supernatant compared with control cells. AA treatment in AngII-treated fibroblasts showing significant decrease in the collagen content in the culture supernatant compared with AngII-treated cells. Control cells treated with either DMSO or AA yielded similar results. AngII-treated cells were also treated with equivalent amounts of DMSO. n = 10 for each experimental group. Results were expressed as ±S.E. of three independent experiments. *, p < 0.05 with respect to control cells; #, p < 0.05 with respect to Ang II-treated cells. D, graphical representation of in vivo hydroxyproline assay showing significantly increased total left ventricular collagen content in ligated rat hearts compared with sham-operated control hearts. AA treatment in ligated rat hearts showed decreased total left ventricular collagen content compared with ligated rat heart. Equivalent amounts of DMSO were administered into sham-operated and renal artery-ligated rat group. n = 7 for each experimental group. Results were expressed as ±S.E. of three independent experiments. **, p < 0.01 with respect to sham-operated control rat group; #, p < 0.05 with respect to renal artery-ligated rat group. E, graphical representation of HW/BW ratio (in milligrams/gram) ratio showing significant increase in renal artery-ligated rat group compared with sham-operated control rat group. AA treatment in ligated animals showed significant down-regulation of HW/BW ratio compared with hypertrophied rat group. Sham-operated and renal artery-ligated rat group were also treated with equivalent amounts of DMSO. n = 7 for each experimental group. Results were expressed as ±S.E. of three independent experiments. **, p < 0.01 with respect to sham-operated control rat group; ##, p < 0.01 with respect to renal artery-ligated rat group. F, graphical representation of CSA measured from H&E-stained heart tissue sections showing significant up-regulation in renal artery-ligated rat heart compared with sham-operated control rat group. AA treatment in ligated animals showed significant down-regulation of CSA compared with ligated rat group. Sham-operated and ligated rats were also treated with equivalent amounts DMSO. n = 7 for each experimental group. Results were expressed as ±S.E. of three independent experiments. ***, p < 0.001 with respect to sham-operated control rat group; ###, p < 0.001 with respect to renal artery-ligated rat group. G, RT-PCR analyses showing significant increase in hypertrophy marker gene expressions, namely anf, β-mhc, and acta1 in renal artery-ligated rat hearts compared with sham-operated control hearts. AA treatment in ligated animals showed significant down-regulation of all these gene expressions compared with ligated rat hearts. Sham-operated control and renal artery-ligated animals were also treated with an equivalent amounts of DMSO. Rpl-32 was used as internal loading control. n = 7 for each experimental group. Results were expressed as ±S.E. of three independent experiments. Representative graphs showing relative alterations in the hypertrophy marker gene expressions among different experimental groups. ***, p < 0.001 with respect to sham-operated control rat group; **, p < 0.01 with respect to sham-operated control rat group; ###, p < 0.001 with respect to renal artery-ligated rat group; ##, p < 0.01 with respect to renal artery-ligated rat group. H, M-mode echocardiographic analyses from parasternal short axis view at papillary muscle level graphically representing decreased %FS and increased LvIDd in renal artery-ligated rat group compared with sham-operated control rat group. AA treatment in ligated animals showed restored %FS and lowered LvIDd in ligated rats compared with the hypertrophied rat group. Sham-operated control and renal artery-ligated animals were also treated with equivalent amounts of DMSO. n = 7 for each experimental group. Results were expressed as ±S.E. of three independent experiments. **, p < 0.01 with respect to sham-operated control rat group; ##, p < 0.01 with respect to renal artery-ligated rat group; #, p < 0.05 with respect to renal artery-ligated rat group.
AA treatment in AngII-pretreated cardiac fibroblasts (Fig. 1A) and renal artery-ligated rat heart (Fig. 1B) showed significant down-regulation of col-1 (2.51 ± 0.025-fold in vitro and 2.20 ± 0.022-fold in vivo) and col-3 (1.77 ± 0.050-fold in vitro and 1.86 ± 0.016-fold in vivo) gene expressions compared with respective DMSO-treated hypertrophy samples in vitro and in vivo (Fig. 1, A and B). Hydroxyproline assay further revealed that AA treatment during hypertrophy leads to significant reduction in total secreted collagen content in fibroblast culture supernatant (301.49 ± 22.191 ng/ml vis à vis 471.99 ± 20.188 ng/ml of culture supernatant; Fig. 1C) as well as total left ventricular collagen content (315.57 ± 28.761 μg/g vis à vis 446.63 ± 12.808 μg/g of wet tissue; Fig. 1D) compared with respective hypertrophy samples in vitro and in vivo (Fig. 1, C and D).
In vitro adult cardiac fibroblasts were also treated with equivalent amounts of AA. There was no significant difference in col-1 and col-3 gene expressions (Fig. 1A) or total secreted collagen content in the culture supernatant (Fig. 1C) of AA-treated control fibroblasts compared with fibroblasts treated with equivalent amounts of DMSO.
Hypertrophy was confirmed in vivo by significantly increased heart weight (in milligrams) to body weight (in grams) ratio (HW/BW; 1.71 ± 0.091-fold; Fig. 1E), increased cardiomyocyte cross-sectional area (CSA) (627.42 ± 21.197 μm2 vis à vis 379.48 ± 10.606 μm2; Fig. 1F), and increased expression of atrial natriuretic factor (anf) (3.27 ± 0.019-fold), β-myosin heavy chain (β-mhc) (2.54 ± 0.092-fold), and skeletal α-actin (acta1) (1.89 ± 0.024-fold) genes (Fig. 1G) in ligated rats compared with sham-operated control rat heart. AA-mediated regression of hypertrophy was confirmed by the significantly reduced HW/BW ratio (1.28 ± 0.067-fold; Fig. 1E), significantly decreased CSA (505.81 ± 20.326 μm2 vis à vis 627.42 ± 21.197 μm2; Fig. 1F), and significant down-regulation in anf (1.69 ± 0.009-fold), β-mhc (2.38 ± 0.086-fold), and acta1 (1.57 ± 0.017-fold) gene expressions (Fig. 1G) in AA-treated hypertrophy samples compared with renal artery-ligated rat heart (Fig. 1, E–G).
Hypertrophic induction significantly reduced cardiac functional efficacy in ligated rats (fractional shortening (%FS): 38.53 ± 1.781%, and left ventricular internal diastolic diameter (LvIDd): 5.65 ± 0.273 mm) compared with sham-operated control rats (%FS: 57.51 ± 1.026%, and LvIDd: 3.56 ± 0.062 mm) as observed by M-mode echocardiography in transthoracic parasternal short axis view at papillary muscle level. Furthermore, significant restoration of such compromised cardiac function was evident in AA-treated ligated rats showing increased %FS (46.67 ± 1.504%) and decreased LvIDd (4.01 ± 0.096 mm) compared with the aforementioned hypertrophy samples in vivo (Fig. 1H).
The viability measured was more than 90% after AA treatment in all hypertrophied fibroblasts as checked by cell viability assay (data not shown). The effective doses of AA for both in vitro and in vivo experiments were determined by maximal regression of collagen during hypertrophy via dose-dependent study (data not shown).
AA directly interacts with PPARα
Fluorescence and circular dichroism (CD) titrations of AA with PPARα
Steady-state fluorescence titrations of PPARα with increasing concentrations of AA showed no apparent shifts around 330 nm (λmax), indicative of unaltered tertiary architecture. However, the corresponding hypochromic shifts observed indicated binding of AA to PPARα (Fig. 2A, panel i). The binding was also apparent when monitored through CD analyses where similar titrations induced marginal secondary structure perturbations (near 55 μm) in this predominantly α-helical protein. (Fig. 2A, panel ii).
Figure 2.
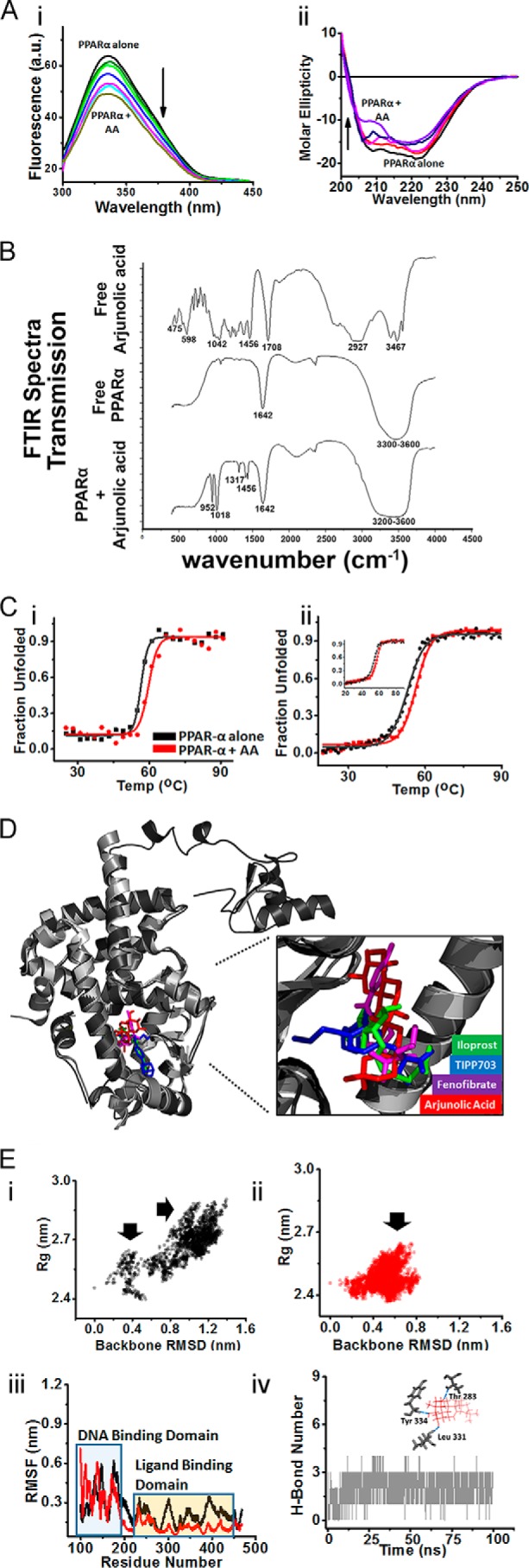
Analyses of interaction between AA and PPARα. A, fluorescence and CD titrations of AA with PPARα. Panel i, titration of 8 μm PPARα with increasing AA concentrations up to 55 μm, monitored by fluorescence spectroscopy. Panel ii, titration of 8 μm PPARα with increasing AA concentrations up to 55 μm, monitored by CD spectroscopy. All titrations were carried out in 20 mm Na3(PO)4, 150 nm NaCl, pH 8.0, buffer at 298 K. B, overlaid FTIR spectra of free AA, free PPARα, and AA-PPARα complex. C, thermal melting study. Panel i, FTS assay to determine melting temperature of PPARα alone (black) and in the presence of AA (red). Panel ii, CD thermal melting of PPARα alone (black) and in presence of AA (red) probed by monitoring changes θ218 (α-helical transitions) and θ222 (inset, β-transitions). Normalized data are plotted as percent unfolding induced by temperature increments. All experiments were carried out in 20 mm Na3(PO)4, 150 nm NaCl, pH 8.0, buffer at 298 K. D, tertiary structure alignment of PPARα (modeled, black), bound to AA (red), iloprost (green), TIPP-703 (blue), and fenofibrate (purple). Zoomed part shows superposition of AA with other agonists occupying identical ligand-binding pocket in PPARα. E, projections of MD simulations and thermal melting profiles for PPARα alone (black) and in bound state with AA (red). Panels i–ii, distributions of backbone RMSD and Rg values averaged over whole 100-ns trajectory for PPARα alone (panel i) and in the presence of AA (panel ii). Arrows indicate major areas of distribution over two components. Panel iii, RMSF analyses of PPARα alone and in the presence of AA averaged over last 10 ns of trajectory. Panel iv, H-bond network for whole trajectory showing persistence of H-bonds between PPARα and AA. Inset shows three H-bonds between AA and Thr-283, Leu-331, and Tyr-334 residues.
Fourier transform infrared spectroscopy (FTIR) analysis
FTIR spectrum of AA supported previous reports (14) showing characteristic –OH, C–H, and C=O stretches at 3467, 2927, and 1708 cm−1 positions, respectively. Characteristic C–O stretching vibrations were also obtained at 475, 598, 1042, and 1456 cm−1. In vitro translated and purified PPARα revealed intense absorption characteristic of amide-I and amide A bands of the secondary structure of a protein (33, 34). The peak at 1642 cm−1 position represented the amide-I bond. The amide-A band (at 3300–3600 cm−1), representing the N–H stretching vibrations, was also obtained. Successful interaction between PPARα and AA by in vitro interaction assay was shown by the absence of peaks at 3467, 2927, and 1708 cm−1 in the complex compared with free AA. Peaks characteristic of free AA at 952, 1018, 1317, 1412, and 1456 cm−1 and those characteristic of free in vitro translated PPARα at 1642 cm−1 and in the range of 3300–3600 cm−1 were maintained in the complex when compared with free AA or free in vitro translated protein, respectively (Fig. 2B).
AA stabilizes tertiary and secondary architecture of PPARα: Thermal melting studies
The stabilization of the tertiary and secondary architecture of PPARα by AA was assessed by monitoring changes in melting temperature (Tm) (Fig. 2C, panels i and ii). In fluorescence thermal shift (FTS) assays, PPARα, bound to AA, showed higher Tm (59.5 ± 0.5 °C) compared with PPARα alone (56.4 ± 0.2 °C) (Fig. 2C, panel i). A similar trend was observed through CD studies where changes in both θ218 (α-helical content, Fig. 2C, panel ii) and θ222 (β content, inset Fig. 2C, panel ii) with respect to temperature clearly showed a higher Tm (56.4 ± 0.1 °C) upon AA binding compared with control (53 ± 0.2 °C).
AA binds to the ligand-binding domain (LBD) of PPARα: Molecular modeling and molecular docking studies
Rattus norvegicus PPARα protein model was built, and the integrity of the model was confirmed through all atomistic molecular dynamics simulation. To gain atomistic details, binding mode of AA with rat-PPARα (UniProtKB-P37230) was compared with the other known agonists, namely iloprost, TIPP-703, and fenofibrate.
