

Abstract
Triplophysa fishes are the primary component of the fish fauna on the Tibetan Plateau and are well adapted to the high-altitude environment. Despite the importance of Triplophysa fishes on the plateau, the genetic mechanisms of the adaptations of these fishes to this high-altitude environment remain poorly understood. In this study, we generated the transcriptome sequences for three Triplophysa fishes, that is, Triplophysa siluroides, Triplophysa scleroptera, and Triplophysa dalaica, and used these and the previously available transcriptome and genome sequences from fishes living at low altitudes to identify potential genetic mechanisms for the high-altitude adaptations in Triplophysa fishes. An analysis of 2,269 orthologous genes among cave fish (Astyanax mexicanus), zebrafish (Danio rerio), large-scale loach (Paramisgurnus dabryanus), and Triplophysa fishes revealed that each of the terminal branches of the Triplophysa fishes had a significantly higher ratio of nonsynonymous to synonymous substitutions than that of the branches of the fishes from low altitudes, which provided consistent evidence for genome-wide rapid evolution in the Triplophysa genus. Many of the GO (Gene Ontology) categories associated with energy metabolism and hypoxia response exhibited accelerated evolution in the Triplophysa fishes compared with the large-scale loach. The genes that exhibited signs of positive selection and rapid evolution in the Triplophysa fishes were also significantly enriched in energy metabolism and hypoxia response categories. Our analysis identified widespread Triplophysa-specific nonsynonymous mutations in the fast evolving genes and positively selected genes. Moreover, we detected significant evidence of positive selection in the HIF (hypoxia-inducible factor)-1A and HIF-2B genes in Triplophysa fishes and found that the Triplophysa-specific nonsynonymous mutations in the HIF-1A and HIF-2B genes were associated with functional changes. Overall, our study provides new insights into the adaptations and evolution of fishes in the high-altitude environment of the Tibetan Plateau and complements previous findings on the adaptations of mammals and birds to high altitudes.
Keywords: Tibetan Plateau, adaptation, transcriptome, accelerated evolution, Triplophysa fishes
Introduction
The cold climate on the Tibetan Plateau, with hypoxic conditions and strong ultraviolet radiation, creates environments that are extremely hostile to most organisms (Bickler and Buck 2007; Cheviron and Brumfield 2012). Nevertheless, several animals living in the Tibetan Plateau are well adapted to these harsh environmental conditions (Gansser 1964). Determination of the selective and neutral forces that drive molecular evolution is fundamental to understanding the genetic bases for the adaptations to the high-altitude environments.
Recent literature has primarily focused on the use of genomic approaches to study the genetic mechanisms of high-altitude adaptations in terrestrial endothermic vertebrates. A set of candidate genes and the expansions of gene families related to energy metabolism and hypoxia response were detected in Tibetan people (Beall et al. 2010; Bigham et al. 2010; Xu et al. 2011), yak (Qiu et al. 2012), Tibetan antelope (Ge et al. 2013), ground tit (Cai et al. 2013; Qu et al. 2013), Tibetan mastiff (Gou et al. 2014; Li et al. 2014; Wang et al. 2014), and Tibetan grey wolf (Zhang et al. 2014), demonstrating the genetic mechanisms underlying highland adaptations. However, to date, only limited amounts of genetic or other information are available on the adaptations of poikilothermic species, such as reptiles, amphibians, and fishes, to the cold, hypoxic conditions of high-altitude habitats. Yang et al. (2012, 2014) compared the transcriptomes of two frogs on the Tibetan Plateau, from a high-elevation and a low-elevation site, to identify candidate genes involved in the adaptations to high elevations, and a similar study was conducted with lizards. Although these observations provided important insights into the evolutionary constraints of adaptations to extreme conditions, our understanding of the genetic bases of such adaptations in fish remains limited. A comprehensive transcriptome analysis of a Tibetan fish, Gymnodiptychus pachycheilus, revealed accelerated genic evolution at the genomic scale (Yang et al. 2015); however, many species of Schizothoracinae, to which G. pachycheilus belongs, are polyploid, as reported in previous studies (Zan et al. 1985; Yu et al. 1990). In polyploidy, typically from whole-genome duplication, one copy is free to evolve neutrally and thereby accumulates random mutations more frequently (Ohno 1970; Selmecki et al. 2015). Because of the polyploidy in these species, whether the accelerated genic evolution of G. pachycheilus is the result of adaptation to the Tibetan Plateau or just the effect of gene duplications is unclear.
Uplift of the Tibetan Plateau contributed to the speciation of the Triplophysa fishes (Chen and Zhu 1984). The Triplophysa fishes are species with distributions at the highest altitudes of the entire Tibetan Plateau ichthyofauna. The genus Triplophysa is a strongly diverged fish group, with 137 currently recognized species, most of which occur on the Tibetan Plateau and adjacent regions (Zhu 1989; Froese and Pauly 2014). As the primary component of the fish fauna on the Tibetan Plateau, Triplophysa fishes evolved specific morphological characteristics for adaptation to the highland environment, such as gradually disappeared scales (scaleless) for withstanding cold, abundant blood vessels in gill filament and pituitary for efficient breathing, black peritoneum for responding to ultraviolet radiation, and secondary sexual characters of male for reproduction (Zhu 1989; Wu Y and Wu C 1992; He et al. 2006; Liu et al. 2009; Ren et al. 2011). Therefore, these plateau-dwelling loaches are an iconic symbol for studies of highland adaptations in fishes; however, the genetic bases for those adaptations remain unknown.
