

Abstract
Locked nucleic acid is a prominent nucleic acid analog with unprecedented target binding affinity to cDNA and RNA oligonucleotides and shows remarkable stability against nuclease degradation. Incorporation of locked nucleic acid nucleotides into an antisense oligonucleotide (AO) sequence can reduce the length required without compromising the efficacy. In this study, we synthesized a series of systematically truncated locked nucleic acid-modified 2′-O-methyl AOs on a phosphorothioate (PS) backbone that were designed to induce skipping exon 23 from the dystrophin transcript in H-2Kb-tsA58 mdx mouse myotubes in vitro. The results clearly demonstrated that shorter AOs (16- to 14-mer) containing locked nucleic acid nucleotides efficiently induced dystrophin exon 23 skipping compared with the corresponding 2′-O-methyl AOs. Our remarkable findings contribute significantly to the existing knowledge about the designing of short LNA-modified oligonucleotides for exon-skipping applications, which will help reduce the cost of exon-skipping AOs and potential toxicities, particularly the 2′-OMe-based oligos, by further reducing the length of AOs.
Keywords: locked nucleic acid, modified nucleotides, antisense oligonucleotide, chemically-modified nucleotides, nucleic acid chemistry, exon-skipping, Duchenne muscular dystrophy
Introduction
Locked nucleic acid (LNA) is one of the prominent nucleic acid analogs with excellent bio-physical properties and has attracted extensive interest in the development of therapeutics as well as diagnostics.1, 2, 3 LNA-modified oligonucleotides show very high binding affinity to both DNA and RNA oligonucleotides and increased stability against nuclease degradation compared with other chemistries, such as 2′-O-methyl (2′-OMe).4, 5 Introduction of LNA nucleotide monomers (Figure 1) improves the potency of therapeutic oligonucleotides in vitro and in vivo over other nucleic acid chemistries when applied as antisense constructs, siRNAs, and anti-miRs and are also found to be well tolerated in animals.6, 7, 8, 9, 10, 11, 12
Figure 1.
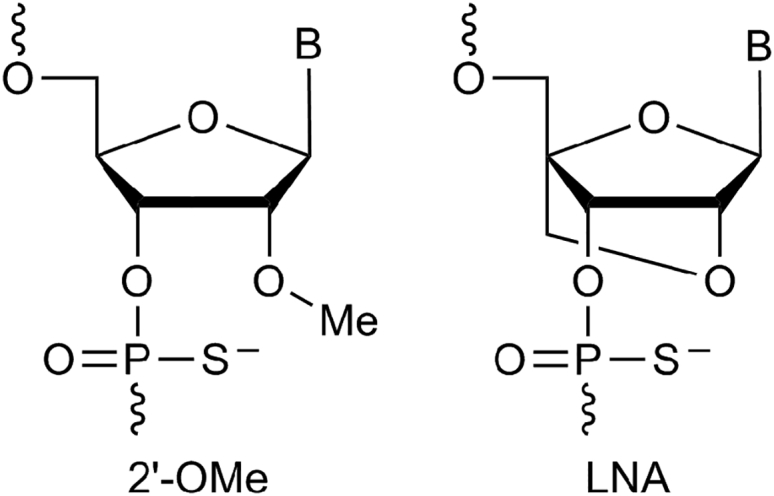
Structure Representation of 2′-OMethyl and LNA Nucleotide Monomers Used in This Study
Duchenne muscular dystrophy (DMD) is a muscle-wasting genetic disease caused by protein-truncating mutations, commonly frameshifting deletions of one or more exons in the dystrophin gene that abolish functional dystrophin expression.13, 14, 15 Splice-switching AOs to induce specific exon-skipping and restore the reading frame and expression of a partially functional dystrophin protein has been explored as a treatment for DMD.16, 17, 18, 19, 20, 21 This most promising approach has been shown to slow the progression of the disease over several years.22 Two such drug molecules, Exondys 5122, 23 (Sarepta Therapeutics) and Drisapersen (BioMarin Pharmaceutical) targeting exon 51, were evaluated in phase 3 clinical trials. The drisapersen trials were halted in September 2013 after primary and secondary endpoints were not met and life-threatening side effects were found.24 In contrast, Exondys 51, a phosphorodiamidate morpholino oligomer (PMO), has been given accelerated approval by the US Food and Drug Administration (FDA) for the treatment of DMD.25 Exondys 51 has been shown to produce modest increases in dystrophin protein and showed an excellent safety profile in the trial participants.26 However, PMO monomers are not compatible with the standard phosphoramidite chemistry, and the current PMO synthesis chemistries are costly.
