

The final efficacy and safety results are reported for the VELVET Cohort 2 trial, which investigated the coinfusion of pertuzumab and trastuzumab in a single infusion bag, followed by vinorelbine.
Keywords: Trastuzumab, Pertuzumab, Vinorelbine, Metastatic breast cancer, HER2-positive
Abstract
Background.
VELVET Cohort 1 demonstrated the applicability of pertuzumab, trastuzumab, and vinorelbine as an alternative first‐line treatment regimen for patients with HER2‐positive locally advanced or metastatic breast cancer (MBC) who cannot receive docetaxel. Co‐infusion of pertuzumab and trastuzumab may reduce clinic time and medical resource utilization. We report results from Cohort 2, in which pertuzumab and trastuzumab were co‐infused, followed by vinorelbine.
Patients and Methods.
During cycle 1, patients with HER2‐positive locally advanced or MBC received loading doses of pertuzumab (840 mg) and trastuzumab (8 mg/kg) on consecutive days, followed by vinorelbine (25 mg/m2) on days two and nine. From cycle 2 onwards, patients received a co‐infusion of pertuzumab (420 mg) and trastuzumab (6 mg/kg) on day one, followed by vinorelbine (30–35 mg/m2) on days one and eight (or days two and nine). The primary endpoint was objective response rate (ORR) in patients with measurable disease. Secondary endpoints included progression‐free survival (PFS) and safety.
Results.
Cohort 2 enrolled 107 patients. The ORR was 63.7% (95% confidence interval [CI] 53.0–73.6) in patients with measurable disease (91/107; 85.0%). Median PFS was 11.5 months (95% CI 10.3–15.8). The most common adverse events [AEs] were diarrhea (57.9%), neutropenia (57.0%), and nausea (41.1%). Grade ≥3 AEs occurred in 85 patients (79.4%) and serious AEs in 44 patients (41.1%). Eighteen patients (16.8%) had AEs suggestive of congestive heart failure.
Conclusion.
These results support the feasibility of pertuzumab and trastuzumab co‐infusion from a safety perspective and support Cohort 1 conclusions that vinorelbine offers an alternative chemotherapy companion for pertuzumab and trastuzumab. The Oncologist 2017;22:1160–1168
Implications for Practice.
Combined treatment with pertuzumab, trastuzumab, and docetaxel is the standard of care for first‐line HER2‐positive metastatic breast cancer. However, some patients cannot, or choose not to, receive docetaxel. VELVET Cohort 2 results support the results from Cohort 1 that suggest that pertuzumab plus trastuzumab and vinorelbine is a suitable alternative for these patients. In addition to this, results from Cohort 2 support the feasibility of administering pertuzumab and trastuzumab together in a single infusion bag, which has the potential to offer greater patient convenience and reduce active health care professional time and medical resource utilization compared with administering them separately.
摘要
背景. VELVET队列1证明, 无法接受多西他赛的HER2阳性局部晚期或转移性乳腺癌(MBC)患者可以使用帕妥珠单抗、曲妥珠单抗和长春瑞滨作为替代一线治疗方案。帕妥珠单抗和曲妥珠单抗联合输注可节约门诊时间并减少医疗资源利用。本文报告了队列2的结果;该队列联合输注帕妥珠单抗与曲妥珠单抗, 随后注射长春瑞滨。
患者和方法. 在第1周期, HER2阳性局部晚期或MBC患者在连续2天内先后接受负荷剂量的帕妥珠单抗(840 mg)和曲妥珠单抗(8 mg/kg), 随后分别于第2天和第9天接受长春瑞滨(25 mg/m2)。从第2周期开始, 患者在第1天联合输注帕妥珠单抗(420 mg)和曲妥珠单抗(6 mg/kg), 随后分别于第1天和第8天(或第2天和第9天)接受长春瑞滨(30–35 mg/m2)。主要终点为病灶可测量患者中的客观缓解率(ORR)。次要终点包括无进展生存期(PFS)和安全性。
结果. 队列2入组了107例患者。病灶可测量患者(91/107;85.0%)的ORR为63.7%[95%置信区间(CI):53.0‐73.6]。中位PFS为11.5个月(95%CI:10.3‐15.8)。最常见的不良事件(AE)为腹泻(57.9%)、中性粒细胞减少症(57.0%)和恶心(41.1%)。85例患者(79.4%)发生≥3级AE, 44例患者(41.1%)发生严重AE。18例患者(16.8%)发生提示充血性心力衰竭的AE。
结论. 上述结果从安全性角度支持了帕妥珠单抗与曲妥珠单抗联合输注的可行性, 并且印证了队列1的结论, 即长春瑞滨可作为帕妥珠单抗与曲妥珠单抗的候选联合化疗药物
Introduction
The combination of pertuzumab, trastuzumab, and docetaxel is the standard of care for first‐line HER2‐positive metastatic breast cancer [1], [2], [3], based on the results of the pivotal Clinical Evaluation of Pertuzumab and Trastuzumab (CLEOPATRA) study [4], [5]. Currently, all three drugs are administered separately. Co‐infusion of drugs would potentially reduce clinic time for patients through reduced patient chair time and reduced post‐infusion observation time. In addition to patient benefits, co‐infusion may also reduce active health care professional time, medical resource utilization per patient, and increase patient flow through the clinic.
While several small‐molecule drugs, including chemotherapy drugs, are routinely co‐infused in single infusion bags (based on supportive stability and compatibility data), little is known about the feasibility of administering multidrug combinations containing monoclonal antibodies (mAbs) [6]. Biophysical and analytical assays have demonstrated the physical and chemical stability of an admixture of pertuzumab and trastuzumab in a single infusion bag; no measurable changes in the admixture occurred for up to 24 hours at 5°C or 30°C [6].
The VELVET study was designed to explore the efficacy and safety of vinorelbine as a chemotherapy partner for pertuzumab and trastuzumab for the first‐line treatment of HER2‐positive locally advanced or metastatic breast cancer. In addition, VELVET is the first study to prospectively investigate the feasibility of co‐infusing the two mAbs in the same infusion bag with the aim of improving convenience for patients.