PPARα-AA complex obtained through docking via “binding site prediction and shape-based ligand matching” (BSP-SLIM) tool showed binding score close to previously studied synthetic agonists such as iloprost, TIPP-703, and fenofibrate. Additionally, the calculated binding free energy (−6.3 kcal/mol) matched with its experimentally determined energy obtained from fluorescence and CD titrations (−6.2 kcal/mol). Interestingly, AA was found to bind to the identical pocket (formed of Met-220, Cys-276, Ser-280, Thr-283, Phe-318, Leu-331, Ile-332, Leu-324, Tyr-334, and His-440) for which binding of all other PPARα agonists have been reported, including the predocked complex with fenofibrate. With root mean square deviation (RMSD) of ∼0.62 Å among all aligned structures, it showed structural superposition of AA with other agonists, confined to the same ligand-binding site. This was further confirmed through tertiary structure alignment of modeled PPARα (bound to AA and fenofibrate) with known crystal structures of PPARα in complex with agonists such as iloprost and TIPP-703 (Fig. 2D and supplemental Table 1).
AA stabilizes the LBD of PPARα
For all atomistic molecular dynamics (MD) simulations, the molecular dynamics simulation trajectories of both free and AA-bound PPARα were analyzed for changes in backbone RMSD, gyration radius (Rg), root mean square fluctuations in Cα atoms (RMSF), as well as protein–ligand H-bond network occupancy. In simulations of PPARα alone, projection of variations in backbone RMSD versus Rg showed dual distribution of species along both components (Fig. 2E, panel i). The projections were, however, dominated by the species with high RMSD and Rg compared with initial model. However, I the presence of AA, only moderate variations were observed along both components (Fig. 2E, panel ii). This system showed single distribution with dominant population close to initial starting conformation (low RMSD and marginal variations in Rg). These variations were identically reflected in RMSF analysis of both systems (Fig. 2E, panel iii). Marginal changes in Cα fluctuations were observed in residues of the DNA-binding domain (DBD) of PPARα. However, the LBD showed much reduced Cα fluctuations in AA-bound PPARα complexes compared with PPARα alone, indicating stabilization of LBD by AA. H-bond network analysis showed persistence of at least three H-bonds per time frame between AA and Thr-283, Leu-331, and Tyr-334 of PPARα (Fig. 2E, panel iv).
AA-induced PPARα transcriptional activation results in increased pparα gene expression in an autoregulatory loop
Effect of AA upon PPARα-mediated autoregulation of PPARα transcription was analyzed by chromatin immunoprecipitation (ChIP) using anti-PPARα antibody coupled to qRT-PCR analyses of PPAR-response element (PPRE) within the PPARα promoter, both in vitro and in vivo. DMSO and non-specific siRNA (NS siRNA)-treated hypertrophy samples showed significant down-regulation in binding of PPARα upon PPRE sequence within the PPARα promoter compared with DMSO and NS siRNA-treated control samples (1.99 ± 0.078-fold in vitro and 2.75 ± 0.199-fold in vivo). However, AA treatment in NS siRNA-treated hypertrophied groups showed significant fold enrichment in binding of PPARα to the PPRE (3.00 ± 0.075-fold in vitro and 3.27 ± 0.237-fold in vivo) compared with DMSO and NS siRNA-treated hypertrophy samples. PPARα knockdown in AA-treated hypertrophied groups showing significantly lowered binding (6.49 ± 0.520-fold in vitro and 8.17 ± 0.749-fold in vivo) compared with AA-treated hypertrophy samples pretreated with NS siRNA were used as negative controls (Fig. 3A). Also, qRT-PCR analyses both in vitro and in vivo indicated significant down-regulation in pparα gene expression in the hypertrophy samples compared with the respective control samples (1.81 ± 0.026-fold in vitro and 3.37 ± 0.154-fold in vivo) and a significant increase in pparα gene expression in AA-treated hypertrophy samples compared with the respective hypertrophied groups (3.45 ± 0.131-fold in vitro and 3.94 ± 0.179-fold in vivo). PPARα knockdown in AA-treated hypertrophied groups showing significantly down-regulated pparα gene expression (5.49 ± 0.208-fold in vitro and 7.04 ± 0.609-fold in vivo) compared with the respective AA-treated hypertrophy samples were used as negative controls (Fig. 3A).
Figure 3.

AA treatment during hypertrophy increases PPARα expression in an autoregulatory loop. A, ChIP assay with anti-PPARα antibody followed by qRT-PCR analyses of the PPRE showing relative binding of PPARα to the PPRE within PPARα promoter among different experimental groups both in vitro and in vivo are represented graphically on logarithmic scale. Hypertrophy samples showed significantly down-regulated-fold enrichment in binding of PPARα to PPRE within the PPARα promoter compared with respective controls. AA treatment in hypertrophy samples further showed significantly increased-fold enrichment of the same, compared with hypertrophy samples. PPARα siRNA treatment in AA-treated hypertrophy groups showing down-regulated-fold enrichment in binding of PPARα to the PPRE compared with the AA-treated hypertrophy samples were used as negative controls. Control and hypertrophy samples in vitro and in vivo were treated with equivalent amounts of DMSO and non-specific (NS) siRNA. AA-treated hypertrophy samples were treated with equivalent amounts of NS siRNA. Chromatin from each experimental group immunoprecipitated with anti-IgG antibody were used for normalization. n = 3 both in vitro and in vivo. Results were analyzed by one-way ANOVA followed by Tukey's post hoc test and expressed as ±S.E. of three independent experiments. **, p < 0.01 with respect to control samples; ###, p < 0.001 with respect to hypertrophy samples; ↑↑, p < 0.01 with respect to AA-treated hypertrophy samples in vitro and/or in vivo. Corresponding changes in pparα mRNA expressions between different experimental groups as observed by qRT-PCR are represented graphically on logarithmic scale. Rpl-32 was used as internal loading control. n = 3 both in vitro and in vivo. Results were analyzed by ANOVA followed by Tukey's post hoc test and expressed as ±S.E. of three independent experiments. **, p < 0.01 with respect to control samples; #, p < 0.05 with respect to hypertrophy samples; ##, p < 0.01 with respect to hypertrophy samples; ↑, p < 0.05 with respect to AA-treated hypertrophy samples; ↑↑, p < 0.01 with respect to AA-treated hypertrophy samples in vitro and/or in vivo. B, Western blot analyses revealed significant decrease in PPARα protein expression during hypertrophy compared with respective control groups that again showed significant recovery in AA-treated hypertrophy samples compared with respective hypertrophy groups in vitro and in vivo. Successful knockdown of PPARα protein expression was also confirmed by PPARα siRNA pretreatment in AA-treated hypertrophy samples. GAPDH was used as internal loading control. Control and hypertrophy samples were treated with equivalent amounts of DMSO and NS siRNA. AA-treated hypertrophy samples were also treated with NS siRNA. Results were analyzed by ANOVA followed by Tukey's post hoc test and expressed as ±S.E. of three independent experiments. n = 10 in vitro, n = 7 in vivo for each experimental group. Representative graphs showing relative changes in expression of PPARα among different experimental groups on logarithmic scale. **, p < 0.01 with respect to control samples; #, p < 0.05 with respect to hypertrophy samples; ##, p < 0.01 with respect to hypertrophy samples; ↑↑, p < 0.01 with respect to AA-treated hypertrophy samples in vitro and/or in vivo. C, Western blot analyses showing time-dependent increase in PPARα expression in AA-treated hypertrophied fibroblasts compared with AngII-treated cells at respective time points under study. GAPDH was used as internal loading control. Results were analyzed by ANOVA followed by Tukey's post hoc test and expressed as ±S.E. of three independent experiments. n = 5 for each experimental group. Representative graphs showing relative changes in PPARα expressions at respective time points among different groups under study. C, control fibroblasts; A, AngII-treated fibroblasts at different time points; A + AA, AngII-treated fibroblasts at different time points, treated along with AA. C and A cells were also treated with equivalent concentration of DMSO. ρρ, <0.01 with respect to A samples at the 3-h time point; øø, <0.01 with respect to A + AA samples at the 3-h time point.
AA increases PPARα protein expression during cardiac hypertrophy
Significant reduction of PPARα protein expression was observed in AngII-treated fibroblasts (2.05 ± 0.088-fold in vitro) and renal artery-ligated rat heart (1.98 ± 0.059-fold in vivo) compared with the respective control groups as analyzed by Western blot. The AA-treated hypertrophy samples showed significant up-regulation in PPARα expression (2.12 ± 0.127-fold in vitro and 2.05 ± 0.031-fold in vivo) compared with the hypertrophy groups (Fig. 3B). Successful knockdown of PPARα expression by PPARα siRNA treatment in AA-treated hypertrophy samples was also confirmed by Western blot analyses (3.18 ± 0.191-fold down-regulation in vitro and 1.67 ± 0.025-fold down-regulation in vivo compared with respective AA-treated hypertrophy samples) (Fig. 3B). Moreover, time-point analyses showed significant up-regulation of PPARα expression in AngII-treated fibroblasts when treated along with AA compared with only AngII treatment at the respective time points under study (Fig. 3C).
AA-mediated up-regulation of PPARα results in regression of cardiac hypertrophy-associated fibrosis
AA-treated hypertrophy samples pretreated with PPARα siRNA resulted in significantly increased expression of col-1 (2.48 ± 0.137-fold in vitro and 2.58 ± 0.058-fold in vivo) and col-3 (3.19 ± 0.06-fold in vitro and 3.42 ± 0.103-fold in vivo) genes compared with respective AA-treated hypertrophy groups as revealed by qRT-PCR (Fig. 4A).
Figure 4.

AA-mediated up-regulation of PPARα results in regression of cardiac fibrosis and improvement of cardiac function. A, graphical representation of qRT-PCR data showing significant down-regulation of col-1 and col-3 gene expressions in AA-treated hypertrophy samples compared with hypertrophy groups. PPARα siRNA-pretreated hypertrophied groups treated along with AA showed significant recovery in these gene expressions compared with AA-treated hypertrophy samples both in vitro and in vivo. Control and hypertrophy samples were treated with equivalent amounts of DMSO and NS siRNA. AA-treated hypertrophy samples were also treated with equivalent amounts of NS siRNA. Rpl-32 was used as internal reference control. Results were analyzed by ANOVA followed by Tukey's post hoc test and expressed as ±S.E. of three independent experiments. n = 10 in vitro and n = 7 in vivo for each experimental group. **, p < 0.01 with respect to control samples; ***, p < 0.001 with respect to control samples; ##, p < 0.01 with respect to hypertrophy samples; ↑, p < 0.05 with respect to AA-treated hypertrophy samples; ↑↑↑, p < 0.001 with respect to AA-treated hypertrophy samples in vitro and/or in vivo. B, graphical representation of in vitro hydroxyproline assay showing significantly restored collagen content in the culture supernatant of PPARα siRNA-pretreated hypertrophied fibroblasts treated along with AA compared with AA-treated hypertrophied cells in vitro. Control and AngII-treated cells were treated with equivalent amounts of DMSO and NS siRNA. AA-treated hypertrophied cells were also treated with equivalent amounts of NS siRNA. Results were analyzed by ANOVA followed by Tukey's post hoc test and expressed as ±S.E. of three independent experiments. n = 10 for each experimental group. **, p < 0.01 with respect to control cells; #, p < 0.05 with respect to AngII-treated cells; ↑, p < 0.05 with respect to AngII-treated cells treated along with AA. C, graphical representation of in vivo hydroxyproline assay showing significant recovery in total left ventricular collagen content in PPARα siRNA-pretreated AA-infused hypertrophied heart compared with AA-treated renal artery-ligated rat heart in vivo. Sham-operated control rats and renal artery-ligated rats were also treated with equivalent amounts of DMSO and NS siRNA. AA-treated ligated rats were also treated with equivalent amounts of NS siRNA. Results were analyzed by ANOVA followed by Tukey's post hoc test and expressed as ±S.E. of three independent experiments. n = 7 for each experimental group. **, p < 0.01 with respect to sham-operated control rat group; ##, p < 0.01 with respect to renal artery-ligated rat group; ↑, p < 0.05 with respect to AA-treated renal artery-ligated rat group. D, micrographs of Massons' trichrome staining showing significantly decreased %CVF in AA-treated hypertrophy samples compared with renal artery-ligated rat heart, which again showed significant recovery in AA-treated ligated samples pretreated with PPAR siRNA. S, sham-operated control group; L, ligated rat group; L + AA, AA-treated ligated rat group; L + AA + PPARα si, PPARα siRNA-infused AA-treated ligated rat group. S and L groups were treated with equivalent amounts of DMSO and NS siRNA. L + AA group animals were also treated with equivalent amounts of NS siRNA. Results were analyzed by ANOVA followed by Tukey's post hoc test and expressed as ±S.E. of three independent experiments. n = 7 for each group. (Scale bar, 40 μm, magnification = ×60). **, p < 0.01 with respect to sham-operated control samples; ##, p < 0.01 with respect to renal artery-ligated hypertrophy samples; ↑↑, p < 0.01 with respect to renal artery-ligated samples treated with AA. E, M-mode echocardiographic analyses of PPARα siRNA-pretreated ligated rats treated with AA showing significant decrease in %FS and significant increase in LvIDd compared with AA-treated ligated rats. Sham controls and renal artery-ligated rats were treated with equivalent amounts of DMSO and NS siRNA. AA-treated ligated rats were also treated with equivalent amounts of NS siRNA. n = 7 for each group. Results were analyzed by ANOVA followed by Tukey's post hoc test and expressed as ±S.E. of three independent experiments. **, p < 0.01 with respect to sham-operated control rat group; ##, p < 0.01 with respect to renal artery-ligated rat group; ↑↑, p < 0.01 with respect to AA-treated renal artery-ligated rat group.
Hydroxyproline assay also revealed significant recovery of secreted collagen content in fibroblast culture supernatant (420.72 ± 28.943 ng/ml vis à vis 296.84 ± 20.981 ng/ml of culture supernatant; Fig. 4B) as well as total left ventricular collagen content (418.82 ± 21.927 μg/g vis à vis 262.29 ± 18.828 μg/g of wet tissue; Fig. 4C) during PPARα siRNA treatment in AA-treated hypertrophied groups compared with respective AA-treated hypertrophy samples in vitro and in vivo. Collagen volume fraction (%CVF) showed significant down-regulation (1.82 ± 0.153-fold) during AA treatment in hypertrophied heart compared with ligated heart tissue. PPARα knockdown in AA-treated ligated rats showed significant increase in %CVF (1.57 ± 0.133-fold) compared with AA-treated in vivo hypertrophy samples (Fig. 4D).