One approach to possibly limit toxicity of the phosphorothioate (PS) AOs that were investigated in previous studies,27, 28, 29, 30, 31 and further reduce costs, would be to truncate oligonucleotide length. Introduction of LNA nucleotide monomers could be one approach, allowing AO length to be truncated by relying on properties such as high target binding affinity, mismatch recognition capability, and increased resistance to nuclease degradation. Because the current PMO chemistry is not compatible with the standard solid-phase oligonucleotide synthesis procedures, 2′-OMePS AOs would be suitable to investigate the impact of LNA nucleotides to develop shorter exon-skipping AOs. Herein, we report the systematic truncation of 2′-OMePS-modified AOs using LNA nucleotides by constructing 2′-OMePS/LNA mixmers and 2′-OMePS/LNA gapmer-like AOs to induce exon 23 skipping in the mouse dystrophin gene transcript in vitro.
Results
In this study, we used a previously reported 20-mer 2′-OMe AO on a PS backbone designed to induce exon 23 skipping in the mouse dystrophin gene transcript.32 The systematic truncation of the 20-mer 2′-OMePS AO (AO1) was performed by removing one to four nucleotides from both 5′ and 3′ ends simultaneously, and their corresponding LNA-modified variants were synthesized as mixmers and gapmers to generate 18-mer (AO2–AO5), 16-mer (AO6–AO9), 14-mer (AO10–AO13), and 12-mer (AO14–AO17) candidate AOs (Table 1). All AOs were synthesized in house on a GE ÄKTA oligopilot plus 10 synthesizer at 1 μmol scale on a PS backbone. The efficacies of all AOs were evaluated for their ability to induce exon 23 skipping in H2K mdx mouse myotubes in vitro. For this purpose, H2K mdx mouse myoblasts were plated on a 24-well plate, differentiated over 24 hr, and transfected with the AOs in complex with the cationic lipid Lipofectin, using a 2:1 (w/w) ratio initially at 100, 200, and 400 nM concentrations, to screen exon-skipping efficiency for all AOs, and later at 12.5, 25, 50, and 100 nM concentrations for the best-performing AOs. The cells were collected after 24 hr, RNA was isolated, and RT-PCR analysis was performed by amplifying across exons 20–26 to evaluate the skipping efficiency of the AOs as previously reported.33 The PCR products were then analyzed using 2% agarose gel electrophoresis, and densitometry analysis of the gel images was performed using ImageJ software. The actual exon-skipping efficiency was determined by expressing the amount of exon 23 skipped RT-PCR product as a percentage of total dystrophin transcript products.
Table 1.
AO Names and Their Sequences Used in This Study
| AO No. | AO Name | Sequence, 5′→ 3′ Direction |
|---|---|---|
| AO1 | DmdE23D (+2–18) | GGCCAAACCUCGGCUUACCU |
| AO2 | DmdE23D (+1–17) | GCCAAACCUCGGCUUACC |
| AO3 | DmdE23D (+1–17) | GCCAAAClCTlCGGCTlUACCl |
| AO4 | DmdE23D (+1–17) | GCCAAAClCUCGGCTlUAlCCl |
| AO5 | DmdE23D (+1–17) | GlClCAAACCUCGGCUUAClCl |
| AO6 | DmdE23D (−1–16) | CCAAACCUCGGCUUAC |
| AO7 | DmdE23D (−1–16) | CCAAAClCTlCGGCTlUACl |
| AO8 | DmdE23D (−1–16) | CCAAAClCUCGGClUTlACl |
| AO9 | DmdE23D (−1–16) | ClClAAACCUCGGCUUAlCl |
| AO10 | DmdE23D (−2–15) | CAAACCUCGGCUUA |
| AO11 | DmdE23D (−2–15) | CAAAClCTlCGGCTlUAl |
| AO12 | DmdE23D (−2–15) | CAAAClCTCGGlClUUAl |
| AO13 | DmdE23D (−2–15) | ClAlAACCUCGGCUTlAl |
| AO14 | DmdE23D (−3–14) | AAACCUCGGCUU |
| AO15 | DmdE23D (−3–14) | AAAClCTlCGGClUTl |
| AO16 | DmdE23D (−3–14) | AAAClCUCGGlClUTl |
| AO17 | DmdE23D (−3–14) | AlAlACCUCGGCTlTl |
2′-OMePS nucleotides are represented as uppercase letters; LNA nucleotide monomers are represented as underlined letters with superscript l.
Evaluation of Systematically Truncated 2′-OMePS AOs to Induce Exon 23 Skipping in mdx Mouse Myotubes In Vitro
First, we tested the potential of the truncated 2′-OMePS AOs including AO2 (18-mer), AO6 (16-mer), AO10 (14-mer), and AO14 (12-mer) in parallel with the 20-mer control AO1. As previously reported,32, 33 AO1 showed efficient exon 23 (full-length product size 901 bp) skipping in vitro by yielding the deletion product of 688 bp at all concentrations (52% at 100 nM, 57% at 200 nM, and 65% at 400 nM; Figures 2A and 2B). The 18-mer AO2 with the two-nucleotide truncation also showed similar efficacy as the 20-mer control AO1 and induced exon-23 skipping levels of 49% at 100 nM, 53% at 200 nM, and 64% at 400 nM. However, a significant drop in the amount of 688 bp product was observed after transfection with the 16-mer AO6 (23% at 100 nM, 30% at 200 nM, and 39% at 400 nM). Notably, the 14-mer AO10 and the 12-mer AO14 failed to show any skipped product of 688 bp (Figure 2A). In all AO transfections (all concentrations), an additional product band at 542 bp was also observed on the gel (Figure 2A) that corresponds to an out-of-frame exon 22/23 dual skipping product, in line with previous reports.32, 33, 34, 35, 36 We found a similar trend of increasing amount of 542 bp band at 100 and 200 nM concentrations (Figure 2B) and a decrease in yield at 400 nM for AO1, AO2, and AO6, but the 14-mer AO10 only showed the product at 400 nM.