VELVET used a two‐cohort study design; results for Cohort 1, in which pertuzumab and trastuzumab were administered separately, followed by vinorelbine, were reported recently [7]. VELVET Cohort 1 showed that vinorelbine is an active and reasonably well‐tolerated chemotherapy partner (no new or unexpected safety signals) for pertuzumab and trastuzumab for the first‐line treatment of patients with HER2‐positive metastatic breast cancer and offers an alternative for patients who cannot, or choose not to, receive docetaxel in this setting [7]. Here we report the final efficacy and safety results for VELVET Cohort 2, in which the co‐infusion of pertuzumab and trastuzumab in a single infusion bag, followed by vinorelbine, was investigated.
Subjects, Materials, and Methods
Eligibility Criteria
Patients were ≥18 years old with HER2‐positive locally advanced (not amenable to curative resection) or metastatic breast cancer. HER2‐positivity was defined as immunohistochemistry (IHC) 3+ or HER2 gene amplification by in situ hybridization (HER2:chromosome 17 ratio of ≥2) and was assessed by local laboratories on primary or metastatic tumor samples with subsequent central analysis (Targos Molecular Pathology GmbH, Kassel, Germany; central results were not required prior to study enrollment). All patients had at least one measurable lesion and/or nonmeasurable disease evaluable according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. An Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, a left ventricular ejection fraction (LVEF) of at least 55% at baseline, and a life expectancy of at least 12 weeks were also required for enrollment. Patients were excluded if they had received prior systemic nonhormonal anticancer therapy in the metastatic or locally advanced setting, although up to two lines of hormonal therapy, one of which could have been in combination with everolimus, were permitted. Other exclusion criteria were prior breast cancer treatment with anti‐HER2 drugs, except trastuzumab and/or lapatinib in the neoadjuvant or adjuvant setting, disease progression while receiving neoadjuvant or adjuvant trastuzumab and/or lapatinib, a disease‐free interval of <6 months from completion of neoadjuvant or adjuvant nonhormonal therapy to time of disease recurrence, uncontrolled central nervous system metastases, and uncontrolled hypertension or clinically significant cardiovascular disease.
Study Design
VELVET was a two‐cohort, open‐label, multicenter, phase II, proof‐of‐concept trial evaluating the safety and efficacy of vinorelbine in combination with pertuzumab and trastuzumab for the first‐line treatment of HER2‐positive locally advanced or metastatic breast cancer (NCT01565083). In Cohort 1, patients received PERJETA® (pertuzumab; F. Hoffmann‐La Roche Ltd, Basel, Switzerland, www.roche.com) and Herceptin® (trastuzumab; F. Hoffmann‐La Roche Ltd, www.roche.com), administered sequentially in separate infusion bags, followed by Bendarelbin (vinorelbine; Bendalis GmbH, Oberhaching, Germany). In Cohort 2, patients received pertuzumab and trastuzumab co‐infused in a single saline infusion bag (from cycle 2 onwards), followed by vinorelbine (supplemental online Fig. 1). In Cohort 2, for co‐infusion of the mAbs, pharmacists were required to adhere to strict study guidelines on dose preparation to ensure that the products were stable in the final compounded sterile preparation. Cohort 2 began enrolling after Cohort 1 was fully enrolled.
All study drugs were given intravenously on a 3‐week schedule. During cycle 1, patients received 840 mg pertuzumab (loading dose) on day one followed by 8 mg/kg trastuzumab (loading dose) on day two, and 25 mg/m2 of vinorelbine on days two and nine. From cycle 2 onwards, patients in Cohort 2 received 420 mg of pertuzumab (maintenance dose) and 6 mg/kg of trastuzumab (maintenance dose) in a single infusion bag on day one, followed by 30–35 mg/m2 of vinorelbine on days one and eight (or days two and nine). Vinorelbine was administered in line with product labeling. The planned infusion and observation times for concomitant (Cohort 2) versus sequential infusion (Cohort 1) of pertuzumab and trastuzumab are shown in Figure 1.
Figure 1.

Planned maximum infusion times for combined versus sequential pertuzumab and trastuzumab infusion. Pertuzumab and trastuzumab were given as sequential infusions in cycle 1 of Cohort 1 and Cohort 2. In Cohort 1, pertuzumab and trastuzumab were given as sequential infusions from cycle 2 onwards; in Cohort 2, pertuzumab and trastuzumab were given as a co‐infusion from cycle 2 onwards. aPertuzumab time was 0.5–1 hour; maximum time is shown [15], [16], [17].
Study drugs were given until investigator‐assessed disease progression, unacceptable toxicity, withdrawal of consent, predefined study end, or death. If vinorelbine was discontinued, antibody therapy was allowed to be continued; if antibody therapy was discontinued, vinorelbine was allowed to be continued. Dose reductions were not permitted for pertuzumab or trastuzumab. For any grade 3–4 toxicities related to vinorelbine, treatment was delayed until toxicity improved to grade 1, after which the dose was reduced to 80%. In the event of elevated bilirubin (>2× the upper limit of normal) or transaminases (>3× the upper limit of normal), the dose of vinorelbine was reduced to 50% [8]. An independent data monitoring committee conducted safety reviews throughout the study. The final review occurred in March 2015.
VELVET was conducted in accordance with good clinical practice guidelines and the Declaration of Helsinki. Approval for the protocol and for any modifications was obtained from independent ethics committees/institutional review boards at each participating site. Written informed consent was obtained from each participant.
Assessments
Tumor response was evaluated according to RECIST v1.1; assessments were performed at baseline, every three cycles up to 36 months, and then every six cycles until disease progression. ECOG performance status was assessed at baseline, every three cycles, and 28 days after treatment discontinuation. LVEF was assessed by echocardiography (ECHO) or multigated acquisition (MUGA) scan during screening and every three cycles. Computed tomography or magnetic resonance imaging brain scans were performed during the screening period only in patients with clinical suspicion of brain metastases and during the study if clinically indicated. Blood counts and laboratory parameters were assessed at baseline, every cycle, and 28 days after treatment discontinuation. Adverse events (AEs) and serious AEs (SAEs) were monitored continuously until 28 days after treatment discontinuation. Study‐related SAEs were collected until resolved. The AEs were graded according to the Medical Dictionary for Regulatory Activities (MedDRA) version 18.1.