The objective of this study was to identify evolutionary patterns, candidate genes, and lineage-specific nonsynonymous mutations that might have facilitated adaptations to the Tibetan Plateau in Triplophysa fishes. In this study, the transcriptomes of Triplophysa siluroides, Triplophysa scleroptera, and Triplophysa dalaica were determined in our laboratory, and we used these transcriptomes with the transcriptome of large-scale loach (Paramisgurnus dabryanus) (Li et al. 2015) and other previously available genome sequences of zebrafish (Danio rerio) and cave fish (Astyanax mexicanus) to investigate changes in evolutionary rates and to identify the potential genetic bases for high-altitude adaptations of Triplophysa fishes.
Materials and Methods
Sampling, RNA Extraction, and Sequencing
The ethics committee of the Institute of Hydrobiology, Chinese Academy of Sciences, approved all of the experimental procedures. Two wild male Triplophysa fishes (T. siluroides and T. scleroptera) were collected from the upper reach of the Yellow River in Ruoergai County, Sichuan Province. To obtain as many expressed genes as possible, five tissues (heart, liver, brain, spleen, and kidney) were sampled and immediately placed in liquid nitrogen for storage at −80 °C until use. Total RNA isolation, RNA-Seq library construction, and sequencing were described in our previous study (Wang et al. 2015). All sequence reads have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive database (accession numbers: SRR1946837 for T. siluroides and SRR1948020 for T. scleroptera).
Data Filtering, De Novo Assembly, and Annotation
The quality of the raw reads was first checked with FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/), and then the reads were filtered by removing reads that contained adapters, reads that contained poly-N and low-quality reads (<Q20) using the Cutadapt application (version 1.6; http://code.google.com/p/cutadapt/) and TrimGalore software (Barbraham, Boinformatics) (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/).
After reads were filtered, the clean Illumina RNA-Seq reads of T. siluroides and T. scleroptera were de novo assembled using the Trinity software (Grabherr et al. 2011) with default parameters. The program CD-HIT-EST was used to remove redundant transcripts with a sequence identity threshold of 0.95 and a word length of 10 to produce the final assembly (Li and Godzik 2006). The assembled sequences were annotated by searching for sequence homology against the NCBI nonredundant protein (NR) database (released on May 3, 2014, at http://www.ncbi.nlm.nih.gov) using BLASTx (Altschul et al. 1997) with a cutoff E-value set at 1e-5.
Ortholog Identification and Alignment
A set of core-orthologs was constructed from eight teleost genome species including zebrafish (D. rerio), medaka (Oryzias latipes), tetraodon (Tetraodon nigroviridis), fugu (Takifugu rubripes), stickleback (Gasterosteus aculeatus), cod (Gadus morhua), platyfish (Xiphophorus maculates), and tilapia (Oreochromis niloticus). All 25,573 one-to-one core-orthologous annotated proteins were downloaded from Ensembl (release 70) and were selected as “primer taxa” which served as the input to generate the core-ortholog database for the program HaMStR v.13.2.3 (Ebersberger et al. 2009) to search for corresponding orthologs in P. dabryanus, T. scleroptera, T. siluroides, and T. dalaica. Zebrafish was used as the representative in the core-orthologs database. Ensembl-annotated one-to-one orthologous genes of D. rerio and A. mexicanus were retrieved using Biomart (Vilella et al. 2009). Large-scale loach (P. dabryanus) is relatively closely related to Triplophysa fishes, diverged from Triplophysa fishes about 30 Ma (Frickhinger 1991; Nelson 2006). Moreover, zebrafish has rich genome data, which can be used to assign and characterize the function of the identified adaptively evolutionary orthologous genes. When more than one corresponding ortholog was identified, the longest one was chosen using our in-house scripts. HaMStR v.13.2.3 was run with strict parameters (-sequence file, -est, -hmmset, -refspec, -representative, and -ublast). Each corresponding orthologous group of six species was extracted with custom Perl scripts to generate multiple sequence alignments.
All the orthologous gene sets were aligned at the codon level with the option “-codon” using the Prank program (Loytynoja and Goldman 2005) and then trimmed using the Gblocks program with the parameter “-t=c” (Castresana 2000). The Gblocks program was used to remove potentially unreliable regions. Additionally, to eliminate the effect of uncertain bases on the test of positive selection, all positions that had gaps (“-”) and “N” in the alignments were deleted. After the trimming process, alignments shorter than 120 bp were discarded.