Figure 2.

RT-PCR Analysis of RNA Prepared from Myogenic Cells Transfected with Systematically Truncated 2′-OMePS AOs
(A) RT-PCR analysis after transfection with AO1: 20-mer-2′-OMePS AO; AO2: 18-mer 2′-OMePS AO; AO6: 16-mer 2′-OMePS AO; AO10: 14-mer 2′-OMePS AO; and AO14: 12-mer 2′-OMePS AO. The arrowheads above the gel images indicate increasing AO concentration (100, 200, and 400 nM). (B) Densitometry analysis of the transcripts induced by 2′-OMePS AOs. Green, exon 23 skipping; blue, dual exon 22/23 skipping; gray, full-length RT-PCR product.
Evaluation of Systematically Truncated LNA/2′-OMePS AO Mixmers and Gapmers to Induce Dystrophin Exon 23 Skipping in mdx Mouse Myotubes In Vitro
We evaluated the impact of LNA nucleotides in truncated 2′-OMePS AOs for which three different types, two mixmers and one gapmer-like AOs, were designed and synthesized by incorporating four LNA nucleotides into each sequence (Table 1). We evaluated the dystrophin exon 23 skipping efficiencies of truncated LNA/2′-OMePS AOs of sizes: 18-mers (AO3–AO5), 16-mers (AO7–AO9), 14-mers (AO11–AO13), and 12-mers (AO15–AO17) in vitro compared with the corresponding 2′-OMePS unmodified AOs (Table 1).
All 18-mer LNA/2′-OMePS AOs, AO3, AO4 (mixmer 1 and 2), and AO5 (gapmer-like) showed efficient exon 23 skipping at all concentrations, similar to that induced by the unmodified AO2 (Figure 3A). The mixmer construct AO4 induced the highest level of exon 23 skipping by yielding 66% and 72% of the 688 bp skipped transcript product at 200 and 400 nM concentrations, respectively, compared with 53% and 64% exon-skipping efficiency induced by the unmodified AO2 (Figures 3A and 3E). All 18-mer LNA/2′-OMePS AOs also induced the exon 22/23 deletion product of 542 bp at varying yields (Figure 3E), but the band intensity was low at 400 nM in the case of AO4.
Figure 3.

RT-PCR Analysis of AO Candidates Inducing Exon 23 Skipping in H2K mdx Mouse Myotubes
(A) 18-mer AOs, (B) 16-mer AOs, (C) 14-mer AOs, and (D) 12-mer AOs. The arrowheads above the gel images indicate increasing AO concentration (100, 200, and 400 nM). (E) Densitometry analysis of exon-skipping efficiency (see text description).
The exon-skipping efficiency of 16-mer LNA/2′-OMePS AO variants, AO7 (mixmer 1), AO8 (mixmer 2), and AO9 (gapmer-like) was tested under the same conditions as above in parallel with the corresponding 2′-OMePS AO6. All LNA-modified truncated AOs induced the exon 23 deletion product of 688 bp at all concentrations tested, but higher exon-skipping levels were achieved at 200 and 400 nM (Figures 3B and 3E). Interestingly, the mixmer AO8 yielded a higher percentage of exon 23 skipping (75%) than any of 18-mer truncated AOs. In contrast, the exon-skipping efficiency of the unmodified 16-mer 2′-OMePS AO was found to be significantly lower at all concentrations, compared with that of LNA-modified AOs (Figures 3B and 3E).
The efficacies of 14-mer LNA-modified 2′-OMePS AO candidates (AO11, mixmer 1; AO12, mixmer 2; and AO13, gapmer-like) were also evaluated together with the unmodified 2′-OMePS AO10. The LNA/2′-OMePS 14-mer-AOs clearly showed the exon 23 deletion product of 688 bp at all concentrations tested, although the highest efficiency was observed at 400 nM (Figures 3C and 3E). Efficient exon 23 skipping was observed with the mixmer AO12 at 400 nM, yielding 66% of the skipped product, in line with the 20-mer 2′-OMePS AO1. However, the 14-mer 2′-OMePS AO10 failed to induce exon 23 skipping at all concentrations. All LNA-modified 14-mer AOs also showed the exon 22/23 dual skipping product of 542 bp as a minor product at all concentrations tested (Figures 3C and 3E).