Study Endpoints
The primary endpoint was investigator‐assessed objective response rate (ORR) according to RECIST v1.1 in patients with measurable disease (target lesion[s] present) at baseline, and was based on the best overall response (BOR), defined as the best response recorded from the start of study treatment until disease progression/recurrence or death. Patients required two consecutive assessments (at least 28 days apart) of partial response or complete response to be defined a responder. Secondary endpoints included time to response, duration of response (in responders), progression‐free survival (PFS; defined as the time from first intake of any study treatment until the first radiographically documented progression of disease or death from any cause), time to progression (TTP; defined as the time from first intake of any study treatment until the first radiographically documented progression of disease or death due to progressive disease only), overall survival (OS; defined as the time from first intake of any study treatment to the date of death, regardless of the cause), safety and tolerability, and quality of life (not reported here). Safety analyses included the incidence and severity of AEs and SAEs, the incidence of congestive heart failure, changes in LVEF from baseline during the study, and laboratory test abnormalities.
Statistical Analysis
Assuming a preferable BOR rate of 70%–80% in each cohort (based on published data [4]), and aiming at a distance from the estimated proportion to the confidence interval (CI) limits of 8%–11%, 95 evaluable patients were required for each cohort. The observed BOR of 70% could be estimated to be between 59%–79% with a probability of 95% (Clopper–Pearson exact CIs), while the observed BOR of 80% could be estimated to be between 71%–88%. Adjusting for a withdrawal rate of approximately 10%, it was planned to enroll 105 patients in each cohort (calculated using analytic software [SAS, version 9.2, SAS, Cary, NC, https://www.sas.com/en_gb/home.html; nQuery, version 6, Statistical Solutions Ltd., Boston, USA, www.statsols.com]). The median time on study was estimated using the reverse censoring Kaplan–Meier method. Efficacy analyses were performed in the intent‐to‐treat (ITT) population (all enrolled patients) at study end (when all patients had been followed up with for at least 2 years after the last patient had enrolled, unless they were lost to follow‐up, withdrew consent, or died). The BOR, PFS, and OS analyses were also conducted for the per‐protocol population (all ITT patients who had received at least one dose of any study treatment and had at least one postbaseline tumor assessment with no major protocol deviations leading to exclusion from the per‐protocol population). The ORR was summarized by the number and percentage of responders, together with two‐sided 95% Clopper–Pearson CIs in patients with measurable disease at baseline. Secondary efficacy endpoints were estimated using the Kaplan–Meier approach. Predefined exploratory subgroup analyses were performed for ORR and PFS, according to prior trastuzumab treatment and by hormone receptor status. AEs were evaluated in the safety population (all patients who received at least one dose of any study treatment). All analyses presented are descriptive. VELVET Cohorts 1 and 2 were not intended to be formally compared, due to the nonrandomized nature of the study and imbalanced baseline characteristics; their data have therefore been published separately [7]. A pertuzumab extension study (NCT02320435) opened in February 2015 to provide continued access to pertuzumab for patients still benefiting from study treatment at the end of the study; this will collect long‐term safety data.
Results
Study Population
Between April and September 2013, 107 patients were enrolled into Cohort 2 at 44 centers across Europe, North America, and South America (Brazil). All patients were included in the ITT and safety populations; 73 (68.2%) were included in the per‐protocol population. Baseline characteristics are shown in Table 1; 58.9% of patients were from Europe, 29.9% from South America (Brazil), and 11.2% from North America. The majority of patients (82.2%) had metastatic disease, 59.8% had hormone receptor positive disease, and 43.0% had received prior nonhormonal systemic cancer therapy for early breast cancer (chemotherapy [32.7%] and/or trastuzumab [17.8%]). The cutoff date for data collection and the study end for Cohort 2 was November 13, 2015.
Table 1. Baseline characteristics (intent‐to‐treat population).
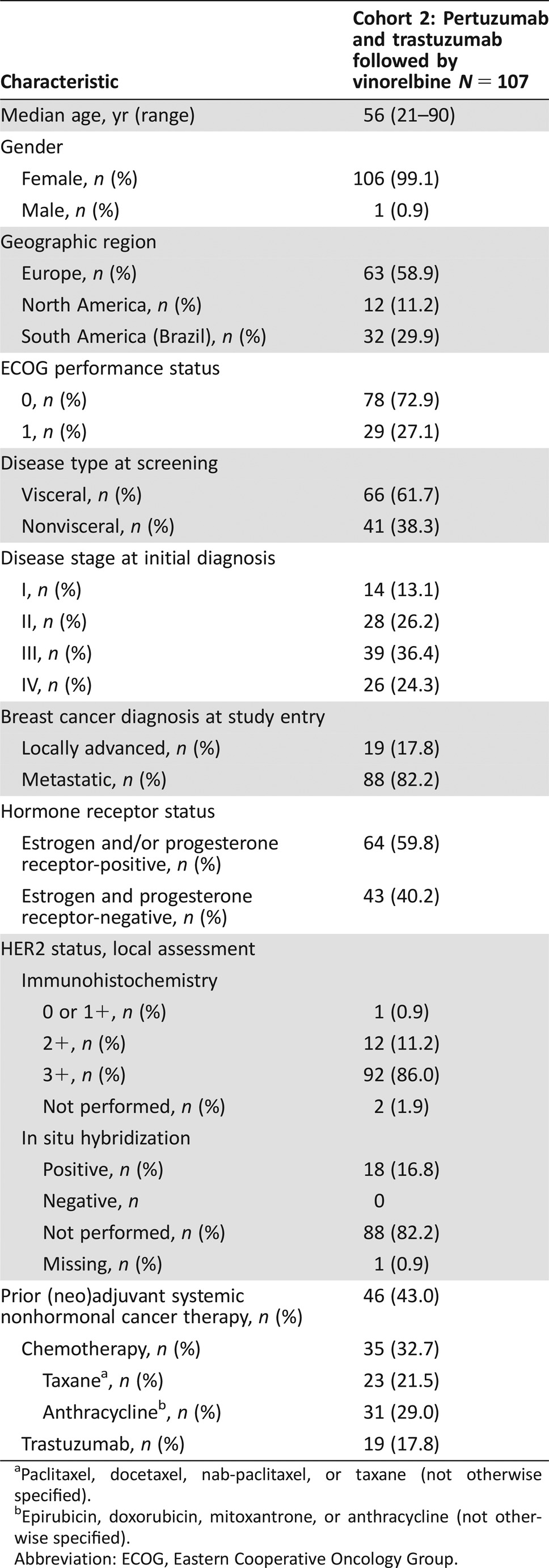
Paclitaxel, docetaxel, nab‐paclitaxel, or taxane (not otherwise specified).