Pretreatment of PPARα siRNA in AA-treated ligated rats also showed deterioration of cardiac function as revealed by decreased %FS and increased LvIDd compared with that of AA-treated hypertrophy groups (%FS: 37.83 ± 1.186% vis à vis 49.39 ± 1.618%, and LvIDd: 5.57 ± 0.144 mm vis à vis 4.79 ± 0.310 mm) by M-mode echocardiography (Fig. 4E). Furthermore, the mean values of the other echocardiographic parameters such as left ventricular anterior and posterior wall diameter both in diastole and systole (LvAWDd, LvAWDs, LvPWDd, LvPWDs), left ventricular volume in diastole and systole (represented by stroke volume (SV)), as well as percentage of left ventricular ejection fraction (EF) measured from all the in vivo experimental groups are summarized in supplemental Fig. S1A with representative images of echocardiographic analyses in supplemental Fig. S1B.
AA-mediated up-regulation of PPARα specifically targets TAK1 to inhibit TGF-β signaling during cardiac hypertrophy
AA treatment in AngII-treated fibroblasts or renal artery-ligated rats significantly regressed expressions of TβRI (1.85 ± 0.007-fold in vitro and 1.94 ± 0.017-fold in vivo), TβRII (1.42 ± 0.026-fold in vitro and 2.09 ± 0.016-fold in vivo), phospho/total SMAD 2 (1.73 ± 0.015-fold in vitro and 2.15 ± 0.025-fold in vivo), phospho/total SMAD 3 (1.36 ± 0.016-fold in vitro and 4.06 ± 0.034-fold in vivo), and phospho/total TAK1 (5.23 ± 0.356-fold in vitro and 7.93 ± 0.372-fold in vivo) compared with hypertrophy samples as revealed by Western blot analyses (Fig. 5A and supplemental Fig. S2A). However, AA treatment in TGF-β-treated cardiac fibroblasts showed no significant alterations in TβRI, TβRII phospho/total SMAD 2 and phospho/total SMAD 3 expression levels, with significantly regressed phospho/total TAK1 level (1.47 ± 0.075-fold) compared with TGF-β-treated cells (Fig. 5B, supplemental Fig. S2B). PPARα siRNA pretreatment in AA-treated hypertrophied fibroblasts or renal artery-ligated rats significantly restored expressions of all these proteins (TβRI, in vitro: 1.39 ± 0.018-fold, and in vivo: 2.76 ± 0.013-fold; TβRII, in vitro: 2.05 ± 0.029-fold, and in vivo: 2.04 ± 0.051-fold; phospho/total SMAD 2, in vitro: 1.53 ± 0.058-fold, and in vivo: 1.37 ± 0.022-fold; phospho/total SMAD 3, in vitro: 1.49 ± 0.016-fold, and in vivo: 3.01 ± 0.047-fold; phospho/total TAK1, in vitro: 4.92 ± 0.309-fold, and in vivo: 10.00 ± 0.280-fold) compared with respective AA-treated hypertrophy samples (Fig. 5A and supplemental Fig. S2A). However, PPARα knockdown in TGF-β-treated fibroblasts during AA treatment showed a significant increase in phospho/total TAK1 level (1.75 ± 0.069-fold) with no significant change in TβRI, TβRII phospho/total SMAD 2, and phospho/total SMAD 3 expression levels compared with TGF-β-infused cells treated along with AA (Fig. 5B and supplemental Fig. S2B).
Figure 5.
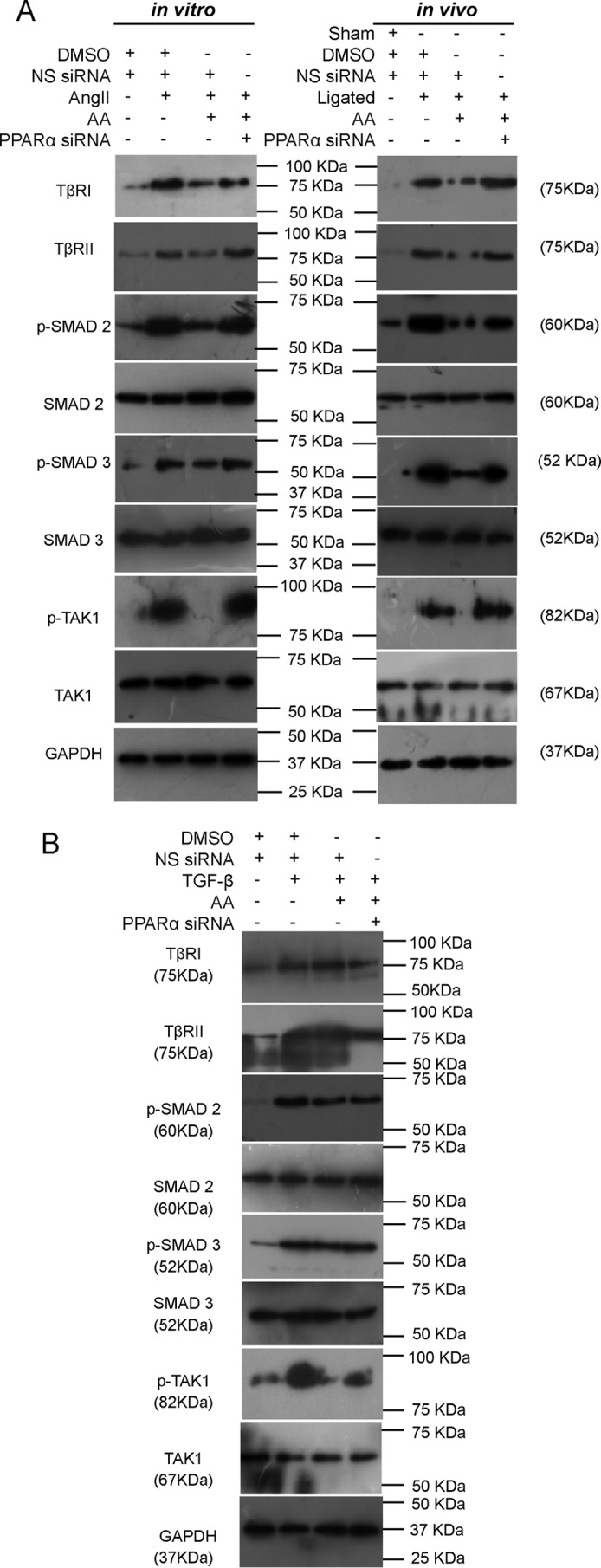
AA-mediated up-regulation of PPARα specifically targets TAK1 for regression of TGF-β signaling during cardiac hypertrophy. A, Western blot analyses showing significantly reduced TβRI, TβRII, phospho/total levels of SMAD 2, SMAD 3, and TAK1 in AA-treated hypertrophy samples compared with hypertrophy groups both in vitro and in vivo. Significantly restored levels of all these proteins were observed in AA-treated hypertrophy groups pretreated with PPARα siRNA compared with AA-treated hypertrophy samples. Control and hypertrophy samples were treated with equivalent amounts of DMSO and NS siRNA. AA-treated hypertrophy samples were also treated with equivalent amounts of NS siRNA. GAPDH was used as internal loading control. n = 10 in vitro and n = 7 in vivo for each experimental group. B, Western blot analyses showing significantly decreased phospho/total TAK1 level during AA treatment in TGF-β-treated cardiac fibroblasts compared with only TGF-β-treated cells. PPARα knockdown in TGF-β- and AA-infused cells showed significant recovery in phospho/total TAK1 level compared with AA-treated fibroblasts pretreated with TGF-β. Expressions of TβRI, TβRII, and phospho/total levels of SMAD 2 and SMAD 3 remained unaltered in these experimental groups. Control and TGF-β-treated fibroblasts were also treated with equivalent amounts of DMSO and NS siRNA. AA-treated fibroblasts pretreated with TGF-β were also treated with equivalent amounts of NS siRNA. GAPDH was used as internal loading control. n = 5 for each group.
AA-mediated up-regulation of PPARα inhibits non-canonical TGF-β axes during hypertrophy
Significant down-regulation of non-canonical TGF-β pathway intermediates, downstream to TAK1, namely phospho/total IKKβ (1.99 ± 0.045-fold in vitro and 1.77 ± 0.041-fold in vivo), phospho/total NF-κBp65 (4.76 ± 0.063-fold in vitro and 2.21 ± 0.096-fold in vivo), phospho/total p38 MAPK (1.57 ± 0.029-fold in vitro and 1.30 ± 0.025-fold in vivo), and phospho/total JNK (1.26 ± 0.050-fold in vitro and 1.21 ± 0.060-fold in vivo), were observed due to AA treatment in hypertrophied groups compared with respective hypertrophy samples as shown by Western blot analyses. However, PPARα siRNA treatment showed significant recovery of phospho/total IKKβ (2.61 ± 0.046-fold in vitro and 1.73 ± 0.014-fold in vivo), phospho/total NF-κBp65 (3.58 ± 0.025-fold in vitro and 3.18 ± 0.119-fold in vivo), phospho/total p38 MAPK (2.23 ± 0.018-fold in vitro and 1.46 ± 0.045-fold in vivo), and phospho/total JNK (1.20 ± 0.050-fold in vitro and 1.21 ± 0.016-fold in vivo) levels compared with AA-treated hypertrophy samples in vitro and in vivo (Fig. 6A and supplemental Fig. S2C).
Figure 6.

AA-mediated regression of collagen gene expression involves PPARα-dependent inactivation of non-canonical TGF-β signaling. A, Western blot analyses showing significantly reduced phospho/total levels of IKKβ, NF-κBp65, p38 MAPK, and JNK in AA-treated hypertrophy samples compared with hypertrophy groups both in vitro and in vivo. Significantly restored levels of all these proteins were observed during PPARα knockdown in AA-treated hypertrophied groups compared with AA-treated hypertrophy samples. Control and hypertrophy groups were also treated with equivalent amounts of DMSO and NS siRNA. AA-treated hypertrophy samples were treated with equivalent amounts of NS siRNA. GAPDH was used as internal loading control. n = 10 in vitro, n = 7 in vivo for each experimental group. B, graphical representation of qRT-PCR analyses showing significant down-regulation of col-1 and col-3 gene expressions in AngII-induced fibroblasts during knockdown of either TAK1 or NF-κBp65 or p38 MAPK via specific siRNA treatments compared with hypertrophied fibroblasts. JNK-specific siRNA treatment in AngII-treated fibroblasts showed no significant regression of collagen gene expression compared with AngII-treated cells. Control and AngII-treated cells were also treated with equivalent amounts of NS siRNA. Rpl32 was used as internal reference control. Results were analyzed by ANOVA followed by Tukey's post hoc test and expressed as ±S.E. of three independent experiments. n = 5 for each group. **, p < 0.01 with respect to control cells; ***, p < 0.001 with respect to control cells; #, p < 0.05 with respect to AngII-treated cells; ##, p < 0.01 with respect to AngII-treated cells. C, dual-luciferase assay showing significant increase in the Col1a1 promoter activity in AngII-treated fibroblasts compared with control fibroblasts. AA treatment in hypertrophied fibroblasts showed significant reduction in Col1a1 promoter activity compared with AngII-treated fibroblasts that again showed significant restoration in AA-treated hypertrophied fibroblasts pretreated with PPARα siRNA. Reduced Col1a1 promoter activity shown in NF-κBp65 siRNA pretreated hypertrophied cells compared with AngII-treated cells was used as negative control. Control and AngII-treated cells were also treated with NS siRNA with or without DMSO. AA-treated hypertrophy samples were also treated with equivalent amounts of NS siRNA. Results were normalized by Renilla luciferase activity in all the treatment groups. Results were analyzed by ANOVA followed by Tukey's post hoc test and expressed as ±S.E. of three independent experiments. n = 5 for each group. **, p < 0.01 compared with DMSO and NS siRNA-treated control cells; ρρ, p < 0.01 compared with NS siRNA-treated control cells; ##, p < 0.01 compared with DMSO and NS siRNA-infused AngII-treated cells; ↑↑, p < 0.01 with respect to AA-treated hypertrophied cells pretreated with NS siRNA; йй, p < 0.01 with respect to AngII-treated cells pretreated with NS siRNA.
Inhibition of either TAK1 or its downstream signaling intermediates results in regression of collagen gene expression in AngII-treated cardiac fibroblasts
Significant down-regulation of collagen gene expressions was observed due to siRNA-mediated knockdown of TAK1 (3.71 ± 0.141-fold for col-1 and 2.24 ± 0.072-fold for col-3), NF-κBp65 (3.46 ± 0.063-fold for col-1 and 2.06 ± 0.125-fold for col-3), or p38 MAPK (1.92 ± 0.094-fold for col-1 and 1.57 ± 0.056-fold for col-3) in AngII-treated fibroblasts compared with NS siRNA-treated hypertrophied cells as revealed by qRT-PCR analyses (Fig. 6B). JNK-specific siRNA, however, had no inhibitory effect upon AngII-induced collagen gene expression. Successful knockdown of TAK1, NF-κBp65, p38 MAPK, and JNK by pretreatment with the respective siRNAs in AngII-treated fibroblasts was confirmed by Western blot analyses (supplemental Fig. S3).
NF-κBp65 activity on the collagen1 (Col1a1) promoter was analyzed by dual-luciferase assay showing a significant increase in luciferase activity (2.08 ± 0.120-fold) of the Col1a1 promoter in AngII-treated cardiac fibroblasts compared with respective control cells. Furthermore, NF-κBp65-driven Col1a1 promoter activity was found to be significantly down-regulated (2.03 ± 0.032-fold) by AA treatment in AngII-treated cells compared with hypertrophied fibroblasts. PPARα knockdown in AA-infused AngII-treated fibroblasts significantly restored (1.92 ± 0.058-fold) such promoter activity compared with AA-treated hypertrophied cells. NF-κBp65 knockdown in AngII-treated cells showing down-regulated NF-κBp65 activity (2.14 ± 0.081-fold) on the Col1a1 promoter compared with AngII-treated cells pretreated with NS siRNA was used as negative control (Fig. 6C).