The 12-mer AOs, AO15 (mixmer 1), AO16 (mixmer 2), and AO17 (gapmer-like) also induced the exon 23 deletion product of 688 bp, but the yield was found to be low compared with the other longer AOs. The LNA/2′-OMePS mixmer AO16 induced a higher percentage of exon 23 skipping at 400 nM (29%; Figures 3D and 3E) than any of the other 12-mer AOs. Not surprisingly, the unmodified 12-mer 2′-OMePS AO14 did not yield any exon 23 skipping, other than approximately 7% of the exon 22/23 dual skipping product (Figures 3D and 3E).
Exon-Skipping Efficacy Analyses of Promising LNA/2′-OMePS AO Candidates at Lower Concentrations
To further investigate the efficacy of truncated LNA-modified 2′OMePS AOs, we then analyzed exon-skipping at lower concentrations (12.5, 25, 50, and 100 nM). For this purpose, the best-performing four LNA/ 2′-OMePS AO candidates for each size (AO4, AO8, AO12, and AO16) out of 12 candidates were evaluated along with their corresponding unmodified 2′-OMePS AOs (AO2, AO6, AO10, and AO14). The 18-mer AO4 showed efficient exon 23 skipping at 25, 50, and 100 nM compared with AO2, for which good skipping was observed at 50 nM and above (Figure 4A). The 16-mer LNA/2′-OMePS AO8 also showed high exon 23 skipping efficacy at 25, 50, and 100 nM, whereas the control AO6 yielded significantly less exon 23 skipped product (Figure 4B). The 14-mer LNA/2′-OMePS AO12 was also induced exon 23 skipping at 50 and 100 nM concentrations, but the 2′-OMePS AO10 failed to induce any exon 23 skipping (Figures 4C and 4E). The 12-mer LNA/2′-OMePS AO16 did not exhibit good skipping efficacy, but the exon 23 skipped product was visible at low levels (up to 20%; Figures 4D and 4E), and the unmodified 12-mer AO14 failed to induce any exon 23 skipping at all concentrations tested. Notably, there were also bands detected at 542 bp corresponding to dual exon 22/23 skipping, and this product was observed at low yields in all cases, except for the 14-mer and 12-mer unmodified AOs (Figures 4C and 4D).
Figure 4.

RT-PCR Analysis of AO Candidates Inducing Exon 23 Skipping in mdx Mouse Myotubes at Low Concentrations
(A) 18-mer AOs, (B) 16-mer AOs, (C) 14-mer AOs, and (D) 12-mer AOs. The arrowheads above the gel images indicate increasing AO concentration (12.5, 25, 50, and 100 nM). (E) Densitometry analysis of exon skipping (see text description).
Discussion
First described in 1993 by Dominski and Kole,37 AO-mediated splice modulation of pre-mRNA is becoming a well-established therapeutic approach. In 2016, two AO drugs were approved by the FDA for the treatment of DMD (Exondys 51; PMO chemistry)25 and spinal muscular atrophy (Spinraza; 2′-O-methoxyethyl PS chemistry).38 The 2′-OMePS-based drug candidate, Drisapersen, also entered phase 3 clinical trials for the treatment of DMD, but the application was rejected by the FDA because of failure to meet primary or secondary endpoints and life-threatening toxicity.24 Alternative strategies are required to further improve the efficacy and limit the toxicity of 2′-OMePS AOs if they are to be used for clinical application. Incorporation of LNA nucleotides to construct shorter AOs with high affinity to the target sequence could be one approach to circumvent this problem. Aartsma-Rus et al.39 reported the use of LNA-fully modified AOs to induce dystrophin exon 46 skipping in DMD patient cells and showed that the LNA AOs yielded higher levels of the exon-skipping product, but the sequence specificity was low when tested in mismatch experiments. This is not surprising, as higher numbers of LNA nucleotides could increase the possibility of off-target binding, and shorter molecules would anneal to off-target sites in the genome/transcriptome. However, regarding the specificity, it is worth mentioning that a short single-stranded 15-mer LNA-modified PS antisense oligonucleotide (AO) targeting the microRNA-122 called Miravirsen has advanced to phase 2A clinical trials. Administration of this short LNA oligo by subcutaneous injection in patients at the highest dose (7 mg/kg) for 4 weeks was found to be safe and exhibited significant decrease in hepatitis C virus RNA, indicating the target specificity of short LNA-modified oligos.40 Later, Shimo et al.41 also reported the design of a series of LNA-modified AOs targeting DMD transcripts and evaluated their efficacy in inducing efficient exon 58 skipping in vitro.