Epirubicin, doxorubicin, mitoxantrone, or anthracycline (not otherwise specified).
Abbreviation: ECOG, Eastern Cooperative Oncology Group.
Treatment Summary
The median time on study was 26.5 months (95% CI 25.6–26.9). The median number of treatment cycles received was 15 (range 2–39) for pertuzumab, 15 (range 2–39) for trastuzumab, and 9 (range 2–39) for vinorelbine. One patient did not receive vinorelbine at cycle 1. The median vinorelbine dose intensity during the first six cycles was 17.34 mg/m2 per week (range 8.4–21.3). Ninety‐two (86.0%) patients discontinued all study treatment; the remaining 15 (14.0%) were still receiving at least one study treatment at the time of study closure. Progressive disease was the main reason for permanent discontinuation of all study treatment (Fig. 2).
Figure 2.

VELVET Cohort 2 study profile. aThe 15 patients ongoing with any study treatment at time of study closure are also counted under administrative/other reasons.
Abbreviation: ITT, intent‐to‐treat.
Efficacy
In the 91 patients (85.0%) with measurable disease at baseline, investigator‐assessed ORR was 63.7% (58/91 patients; 95% CI 53.0–73.6) with seven (7.7%) complete and 51 (56.0%) partial responses observed (Table 2). The median time to response for all 91 patients was 2.2 months (95% CI 2.1–4.4). The median duration of response in responders (58/91 patients) was 11.8 months (95% CI 7.5–17.9; supplemental online Fig. 2). At study end, 72 patients (67.3%) had progressed or died and the median PFS was 11.5 months (95% CI 10.3–15.8; Table 2; Fig. 3). The median TTP was 12.8 months (95% CI 10.4–17.1). Median OS was not reached by study end (data not shown), at which time 23 (21.5%) patients had died and 84 (78.5%) were censored. Per‐protocol analyses for BOR, PFS, and OS were in line with the ITT analyses (data not shown).
Table 2. Investigator‐assessed BOR and PFS for all patients and for predefined subgroup analyses stratified by prior trastuzumab treatment and hormone receptor status (intent‐to‐treat population).
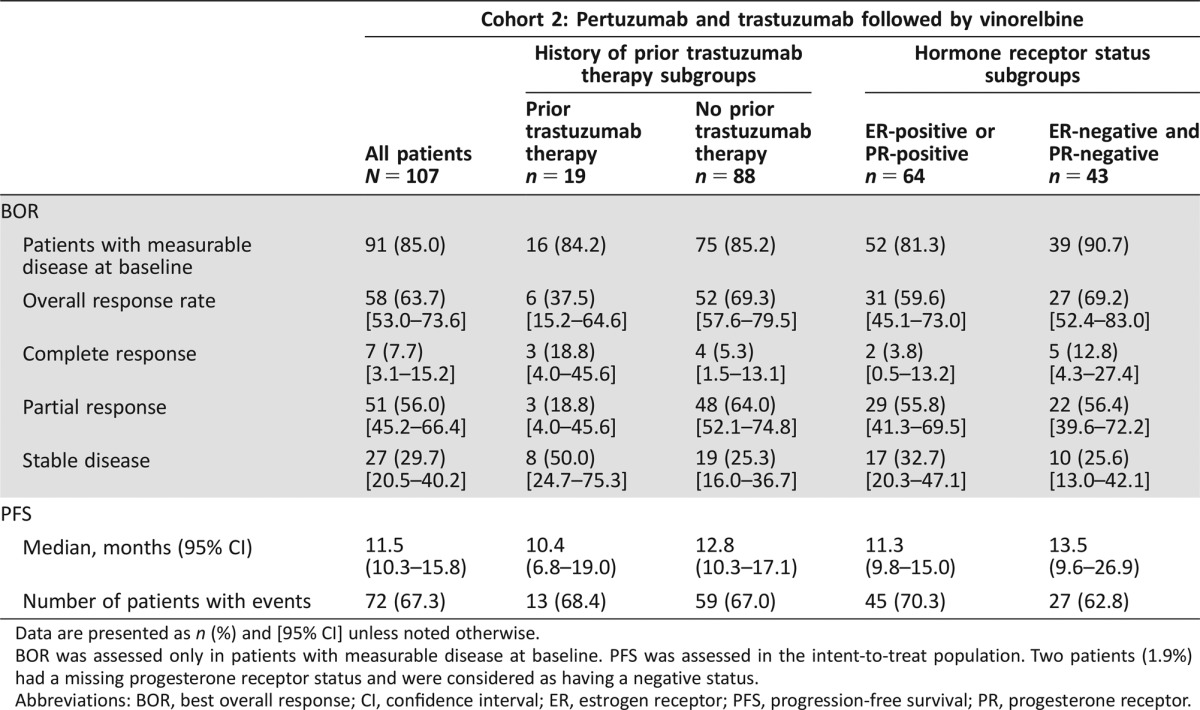
Data are presented as n (%) and [95% CI] unless noted otherwise.
BOR was assessed only in patients with measurable disease at baseline. PFS was assessed in the intent‐to‐treat population. Two patients (1.9%) had a missing progesterone receptor status and were considered as having a negative status.
Abbreviations: BOR, best overall response; CI, confidence interval; ER, estrogen receptor; PFS, progression‐free survival; PR, progesterone receptor.
Figure 3.

Progression‐free survival (intent‐to‐treat population).
Abbreviation: CI, confidence interval.
Exploratory Analyses
In predefined exploratory subgroup analyses, a higher ORR and a longer median PFS were observed in trastuzumab‐naïve patients compared with patients with prior trastuzumab treatment, and in patients with hormone receptor negative disease compared with patients with hormone receptor positive disease (Table 2). However, results should be interpreted with caution due to the small numbers of patients in subgroups and the noncomparative nature of the analysis.