Analyses of interaction between PPARα and TAK1
Molecular modeling study
R. norvegicus TAK1 protein model was built, and integrity of the model was confirmed through all atomistic molecular dynamics simulations (supplemental Fig. S4).
Molecular docking study
For docking between PPARα and TAK1, the interfacial interacting residues between these two proteins were identified using CPORT to limit the plausible protein-protein docking landscape. Using HADDOCK for docking, 23 clusters of the 296 predicted structures were found that represented 70.4% of the water-refined models. The best conformation in the top cluster according to best fit HADDOCK score revealed not only that PPARα and TAK1 extensively interact via inter-protein hydrogen bonds, but that it also gives an insight into the possible binding interface. The model with the best HADDOCK score revealed maximum contribution of N-terminal transactivation domain (AF-1) of PPARα for hydrogen bonding with TAK1 with added contribution of a few hydrogen bonds through the hinge region and LBD. Only one residue at position 107 from DBD has been shown to interact with TAK1 (Fig. 7A). Status of interaction between PPARα and TAK1 is summarized in supplemental Table 2, A and B.
Figure 7.

Analyses of interaction of PPARα with TAK1. A, overall schematic representation of the docking simulation between predicted structures of full-length rat TAK1 (silver) and different domains of rat-PPARα (cyan, AF-1; orange, DBD; green, H + LBD) based on the best fit HADDOCK score. B, FRAP analysis showed a positive FRET efficiency between endogenous PPARα and TAK1 in cardiac fibroblasts. Cells were probed for endogenous PPARα and TAK1 expressions with respective primary antibodies and stained with PPARα-FITC (green) and TAK1-TRITC (red). TRITC was subjected to 50% photobleaching. n = 5. Panel i, prebleach donor; panel ii, postbleach donor; panel iii, delta donor; panel iv, prebleach acceptor; panel v, postbleach acceptor; panel vi, FRET efficiency. C, co-IP experiments were done by immunoprecipitating proteins with anti-PPARα antibody followed by immunoblotting with anti-TAK1 antibody in vitro. PPARα-overexpressed AngII-treated fibroblasts were used as a positive control. Normalization was done by Western blot with anti-PPARα antibody in the same samples. Control and AngII-treated cells were also treated with either DMSO or empty pCDNA6/V5-HisB vector yielding similar results. n = 5 for each group. Results were analyzed by ANOVA followed by Tukey's post hoc test and expressed as ±S.E. of three independent experiments. Graph showing relative changes in the level of interaction between PPARα and TAK1 between different experimental groups. C, control fibroblasts; A, AngII-treated fibroblasts; A + AA, AA co-treated AngII infused fibroblasts; A + pOV, PPARα overexpressed AngII-treated fibroblasts. *, p < 0.01 with respect to control fibroblasts; ##, p < 0.01 with respect to AngII-treated fibroblasts.
FRET assay
FRET assay measured fluorescence recovery after photobleaching (FRAP) showing positive FRET efficiency (20.87 ± 1.151), which revealed direct interaction between endogenous PPARα and TAK1 in control fibroblasts in vitro (Fig. 7B).
Co-immunoprecipitation (Co-IP) assay
AA treatment in hypertrophied fibroblasts resulted in significant up-regulation (3.63 ± 0.334-fold) of PPARα-bound TAK1 expression compared with respective hypertrophy samples in vitro as revealed by immunoprecipitation with anti-PPARα antibody followed by Western blot with anti-TAK1 antibody. PPARα overexpression in AngII-treated fibroblasts showing significantly increased binding between PPARα and TAK1 (2.73 ± 0.177-fold) compared with hypertrophied cells pretreated with empty pCDNA6/V5 His B mammalian expression vector were used as positive control (Fig. 7C).
Study of the effect of PPARα domains upon PPARα-TAK1 interaction
For His-tagged plasmids with inserts of full-length PPARα (F), N-terminal transactivation domain (AF-1), DNA-binding domain (DBD), or hinge region along with the ligand-binding domain (H + LBD) were overexpressed into AngII-treated cardiac fibroblasts. Immunoprecipitation with anti-His antibody followed by Western blot analyses with anti-TAK1 antibody revealed the strongest binding of TAK1 with AF-1 (96.97 ± 1.829%), a moderate interaction with H + LBD (35.78 ± 0.975%), and an almost negligible interaction with DBD (10.59 ± 0.887%) in comparison with TAK1 interaction with F (100%), which was used as a positive control (Fig. 8A). Thus, DBD (9.03 ± 0.683-fold) and H + LBD (2.64 ± 0.076) showed significantly lower binding with TAK1, compared with AF-1 in AngII-treated cells (Fig. 8A and supplemental Fig. S5A).
Figure 8.
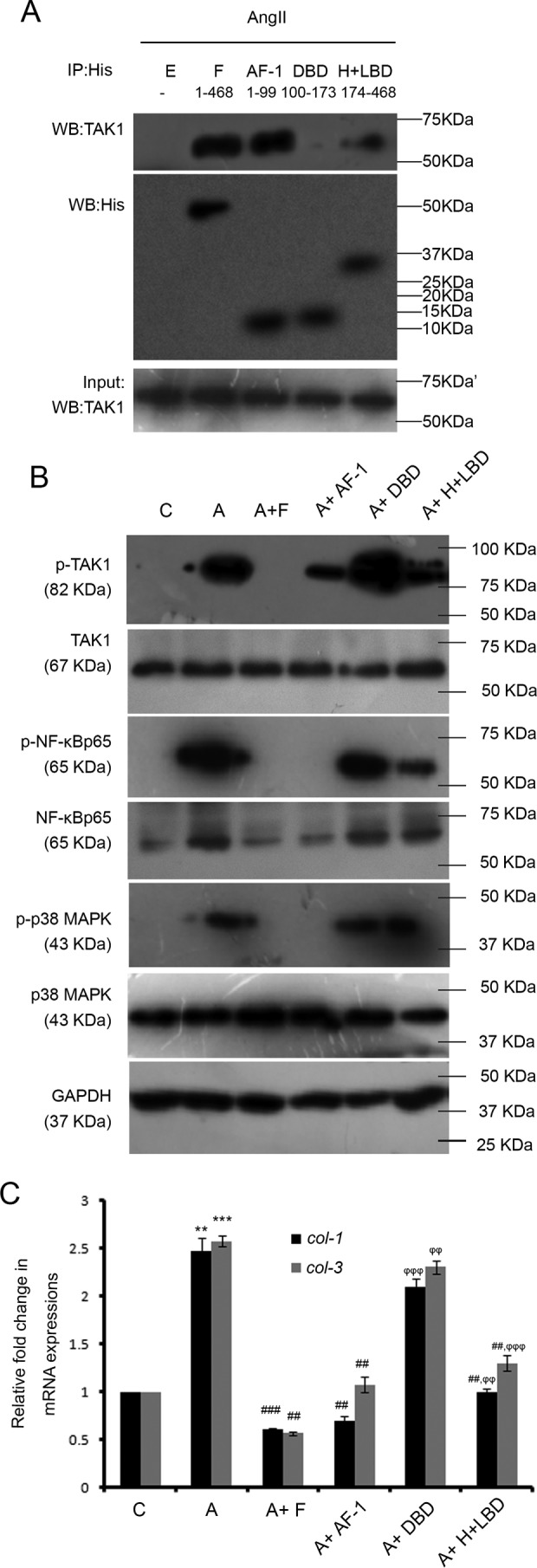
Study of the effect of different PPARα domains upon PPARα-TAK1 interaction and their roles in modulation of non-canonical TGF-β pathway-induced collagen synthesis in hypertrophied fibroblasts. A, co-IP experiments were done by immunoprecipitating proteins with anti-His antibody followed by immunoblotting with anti-TAK1 antibody in vitro. Normalization was done by immunoblotting with anti-His antibody. Results were analyzed by ANOVA followed by Tukey's post hoc test and expressed as ±S.E. of three independent experiments. n = 5 for each group. E, empty pCDNA6/V5 HisB vector; F, full-length PPARα plasmid; AF-1, PPARα N-terminal transactivation domain plasmid; DBD, PPARα DNA-binding domain; H + LBD, PPARα combined Hinge region+ C-terminal ligand-binding domain plasmid. All the plasmids were transfected into AngII-treated fibroblasts. n = 5 for each group. B, Western blot (WB) analyses showing alterations in expression levels of phospho/total TAK1, NF-κBp65, and p38 MAPK in AngII-treated cells transfected with full-length or individual domains of PPARα plasmids compared with AngII-treated fibroblasts. GAPDH was used as internal loading control. C, control fibroblasts; A, AngII-treated fibroblast; A + F, full-length PPARα transfected AngII-treated fibroblasts; A + AF-1, PPARα N-terminal transactivation domain transfected AngII-treated cells; A + DBD, PPARα DNA-binding domain transfected AngII-treated cells; A + H + LBD, PPARα combined Hinge region+ C-terminal Ligand-binding domain transfected AngII-treated cells. C and A cells were also treated with empty pCDNA6/V5 HisB vector. n = 5 for each group. C, graphical representation of qRT-PCR analyses showing changes in levels of col-1 and col-3 gene expressions in AngII-treated cells transfected with full-length or individual domains of PPARα plasmids compared with AngII-treated fibroblasts. Rpl-32 was used as internal loading control. Results were analyzed by ANOVA followed by Tukey's post hoc test and expressed as ±S.E. of three independent experiments. C, control fibroblasts; A, AngII-treated fibroblast; A + F, full-length PPARα transfected AngII-treated cells; A + AF-1, PPARα N-terminal transactivation domain transfected AngII-treated cells; A + DBD, PPARα DNA-binding domain transfected AngII-treated cells; A + H + LBD, PPARα combined Hinge region+ C-terminal ligand-binding domain transfected AngII-treated cells. C and A cells were also treated with empty pCDNA6/V5 HisB vector. n = 5 for each group. **, p < 0.01 with respect to control cells; ***, p < 0.001 with respect to C; ##, p < 0.01 with respect to A; ###, p < 0.001 with respect to A; ϕϕ, p < 0.01 with respect to A + F; ϕϕϕ, p < 0.001 with respect to A + F.
Study of the effect of PPARα domains in AngII-treated fibroblasts upon modulation of non-canonical TGF-β pathway and collagen synthesis
The F plasmid overexpressed AngII-treated cardiac fibroblasts showed significant reduction in the level of phospho/total TAK1 (3.68 ± 0.065-fold), phospho/total NF-κBp65 (5.13 ± 0.109-fold), and phospho/total p38 MAPK (3.69 ± 0.141-fold) compared with empty pCDNA6/V5 His B mammalian expression plasmid-infused AngII-treated cells. AF-1 and H + LBD overexpression into AngII-treated cells also showed significant regression of phospho/total TAK1 (2.07 ± 0.144-fold for AF-1 and 1.49 ± 0.117-fold for H + LBD), phospho/total NF-κBp65 (5.16 ± 0.165-fold for AF-1 and 1.36 ± 0.066-fold for H + LBD), and phospho/total p38 MAPK (3.56 ± 0.185-fold for AF-1 and 1.54 ± 0.049-fold for H + LBD) levels compared with AngII-treated cells. The overexpression of DBD in AngII-treated cells showed no significant difference in the expression level of such proteins compared with the hypertrophied cells. Moreover, AF-1 overexpression in AngII-treated cells showed significantly higher levels of regression of the aforementioned proteins (1.38 ± 0.028-fold for phospho/total TAK1, 3.79 ± 0.199-fold for phospho/total NF-κBp65, and 2.31 ± 0.183-fold for phospho/total p38 MAPK) compared with H + LBD-overexpressed AngII-infused fibroblasts (Fig. 8B and supplemental Fig. S5B).
The qRT-PCR analyses showed significant down-regulation in col-1 and col-3 gene expressions (3.48 ± 0.17-fold for col-1 and 3.47 ± 0.249-fold for col-3) in F-overexpressed AngII-treated fibroblasts compared with AngII-treated cells pretreated with empty plasmid. A similar trend of regression was also observed by AF-1 and H + LBD overexpression in AngII-treated fibroblasts (2.66 ± 0.233-fold and 1.478 ± 0.017-fold for col-1; 2.35 ± 0.202-fold and 1.875 ± 0.073-fold for col-3 respectively) compared with AngII-treated cells. However, AF-1 overexpression caused significantly higher levels of regression in collagen gene expression (1.77 ± 0.081-fold for col-1 and 1.24 ± 0.025-fold for col-3) compared with H + LBD overexpression in hypertrophied fibroblasts. In contrast, DBD overexpression in AngII-treated cells showed no significant change in collagen gene expressions compared with hypertrophied fibroblasts (Fig. 8C).
Discussion
Phytomedicines are promising candidates for unraveling novel strategies to combat maladaptive cardiac remodeling. This study focuses on the precise mechanism of protection conferred by the phytochemical AA during pressure overload cardiac hypertrophy and fibrosis. Our investigation showed that AA significantly inhibits excess synthesis of collagen and its subsequent deposition in ECM during cardiac hypertrophy both in vitro and in vivo. Improvement in cardiac function was also observed with AA treatment in renal artery-ligated rats along with significant regression of collagen synthesis and redemption from hypertrophic load (Fig. 1, A–H, and supplemental Fig. S1, A and B). This encouraged us to probe into the specific molecular mechanism involved in AA-mediated amelioration of fibrosis during cardiac hypertrophy.
Arjunolic acid (2,3,23-trihydroxyolean-12-en-28-oic acid) is a pentacyclic triterpenoid monocarboxylic acid substituted by three hydroxy groups at positions 2, 3, and 23 (CHEBI: 68381). Two hydroxyl groups and one hydroxymethyl group are attached to the “A” ring. The carboxyl group is attached at the ring junction of the cis-fused “D” and “E” rings (35, 36). Triterpenoids have long been known for their ability to suppress inflammatory pathways (37) and have been recognized to activate different PPARs (38, 39). As PPARα is the predominant cardiac isoform of the PPAR family (15), interaction of PPARα with AA was studied in detail.