In this study, we explored the ability of LNA nucleotides to fine-tune the performance of 2′-OMePS AOs by screening systematically truncated LNA-modified 2′-OMePS mixmers and gapmer-like AOs to induce exon 23 skipping in the H2K mdx mouse myotubes in vitro. We designed and synthesized systematically truncated series of LNA/2′-OMePS AOs (from 18-mer to 12-mer) and their corresponding 2′-OMePS sequences by systematically removing one nucleotide each from the 3′ and 5′ ends simultaneously, beginning with a previously reported 20-mer sequence32, 33 (Table 1). Two mixmer types and one gapmer-like LNA/2′-OMePS AOs were synthesized containing four LNA nucleotides in each of the sequences. In one of the mixmers, two LNA nucleotides were placed toward the 3′ end and in the middle region, while in the second mixmer design, three LNA nucleotides were placed toward the 3′ end and one LNA in the middle region. All gapmer-like designs had two LNA nucleotides positioned consecutively at both 3′ and 5′ ends. In line with our previous reports,34, 35, 36 the efficacy of each AO was first tested in H2K mdx mouse myotubes in vitro at 100, 200, and 400 nM concentrations.
The results demonstrated that the 18-mer AO2 with two nucleotide truncations maintained similar efficacy to the 20-mer AO1, but further truncation of the AO2 sequence by additional two nucleotides (AO6) resulted in a significant drop in exon 23 skipping efficiency. Truncation of 2′-OMePS AOs clearly affected the efficacy, as the 20-mer AO1 was found to be one of the best-performing AOs (but less efficient than the 25-mer AO)32 in inducing exon 23 skipping in vitro, compared with the truncated variants, while the introduction of LNA nucleotides in the truncated 2′-OMePS AOs improved the efficacy of exon 23 skipping. All 18-mer LNA/2′-OMePS AOs induced exon 23 skipping. Interestingly, one of the mixmers AO4 showed higher skipping efficacy than the 20-mer 2′-OMePS AO1, and this may suggest that the positioning of LNA nucleotides could be better toward the 3′ end of the AOs. The impact of LNA nucleotide inclusion is very evident when analyzing the 16-mer AOs, as all LNA/2′-OMePS AO variations induced exon 23 skipping at 2-fold higher efficiency compared with 2′-OMePS AO.
The best-performing LNA-modified 2′-OMePS AOs of each size identified above, 18-mer AO4, 16-mer AO8, 14-mer AO12, and AO16, along with the corresponding unmodified 2′-OMePS AOs, were further evaluated for exon 23 skipping efficacy at lower concentrations. In general, the AOs showed a similar trend in skipping efficiencies as discussed for those AOs at higher concentrations. The results clearly demonstrated that all LNA/ 2′-OMePS AOs were capable of inducing exon 23 skipping at lower concentrations, in a dose-dependent manner, whereas the exon 23 skipping efficiencies of the 2′-OMePS control AOs were significantly lower and proportional to the length of the AOs. The 18-mer, 16-mer, and 14-mer LNA/2′OMePS AO4, AO8, and AO12 showed higher exon 23 skipping efficacy at 25, 50, and 100 nM concentrations, in a dose-dependent manner, whereas the 12-mer AO16 was not effective. Although the unmodified 18-mer 2′-OMePS AO showed similar efficacy as the LNA/2′-OMePS variant, further reduction in size to a 16-mer significantly affected the yield of exon 23 skipped transcript product. The 14-mer and 12-mer truncated sequences failed to induce any skipping, which may suggest that the 2′-OMePS chemistry requires a minimum length of 16 nucleotides in order to act as an exon-skipping AO sequence. The RT-PCR product of 542 bp corresponding to exon 22/23 dual skipping was also observed in all cases, except for the unmodified AO10 and AO14, but the product yield was less at lower concentrations. To validate any potential off-target exon-skipping with the truncated LNA-modified short AOs, we have performed a quick experiment to see any possible changes on the widely expressed gene transcript (SMN1) using the total RNA isolated after the treatment with truncated LNA-modified 2′-OMePs AOs (we used the RNA sample of AO8 transfection at 100 nM). We did not observe any changes in the level of SMN1 RNA with and without the AO8 treatment (Figure S1). This experiment provides an indication that our AOs may be target specific.
In conclusion, truncation of AOs could substantially reduce cost and AO chemistry-mediated side effects. Our results highlight that shorter exon-skipping AOs with high efficacy can be constructed using LNA nucleotides in combination with other chemistries, such as 2′-OMePS AOs. Short LNA-modified 2′-OMePS mixmer AOs were found to be more effective in inducing dystrophin exon 23 skipping in vitro at very low doses, compared with the unmodified 2′-OMePS AOs. We suggest that shorter LNA-modified 2′-OMePS AOs may offer great potential in improving efficacy and utility of 2′-OMe AOs for therapeutic applications.
Materials and Methods
Design and Synthesis of Chemically Modified AOs
All AOs (Table 1) were synthesized in house on GE ÄKTA oligopilot plus 10 (GE Healthcare Life Sciences) oligonucleotide synthesizer using standard phosphoramidite chemistry in 1 μmol scale. All synthesis reagents were purchased from Merck Millipore. Synthesized oligonucleotides were deprotected and cleaved from the solid support by treatment with NH4OH (Merck Millipore) at 55°C overnight, and the crude oligonucleotides were then purified by desalting through the illustra NAP-10 columns (GE Healthcare Life Sciences).