Phylogenetic Analyses and Evolutionary Rate Estimation
Concatenated alignments constructed from all orthologs were used to construct a phylogenetic tree for these six species. The concatenate data were first partitioned by genes due to the different evolutionary rates. PartitionFinder software was used to determine the optimal partition scheme and corresponding best-fitting nucleotide substitution model under the Bayesian information criterion (Lanfear et al. 2012). The maximum-likelihood (ML) analysis was implemented in RAxML 7.0.3 (Stamatakis 2006). A total of 1,000 nonparametric bootstrap replicates were performed with GTRGAMMA substitution model applied to all partitions. The best-coring ML tree with branch lengths and bootstrap support values was obtained and used in the subsequent analyses.
The codeml program in the PAML package (Yang 2007) with the free ratio model (model = 1) was used to estimate the evolutionary rate along each lineage for the six species. The lineage-specific mean Ka/Ks ratios (the ratios of the number of nonsynonymous substitutions per nonsynonymous site [Ka] to the number of synonymous substitutions per synonymous site [Ks]) were estimated for each ortholog, the concatenated alignments constructed from all orthologs, and the 1,000 concatenated alignments constructed from ten randomly chosen orthologs. The resulting codeml data (such as the N, S, dN, dS, and dN/dS values) for the genes of each lineage were calculated. Genes with N*dN or S*dS < 1 or dS > 2 were filtered following the approach used in a previous study (Goodman et al. 2009). Comparisons of the evolutionary rates along each lineage of the six species were conducted using the Wilcoxon rank sum test.
To identify rapidly evolving GO (Gene Ontology) categories in Triplophysa fishes relative to P. dabryanus, the average dN/dS values for each GO category were calculated, with only GO categories containing more than ten orthologous genes included in these analyses. The Wilcoxon rank sum test was used to identify GO categories with significantly higher dN/dS values in Triplophysa fishes or in the P. dabryanus lineage.
Identification of Fast Evolving Genes and Positively Selected Genes
The branch model was used to identify the fast evolving genes (FEGs) in the codeml program in the PAML package (Yang 2007). The null model assumed that all branches of the tree evolved at the same rate (the same ω), and the alternative model allowed for a specifically tested branch (foreground branch) to evolve under a different rate. A likelihood ratio test (LRT) (df = 1) was used to discriminate between alternative models for each ortholog in the gene set. We corrected for multiple testing by applying the false discovery rate method (FDR < 0.05) (Benjamini and Hochberg 1995) as implemented in R (http://www.R-project.org). Genes were considered FEGs if they had an FDR-adjusted P value < 0.05 and a higher ω value in the foreground branch than in the background branches.
To find genes that potentially experienced positive selection, the improved branch-site model (model = 2, Nsites = 2) in the codeml program in the PAML package was used to detect signatures of positive selection on individual codons in a specific branch (Zhang et al. 2005). By setting the Triplophysa branches as the foreground branch, we compared a selection model that allowed a class of codons on that branch to have dN/dS > 1 (ModelA2, fix_omega = 0, omega = 1.5) with a neutral model that constrained this additional class of sites to have dN/dS = 1 (ModelA1, fix_omega = 1, omega = 1). An LRT compared a model with positive selection on the foreground branch to a null model in which no positive selection occurred on the foreground branch and calculated the statistic (2Δln) to obtain a P value. As noted above, we performed a correction for multiple testing using an FDR criterion and reported genes with an adjusted P value < 0.05 as positively selected genes (PSGs).
After FEGs and PSGs were detected, the DAVID Functional Annotation tool was used to perform GO functional enrichment analyses (Dennis et al. 2003; Huang et al. 2009). Within each annotation cluster, DAVID listed the GO terms that were significantly enriched.
Triplophysa Lineage-Specific Nonsynonymous Mutation Identification and Analysis
Using our in-house scripts, Triplophysa lineage-specific nonsynonymous mutations were identified in amino acid sites of all orthologous genes, FEGs and PSGs. Using the lineage-specific nonsynonymous mutations of all the orthologous genes as the background, the nonsynonymous substitution rates of the FEGs and the PSGs were compared with that of all orthologous genes with chi-square tests. Additionally, we compared the numbers of Triplophysa lineage-specific nonsynonymous substitutions in the FEGs and non-FEGs, in the PSGs and non-PSGs, and in the overlap and nonoverlap of the FEGs and PSGs and then performed Wilcoxon rank sum tests to determine the significance of the differences.
To further verify the importance of the lineage-specific nonsynonymous mutations, we also focused on several important functional genes related to hypoxia, namely, the HIF (hypoxia-inducible factor) alpha paralogs (HIF-1A/B and HIF-2A/B). Orthologs of the HIF alpha paralogs in representative fishes were retrieved from Ensembl (Cunningham et al. 2015) and were aligned with MEGA4.0 (Tamura et al. 2007). The codeml branch-site model (model = 2, Nsites = 2) in the PAML package (version 4.7) was used to detect positive selection, and positively selected sites (PSSs) were identified using the Bayes empirical Bayes (BEB) method. Using the software SIFT (Kumar et al. 2009), the functional consequences of the Triplophysa lineage-specific nonsynonymous mutations were predicted, and then the positive selected genes were searched against the PDB database (Rose et al. 2013) to identify suitable templates for homology modeling. According to the zebrafish HIF alpha paralogs reference, the 3D-structural models were predicted using the I-TASSER software (Roy et al. 2010), visualized with PyMOL v1.5 (Schrödinger, LLC, New York). Structure-based energy calculation was measured with the Eris server (Yin et al. 2007) with the flexible backbone method and pre-relaxation options.