Stabilizing interaction between AA and PPARα was confirmed by fluorescence, CD titrations, and thermal shift assays (Fig. 2, A and C). FTIR spectrum of the conjugate in a cell-free system also indicated successful docking via masking of the hydrogen bond-forming groups of AA. However, amide-I and amide-A peaks of PPARα remained intact in the conjugate indicating maintenance of protein architecture (Fig. 2B). Further evidence for AA as a potential agonist of PPARα came through molecular docking studies where the former occupied an identical binding pocket as reported for established agonists such as iloprost, TIPP-703 (40, 41), and fenofibrate (42), which is a well-known PPARα activator exerting its effect in cardiac fibrosis (Fig. 2D) (18, 19). Moreover, higher affinity of PPARα to AA compared with the other agonists as estimated by their free energy of binding suggests that AA might act as an even better agonist (supplemental Table 1). Simulation outcomes further confirmed AA-driven stabilization of PPARα via persistence of strong hydrogen bonding network between AA and the LBD of PPARα (Fig. 2E). In many previous reports, the agonistic potential had been attributed to stabilization of helices in this region (31, 43, 44), which is also observed in our case through RMSF analysis (Fig. 2E, panel iii). Altogether, these data suggested that AA-driven stabilization of PPARα could shift the equilibrium of PPARα toward the active configurations resulting in higher interactions with different co-regulators, thus augmenting activation of PPARα-driven transcriptional machinery (45, 46).
The presence of PPRE, the putative PPARα-binding site on the PPARα promoter, suggests that PPARα might regulate its own transcription (47). ChIP analyses coupled to qRT-PCR revealed AA induced increased binding of PPARα upon PPRE within the PPARα promoter during hypertrophy, and the resultant PPARα transcriptional activation increased PPARα mRNA and protein expression during AA treatment in hypertrophy in an autoregulatory loop (Fig. 3, A and B). Time-dependent rise in PPARα expression in AA-treated hypertrophied cells further confirmed AA-mediated PPARα agonism during hypertrophy (Fig. 3C).
A significant increase in collagen transcription and its extracellular accumulation during PPARα knockdown in AA-infused AngII-treated fibroblasts compared with AA-treated hypertrophied cells implied the importance of PPARα in modulation of collagen transcription in cardiac fibroblasts. Moreover, PPARα knockdown in AA-treated ligated rats resulted in increased synthesis and accumulation of collagen with subsequent deterioration of cardiac function that altogether confirms the influence of PPARα in AA-mediated cardioprotection during hypertrophy and associated fibrosis (Fig. 4, A–E, and supplemental Fig. S1, A and B). PPARα activation during AA treatment caused inhibition of AngII-induced TGF-β signaling by affecting both canonical and non-canonical branches (Fig. 5A and supplemental Fig. S2A). However, this effect might result from reduction in the TGF-β level as a secondary response to minimized hypertrophic load. Therefore, fibroblasts were treated with TGF-β instead of AngII in the presence or absence of AA, and the results proclaimed that AA predominantly affects the non-canonical branch of the TGF-β signaling pathway via selectively inactivating TAK1 and not TβRI, TβRII, or SMADs. PPARα knockdown in TGF-β-treated fibroblasts during AA treatment restored TAK1 phosphorylation compared with AA-infused TGF-β-treated fibroblasts further confirming the specificity of AA toward PPARα-mediated deactivation of non-canonical TGF-β signaling (Fig. 5B and supplemental Fig. S2B).
AA-mediated inactivation of TAK1 and downstream proteins, namely IKKβ/NF-κBp65, p38 MAPK, and JNK, also suggested significant involvement of PPARα during AA treatment in hypertrophy groups (Fig. 6A and supplemental Fig. S2C). Knockdown experiments revealed remarkable contribution of TAK1 as well as NF-κBp65 and p38 MAPK in promoting collagen transcription in AngII-treated fibroblasts in vitro. In contrast, JNK showed no effect upon collagen synthesis at hypertrophic stimulus to cardiac fibroblasts in vitro (Fig. 6B). Our group and others have previously shown the involvement of p38 MAPK (48, 49) and NF-κBp65 (50, 51) in cardiac collagen biosynthesis. In addition to that, analysis of luciferase activity of the Col1a1 promoter containing the NF-κBp65-binding site also revealed the role of NF-κBp65 as a transcriptional activator of collagen promoter during hypertrophy, which could be targeted for inhibition by AA-driven PPARα activation (Fig. 6C). Interestingly, PPARα-mediated down-regulation of non-canonical TGF-β axes during AA treatment in hypertrophy samples was associated with decreased phosphorylation of TAK1, without any alteration in its total level (Fig. 5, A and B). This clearly indicates that changes in TAK1 activation during AA treatment in hypertrophy samples result from post-translational modifications and are not related to PPARα-mediated regulation of gene transcription. Reports stating the roles of PPARs in modulating signaling pathways by interacting with other signal intermediates (52–54) prompted us to check whether PPARα directly interacts with TAK1. Successful docking between PPARα and TAK1 as revealed by in silico analyses (Fig. 7A) was further validated by FRET–FRAP assay in fibroblasts (Fig. 7B). Additionally, co-IP data showing significantly higher interaction between PPARα and TAK1 in AA-treated hypertrophied cells as well as in PPARα-overexpressed hypertrophied cells compared with AngII-treated cells also suggested that the level of PPARα-bound TAK1 is directly proportional to the availability of PPARα (Fig. 7C).
Analyses of domain specificity for PPARα and TAK1 interaction in AngII-treated fibroblasts revealed that the AF-1 domain of PPARα contributes maximally to the binding strength of PPARα with TAK1 when compared with that of full-length PPARα (F) (Fig. 8A and supplemental Fig. S5A). The best fit cluster of PPARα-TAK1 interaction model in our bioinformatic analyses also identified the highest number of interacting amino acids from the AF-1 domain of PPARα (supplemental Table 2A). Interestingly, in silico data suggest that TAK1 interacts with PPARα via amino acids belonging to its kinase domain (36–291 amino acids in rat-TAK1), which also contains TAK1 phosphorylation sites (Thr-184/187) (Fig. 7A and supplemental Table 2A). This further indicates that PPARα-TAK1 interaction inactivates TAK1 possibly by preventing its phosphorylation.
Wet lab experiments confirmed that the higher the binding between different PPARα domains and TAK1 in AngII-treated fibroblasts, the greater is the amount of regression in TAK1 phosphorylation with subsequent down-regulation of non-canonical TGF-β axes (Fig. 8B and supplemental Fig. S5B). This finding also corroborates the inactivation of the non-canonical branch of TGF-β signaling (Figs. 5A and 6A) during AA treatment in hypertrophy samples resulting from AA-induced PPARα up-regulation (Fig. 3, A and B) and increased interaction between PPARα and TAK1 (Fig. 7C). The degree of regression of collagen synthesis as observed by overexpression of different PPARα domains into hypertrophied fibroblasts also followed the similar trend revealing maximum regression by AF-1 overexpression (Fig. 8C).
An earlier report suggested that deletion of the AF-1 region of PPARα did not affect its function keeping its DNA-binding activity unaltered (55). However, our study shows that the AF-1 region of PPARα promotes PPARα–TAK1 protein-protein interaction resulting in suppression of the non-canonical TGF-β pathway and reduced collagen gene transcription during cardiac hypertrophy. The H region (helix-1) of PPAR-γ has been reported to bind with cytosolic kinases such as ERK5 (52) and PKC-α (53) in other cell types. Although significantly lower than AF-1, interaction of H + LBD of PPARα with TAK1 also had a significant role in regression of non-canonical TGF-β pathway-induced collagen synthesis during hypertrophy.
In summary, our work for the first time shows a precise mechanism of AA action in regression of cardiac hypertrophy-associated fibrosis and subsequent improvement of cardiac function. This work assigns a new role of AA as a PPARα agonist, and such transcriptional activation and increased expression of PPARα during the abovementioned pathophysiological condition inactivates the non-canonical TGF-β axes (Fig. 9A). Specific molecular mechanism involves direct interaction of PPARα, predominantly by its N-terminal AF-1 domain with TAK1 kinase domain, leading to decreased phosphorylation of TAK1 and its downstream signal intermediates ultimately inhibiting excess collagen transcription during hypertrophy (Fig. 9B).
Figure 9.
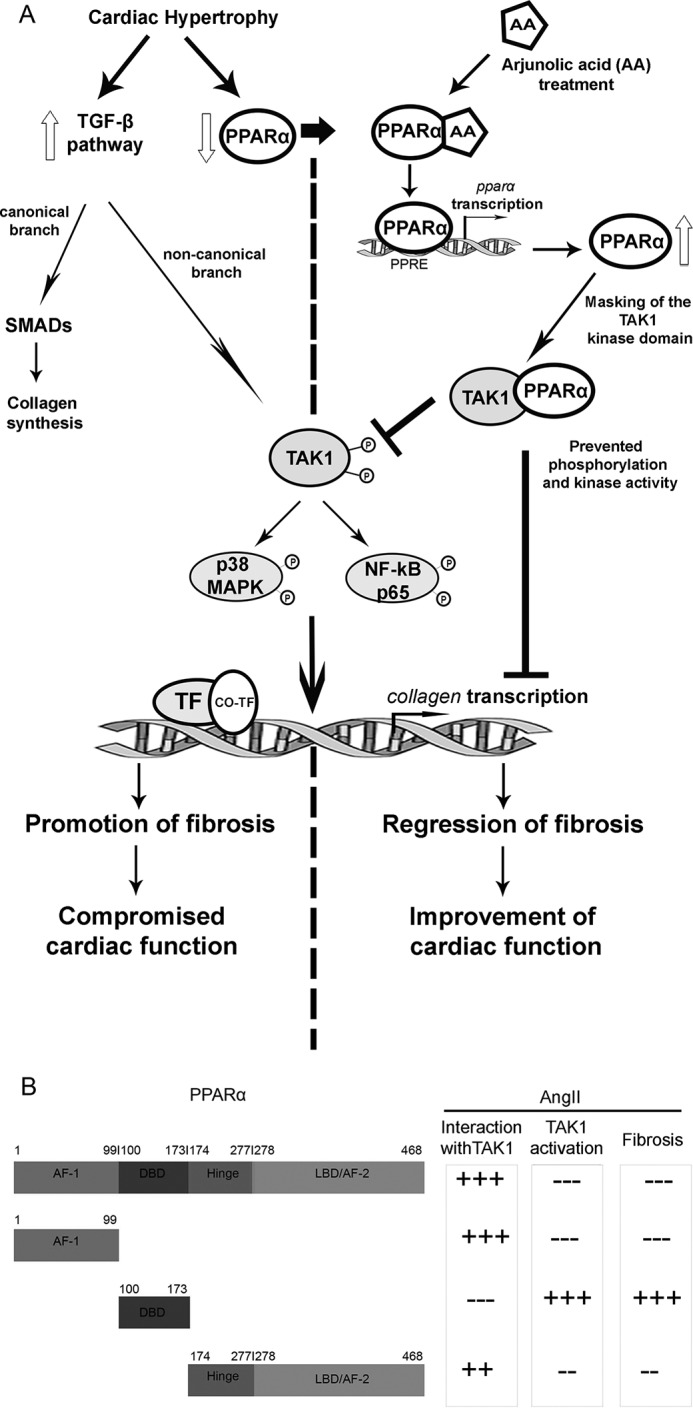
Schematic representation of the molecular mechanism of AA action upon cardiac hypertrophy-associated fibrosis. A, during cardiac hypertrophy TGF-β signaling pathway action is promoted leading to excess collagen synthesis with down-regulated PPARα expression. Treatment with AA in hypertrophy samples increases PPARα expression in an autoregulatory loop leading to increased binding of PPARα to TAK1 thereby ameliorating TAK1-driven non-canonical TGF-β axes with subsequent regression of collagen synthesis. B, schematic representation of different PPARα domains interacting with TAK1 and the role of PPARα-TAK1 interaction in prevention of phosphorylation-dependent activation of TAK1 for subsequent regression of collagen synthesis in AngII-treated adult cardiac fibroblasts.
TGF-β signaling is one of the key mediators for maintenance of normal cellular function and homeostasis of cardiac fibroblasts (56). As TAK1 resides at a moderately downstream position in TGF-β axes, inhibiting TAK1 by using AA as an anti-fibrotic agent might be therapeutically advantageous. Further detailing of PPARα-binding surface in TAK1 or the influence of TAK-binding proteins in such interaction would be necessary for better understanding of the regulation of TAK1 autoactivation and its role in ECM turnover. Not only that, but transition of such a natural compound from initial screening through preclinical and clinical trials, and finally, to a marketable drug form is associated with challenging demands for the plant source. Therefore, organic synthesis of AA bio-mimetics might be beneficial considering the wide range of protective effects exerted by AA with enhanced clinical significance against such deadly diseases.
Experimental procedures
Animals used
28-Week old male Wistar rats (R. norvegicus; Taxonomy ID: 10116) (n = 7 per experimental group) used in this study were procured from National Institute of Nutrition, Hyderabad, AP, India. The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the National Institute of Health (NIH Publication No. 85-23, revised 1996) and was also approved by the Institutional Animal Ethics Committee, University of Calcutta (Registration no. 885/ac/05/CPCSEA), registered under “Committee for the Purpose of Control and Supervision of Experiments on Laboratory Animals” (CPCSEA), Ministry of Environment and Forests, Government of India.
Isolation and culture of adult cardiac fibroblasts
Adult cardiac fibroblast cells were isolated from 28-week-old male Wistar rat hearts by the collagenase dispersion method. Briefly, animals were euthanized in a prefilled carbon dioxide (CO2) chamber with 100% concentration of CO2, and the hearts were chopped and digested by collagenase (80 units/ml DMEM; Worthington). The cells were pelleted by centrifugation and resuspended in fresh complete DMEM supplemented with 10% fetal bovine serum and plated in cell culture flask. Cells were maintained at 37 °C with 5% CO2 and were subsequently passaged (49).
Generation of cardiac hypertrophy in vitro and in vivo
75–80% confluent serum-starved adult cardiac fibroblasts were treated with 10−8 mol/liter (Sar1)-AngII (Bachem, Torrance, CA) for 24 h to generate cardiac hypertrophy model in vitro. AngII was replenished every 6 h throughout the incubation period (49).
In vivo cardiac hypertrophy model was generated by ligating the right renal artery of 28-week-old male Wistar rats (250–300 g) as described earlier (49). Rats were anesthetized with an intraperitoneal injection of a mixture of ketamine (80 mg/kg) and xylazine (12 mg/kg). Sham-operated control group underwent a similar procedure without renal artery ligation. Animals were maintained in optimum condition for 14 days and were sacrificed on the 15th day after surgery. Hearts were removed, collected in liquid N2, and stored in −80 °C for future use.