The ORR and PFS were also assessed in a post hoc exploratory manner according to centrally assessed HER2 status. Seventy‐four of 107 patients (69.2%) had HER2‐positive disease (of whom 67 [90.5%] were scored IHC 3+) and 15 (14.0%) were found to have HER2‐negative disease by central assessment. The remaining 18 patients (16.8%) did not undergo central assessment for HER2‐positivity. In the centrally confirmed HER2‐positive patients with measurable disease at baseline (67 of 74; 90.5%), the ORR was 71.6% (48 of 67 patients; 95% CI 59.3–82.0) and the median PFS was 12.9 months (95% CI 9.8–21.9). In the IHC 3+ subgroup of patients with measurable disease at baseline (60 of 67; 89.6%), the ORR was 71.7% (43 of 60 patients; 95% CI 58.6–82.5). The median PFS in this subgroup was 15.8 months (95% CI 9.8–26.9).
Safety
All 107 patients were included in the safety population. AEs were reported in 106 patients (99.1%). The most frequently reported AEs were diarrhea (57.9%), neutropenia (57.0%), nausea (41.1%), fatigue (38.3%), and constipation (32.7%). AEs of any grade with an incidence of ≥20% are shown in Table 3. Grade ≥3 AEs were reported in 85 patients (79.4%; Table 3); neutropenia (31.8%) and hypertension (14.0%) were most commonly reported. Granulocyte‐colony stimulating factor was administered concomitantly in 19 (17.8%) patients for neutropenia management. SAEs were experienced by 44 patients (41.1%), with pyrexia (5.6%), pneumonia (3.7%), neutropenia (2.8%), and febrile neutropenia (2.8%) reported in more than two patients (Table 3). Of note, the proportion of patients with an AE of hypertension was higher in South America (Brazil) than in Europe or North America (any grade: 43.8% vs. 22.2% vs. 25.0%; grade 3: 34.4% vs. 4.8% vs. 8.3%).
Table 3. AEs, grade ≥3 AEs (based on AEs of any grade with an incidence of ≥20%), and SAEs (based on SAEs of any grade with an incidence of >1 patient) (safety population).
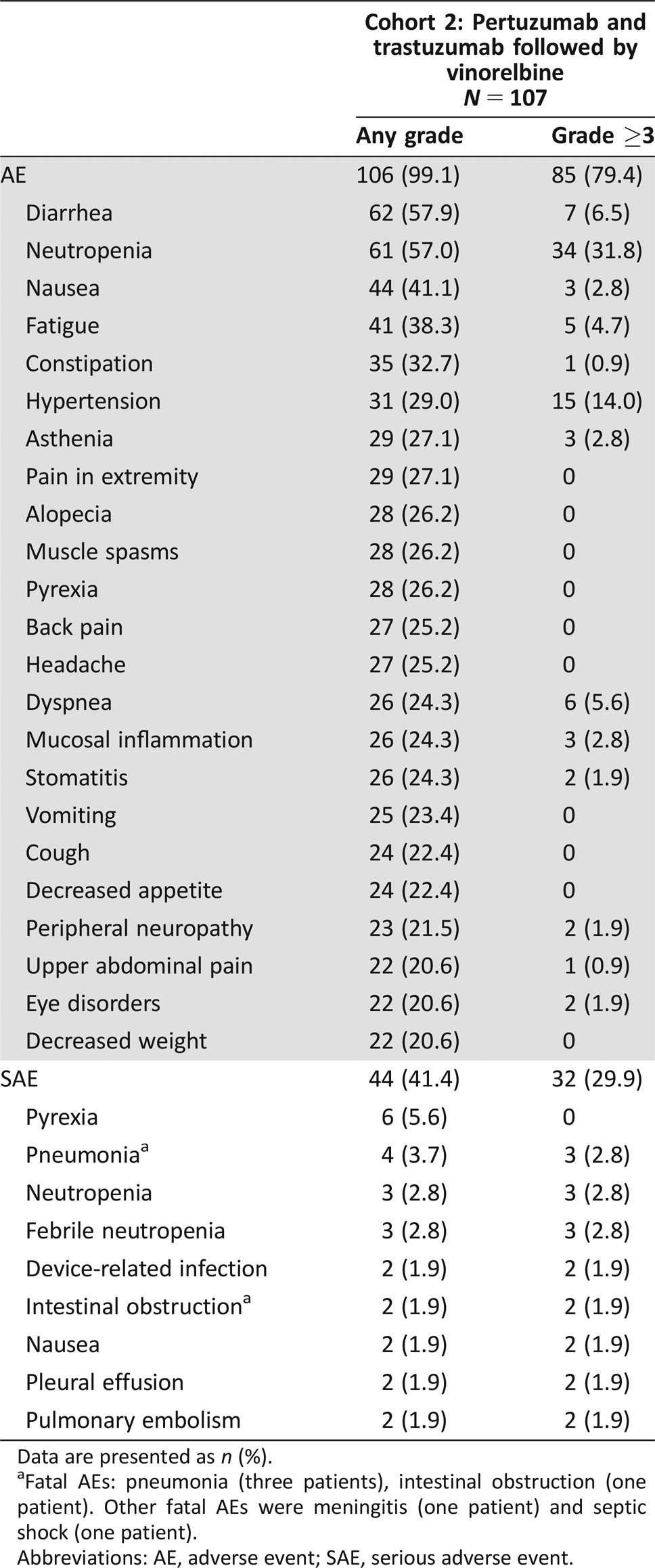
Data are presented as n (%).
Fatal AEs: pneumonia (three patients), intestinal obstruction (one patient). Other fatal AEs were meningitis (one patient) and septic shock (one patient).
Abbreviations: AE, adverse event; SAE, serious adverse event.
AEs were considered related to treatment in 81 patients (75.7%) treated with pertuzumab, 82 patients (76.6%) treated with trastuzumab, and 105 patients (98.1%) treated with vinorelbine. AEs led to study drug interruption in 85 patients (79.4%) and discontinuation in 25 patients (23.4%). The most commonly discontinued drug was vinorelbine, which was discontinued in 23 patients (21.5%), while pertuzumab and trastuzumab were discontinued in nine patients (8.4%) each. Of the 25 patients who discontinued study treatment, six (5.6%) discontinued due to neutropenia, two (1.9%) due to general physical health deterioration, two (1.9%) due to peripheral neuropathy, and 15 (14.0%) due to other AEs.