Results
A total of 26,362,264 raw 101-bp paired-end reads of T. siluroides and 48,836,596 raw 101-bp paired-end reads of T. scleroptera were generated. After trimming of the adapter sequences and removal of sequences of low quality, the de novo assemblies of the clean reads produced 58,911 and 119,246 unigenes for T. siluroides and T. scleroptera, respectively. Detailed assembly results are summarized in supplementary table S1Supplementary Material online, and the T. dalaica transcriptome was previously available (Wang et al. 2015). A total of 31,135 (52.8%) unigenes of T. siluroides and 38,825 unigenes (32.6%) of T. scleroptera had significant matches to currently known proteins in the NR database. A BLASTx top-hit species distribution of the gene annotations from the NR database showed the highest homology to the zebrafish. Of the BLASTx top-hits, 31,091 (80.1%) matched zebrafish protein sequences in T. scleroptera, whereas 23,022 (73.9%) matched zebrafish protein sequences in T. siluroides.
Accelerated Evolution of the Triplophysa Lineages
A total of 3,217 1:1:1:1:1:1 putative orthologous genes were first identified for A. mexicanus, D. rerio, P. dabryanus, T. siluroides, T. scleroptera, and T. dalaica. After our alignment treatments and trimming for quality control, 2,269 orthologous genes were advanced to the subsequent evolutionary analyses.
The phylogeny of the six species was constructed from the concatenated 2,269 orthologous genes (fig. 1A) with the partitioned ML method. Based on the above constructed phylogenetic tree, the ratio of nonsynonymous to synonymous substitutions (Ka/Ks) for each ortholog were evaluated in the different branches at the gene level. The free ratio model (model = 1) in PAML was used (Yang 2007), which allowed for a separate Ka/Ks ratio for each branch. Averaged for all 2,269 orthologous genes, all three Triplophysa fish branches (T. siluroides, T. scleroptera, and T. dalaica) had significantly higher ratios of nonsynonymous to synonymous substitutions (Ka/Ks) than the remaining branches of the tree (Wilcoxon rank sum test, P < 2.2 e-16), which indicated accelerated evolution in the Triplophysa lineages (fig. 1B). Furthermore, we calculated the Ka/Ks ratio for each branch for a concatenated alignment of 2,269 orthologs and 1,000 concatenated alignments constructed from ten randomly chosen orthologs, which also revealed that all the Triplophysa fishes had significantly higher Ka/Ks ratios than the other fish branches (Wilcoxon rank sum test, P < 2.2 e-16) (fig. 1C and D). Moreover, we examined the Ka/Ks ratio for each gene and found a larger number of genes with higher Ka/Ks ratios in the Triplophysa fishes (T. siluroides, T. scleroptera, and T. dalaica) than in the P. dabryanus lineage (1,153 vs. 414, 659 vs. 386, and 792 vs. 410, respectively).
Fig. 1.—

Phylogenetic tree used in this study (A) and the Ka/Ks ratios for the terminal branches obtained from each ortholog (B), concatenated alignments constructed from all orthologs (C), and 1,000 concatenated alignments constructed from ten randomly chosen orthologs (D). The blue line in (A) represents the Triplophysa fishes, which are highlighted in light blue. Tda, T. dalaica; Tsc, T. scleroptera; Tsi, T. siluroides; Pda, P. dabryanus; Dre, D. rerio; Ame, A. mexicanus.
To identify lineage-specific accelerated GO categories, the mean Ka/Ks ratios for the different GO categories were calculated for each of the Triplophysa fishes and the P. dabryanus branch. The number of GO categories with higher mean Ka/Ks ratios in the T. siluroides, T. scleroptera, and T. dalaica lineages was significantly greater than that in the P. dabryanus lineage (406 vs. 25, 383 vs. 45 and 383 vs. 48, respectively). The statistical analysis found a total of 188, 156, and 150 GO categories with relatively higher evolutionary rates in the T. siluroides, T. scleroptera, and T. dalaica lineages, respectively, compared with that for the P. dabryanus lineage (Wilcoxon rank sum test, P < 0.05). The comparative analysis between Ka/Ks ratios in the lineage of each of the Triplophysa fishes and in the P. dabryanus lineage revealed that many GO categories with significantly higher Ka/Ks ratios in Triplophysa fishes were associated with hypoxia response and energy metabolism: “ATP binding,” “mitochondrion,” “blood coagulation,” “cellular response to hypoxia,” “response to hypoxia,” and “oxidation-reduction process” (fig. 2A–C and supplementary tables S2–S4Supplementary Material online).
Fig. 2.—

Mean Ka/Ks ratios for each GO category with more than ten orthologs in each of the Triplophysa fishes and P. dabryanus (A–C, Tda, T. dalaica; Tsc, T. scleroptera; Tsi, T. siluroides). GO categories with statistically significantly higher Ka/Ks ratios in Triplophysa fish (red) and P. dabryanus (blue) are highlighted. Points with light red and light blue represent GO categories with higher but statistically not significant Ka/Ks ratios in each of the Triplophysa fishes and P. dabryanus.