Hypertrophy was measured by the HW (in milligrams) to BW (in grams) ratio, and activation of hypertrophy marker genes (anf, β-mhc, and acta1) was measured by RT-PCR analyses (57).
Measurement of cardiomyocyte CSA
To analyze cardiomyocyte cross-sectional area as a marker of hypertrophy, hearts were excised, washed in 1× PBS, and fixed in Karnovsky's fixative. The samples were then paraffin-embedded and cut into 4–5-μm sections. Tissue sections were processed and stained with hematoxylin and eosin (H&E), and the cardiomyocyte CSA in each sample was quantified (57) using a computer morphometric program (ImageJ, National Institutes of Health).
Evaluation of collagen deposition
Hydroxyproline assay
Hydroxyproline assay was performed to measure total secreted collagen content in fibroblast culture supernatant in vitro and total left ventricular collagen content from rat heart tissues. With the help of standard curve, hydroxyproline content in the unknown samples was calculated. The amount of collagen was estimated by multiplying hydroxyproline content by a factor of 8.2 (49).
Estimation of %CVF
Heart tissues from each group of animals were taken for the analysis of %CVF. Coronal sections obtained from the equator of the left ventricle were fixed and stained with Massons' trichrome (Sigma) to analyze collagen deposition under microscope (Nikon NIS BR) (Nikon, Shinagawa, Tokyo, Japan) following standard protocol. Fifty sections were scanned, and at least 10 images were captured from each section. Images were digitized and processed by ImageJ, a computer morphometric program. CVF was calculated as the sum of all collagen-stained tissue areas of the coronal sections represented as percentage (%) of the total surface of the section. Color segmentation was applied to separate the stained tissues from other nonspecific objects (58).
Determination of cardiac function
Cardiac function of lightly sedated animals from sham-operated control group, renal artery-ligated group, AA-treated hypertrophied rat group, and PPARα siRNA-infused AA-treated ligated rat group was measured by M-mode echocardiographic analysis by a transthoracic study in a short axis view at a papillary muscle level on the 15th day before euthanization. In vivo ventricular PPARα siRNA injection was also guided by echocardiography. Digitized images were obtained using an ultrasound system (Vivid S5 system, GE Healthcare) for the calculation of %FS, LvIDd, LvAWDd. LvAWDs, LvPWDd, LvPWDs, SV, and EF following standard guidelines (58).
Treatment of fibroblasts with TGF-β
Another set of adult cardiac fibroblasts was treated with active TGF-β-2 (Life Technologies, Inc.) at a dose of 8 ng/ml serum-free media for 16 h, as described previously (59), to induce hypertrophic effect. TGF-β-treated cells were pretreated with AA with or without simultaneous treatment of PPARα siRNA following standard protocol.
Treatment with AA in vitro and in vivo
AA was extracted and purified from core wood of the plant T. arjuna (14). In vitro hypertrophied fibroblasts were treated with 20 μm AA (dissolved in DMSO) 6 h after first AngII treatment. Effective dose was chosen from a dose-dependent study (5–100 μm AA) based on optimal results with minimum mortality. A group of control fibroblasts was also treated with 20 μm AA. All other control and AngII-treated cells were also treated with equivalent amounts of DMSO. Time-dependent changes upon AA treatment were analyzed in AngII-treated cells at 3 and 6 h after induction with AA. A set of TGF-β-treated cells were also treated with 20 μm AA 6 h after TGF-β treatment.
In vivo renal artery-ligated rats were treated with AA and dissolved in DMSO by intraperitoneal injection on every alternate day from day 6 to day 14 at a dose of 10 mg/kg/day after ligation. Effective dose was chosen after a dose-dependent study (5 mg to 50 mg/kg/day as described above) based on optimal results. Untreated sham-operated rats and renal artery-ligated rats were also treated with equivalent amounts of DMSO. Animals were sacrificed after 14 days. Commercially available arjunolic acid (SMB00119; Sigma) was also used at similar dosage showing uniform results (data not shown).
Cell viability assay
Cell viability and cytotoxicity in the presence or absence of AA in AngII-treated fibroblasts were determined colorimetrically by Cell titer 96® AQueous one solution cell proliferation assay (Promega, Madison, WI) following the manufacturer's protocol.
Administration of siRNAs
For in vitro experiments, cardiac fibroblasts were treated with 100 nm PPARα siRNA (S130650, Silencer® select siRNA, Ambion; Thermo Fisher Scientific, Waltham, MA), TAK1 siRNA (S10905939, Qiagen, Hilden, Germany), NF-κBp65 siRNA (S103097885, Qiagen, Hilden, Germany), p38 MAPK siRNA (6243, Cell Signaling Technology, Danvers, MA), JNK siRNA (6233, Cell Signaling Technology), or NS siRNA (All Stars Negative Control siRNA, S103650325, Qiagen, Hilden, Germany) using HiPerfect transfection reagent (Qiagen, Hilden, Germany) as per the manufacturer's protocol. 18 h after transfection, cells were used for AngII treatment with or without AA treatment.
For in vivo experiments, PPARα siRNA (S130650, Ambion® In vivo siRNA, Thermo Fisher Scientific) in RNase-free sterile PBS was injected directly into ventricles of ligated animals (49) that are to be treated with AA with simultaneous echocardiographic guidance, following the manufacturer's instructions at a concentration of 10 nmol on every alternate day from the 5th day of ligation before sacrifice. Sham-operated and renal artery-ligated rats were also treated with equivalent amounts of DMSO along with in vivo NS siRNA (4457287, Ambion® In vivo Negative Control siRNA, Thermo Fisher Scientific). Ligated rats to be treated with AA were also pretreated with equivalent amounts of NS siRNA.
Structural modeling of PPARα and TAK1
The amino acid sequence of R. norvegicus PPARα (P37230) and TAK1 (P0C8E4) were retrieved from the UniProtKB database (http://www.uniprot.org/).5 The retrieved amino acid sequences of PPARα and TAK1 were subjected to a protein-protein BLAST (BLASTp) search against the PDB to identify a suitable template structure for comparative modeling. The 3D structure predictions of these two proteins were then performed using I-TASSER (60). For rat-PPARα, PDB code 4BCR, chain A, was selected as the best suited template with 99% identity and 45% sequence coverage (E value 0.0). For rat-TAK1, PDB code 5JGA, chain A, was selected as a suitable template with 93% identity and 58% sequence coverage (E value 5e−170). The best I-TASSER outputs were refined using ModRefiner (61). The I-TASSER Z-scores were analyzed to check the likelihood that the domains had correct folds and correct overall tertiary structure. The final model was also checked for its quality using ProQ2 (62). Furthermore, refined structures were subjected for MD simulation.
All atomistic MD simulation
To check for conformational stability, MD simulation was done using GROMACS 4.6.1 (63). All atomistic simulations were carried out using the CHARMM36 all-atom force field (64–66) using periodic boundary condition. The starting model was solvated in a periodic box with TIP3 water model. Nine Na+ ions were added to the solvent to neutralize electrical net charge of the protein. The system was then minimized for 50,000 steps using a steepest descent algorithm with emtol of 200 kJ/mol after a minimization with emtol of 100 kJ/mol. This was followed by an equilibration run of 100 ps in NVT ensemble with restraints on the protein atoms. The NPT ensemble was used for production simulation. Systems were simulated at 310 K and maintained by a Berendsen thermostat with a time constant of 1 ps with the protein and non-protein molecules coupled separately. Pressure coupling was done employing a Parrinello-Rahman barostat using a 1 bar reference pressure and a time constant of 2.0 ps with compressibility of 4.5e−5 bar using isotropic scaling scheme. Electrostatic interactions were calculated using the Particle Mesh Ewald (PME) summation with a 2-fs time step for each run. For simulations of PPARα-AA complex, identical parameters were employed. Topology for AA (PubChem ID: CID73641) and other parameters were assigned using SwissParam (67).
Three-dimensional structural modeling of the chemical compounds
The 2D structures of AA (PubChem ID: CID73641) and fenofibrate (PubChem ID: CID3339) were retrieved from the NCBI PubChem database. The compounds were drawn using ChemDraw 8 software. The two-dimensional molecules were then converted to three-dimensional structures using OpenBabel (68). The energy minimization was performed using Maestro 9.8 (Schrödinger, LLC, New York).
Molecular docking
Docking of AA to PPARα was performed using the BSP-SLIM tool (69). Briefly, the holo-structures with similar global topology to PPARα were identified, and the geometric centers of bound ligands in the holo-forms were clustered to identify putative AA-binding sites. Subsequently, shape and chemical feature comparison of multiple target ligand conformers with all negative images of binding pockets was done employing the OMEGA program. Best overlay for each ligand conformer with the negative images was carried out using OEChem toolkit version 1.7, whereas the chemical features of AA were assigned using the Implicit-MillsDean color force field (70). Finally, all ligand conformations were sorted by docking scores with a RMSD tolerance limit of 4 Å.
The top lowest energy binding complex was re-ranked by performing flexible docking using Molecular Operating Environment (version 2009.10). The triangular matching placement method with the Affinity dG scoring function was employed to generate most favorable poses of ligand conformations by aligning ligand triplets of atoms with triplets of receptor site points. AA-PPARα docking scores were compared with the score obtained for known agonists of PPARα, i.e. iloprost (PDB code 3SP6) and TIPP-703 (PDB code 2ZNN) and fenofibrate (obtained through pre-docking by BSP-SLIM). In contrast, in silico binding affinities were calculated using AutoDock Vina 1.1.2 (71) extension of UCSF chimera (72).
For protein-protein docking, residues forming the interface surfaces were predicted. CPORT program drew a consensus for the interface residues using PIER, cons-PPISP, SPPIDER, and PINUP. The consensus residues were mapped onto the model. The predicted interface residues were submitted to the PPARα and TAK1 monomer model for protein-protein docking to build the heterodimer model, using HADDOCK web server (73, 74). Molecular graphics and analyses were performed with the UCSF Chimera (72).
Fluorescence and CD spectroscopy
Fluorometric titrations were carried out in LS55 fluorescent spectrometer (PerkinElmer Life Sciences). At constant PPARα (in vitro translated and purified) protein concentration of 8 μm, titrations were carried with increasing concentrations of AA up to 100 μm with a pre-scan delay of 300 s. Incubated samples were excited at 274 nm due to absence of tryptophan, and emission spectra were recorded from 290 to 400 nm with excitation/emission slits set 10/10.
CD titrations were carried out in J-1500 circular dichroism spectrophotometer (Jasco, Easton, MD). Far-UV CD titrations were carried out following similar protocol in a 2-mm path length quartz cuvette and scanned from 250 to 200 nm at scan rate of 200 nm per min.
Fluorescence-based thermal shift (FTS) assay
FTS assay for PPARα was done to assess tertiary structure stabilization by monitoring change in Tyr emission at 330 nm over the temperature range from 25 to 90 °C with step size of 2 °C. Excitation and emission slits were kept at 5/10 with an equilibration time of 1 min at each step.
Thermal denaturation of PPARα (8 μm) was done both in the absence or presence of AA (100 μm). AA incubation with PPARα was done 30 min prior to the experiment. To assess secondary structure stabilization of PPARα by AA, changes in both θ218 and θ222 were monitored against temperature range of 25 to 90 °C at ramping rate of 1 °C per min.
In vitro coupled transcription-translation and in vitro interaction assay
Full-length PPARα, cloned in pCDNA6/V5 His B mammalian expression plasmid (Invitrogen), was subjected to in vitro coupled transcription-translation using TnT® Quick-coupled Transcription/Translation system (Promega, Madison, WI) according to the manufacturer's specifications. Briefly, 2 μg circular plasmids were added to the rabbit reticulocyte lysates in 50-μl reactions for 60–90 min at 30 °C. These His6-tagged products were then purified using nickel-nitrilotriacetic acid spin kit (Qiagen, Hilden, Germany) in native condition and analyzed by SDS-PAGE followed by Coomassie Blue staining to check the purity (data not shown). Purified products were then subjected to in vitro interaction in binding buffer overnight on a rocker at 4 °C with AA (59). This complex along with the purified protein was used for further assay.
FTIR spectroscopy
FTIR spectra were obtained with a Jasco FTIR 6300 spectrometer (Jasco, Easton, MD). The spectra presented were obtained from an average of 64 scans. The resolution of the spectra was 4 cm−1. The spectra were acquired at room temperature. All data analyses were performed with the data analysis software Origin (OriginLab, Northampton, MA) provided with the FTIR instrument.
RNA isolation
Total RNA from cells (24-h treatment) and tissues (on the 15th day after surgery) was isolated using TRIzol reagent (Invitrogen) following the manufacturer's instructions.
RT-PCR and qRT-PCR
1 μg of total RNA was used to make cDNA using the cloned avian myeloblastosis virus first-strand cDNA synthesis kit (Invitrogen). RT-PCR was done for col-1, col-3, anf, β-mhc, acta1, and ribosomal protein L32 (rpl 32). rpl32 was used as loading control.
Relative quantification of PCR-amplified products was also done by qRT-PCR with Power SYBR GreenTM PCR master mix using Applied Biosystems StepOne® real-time PCR system (4369074, Applied Biosystems) for col-1, col-3, pparα, and rpl32, where rpl32 was used as a reference gene to normalize expression of other genes. Relative gene expression was quantified by comparative “Ct” (2−ΔΔCt) method (49). Both RT-PCR and qRT-PCR primer sequences are summarized in supplemental Table 3, A and B.