Cell Culture and Transfection
H-2Kb-tsA58 mdx myoblasts42, 43 (H2K mdx cells) were cultured and differentiated as described previously.33 Briefly, when 60%–80% confluent myoblast cultures were treated with trypsin (Thermo Fisher Scientific) and seeded on 24-well plates pre-treated with 50 μg/mL poly-D-lysine (Merck Millipore), followed by 100 μg/ml Matrigel (Corning, supplied through In Vitro Technologies) at a density of 2 × 104 cells/well. Cells were differentiated into myotubes in DMEM (Thermo Fisher Scientific) containing 5% horse serum (Thermo Fisher Scientific) by incubating at 37°C in 5% CO2 for 24 hr. AOs were complexed with Lipofectin (Thermo Fisher Scientific) at a ratio of 2:1 (w/w) (Lipofectin/AO) and used in a final transfection volume of 500 μL/well in a 24-well plate as per the manufacturer’s instructions, except that the solution was not removed after 3 hr.
RNA Extraction and RT-PCR
RNA was extracted from transfected cells using Direct-zol RNA MiniPrep Plus with TRI Reagent (Zymo Research, supplied through Integrated Sciences) as per the manufacturer’s instructions. The dystrophin transcripts were then analyzed by RT-PCR using SuperScript III Reverse Transcriptase (Thermo Fisher Scientific) across exons 20–26, as described previously.33 PCR products were separated on 2% agarose gels in Tris-acetate-EDTA buffer, and the images were captured on a Fusion Fx gel documentation system (Vilber Lourmat, Marne-la-Vallee, France). Densitometry was performed by ImageJ software.44 The actual exon-skipping efficiency was determined by expressing the amount of exon 23 skipped RT-PCR product as a percentage of total dystrophin transcript products.
Author Contributions
B.T.L. performed the experiments and co-wrote the manuscript. R.N.V. conceived the idea, initiated the research, and co-wrote the manuscript. S.D.W. and S.F. provided suggestions. All authors proofread and corrected the manuscript.
Conflicts of Interest
S.D.W. and S.F. are consultants to Sarepta Therapeutics. B.T.L., A.A., and R.N.V. declare no conflicts of interest.
Acknowledgments
R.N.V. greatly acknowledges financial support from the Department of Health (Merit Award) the Western Australian Government; the McCusker Charitable Foundation; and the Perron Institute for Neurological and Translational Science. B.T.L. thanks the Murdoch International Postgraduate Scholarship scheme of Murdoch University.
Footnotes
Supplemental Information includes one figure and can be found with this article online at https://doi.org/10.1016/j.omtn.2017.09.002.
Supplemental Information
References
- 1.Veedu R.N., Wengel J. Locked nucleic acids: promising nucleic acid analogs for therapeutic applications. Chem. Biodivers. 2010;7:536–542. doi: 10.1002/cbdv.200900343. [DOI] [PubMed] [Google Scholar]
- 2.Veedu R.N., Wengel J. Locked nucleic acid as a novel class of therapeutic agents. RNA Biol. 2009;6:321–323. doi: 10.4161/rna.6.3.8807. [DOI] [PubMed] [Google Scholar]
- 3.Imanishi T., Obika S. BNAs: novel nucleic acid analogs with a bridged sugar moiety. Chem. Commun. (Camb.) 2002;(16):1653–1659. doi: 10.1039/b201557a. [DOI] [PubMed] [Google Scholar]
- 4.Elmén J., Thonberg H., Ljungberg K., Frieden M., Westergaard M., Xu Y., Wahren B., Liang Z., Ørum H., Koch T., Wahlestedt C. Locked nucleic acid (LNA) mediated improvements in siRNA stability and functionality. Nucleic Acids Res. 2005;33:439–447. doi: 10.1093/nar/gki193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kierzek E., Pasternak A., Pasternak K., Gdaniec Z., Yildirim I., Turner D.H., Kierzek R. Contributions of stacking, preorganization, and hydrogen bonding to the thermodynamic stability of duplexes between RNA and 2′-O-methyl RNA with locked nucleic acids. Biochemistry. 2009;48:4377–4387. doi: 10.1021/bi9002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petersen M., Wengel J. LNA: a versatile tool for therapeutics and genomics. Trends Biotechnol. 2003;21:74–81. doi: 10.1016/S0167-7799(02)00038-0. [DOI] [PubMed] [Google Scholar]
- 7.Jepsen J.S., Sørensen M.D., Wengel J. Locked nucleic acid: a potent nucleic acid analog in therapeutics and biotechnology. Oligonucleotides. 2004;14:130–146. doi: 10.1089/1545457041526317. [DOI] [PubMed] [Google Scholar]