Positively Selected Genes and FEGs
To identify genes that might be candidates for Triplophysa lineage-specific adaptations, we applied eight different branch-site and branch LRTs: Three terminal branches (each branch of T. dalaica, T. scleroptera, and T. siluroides); the internal branch ancestral to T. dalaica and T. scleroptera; the whole clade of T. dalaica and T. scleroptera; the internal branch ancestral to T. dalaica, T. scleroptera, and T. siluroides; the whole clade of T. dalaica, T. scleroptera, and T. siluroides; and the P. dabryanus branch. The whole clade represented both the ancestral branch and the terminal branch for a specific lineage. In total, we identified 278 PSGs and 833 FEGs from all the Triplophysa branches and 50 PSGs and 112 FEGs in the P. dabryanus branch after conservatively correcting for multiple testing (supplementary tables S5 and Supplementary Data, Supplementary Material online). To identify genes that might directly contribute to adaptations to the Tibetan Plateau, we found a subset (n = 196) of genes that were both FEGs and PSGs from all the Triplophysa branches, and among these, identified 12 candidate genes related to the hypoxia response: ADAR, LSP1, HMOX1A, IL6ST, HDAC3, ANGPTL3, CAST, EPRS, SMOC1, FOXO4, ADAM8B, and TNFRSF1B (table 1 and supplementary table S7, Supplementary Material online).
Table 1.
Genes Identified as both FEGs (Branch Model) and PSGs (Branch-Site Model) Involved in the Hypoxia Response in Specific Triplophysa Fishes Lineages
| Gene ID (Gene Name) | Description | Branch along Which PSGs Was Detected | Branch along Which FEGs Was Detected |
|---|---|---|---|
| ENSDARG00000012389 (ADAR) | Adenosine deaminase, RNA-specific | Ancestral branch of Tda, Tsc, and Tsi | Whole clade of Tda, Tsc, and Tsi |
| ENSDARG00000027310 (LSP1) | Lymphocyte-specific protein 1 | Terminal branch of Tda and whole clade of Tda and Tsc | Whole clade of Tda, Tsc, and Tsi |
| ENSDARG00000027529 (HMOX1A) | Heme oxygenase (decycling) 1a | Ancestral branch of Tda and Tsc | Whole clade of Tda, Tsc, and Tsi |
| ENSDARG00000030498 (IL6ST) | Interleukin 6 signal transducer | Terminal branch of Tda and whole clade of Tda, Tsc, and Tsi | Terminal branch of Tsi, whole clade of Tda and Tsc, and whole clade of Tda, Tsc and Tsi |
| ENSDARG00000037514 (HDAC3) | Histone deacetylase 3 | Terminal branch of Tsi | Terminal branch of Tsi and whole clade of Tda, Tsc and Tsi |
| ENSDARG00000044365 (ANGPTL3) | Angiopoietin-like 3 | Terminal branch of Tda and whole clade of Tda and Tsc | Terminal branch of Tda, terminal branch of Tsi, whole clade of Tda and Tsc, and whole clade of Tda, Tsc and Tsi |
| ENSDARG00000058693 (CAST) | Calpastatin | Terminal branch of Tda, terminal branch of Tsc, terminal branch of Tsi, and ancestral branch of Tda and Tsc | Whole clade of Tda, Tsc and Tsi |
| ENSDARG00000060494 (EPRS) | Glutamyl-prolyl-tRNA synthetase | Terminal branch of Tda, terminal branch of Tsc, terminal branch of Tsi, and ancestral branch of Tda and Tsc | Terminal branch of Tda, ancestral branch of Tda and Tsc, whole clade of Tda and Tsc, and whole clade of Tda, Tsc and Tsi |
| ENSDARG00000089188 (SMOC1) | SPARC-related modular calcium binding 1 | Terminal branch of Tda | Whole clade of Tda and Tsc and whole clade of Tda, Tsc and Tsi |
| ENSDARG00000055792 (FOXO4) | Forkhead box O4 | Ancestral branch of Tda and Tsc, whole clade of Tda and Tsc, and whole clade of Tda, Tsc and Tsi | Ancestral branch of Tda and Tsc |
| ENSDARG00000057644 (ADAM8B) | A disintegrin and metalloproteinase domain 8b | Terminal branch of Tda, whole clade of Tda and Tsc, and whole clade of Tda, Tsc and Tsi | Terminal branch of Tda, whole clade of Tda and Tsc, and whole clade of Tda, Tsc and Tsi |
| ENSDARG00000070165 (TNFRSF1B) | Tumor necrosis factor receptor superfamily, member 1B | Terminal branch of Tda, terminal branch of Tsc, terminal branch of Tsi, whole clade of Tda and Tsc, and whole clade of Tda, Tsc and Tsi | Terminal branch of Tsi, whole clade of Tda and Tsc, and whole clade of Tda, Tsc and Tsi |
Note.—Gene identifier from Ensembl (gene ID from zebrafish) and gene description (Description) are provided. Whole clade represents both the ancestral branch and the terminal branch for specific lineages. Tda, Triplophysa dalaica; Tsc, Triplophysa scleroptera; Tsi, Triplophysa siluroides.