ChIP assay
ChIP analysis was performed with modifications from the methodology described by Kouskouti and Talianidis (75). Briefly, cardiac fibroblasts and cardiac tissue sections were treated with 37% formaldehyde (final concentration 1%) for cross-linking proteins to DNA. Cross-linking was stopped by adding 10× glycine (final concentration 0.125 m) after 10 min and mixed at room temperature. Cardiac fibroblasts were now scraped out in ice-cold 1× PBS with protease inhibitor cocktail (PIC; P8340; Sigma). Cardiac tissue samples with cross-linked chromatin were also taken in ice-cold 1× PBS with PIC and were thoroughly minced with a Dounce homogenizer. Both in vitro and in vivo samples were now centrifuged at 1500 rpm for 5 min at 4 °C, and the pellets were resuspended in sonication buffer (50 mm HEPES, 140 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, and 0.1% SDS, pH 7.9). The samples were now sonicated and centrifuged to obtain supernatants containing 500–800-bp chromatin fragments. For each reaction, 400 μg of purified chromatin was precleared with protein G-Sepharose bead (P3296; Sigma) and immunoprecipitated overnight at 4 °C either by anti-PPARα antibody or anti-IgG antibody (used as a negative control) (antibodies listed in supplemental Table 4). Furthermore, antibody-bound chromatin complexes were washed using low- and high-salt wash buffers followed by Tris/EDTA (TE) buffer wash, all at 4 °C. Protein-DNA complexes were eluted from beads by elution buffer (50 mm NaHCO3, 50 mm Tris, 1 mm EDTA, and 1% SDS) for 10 min at 65 °C. To separate protein from DNA, samples were treated with 21 μl of 4 m NaCl at 65 °C overnight. Further purification of DNA was done by RNase A (R4875, Sigma) incubation at 37 °C for 1 h followed by addition of proteinase K (P6556, Sigma), EDTA, and Tris, pH 6.5, for 2 h at 42 °C. DNA extraction was done following phenol/chloroform isoamyl alcohol method, and the purity and concentration were analyzed. 2 μl of DNA from each sample were used for ChIP-coupled qRT-PCR (primers listed in supplemental Table 3B). Data analysis was done by the “Fold Enrichment Method,” where ChIP signals are normalized by the IgG signals.
Plasmid construction, transfection
Coding DNA sequences of R. norvegicus full-length PPARα (reference sequence NM_013196.1) (F: 1–1406 nucleotides, 1–478 amino acids) along with its domains N-terminal transactivation domain (AF-1: 1–297 nucleotides, 1–99 amino acids), DNA-binding domain (298–520 nucleotides, 100–173 amino acids), and hinge region followed by C-terminal ligand-binding domain (H + LBD: 521–1406 nucleotides, 174–478 amino acids) were amplified from extracted RNA by RT-PCR with respective primers (supplemental Table 3C) and cloned in-frame into pCDNA6/V5 HisB mammalian expression vector (Invitrogen) with C-terminal His tag as described previously (59). All the clones were confirmed by sequencing (Applied BiosystemsTM, 3730 DNA Analyzer, Waltham, MA). Plasmids were transfected to AngII-treated adult cardiac fibroblasts with the help of TurboFect transfection reagent (Thermo Fisher Scientific) as per the manufacturer's protocol. An empty pCDNA6/V5 His-B mammalian expression vector was also transfected into control and AngII-treated cells.
Dual-luciferase assay
2.4-kb collagen 1a1-luc plasmid with insert sequence ranging between −2300 and 100 bp of col1a1 gene (UniProt Q63121) with putative NF-κBp65-binding site was constructed in PGL-3 vector following standard protocol. Primers are listed in supplemental Table 3D. Clones were confirmed by sequencing (Applied BiosystemsTM, 3730 DNA analyzer). Col1a1-luc and pRL-TK (Promega) were used for dual-luciferase assay as per the manufacturer's instruction. Firefly, luciferase activity of Col1a1 promoter containing PGL-3 vector, and Renilla luciferase activity of PRL-TK were measured in 20 μl of cell lysate using the Dual-Luciferase Reporter Assay System (Promega) using a luminometer (VarioskanTM LUX multimode microplate reader, VL0LA0D0, Thermo Fisher Scientific). Relative activity was defined as the ratio of firefly luciferase activity to Renilla luciferase activity (76).
Protein isolation
Protein from adult cardiac fibroblasts was isolated using M-PER® mammalian protein extraction reagent (Thermo Fisher Scientific) according to the manufacturer's protocol. Protein extracts were prepared from ventricular tissue following the procedure described previously (77).
Western blot assay
Western blot analyses were done to study protein expressions both in vitro and in vivo as described previously (77). Briefly, 30-μg protein samples were separated in 10–12.5% SDS-PAGE and transferred to PVDF membrane. After blocking with 5% nonfat dry milk, membranes were incubated with polyclonal antibodies to the following: TβRI and TβRII (Santa Cruz Biotechnology, Dallas); total TAK1 (Abcam, Cambridge, UK); phospho (Thr-184/187)-TAK1, phospho (Thr-180/Tyr-182), and total p38 MAPK, total SMAD 3, and phospho (Thr-183/Tyr-185)-JNK (Cell Signaling Technology); monoclonal antibodies to PPARα (Abcam, Cambridge, UK); phospho (Ser 465/467) and total SMAD 2, phospho (Ser-423/425) SMAD 3, phospho (Ser-536), and total NF-κBp65, total JNK, and GAPDH (Cell Signaling Technology); phospho (Tyr-199) and total IKKβ (Abcam, Cambridge, UK); and HRP-conjugated secondary antibodies (Pierce). Specifications of the antibodies used are summarized in supplemental Table 4. Immunoreactive bands were visualized using ImmobilonTM Western chemiluminescence HRP substrate (Millipore). A list of antibodies are summarized in supplemental Table 4. The blots were scanned and quantitated using GelDoc XR system and Quantity One® software version 4.6.3 (Bio-Rad).
Co-IP assay
Co-IP was done following the manufacturer's protocol (Pierce Co-IP kit, Thermo Fisher Scientific). Briefly, proteins were incubated with fast-flow protein G- or protein A-Sepharose beads and centrifuged to eliminate non-specifically bound proteins (59). 200 μg of precleared protein were immunoprecipitated using primary antibody. Immunoprotein complex was again incubated with protein G (P3296, Sigma) or protein A (P3391; Sigma)-Sepharose beads. Attached proteins were then eluted from beads in 1% SDS buffer followed by immunoblotting. In this study, immunoprecipitation was done using primary antibodies for anti-PPARα or anti-His (antibodies are listed in supplemental Table 4). For immunoprecipitation with anti-His, proteins were purified in nickel column, as described previously for in vitro protein purification above. Western blot analysis was done using monoclonal antibody against TAK1. Normalization was done by Western blot analysis using the same antibodies used to immunoprecipitate the proteins.
FRAP
For endogenous PPARα and TAK1 interaction, cells were treated with PPARα-TRITC and TAK1-FITC. As TRITC and FITC work as a donor-acceptor in FRAP assay, TRITC was subjected to 50% bleaching, and FITC intensity in a single cell was measured. FRET efficiency was calculated in confocal microscope (Olympus FV1200, Olympus, Singapore) (76).
Statistical analysis
All the results were expressed as means ± S.E. of three independent experiments. Data were analyzed by independent sample t test by SPSS (version 14.0). For multiple comparison experiments, one-way analysis of variance (ANOVA) was conducted followed by Tukey's post hoc test. Results with a p value <0.05 were considered significant.
Author contributions
S. Sarkar conceived the idea of the project. T. B. designed and conducted most of the experiments, analyzed the results, and wrote the paper with S. Sarkar. E. C. assisted in most experiments with animals. J. S. and B. K. did most of the bioinformatic and biophysical studies and analyzed the data. A. R. conducted protein modeling and protein-protein docking, and analyzed the data with S. Sengupta. V. T. did extraction and purification of arjunolic acid used in the study.
Supplementary Material
Acknowledgment
We thank the Indian Institute of Technology (IIT)-Delhi High Performance Computing (HPC) facility for computational resources.
This work was supported in part by Department of Biotechnology, Ministry of Science and Technology, India Grants BT-PR3709/BRB/10/980/2011 and BT/PR7016/NNT/28/641/2012 and Department of Science and Technology, Government of India, Grant SB/SO/HS-148/2013 (to S. Sarkar). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S5 and Tables S1–S4.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- ECM
- extracellular matrix
- PPARα
- peroxisome proliferator-activated receptor α
- AngII
- angiotensin II
- AA
- arjunolic acid
- TAC
- transverse aortic constriction
- TβRI
- TGF-β receptor-I
- TβRII
- TGF-β receptor-II
- SMAD
- suppressor of mothers against decapentaplegic
- IKK
- inhibitor of κB kinase
- col-1
- collagen-1
- col-3
- collagen-3
- anf
- atrial natriuretic factor
- β-mhc
- β-myosin heavy chain
- %FS
- percentage of fractional shortening
- LvIDd
- left ventricular internal diastolic dimension
- LvAWDd
- left ventricular anterior wall diameter during diastole
- LvAWDs
- left ventricular anterior wall diameter during systole
- LvPWDd
- left ventricular posterior wall diameter during diastole
- LvPWDs
- left ventricular posterior wall diameter during systole
- EDV
- left ventricular end diastolic volume
- ESV
- left ventricular end systolic volume
- SV
- stroke volume
- EF
- ejection fraction
- PDB
- Protein Data Base
- NS siRNA
- non-specific siRNA
- CVF
- collagen volume fraction
- DBD
- DNA-binding domain
- LBD
- ligand-binding domain
- PPRE
- PPAR-response element
- BSP-SLIM
- Binding Site Prediction and Shape-based Ligand Matching
- RMSD
- root mean square deviation
- RMSF
- root mean square fluctuation
- qRT-PCR
- quantitative real-time reverse transcription-polymerase chain reaction
- ANOVA
- analysis of variance
- TRITC
- tetramethylrhodamine isothiocyanate
- BW
- body weight
- HW
- heart weight
- F
- full-length PPARα
- CSA
- cross-sectional area
- MD
- molecular dynamics
- FTS
- fluorescence-based thermal shift
- co-IP
- co-immunoprecipitation.
References
- 1. Kong P., Christia P., and Frangogiannis N. G. (2014) The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 71, 549–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Creemers E. E., and Pinto Y. M. (2011) Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc. Res. 89, 265–272 [DOI] [PubMed] [Google Scholar]
- 3. Rosenkranz S. (2004) TGF-β1 and angiotensin networking in cardiac remodeling. Cardiovasc. Res. 63, 423–432 [DOI] [PubMed] [Google Scholar]
- 4. Kurdi M., and Booz G. W. (2011) New take on the role of angiotensin II in cardiac hypertrophy and fibrosis. Hypertension 57, 1034–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brilla C. G., Funck R. C., and Rupp H. (2000) Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation 102, 1388–1393 [DOI] [PubMed] [Google Scholar]
- 6. Chen J. C., Tsai C. C., Chen L. D., Chen H. H., and Wang W. C. (2000) Therapeutic effect of gypenoside on chronic liver injury and fibrosis induced by CCl 4 in rats. Am. J. Chin. Med. 28, 175–185 [DOI] [PubMed] [Google Scholar]
- 7. Fang J., Xu S. W., Wang P., Tang F. T., Zhou S. G., Gao J., Chen J. W., Huang H. Q., and Liu P. Q. (2010) Tanshinone II-A attenuates cardiac fibrosis and modulates collagen metabolism in rats with renovascular hypertension. Phytomedicine 18, 58–64 [DOI] [PubMed] [Google Scholar]
- 8. Dwivedi S., and Chopra D. (2014) Revisiting Terminalia arjuna–an ancient cardiovascular drug. J. Tradit. Complement. Med. 4, 224–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maulik S. K., and Talwar K. K. (2012) Therapeutic potential of Terminalia arjuna in cardiovascular disorders. Am. J. Cardiovasc. Drugs 12 157–163 [DOI] [PubMed] [Google Scholar]
- 10. Amalraj A., and Gopi S. (2017) Medicinal properties of Terminalia arjuna (Roxb.) Wight & Arn.: a review. J. Tradit. Complement. Med. 7, 65–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kumar S., Enjamoori R., Jaiswal A., Ray R., Seth S., and Maulik S. K. (2009) Catecholamine-induced myocardial fibrosis and oxidative stress is attenuated by Terminalia arjuna (Roxb.). J. Pharm. Pharmacol. 61, 1529–1536 [DOI] [PubMed] [Google Scholar]
- 12. Dwivedi S. (2007) Terminalia arjuna Wight & Arn.—a useful drug for cardiovascular disorders. J. Ethnopharmacol. 114, 114–129 [DOI] [PubMed] [Google Scholar]
- 13. Ghosh J., and Sil P. C. (2013) Arjunolic acid: a new multifunctional therapeutic promise of alternative medicine. Biochimie 95, 1098–1109 [DOI] [PubMed] [Google Scholar]
- 14. Ramesh A. S., Christopher J. G., Radhika R., Setty C. R., and Thankamani V. (2012) Isolation, characterisation and cytotoxicity study of arjunolic acid from Terminalia arjuna. Nat. Prod. Res. 26, 1549–1552 [DOI] [PubMed] [Google Scholar]
- 15. Kelly D. P. (2003) PPARs of the heart. Circ. Res. 92, 482–484 [DOI] [PubMed] [Google Scholar]
- 16. Smeets P. J., Planavila A., Van Der Vusse G. J., and Van Bilsen M. (2007) Peroxisome proliferator-activated receptors and inflammation: take it to heart. Acta Physiol. 191, 171–188 [DOI] [PubMed] [Google Scholar]
- 17. Jiao M., Ren F., Zhou L., Zhang X., Zhang L., Wen T., Wei L., Wang X., Shi H., Bai L., Zheng S., Zhang J., Chen Y., Han Y., Zhao C., and Duan Z. (2014) Peroxisome proliferator-activated receptor α activation attenuates the inflammatory response to protect the liver from acute failure by promoting the autophagy pathway. Cell Death Dis. 5, e1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Diep Q. N., Benkirane K., Amiri F., Cohn J. S., Endemann D., and Schiffrin E. L. (2004) PPARα activator fenofibrate inhibits myocardial inflammation and fibrosis in angiotensin II-infused rats. J. Mol. Cell. Cardiol. 36, 295–304 [DOI] [PubMed] [Google Scholar]
- 19. Ogata T., Miyauchi T., Sakai S., Takanashi M., Irukayama-Tomobe Y., and Yamaguchi I. (2004) Myocardial fibrosis and diastolic dysfunction in deoxycorticosterone acetate-salt hypertensive rats is ameliorated by the peroxisome proliferator-activated receptor-α activator fenofibrate, partly by suppressing inflammatory responses associated with the nuclear factor-κB pathway. J. Am. Coll. Cardiol. 43, 1481–1488 [DOI] [PubMed] [Google Scholar]