The incidence of cardiac AEs of all grades was 17.8% (19 patients) and of grade ≥3 AEs was 4.7% (five patients; one grade 3 cardiac failure [0.9%], two grade 3 left ventricular dysfunction [1.9%], one grade 3 tachycardia [0.9%], and one grade 4 supraventricular tachycardia [0.9%]). Additionally, ten patients (9.3%) had nonserious AEs, which were not reported as cardiac disorders but were classified as being suggestive of congestive heart failure according to Standardised MedDRA Queries; the majority had peripheral edema and two patients experienced Grade 2 decreased ejection fraction. During study treatment, the worst LVEF value of most patients (91.6%) remained >50% (supplemental online Table 1). Significant declines in LVEF (defined as a decline of ≥10% points from baseline to 50%–45%, in accordance with the study protocol) were observed in four patients (3.7%), with an additional two patients (1.9%) experiencing a decline to <45% (supplemental online Table 1). To make these findings comparable to the results of other pivotal studies, including CLEOPATRA, the incidence of LVEF declines by ≥10% points from baseline to <50% was also analyzed (n = 4 patients, 3.7%); the findings were expectedly consistent with prior data.
Twenty‐three patients (21.5%) died during the study; most deaths (14.0%, 15 patients) were due to disease progression, with six deaths (5.6%) resulting from AEs while on treatment (three from pneumonia, one from intestinal obstruction, one from meningitis, and one from septic shock). For two deaths (1.9%), the cause was not reported.
Anticancer Therapies After Discontinuation of Study Treatment
Seventy‐six of 107 patients (71.0%) received anticancer therapies after the discontinuation of study treatment (supplemental online Table 2); trastuzumab was received by 45 patients (59.2%) and pertuzumab by 20 patients (26.3%).
Discussion
Changes in drug formulation and/or administration methods have the potential to improve convenience for both patients and health care providers while maintaining safety and efficacy [9], [10], [11], [12], [13], [14]. Cohort 2 of the VELVET study is the first to provide evidence to support the feasibility of administering pertuzumab and trastuzumab together in a single infusion bag, followed by vinorelbine. Co‐infusion is advantageous because it may offer patients greater convenience while also reducing time spent in the clinic. The dosing schedule of vinorelbine used in VELVET was the same as that used in the Herceptin Plus Navelbine or Taxotere (HERNATA) study [8], with the exception of a lower dose during cycle 1 in order to monitor safety with the addition of pertuzumab. Additionally, sequential infusion of pertuzumab and trastuzumab was required during cycle 1 for VELVET Cohort 2 in an effort to appropriately monitor safety during the initial infusion.
In VELVET, the potential time saving for co‐infusion compared with standard sequential infusion of pertuzumab and trastuzumab after cycle 1 was 1–1.5 hours during cycle 2 (2.5 hours vs. 3.5–4.0 hours), and 2–2.5 hours from cycle 3 onwards (1.5 hours vs. 3.5–4.0 hours), including infusion and post‐infusion observation time (Fig. 1) [15], [16], [17]. A time‐and‐motion study would help to confirm the time savings and medical resource utilization for the combined infusion.
The results from VELVET Cohort 2 show that co‐infusion of pertuzumab and trastuzumab followed by vinorelbine is active and reasonably well tolerated for the first‐line treatment of HER2‐positive locally advanced or metastatic breast cancer with an investigator‐assessed ORR of 63.7% (95% CI 53.0–73.6) and a median PFS of 11.5 months (95% CI 10.3–15.8) observed. Efficacy was lower than expected based on the results from Cohort 1 of the VELVET study, in which pertuzumab and trastuzumab were administered sequentially. Of note, a formal exploratory comparison between the cohorts was initially planned but was not performed due to the nonrandomized nature of the trial and important imbalances in baseline characteristics (proportion of patients with prior trastuzumab treatment or with locally advanced disease, and inter‐regional differences with the inclusion of patients from South America [Brazil] in Cohort 2 but not in Cohort 1) [7]. Differences between the baseline characteristics were an anticipated consequence of the sequential recruitment that was conducted for the two cohorts and the different sites involved in each part of the study. Nonetheless, the ORR observed in Cohort 2 was in line with the protocol assumption of a BOR of 70% (95% CI 59–79), albeit at the lower end of the confidence interval.
The lower‐than‐expected efficacy may be due, in part, to discordance between local and central HER2 testing results since 14.0% of patients were found to have HER2‐negative disease and 69.2% of patients to have HER2‐positive disease by the central laboratory; the remaining patients (16.8%) did not have their HER2 status tested centrally. Post hoc exploratory subgroup analyses showed a higher ORR in patients with centrally versus locally confirmed HER2‐positive disease (71.6% [95% CI 59.3–82.0] vs. 63.7% [95% CI 53.0–73.6], respectively) and a longer median PFS (12.9 months [95% CI 9.8–21.9] vs. 11.5 months [95% CI 10.3–15.8], respectively). When considering only those patients with HER2 IHC 3+ disease, the ORR was 71.7% (95% CI 58.6–82.5) vs. 67.1% (95% CI 55.6–77.3) and the median PFS was 15.8 months (95% CI 9.8–26.9) vs. 11.9 months (95% 9.8–16.4) with central and local assessment, respectively. Similar findings concerning improved efficacy in patients with centrally versus locally confirmed HER2‐positive disease were also reported in other studies conducted in the early breast cancer setting [18]. These findings highlight the importance of following the standards for interpretation and reporting of HER2 testing [19], particularly in the context of a clinical trial investigation.
As expected, in exploratory subgroup analyses, trastuzumab‐naïve patients had a higher ORR and longer median PFS than those who had prior trastuzumab treatment, although the number of trastuzumab‐pretreated patients with measurable disease was small (n = 16) and CIs were wide and overlapping, limiting interpretation. Similarly, meaningful observations on efficacy cannot be drawn from the hormone receptor subgroup analyses due to the small numbers in each subgroup and wide and overlapping CIs.