Functional enrichment analyses showed that the PSGs (278) and the FEGs (833) from all Triplophysa branches were significantly enriched in functional categories related to energy metabolism and to adaptation to hypoxia. These categories are biologically relevant for life at high altitudes. The ATPase genes (ATPase activity, coupled and ATPase activity) have a role in providing energy. Additionally, “ATP binding,” “phosphorylation,” “NADP metabolic process” and “cellular carbohydrate catabolic process” are related to energy metabolism (table 2 and supplementary table S8, Supplementary Material online), and the “Oxidoreduction coenzyme” and “Heat shock protein binding” categories might be involved in hypoxia adaptation (table 2 and supplementary table S8, Supplementary Material online).
Table 2.
GO Enrichment Analysis for FEGs and PSGs in all Triplophysa Fishes
| Gene Class | Category/GO ID | GO Term | Gene Number | P Value | Fold Enrichment |
|---|---|---|---|---|---|
| FEGs | BP/GO:0006733 | Oxidoreduction coenzyme metabolic process | 5 | 0.0140 | 5.2111 |
| BP/GO:0044275 | Cellular carbohydrate catabolic process | 7 | 0.0232 | 3.1267 | |
| BP/GO:0006796 | Phosphate metabolic process | 36 | 0.0437 | 1.3748 | |
| BP/GO:0006793 | Phosphorus metabolic process | 36 | 0.0437 | 1.3748 | |
| BP/GO:0006739 | NADP metabolic process | 4 | 0.0081 | 9.0958 | |
| MF/GO:0005524 | ATP binding | 59 | 0.0025 | 1.4580 | |
| MF/GO:0031072 | Heat shock protein binding | 7 | 0.0106 | 3.7084 | |
| PSGs | BP/GO:0006793 | Phosphorus metabolic process | 14 | 0.0268 | 1.9094 |
| BP/GO:0006796 | Phosphate metabolic process | 14 | 0.0268 | 1.9094 | |
| BP/GO:0006468 | Protein amino acid phosphorylation | 10 | 0.0659 | 1.9463 | |
| BP/GO:0016310 | Phosphorylation | 11 | 0.0691 | 1.8402 | |
| MF/GO:0042623 | ATPase activity, coupled | 8 | 0.0032 | 4.0984 | |
| MF/GO:0016887 | ATPase activity | 8 | 0.0090 | 3.3790 | |
| MF/GO:0004674 | Protein serine/threonine kinase activity | 8 | 0.0969 | 2.0231 |
Widespread Triplophysa Lineage-Specific Nonsynonymous Mutations
Lineage-specific nonsynonymous mutations can contribute to lineage-specific adaptations. Among the 2,269 orthologous genes, 1,958 genes possessed Triplophysa lineage-specific nonsynonymous mutations, whereas 257 of the 278 PSGs, 794 of the 833 FEGs, and 183 of the 196 overlaps of both FEGs and PSGs possessed Triplophysa lineage-specific nonsynonymous mutations. Chi-square tests showed that the percentage of genes having Triplophysa lineage-specific nonsynonymous mutations of functional genes (the FEGs, the PSGs, and the overlap of both FEGs and PSGs) was significantly higher than that of background (P < 1.91 e-012, P = 0.004, and P = 0.005, respectively) (fig. 3A). More importantly, in comparisons between the number of Triplophysa lineage-specific nonsynonymous mutations in FEGs and non-FEGs, in PSGs and non-PSGs, and in the overlap and nonoverlap of FEGs and PSGs, the number of Triplophysa lineage-specific nonsynonymous mutations were significantly higher in the FEGs, the PSGs, and the overlap of FEGs and PSGs (Wilcoxon rank sum test, P < 2.2 e-16, P < 6.161 e-10, and P < 5.016 e-11, respectively) (fig. 3B).
Fig. 3.—

Analysis of Triplophysa lineage-specific nonsynonymous mutations. (A) Percentage of genes having Triplophysa lineage-specific nonsynonymous mutations. “All” represents all the orthologous genes. “Overlap” represents a subset of genes that are both FEGs and PSGs. (B) Number of Triplophysa lineage-specific nonsynonymous mutations among FEGs and non-FEGs, PSGs and non-PSGs, and overlap and nonoverlap of FEGs and PSGs. Significant differences are indicated by asterisks, based on chi-square test (A) and Wilcoxon rank-sum test (B), **P < 0.01.