- 20. Seymour E. M., Bennink M. R., Watts S. W., and Bolling S. F. (2010) Whole grape intake impacts cardiac peroxisome proliferator-activated receptor and nuclear factor κB activity and cytokine expression in rats with diastolic dysfunction. Hypertension 55, 1179–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smeets P. J., Teunissen B. E., Willemsen P. H., van Nieuwenhoven F. A., Brouns A. E., Janssen B. J., Cleutjens J. P., Staels B., van der Vusse G. J., and van Bilsen M. (2008) Cardiac hypertrophy is enhanced in PPARα−/− mice in response to chronic pressure overload. Cardiovasc. Res. 78, 79–89 [DOI] [PubMed] [Google Scholar]
- 22. Hata A., and Chen Y. G. (2016) TGF-β signaling from receptors to Smads. Cold Spring Harb. Perspect. Biol. 8, a022061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Divakaran V., Adrogue J., Ishiyama M., Entman M. L., Haudek S., Sivasubramanian N., and Mann D. L. (2009) Adaptive and maladptive effects of Smad3 signaling in the adult heart following hemodynamic pressure overloading. Circ. Heart Fail. CIRCHEARTFAILURE-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kishimoto K., Matsumoto K., and Ninomiya-Tsuji J. (2000) TAK1 mitogen-activated protein kinase kinase kinase is activated by autophosphorylation within its activation loop. J. Biol. Chem. 275, 7359–7364 [DOI] [PubMed] [Google Scholar]
- 25. Zhang Y. E. (2009) Non-Smad pathways in TGF-β signaling. Cell Res. 19, 128–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shim J. H., Xiao C., Paschal A. E., Bailey S. T., Rao P., Hayden M. S., Lee K. Y., Bussey C., Steckel M., Tanaka N., Yamada G., Akira S., Matsumoto K., and Ghosh S. (2005) TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev. 19, 2668–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mao R., Fan Y., Mou Y., Zhang H., Fu S., and Yang J. (2011) TAK1 lysine 158 is required for TGF-β-induced TRAF6-mediated Smad-independent IKK/NF-κB and JNK/AP-1 activation. Cell. Signal. 23, 222–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang D., Gaussin V., Taffet G. E., Belaguli N. S., Yamada M., Schwartz R. J., Michael L. H., Overbeek P. A., and Schneider M. D. (2000) TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat. Med. 6, 556–563 [DOI] [PubMed] [Google Scholar]
- 29. Ono K., Ohtomo T., Ninomiya-Tsuji J., and Tsuchiya M. (2003) A dominant negative TAK1 inhibits cellular fibrotic responses induced by TGF-β. Biochem. Biophys. Res. Commun. 307, 332–337 [DOI] [PubMed] [Google Scholar]
- 30. Guo B., Koya D., Isono M., Sugimoto T., Kashiwagi A., and Haneda M. (2004) Peroxisome proliferator-activated receptor-γ ligands inhibit TGF-β1-induced fibronectin expression in glomerular mesangial cells. Diabetes 53, 200–208 [DOI] [PubMed] [Google Scholar]
- 31. Poleni P. E., Bianchi A., Etienne S., Koufany M., Sebillaud S., Netter P., Terlain B., and Jouzeau J. Y. (2007) Agonists of peroxisome proliferators-activated receptors (PPAR) α, β/δ or γ reduce transforming growth factor (TGF)-β-induced proteoglycans' production in chondrocytes. Osteoarthr. Cartil. 15, 493–505 [DOI] [PubMed] [Google Scholar]
- 32. Subramanian V., Seemann I., Merl-Pham J., Hauck S. M., Stewart F. A., Atkinson M. J., Tapio S., and Azimzadeh O. (2017) Role of TGFβ and PPARα signaling pathways in radiation response of locally exposed heart: integrated global transcriptomics and proteomics analysis. J. Proteome Res. 16, 307–318 [DOI] [PubMed] [Google Scholar]
- 33. Surewicz W. K., Mantsch H. H., and Chapman D. (1993) Determination of protein secondary structure by Fourier transform infrared spectroscopy: a critical assessment. Biochemistry 32, 389–394 [DOI] [PubMed] [Google Scholar]
- 34. Krimm S., and Bandekar J. (1986) Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. Adv. Protein Chem. 38, 181–364 [DOI] [PubMed] [Google Scholar]
- 35. Abdul Ajees A., and Balakrishna K. (2002) Arjunolic acid. Acta Crystallogr. Sect. E Struct. Rep. Online 10.1107/S1600536802006451 [DOI] [Google Scholar]
- 36. Hemalatha T., Pulavendran S., Balachandran C., Manohar B. M., and Puvanakrishnan R. (2010) Arjunolic acid: a novel phytomedicine with multifunctional therapeutic applications. Indian J. Exp. Biol. 48, 238–247 [PubMed] [Google Scholar]
- 37. Yadav V. R., Prasad S., Sung B., Kannappan R., and Aggarwal B. B. (2010) Targeting inflammatory pathways by triterpenoids for prevention and treatment of cancer. Toxins 2, 2428–2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goto T., Takahashi N., Kato S., Egawa K., Ebisu S., Moriyama T., Fushiki T., and Kawada T. (2005) Phytol directly activates peroxisome proliferator-activated receptor α (PPARα) and regulates gene expression involved in lipid metabolism in PPARα-expressing HepG2 hepatocytes. Biochem. Biophys. Res. Commun. 337, 440–445 [DOI] [PubMed] [Google Scholar]
- 39. Lee H. K., Nam G. W., Kim S. H., and Lee S. H. (2006) Phytocomponents of triterpenoids, oleanolic acid and ursolic acid, regulated differently the processing of epidermal keratinocytes via PPAR-α pathway. Exp. Dermatol. 15, 66–73 [DOI] [PubMed] [Google Scholar]
- 40. Jin L., Lin S., Rong H., Zheng S., Jin S., Wang R., and Li Y. (2011) Structural basis for iloprost as a dual peroxisome proliferator-activated receptor α/δ agonist. J. Biol. Chem. 286, 31473–31479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oyama T., Toyota K., Waku T., Hirakawa Y., Nagasawa N., Kasuga J. I., Hashimoto Y., Miyachi H., and Morikawa K. (2009) Adaptability and selectivity of human peroxisome proliferator-activated receptor (PPAR) pan agonists revealed from crystal structures. Acta Crystallogr. D Biol. Crystallogr. 65, 786–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Park M. H., Park J. Y., Lee H. J., Kim D. H., Chung K. W., Park D., Jeong H. O., Kim H. R., Park C. H., Kim S. R., Chun P., Byun Y., Moon H. R., and Chung H. Y. (2013) The novel PPAR α/γ dual agonist MHY 966 modulates UVB–induced skin inflammation by inhibiting NF-κB activity. PLoS ONE 8, e76820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cronet P., Petersen J. F., Folmer R., Blomberg N., Sjöblom K., Karlsson U., Lindstedt E. L., and Bamberg K. (2001) Structure of the PPARα and-γ ligand-binding domain in complex with AZ 242; ligand selectivity and agonist activation in the PPAR family. Structure 9, 699–706 [DOI] [PubMed] [Google Scholar]
- 44. dos Santos J. C., Bernardes A., Giampietro L., Ammazzalorso A., De Filippis B., Amoroso R., and Polikarpov I. (2015) Different binding and recognition modes of GL479, a dual agonist of peroxisome proliferator-activated receptor α/γ. J. Struct. Biol. 191, 332–340 [DOI] [PubMed] [Google Scholar]
- 45. McMullen P. D., Bhattacharya S., Woods C. G., Sun B., Yarborough K., Ross S. M., Miller M. E., McBride M. T., LeCluyse E. L., Clewell R. A., and Andersen M. E. (2014) A map of the PPARα transcription regulatory network for primary human hepatocytes. Chem. Biol. Interact. 209, 14–24 [DOI] [PubMed] [Google Scholar]
- 46. Youssef J., and Badr M. (2015) Peroxisome proliferator-activated receptors: features, functions, and future. Nucl Receptor Res. 2, 1–30 [Google Scholar]
- 47. Tzeng J., Byun J., Park J.Y., Yamamoto T., Schesing K., Tian B., Sadoshima J., and Oka S. (2015) An ideal PPAR response element bound to and activated by PPARα. PLoS ONE 10, e0134996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang S., Weinheimer C., Courtois M., Kovacs A., Zhang C. E., Cheng A. M., Wang Y., and Muslin A. J. (2003) The role of the Grb2–p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J. Clin. Invest. 111, 833–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mir S. A., Chatterjee A., Mitra A., Pathak K., Mahata S. K., and Sarkar S. (2012) Inhibition of signal transducer and activator of transcription 3 (STAT3) attenuates interleukin-6 (IL-6)-induced collagen synthesis and resultant hypertrophy in rat heart. J. Biol. Chem. 287, 2666–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hedhli N., Lizano P., Hong C., Fritzky L. F., Dhar S. K., Liu H., Tian Y., Gao S., Madura K., Vatner S. F., and Depre C. (2008) Proteasome inhibition decreases cardiac remodeling after initiation of pressure overload. Am. J. Physiol. Heart Circ. Physiol. 295, H1385–H1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wei C., Kim I. K., Kumar S., Jayasinghe S., Hong N., Castoldi G., Catalucci D., Jones W. K., and Gupta S. (2013) NF-κB mediated miR-26a regulation in cardiac fibrosis. J. Cell. Physiol. 228, 1433–1442 [DOI] [PubMed] [Google Scholar]
- 52. Akaike M., Che W., Marmarosh N. L., Ohta S., Osawa M., Ding B., Berk B. C., Yan C., and Abe J. (2004) The hinge-helix 1 region of peroxisome proliferator-activated receptor γ1 (PPARγ1) mediates interaction with extracellular signal-regulated kinase 5 and PPARγ1 transcriptional activation: involvement in flow-induced PPARγ activation in endothelial cells. Mol. Cell. Biol. 24, 8691–8704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. von Knethen A., Soller M., Tzieply N., Weigert A., Johann A. M., Jennewein C., Köhl R., and Brüne B. (2007) PPARγ1 attenuates cytosol to membrane translocation of PKCα to desensitize monocytes/macrophages. J. Cell Biol. 176, 681–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stockert J., Wolf A., Kaddatz K., Schnitzer E., Finkernagel F., Meissner W., Müller-Brüsselbach S., Kracht M., and Müller R. (2013) Regulation of TAK1/TAB1-mediated IL-1β signaling by cytoplasmic PPARβ/δ. PLoS ONE 8, e63011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lazennec G., Canaple L., Saugy D., and Wahli W. (2000) Activation of peroxisome proliferator-activated receptors (PPARs) by their ligands and protein kinase A activators. Mol. Endocrinol. 14, 1962–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Leask A. (2015) Getting to the heart of the matter. Circ. Res. 116, 1269–1276 [DOI] [PubMed] [Google Scholar]
- 57. Ray A., Rana S., Banerjee D., Mitra A., Datta R., Naskar S., and Sarkar S. (2016) Improved bioavailability of targeted curcumin delivery efficiently regressed cardiac hypertrophy by modulating apoptotic load within cardiac microenvironment. Toxicol. Appl. Pharmacol. 290, 54–65 [DOI] [PubMed] [Google Scholar]
- 58. Rana S., Datta K., Reddy T. L., Chatterjee E., Sen P., Pal-Bhadra M., Bhadra U., Pramanik A., Pramanik P., Chawla-Sarkar M., and Sarkar S. (2015) A spatio-temporal cardiomyocyte targeted vector system for efficient delivery of therapeutic payloads to regress cardiac hypertrophy abating bystander effect. J. Control. Release 200, 167–178 [DOI] [PubMed] [Google Scholar]
- 59. Datta R., Bansal T., Rana S., Datta K., Chattopadhyay S., Chawla-Sarkar M., and Sarkar S. (2015) Hsp90/Cdc37 assembly modulates TGFβ receptor-II to act as a profibrotic regulator of TGFβ signaling during cardiac hypertrophy. Cell. Signal. 27, 2410–2424 [DOI] [PubMed] [Google Scholar]
- 60. Yang J., and Zhang Y. (2015) I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 43, W174–W181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xu D., and Zhang Y. (2011) Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 101, 2525–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ray A., Lindahl E., and Wallner B. (2012) Improved model quality assessment using ProQ2. BMC Bioinformatics 13, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., and Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 [DOI] [PubMed] [Google Scholar]
- 64. Brooks B. R., Brooks C. L. 3rd., MacKerell A. D. Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., et al. (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Huang J., and MacKerell A. D. (2013) CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Denning E. J., Priyakumar U. D., Nilsson L., and Mackerell A. D. Jr. (2011) Impact of 2′-hydroxyl sampling on the conformational properties of RNA: update of the CHARMM all-atom additive force field for RNA. J. Comput. Chem. 32, 1929–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zoete V., Cuendet M. A., Grosdidier A., and Michielin O. (2011) SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem. 32, 2359–2368 [DOI] [PubMed] [Google Scholar]
- 68. O'Boyle N. M., Banck M., James C. A., Morley C., Vandermeersch T., and Hutchison G. R. (2011) Open Babel: An open chemical toolbox. J. Cheminform. 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lee H. S., and Zhang Y. (2012) BSP-SLIM: A blind low-resolution ligand-protein docking approach using predicted protein structures. Proteins 80, 93–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mills J. E., and Dean P. M. (1996) Three-dimensional hydrogen-bond geometry and probability information from a crystal survey. J. Comput. Aided Mol. Des. 10, 607–622 [DOI] [PubMed] [Google Scholar]
- 71. Trott O., and Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 73. Dominguez C., Boelens R., and Bonvin A. M. (2003) HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 74. de Vries S. J., van Dijk A. D., Krzeminski M., van Dijk M., Thureau A., Hsu V., Wassenaar T., and Bonvin A. M. (2007) HADDOCK versus HADDOCK: new features and performance of HADDOCK2. 0 on the CAPRI targets. Proteins 69, 726–733 [DOI] [PubMed] [Google Scholar]
- 75. Kouskouti A., and Talianidis I. (2005) Histone modifications defining active genes persist after transcriptional and mitotic inactivation. EMBO J. 24, 347–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Datta R., Bansal T., Rana S., Datta K., Datta Chaudhuri R., Chawla-Sarkar M., and Sarkar S. (2017) Myocyte-derived Hsp90 modulates collagen up-regulation via biphasic activation of STAT-3 in fibroblasts during cardiac hypertrophy. Mol. Cell. Biol. 37, e00611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mitra A., Basak T., Datta K., Naskar S., Sengupta S., and Sarkar S. (2013) Role of α-crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis. 4, e582. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.