The safety profile was consistent with the known toxicity profiles of each drug [16], [17], [20] and with VELVET Cohort 1 [7], and no unexpected safety signals were observed. This supports the feasibility of the co‐infusion of pertuzumab and trastuzumab in a single infusion bag. Diarrhea and neutropenia were the most frequently reported AEs; however, treatment discontinuation due to these AEs was low (diarrhea: one patient; neutropenia: six patients). The incidence of hypertension (any grade: 31 patients [29.0%]; grade 3: 15 patients [14.0%]) was higher than expected; however, no patients discontinued treatment because of hypertension. The findings on hypertension are most likely attributable to a protocol and electronic case report form amendment that required vital signs to be assessed post‐infusion. The amendment occurred after Cohort 1 finished enrolling and led to procedural differences between the cohorts, resulting in a much higher percentage of patients in Cohort 2 having post‐infusion blood pressure measurements taken at cycle 1. Importantly, cardiac safety (incidence of cardiac dysfunction and LVEF declines <50%) was in line with previous studies of pertuzumab and trastuzumab (standard sequential infusion) [7], [21], [22].
VELVET Cohort 2 is limited by the absence of a comparator arm and the relatively small number of patients; however, it adds to the large clinical trial datasets already available for each of the study drugs and provides the first efficacy and safety data for concomitant administration of two mAbs, pertuzumab and trastuzumab, in a single infusion bag.
Conclusion
Co‐infusion of pertuzumab and trastuzumab is feasible from a safety standpoint and may offer patients greater convenience and time savings compared with conventional sequential infusion. Consistent with VELVET Cohort 1, the results from VELVET Cohort 2 suggest that vinorelbine offers an alternative chemotherapy companion for pertuzumab and trastuzumab, particularly for those patients who cannot, or choose not to, receive docetaxel for the first‐line treatment of HER2‐positive locally advanced or metastatic breast cancer.
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgments
This study was funded by F. Hoffmann‐la Roche Ltd, Basel, Switzerland. Targos Molecular Pathology GmbH, Kassel, Germany conducted central HER2 testing. Support for third‐party writing assistance for this manuscript, furnished by Debbie Briggs, Ph.D., and Katie Wilson, Ph.D., of Health Interactions, was provided by F. Hoffmann‐La Roche Ltd. We thank the following VELVET study investigators: Brazil (S. Costa, R. Hegg, M. Mano, J. Nunes), Denmark (M. Andersson, A.H. Jacobsen, S. Linnet), France (S. Delaloge, J. Grenier, A.‐C. Hardy Bessard, C. Levy, J. Medioni, C. Moldovan, M. Mousseau, T. Petit, J.‐P. Spano, B. You, L. Zelek), Germany (W. Abenhardt, S. Bauer, B. Flath, S. Fuxius, R. Hansen, E. Ladda, L. Mueller, S. Roesel, C. Salat, C. Schulte, U. Soeling, H. Tesch, H.‐W. Tessen, J. Wamhoff, J. Wiegand, M. Zaiss), Italy (A. Bottini, M. Cazzaniga, L. Del Mastro, A. Falcone, S. Gori, P. Marchetti, F. Roila, S. Spazzapan, C. Zamagni), Spain (N. Batista Lopez, E. Blanco Campanario, Y. Fernandez Perez, I. Garau Llinas, L. Gonzalez Cortijo, J.M. López Vega, R. Perez Carrion, A. Santaballa Bertran), and the U.S. (K. Blackwell, H. Burstein, V. Hansen, R.A. Hirsch, R. Livingston, C. Ma, E. Macrae, E. Mrozek, R.H. Oyola, G. Rodriguez, G. Sledge, J. Specht, E. Tan‐Chiu, C. Vaughn, C. Vogel). These analyses have been presented in part by the following:
(a) E.A. Perez, J.M. López‐Vega, L. del Mastro, T. Petit, L. Mitchell, C. Pelizon, M. Andersson. Trastuzumab‐pertuzumab‐vinorelbine for first‐line treatment of patients with HER2‐positive metastatic breast cancer: a single‐arm, two‐cohort, phase II study (VELVET). American Society of Clinical Oncology (ASCO) Annual Meeting, 1–5 June 2012, Chicago USA. Trials‐in‐progress poster TPS653; (b) M. Andersson, J.M. López‐Vega, L. del Mastro, T. Petit, L. Mitchell, C. Pelizon, E.A. Perez. Pertuzumab and trastuzumab in combination with vinorelbine for first‐line treatment of patients with HER2‐positive locally advanced or metastatic breast cancer: a single‐arm, two‐cohort, Phase II study (VELVET). European Society for Medical Oncology (ESMO), 28 September–2 October 2012, Vienna, Austria. Trials‐in‐progress poster 408TiP; (c) E.A. Perez, J.M. López‐Vega, L. del Mastro, T. Petit, C. Zamagni, U. Freudensprung, L. Bastière‐Truchot, R. Walker, M. Andersson. Interim safety of pertuzumab in combination with trastuzumab and vinorelbine for first‐line treatment of patients with HER2‐positive‐locally advanced or metastatic breast cancer. European Breast Cancer Conference (EBCC), 19–21 March 2014, Glasgow, UK. Poster 037; (d) M. Andersson, J.M. López‐Vega, T. Petit, C. Zamagni, U. Freudensprung, S. Robb, E. Restuccia, E.A. Perez. Interim safety and efficacy of pertuzumab, trastuzumab and vinorelbine for first‐line treatment of patients with HER2‐positive locally advanced or metastatic breast cancer. European Society for Medical Oncology (ESMO), 26–30 September 2014, Madrid, Spain. Poster 361PD; and (e) M. Andersson, J.M. López‐Vega, T. Petit, C. Zamagni, M. Donica, J. Kamber, E.A. Perez. The co‐administration of pertuzumab and trastuzumab as a single infusion, followed by vinorelbine, in first‐line treatment of HER2‐positive locally advanced or metastatic breast cancer patients: VELVET study interim analysis. American Society of Clinical Oncology (ASCO) Annual Meeting, 29 May–5 June 2015, Chicago USA. Poster P586. EAP current affiliation Genentech Inc., South San Francisco, CA, USA and Mayo Clinic, Jacksonville, Florida, USA.