- 8.Veedu R.N., Wengel J. Locked nucleic acid oligonucleotides: toward clinical applications. In: Zhang L.H., Xi Z., Chattopadhyaya J., editors. Medicinal Chemistry of Nucleic Acids. John Wiley & Sons; 2011. pp. 335–348. [Google Scholar]
- 9.Wahlestedt C., Salmi P., Good L., Kela J., Johnsson T., Hökfelt T., Broberger C., Porreca F., Lai J., Ren K. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl. Acad. Sci. U S A. 2000;97:5633–5638. doi: 10.1073/pnas.97.10.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torres A., Kozak J., Korolczuk A., Wdowiak P., Domańska-Glonek E., Maciejewski R., Torres K. In vitro and in vivo activity of miR-92a-Locked Nucleic Acid (LNA)-Inhibitor against endometrial cancer. BMC Cancer. 2016;16:822. doi: 10.1186/s12885-016-2867-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Martino M.T., Gullà A., Gallo Cantafio M.E., Altomare E., Amodio N., Leone E., Morelli E., Lio S.G., Caracciolo D., Rossi M. In vitro and in vivo activity of a novel locked nucleic acid (LNA)-inhibitor-miR-221 against multiple myeloma cells. PLoS ONE. 2014;9:e89659. doi: 10.1371/journal.pone.0089659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallo Cantafio M.E., Nielsen B.S., Mignogna C., Arbitrio M., Botta C., Frandsen N.M. Pharmacokinetics and pharmacodynamics of a 13-mer LNA-inhibitor-miR-221 in mice and non-human primates. Mol. Ther. Nucleic Acids. 2016;5:e336. doi: 10.1038/mtna.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies K.E., Nowak K.J. Molecular mechanisms of muscular dystrophies: old and new players. Nat. Rev. Mol. Cell Biol. 2006;7:762–773. doi: 10.1038/nrm2024. [DOI] [PubMed] [Google Scholar]
- 14.Mercuri E., Muntoni F. Muscular dystrophies. Lancet. 2013;381:845–860. doi: 10.1016/S0140-6736(12)61897-2. [DOI] [PubMed] [Google Scholar]
- 15.Arechavala-Gomeza V., Anthony K., Morgan J., Muntoni F. Antisense oligonucleotide-mediated exon skipping for Duchenne muscular dystrophy: progress and challenges. Curr. Gene Ther. 2012;12:152–160. doi: 10.2174/156652312800840621. [DOI] [PubMed] [Google Scholar]
- 16.Kole R., Krieg A.M. Exon skipping therapy for Duchenne muscular dystrophy. Adv. Drug Deliv. Rev. 2015;87:104–107. doi: 10.1016/j.addr.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 17.Mitrpant C., Fletcher S., Wilton S.D. Personalised genetic intervention for Duchenne muscular dystrophy: antisense oligomers and exon skipping. Curr. Mol. Pharmacol. 2009;2:110–121. doi: 10.2174/1874467210902010110. [DOI] [PubMed] [Google Scholar]
- 18.Fletcher S., Bellgard M.I., Price L., Akkari A.P., Wilton S.D. Translational development of splice-modifying antisense oligomers. Expert Opin. Biol. Ther. 2017;17:15–30. doi: 10.1080/14712598.2017.1250880. [DOI] [PubMed] [Google Scholar]
- 19.Fairclough R.J., Wood M.J., Davies K.E. Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nat. Rev. Genet. 2013;14:373–378. doi: 10.1038/nrg3460. [DOI] [PubMed] [Google Scholar]
- 20.Bao T.L., Veedu R.N., Fletcher S., Wilton S.D. Antisense oligonucleotide development for the treatment of muscular dystrophies. Expert Opin. Orphan Drugs. 2016;4:139–152. [Google Scholar]
- 21.Wilton S.D., Veedu R.N., Fletcher S. The emperor’s new dystrophin: finding sense in the noise. Trends Mol. Med. 2015;21:417–426. doi: 10.1016/j.molmed.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 22.Mendell J.R., Rodino-Klapac L.R., Sahenk Z., Roush K., Bird L., Lowes L.P., Alfano L., Gomez A.M., Lewis S., Kota J., Eteplirsen Study Group Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013;74:637–647. doi: 10.1002/ana.23982. [DOI] [PubMed] [Google Scholar]
- 23.Miceli M.C., Nelson S.F. The case for eteplirsen: paving the way for precision medicine. Mol. Genet. Metab. 2016;118:70–71. doi: 10.1016/j.ymgme.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 24.GlobeNewswire. GSK and Prosensa announce primary endpoint not met in phase III study of drisapersen in patients with Duchenne muscular dystrophy, https://globenewswire.com/news-release/2013/09/20/574726/10049265/en/GSK-and-Prosensa-Announce-Primary-Endpoint-Not-Met-in-Phase-III-Study-of-Drisapersen-in-Patients-With-Duchenne-MuscularDystrophy.html.