Selection analysis of the HIF alpha paralogs revealed that HIF-1A and HIF-2B genes experienced positive selection (P < 1.905 e-13 and 1.141 e-02, respectively). In the HIF-1A genes, PSSs were identified using the BEB method, some of which were also Triplophysa lineage-specific nonsynonymous mutations, although no PSSs were identified in the HIF-2B gene. We examined the nonsynonymous mutations in HIF-1A gene in detail and found that some Triplophysa lineage-specific nonsynonymous mutations, including S329N, A407R, S710P, R743T, and L746Y, were PSSs. Amino acid site L712I was a Triplophysa lineage-specific nonsynonymous mutation but did not undergo positive selection. The S329N and L712I were invariant among all other representative fishes, whereas the remaining variants were variable in the other representative fishes (fig. 4A). Similarly, in HIF-2B gene, R65Q, Q123P, H129Q, H418L, and P462S exhibited Triplophysa lineage-specific nonsynonymous mutations (fig. 4B). Further analyses with more distantly related species confirmed the same patterns (supplementary fig. S1, Supplementary Material online). The prediction of the functional effects of the variants indicated that S329N, S710P, L712I and L746Y in HIF-1A gene and R65Q, Q123P and H129Q in HIF-2B gene should be deleterious, whereas the others should be tolerated (table 3). The suitable templates 4H6J and 1L8C were identified for the homology modeling of the PAC domain and the C-TAD domain of HIF-1A, respectively, whereas no suitable templates were identified for the corresponding domains of HIF-2B. Using the crystal structure of the PAC and C-TAD domains of human HIF-1A as templates, our homology modeling suggested that the S329N mutation occurred in the PAC domain, whereas the S710P, L712I and L746Y mutations occurred in the C-TAD domain (fig. 4C and D). As these mutations were located in functional domains of the protein and predicted functional consequences, the Triplophysa lineage-specific nonsynonymous mutations might be the primary reasons for high-altitude hypoxic adaptations in these fish.
Fig. 4.—
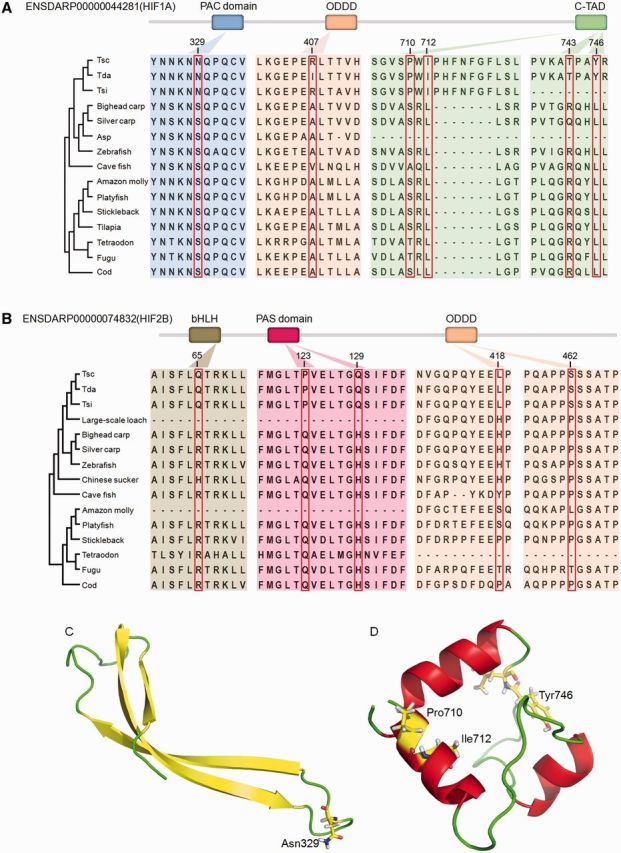
Evolutionary analysis and sequence alignments of Triplophysa lineage-specific nonsynonymous mutations across representative fishes based on positively selected HIF-1A gene (A) and HIF-2B gene (B). The protein coordinates of HIF-1A and HIF-2B referred to the Ensembl ID ENSDARP00000044281 and ENSDARP00000074832, respectively. (C) Structure model of the PAC domain. The mutated site S329N in the PAC domain is indicated and was predicted to decrease the thermodynamic stability of the domain (ΔΔG = 1.42). (D) Structure model of the C-TAD domain of HIF-1A and three mutated sites S710P, L712I, and L746Y in the C-TAD domain are marked and are predicted to decrease the thermodynamic stability of the domain (ΔΔG = 0.96 kcal/mol).
Table 3.
Triplophysa-Specific Nonsynonymous Mutations in the PSGs HIF-1A and HIF-2B
| Gene Name | Amino Acid Variant | SIFT Score | Consequence |
|---|---|---|---|
| HIF-1A | S329N | 0.01 | Deleterious |
| A407R | 0.07 | Tolerated | |
| S710P | 0.00 | Deleterious | |
| L712I | 0.01 | Deleterious | |
| R743T | 0.29 | Tolerated | |
| L746Y | 0.00 | Deleterious | |
| HIF-2B | R65Q | 0.03 | Deleterious |
| Q123P | 0.00 | Deleterious | |
| H129Q | 0.00 | Deleterious | |
| H418L | 0.66 | Tolerated | |
| V420L | 0.80 | Tolerated | |
| D437S | 1.00 | Tolerated | |
| P462S | 1.00 | Tolerated |
Discussion
We sequenced and assembled mixed-tissue transcriptomes from three Triplophysa fishes, that is, T. siluroides, T. scleroptera, and T. dalaica, in combination with previously available data to identify the underlying genetic mechanisms for adaptation of fishes to high-altitudes, including evolutionary patterns, adaptive evolutionary genes, and lineage-specific nonsynonymous mutations.