Author Contributions
Conception/Design: Michael Andersson, Edith A. Perez
Provision of study material or patients: Michael Andersson, José Manuel López‐Vega, Thierry Petit, Claudio Zamagni
Collection and/or assembly of data: Michael Andersson, Julia Kamber, Edith A. Perez
Data analysis and interpretation: Michael Andersson, José Manuel López‐Vega, Claudio Zamagni, Valerie Easton, Julia Kamber, Eleonora Restuccia, Edith A. Perez
Manuscript writing: Michael Andersson, Claudio Zamagni, Julia Kamber, Valerie Easton, Eleonora Restuccia, Edith A. Perez
Final approval of manuscript: Michael Andersson, José Manuel López‐Vega, Thierry Petit, Claudio Zamagni, Valerie Easton, Julia Kamber, Eleonora Restuccia, Edith A. Perez
Disclosures
Michael Andersson: Roche (RF, H, SAB), Pierre Fabre (H, SAB), Novartis (RF, H), Pfizer (SAB); Thierry Petit: Roche, Novartis, Pfizer (C/A, SAB); Claudio Zamagni: Roche (C/A, RF, H, SAB); Valerie Easton: Roche (E); Julia Kamber: Roche (E, OI); Eleonora Restuccia: Roche (E); Edith A. Perez: Genentech (E). José M. López‐Vega indicates no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Giordano SH, Temin S, Kirshner JJ et al. Systemic therapy for patients with advanced human epidermal growth factor receptor 2‐positive breast cancer: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol 2014;32:2078–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cardoso F, Costa A, Norton L et al. ESO‐ESMO 2nd international consensus guidelines for advanced breast cancer (ABC2). Breast 2014;23:489–502. [DOI] [PubMed] [Google Scholar]
- 3.National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Gastric Cancer. Version 3. 2016. Available at http://www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed August, 2016. [DOI] [PubMed]
- 4. Baselga J, Cortés J, Kim SB et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med 2012;366:109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Swain SM, Baselga J, Kim SB et al. Pertuzumab, trastuzumab, and docetaxel in HER2‐positive metastatic breast cancer. N Engl J Med 2015;372:724–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Glover ZW, Gennaro L, Yadav S et al. Compatibility and stability of pertuzumab and trastuzumab admixtures in i.v. infusion bags for coadministration. J Pharm Sci 2013;102:794–812. [DOI] [PubMed] [Google Scholar]
- 7. Peréz EA, López‐Vega JM, Petit T et al. Safety and efficacy of vinorelbine in combination with pertuzumab and trastuzumab for first‐line treatment of patients with HER2‐positive locally advanced or metastatic breast cancer: VELVET Cohort 1 final results. Breast Cancer Res 2016;18:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Andersson M, Lidbrink E, Bjerre K et al. Phase III randomized study comparing docetaxel plus trastuzumab with vinorelbine plus trastuzumab as first‐line therapy of metastatic or locally advanced human epidermal growth factor receptor 2‐positive breast cancer: The HERNATA study. J Clin Oncol 2011;29:264–271. [DOI] [PubMed] [Google Scholar]
- 9. Ismael G, Hegg R, Muehlbauer S et al. Subcutaneous versus intravenous administration of (neo)adjuvant trastuzumab in patients with HER2‐positive, clinical stage I‐III breast cancer (HannaH study): A phase 3, open‐label, multicentre randomised trial. Lancet Oncol 2012;13:869–878. [DOI] [PubMed] [Google Scholar]
- 10. Launay‐Vacher V. An appraisal of subcutaneous trastuzumab: A new formulation meeting clinical needs. Cancer Chemother Pharmacol 2013;72:1361–1367. [DOI] [PubMed] [Google Scholar]
- 11. Pivot X, Gligorov J, Müller V et al. Preference for subcutaneous or intravenous administration of trastuzumab in patients with HER2‐positive early breast cancer (PrefHer): An open‐label randomised study. Lancet Oncol 2013;14:962–970. [DOI] [PubMed] [Google Scholar]
- 12. Pivot X, Gligorov J, Müller V et al. Patients' preferences for subcutaneous trastuzumab versus conventional intravenous infusion for the adjuvant treatment of HER2‐positive early breast cancer: Final analysis of 488 patients in the international, randomized, two‐cohort PrefHer study. Ann Oncol 2014;25:1979–1987. [DOI] [PubMed] [Google Scholar]
- 13. De Cock E, Pivot X, Hauser N et al. A time and motion study of subcutaneous versus intravenous trastuzumab in patients with HER2‐positive early breast cancer. Cancer Med 2016;5:389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.European Medicines Agency. Herceptin SC Summary of Product Characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000278/WC500074922.pdf. Accessed March, 2016.
- 15. Andersson M, López‐Vega JM, Petit T et al. The co‐administration of pertuzumab (P) and trastuzumab (T) as a single infusion, followed by vinorelbine (V), in first‐line (1L) treatment of HER2‐positive locally advanced or metastatic breast cancer (MBC) patients (pts): VELVET study interim analysis. J Clin Oncol 2015;33(suppl abstr 586). [Google Scholar]
- 16.Herceptin® (trastuzumab). Summary of Product Characteristics. Roche Registration Ltd. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000278/WC500074922.pdf. Accessed September, 2016.
- 17.PERJETA® (pertuzumab). Summary of Product Characteristics. Roche Registration Ltd. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002547/WC500140980.pdf. Accessed August, 2016.
- 18. Chan A, Delaloge S, Holmes FA et al. Neratinib after trastuzumab‐based adjuvant therapy in patients with HER2‐positive breast cancer (ExteNET): A multicentre, randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol 2016;17:367–377. [DOI] [PubMed] [Google Scholar]
- 19. Wolff AC, Hammond ME, Hicks DG et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 2013;31:3997–4013. [DOI] [PubMed] [Google Scholar]
- 20.Vinorelbine. Summary of Product Characteristics. Actavis UK Ltd. Available at https://www.medicines.org.uk/emc/medicine/24189. Accessed August, 2016.
- 21. Lenihan D, Suter T, Brammer M et al. Pooled analysis of cardiac safety in patients with cancer treated with pertuzumab. Ann Oncol 2012;23:791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Swain SM, Ewer MS, Cortés J et al. Cardiac tolerability of pertuzumab plus trastuzumab plus docetaxel in patients with HER2‐positive metastatic breast cancer in CLEOPATRA: A randomized, double‐blind, placebo‐controlled phase III study. The Oncologist 2013;18:257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.