- 25.Syed Y.Y. Eteplirsen: first global approval. Drugs. 2016;76:1699–1704. doi: 10.1007/s40265-016-0657-1. [DOI] [PubMed] [Google Scholar]
- 26.Mendell J.R., Goemans N., Lowes L.P., Alfano L.N., Berry K., Shao J., Kaye E.M., Mercuri E., Eteplirsen Study Group and Telethon Foundation DMD Italian Network Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016;79:257–271. doi: 10.1002/ana.24555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agrawal S. Antisense oligonucleotides: towards clinical trials. Trends Biotechnol. 1996;14:376–387. doi: 10.1016/0167-7799(96)10053-6. [DOI] [PubMed] [Google Scholar]
- 28.Sarmiento U.M., Perez J.R., Becker J.M., Narayanan R. In vivo toxicological effects of rel A antisense phosphorothioates in CD-1 mice. Antisense Res. Dev. 1994;4:99–107. doi: 10.1089/ard.1994.4.99. [DOI] [PubMed] [Google Scholar]
- 29.Galbraith W.M., Hobson W.C., Giclas P.C., Schechter P.J., Agrawal S. Complement activation and hemodynamic changes following intravenous administration of phosphorothioate oligonucleotides in the monkey. Antisense Res. Dev. 1994;4:201–206. doi: 10.1089/ard.1994.4.201. [DOI] [PubMed] [Google Scholar]
- 30.Agrawal S., Rustagi P.K., Shaw D.R. Novel enzymatic and immunological responses to oligonucleotides. Toxicol. Lett. 1995;82-83:431–434. doi: 10.1016/0378-4274(95)03573-7. [DOI] [PubMed] [Google Scholar]
- 31.Agrawal S., Jiang Z., Zhao Q., Shaw D., Cai Q., Roskey A., Channavajjala L., Saxinger C., Zhang R. Mixed-backbone oligonucleotides as second generation antisense oligonucleotides: in vitro and in vivo studies. Proc. Natl. Acad. Sci. U S A. 1997;94:2620–2625. doi: 10.1073/pnas.94.6.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilton S.D., Lloyd F., Carville K., Fletcher S., Honeyman K., Agrawal S., Kole R. Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscul. Disord. 1999;9:330–338. doi: 10.1016/s0960-8966(99)00010-3. [DOI] [PubMed] [Google Scholar]
- 33.Mann C.J., Honeyman K., Cheng A.J., Ly T., Lloyd F., Fletcher S., Morgan J.E., Partridge T.A., Wilton S.D. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc. Natl. Acad. Sci. U S A. 2001;98:42–47. doi: 10.1073/pnas.011408598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le B.T., Filichev V.V., Veedu R.N. Investigation of twisted intercalating nucleic acid (TINA)-modified antisense oligonucleotides for splice modulation by induced exon-skipping in vitro. RSC Advances. 2016;6:95169–95172. [Google Scholar]
- 35.Le B.T., Chen S., Abramov M., Herdewijn P., Veedu R.N. Evaluation of anhydrohexitol nucleic acid, cyclohexenyl nucleic acid and d-altritol nucleic acid-modified 2′-O-methyl RNA mixmer antisense oligonucleotides for exon skipping in vitro. Chem. Commun. (Camb.) 2016;52:13467–13470. doi: 10.1039/c6cc07447b. [DOI] [PubMed] [Google Scholar]
- 36.Chen S., Le B.T., Rahimizadeh K., Shaikh K., Mohal N., Veedu R.N. Synthesis of a morpholino nucleic acid (MNA)-uridine phosphoramidite, and exon skipping using MNA/2′-O-methyl mixmer antisense oligonucleotide. Molecules. 2016;21:1582. doi: 10.3390/molecules21111582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dominski Z., Kole R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc. Natl. Acad. Sci. U S A. 1993;90:8673–8677. doi: 10.1073/pnas.90.18.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corey D.R. Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat. Neurosci. 2017;20:497–499. doi: 10.1038/nn.4508. [DOI] [PubMed] [Google Scholar]
- 39.Aartsma-Rus A., Kaman W.E., Bremmer-Bout M., Janson A.A., den Dunnen J.T., van Ommen G.J., van Deutekom J.C. Comparative analysis of antisense oligonucleotide analogs for targeted DMD exon 46 skipping in muscle cells. Gene Ther. 2004;11:1391–1398. doi: 10.1038/sj.gt.3302313. [DOI] [PubMed] [Google Scholar]
- 40.Gebert L.F.R., Rebhan M.A.E., Crivelli S.E.M., Denzler R., Stoffel M., Hall J. Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res. 2014;42:609–621. doi: 10.1093/nar/gkt852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimo T., Tachibana K., Saito K., Yoshida T., Tomita E., Waki R., Yamamoto T., Doi T., Inoue T., Kawakami J., Obika S. Design and evaluation of locked nucleic acid-based splice-switching oligonucleotides in vitro. Nucleic Acids Res. 2014;42:8174–8187. doi: 10.1093/nar/gku512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bulfield G., Siller W.G., Wight P.A., Moore K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. U S A. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rando T.A., Blau H.M. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J. Cell Biol. 1994;125:1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.