The determination of how species adapt to extreme environments is a central theme for research in evolutionary biology (Smith and Eyre-Walker 2002). Generally, the signals of adaptive evolution could be detected by an increased ratio of nonsynonymous to synonymous substitutions (Bakewell et al. 2007). Our results that Tibetan loaches showed genome-wide accelerated evolution (higher Ka/Ks ratio) relative to other low-altitude fishes suggest that Tibetan loaches may have undergone adaptive evolution that allows them to cope with their extremely inhospitable high-altitude environments. In addition, there are many GO categories involved in the hypoxia response and energy metabolism which were found to have evolved faster in each of the three Triplophysa fishes than the P. dabryanus lineage. The above evidence of accelerated evolution provided important insights into the mechanisms of high altitude adaptation of Triplophysa fishes, which were also observed in endothermic animals, such as yak (Qiu et al. 2012) and poikilothermic species, such as lizards (Yang et al. 2014) and schizothoracine fishes (Yang et al. 2015).
It is well-known that the environmental conditions are extremely adverse in high-altitude habitats. The extreme environments necessitate high energy metabolism and hypoxia adaptation in indigenous species living in Tibetan plateau. Our analyses yielded a list of adaptively evolving genes that included 278 PSGs and 833 FEGs in the Triplophysa lineages, which are indeed significantly enriched in functional categories associated with energy metabolism and hypoxia response. This observation indicated that the genes are influenced by natural selection during the evolution of Triplophysa lineages. Notably, the hypoxic environment is a vital factor that imposes severe physiological challenges on species living at high altitude (Storz, Scott, et al. 2010). Nevertheless, Triplophysa fishes that lived in the Tibetan rivers and lakes have well adapted to these harsh environmental conditions. Previous studies of the physiological mechanisms of acclimation to hypoxia have been investigated in hypoxia tolerant fishes, such as Amazonian fish Prochilodus nigricans (Val et al. 2015) and the scaleless fish Galaxias maculates (Urbina and Glover 2012). Hypoxia survival in fish requires a well-coordinated response to either increase the capacity of O2 uptake and delivery to the tissues through blood adjustments, especially blood oxygen affinity, or reduce oxygen consumption through strong metabolic rate depression (Richards 2011; Urbina and Glover 2012; Val et al. 2015). These adaptive physiological responses of acclimation to chronic hypoxia principally result from changes in gene expressions and gene mutations (Semenza 2000; Xiao 2015), which can alter the capacity of fish to endure hypoxia. In particular, HIF is a master regulator in the hypoxia signaling pathway, which largely mediated the regulation and coordination of changes in gene expression in response to hypoxia in fishes (Xiao 2015). Recent studies in several fishes have shed light on the molecular and genetic basis underlying their response to hypoxia with cDNA microarrays, transcriptome sequencing, and proteomics technologies (Gracey et al. 2001; Chen et al. 2013; Liao et al. 2013; Ao et al. 2015). Through comparative genomic analyses, we identified 12 candidate genes specifically linked to hypoxia response. For example, HMOX1A (heme oxygenase (decycling) 1a) is induced by a wide variety of oxidative stresses (Tomaro et al. 1991), which is cloned in the crucian carp (Carassius carassius) and plays an important role in preventing hypoxia-induced cell death (Wang et al. 2008). And HDAC3 (HIF-1α-induced histone deacetylase 3) is essential in response to hypoxia and serves as an essential corepressor to repress epithelial gene expression (Wu et al. 2011). Additionally, LSP1 (lymphocyte-specific protein 1) and CAST (calpastatin) appear to be unregulated in response to hypoxia (Blomgren et al. 1999; Martin-Rendon et al. 2007). SMOC1 (SPARC-related modular calcium binding 1) is potentially linked to altered vascular structure (Sherva et al. 2007) and promotes angiogenesis (Awwad et al. 2015). ADAM8B has a potential role as a hypoxia-dependent protein in the pathogenesis and evolution of pancreatic cancer (Valkovskaya 2008). IL6ST and EPRS are listed in the HypoxiaDB (a database of hypoxia regulated proteins) and are associated with hypoxia response (Khurana et al. 2013). These results indicate that hypoxia-regulated genes which have undergone adaptive evolution in Tibetan loaches represent an adaptation to the extreme environments on the Tibetan Plateau.
As previously demonstrated with theoretical models, two populations living in identical environments have an increased probability of fixing identical mutation by natural selection (Orr 2005). Therefore, to further explore the implications for Triplophysa lineage-specific adaptations to high-altitude environments, we focused on lineage-specific nonsynonymous mutations that occurred in functional genes extensively (PSGs, FEGs, and the overlap of the two). More recently, lineage-specific mutations were employed to study convergent evolution in marine mammals. Comparative genomic analysis in that study revealed 15 of the 44 identical nonsynonymous amino acid substitutions were in genes evolving under positive selection along the marine mammal lineages, which had known functional associations with the phenotypic adaptations to swimming, buoyancy and oxygen store in the aquatic environment (Foote et al. 2015). This is consistent with our assumption that lineage-specific mutations are responsible for adaptation to the environments where they lived. Additionally, lineage-specific mutations in several important functional genes, including EPAS1 and HBB, were used to account for adaptation to high-altitude hypoxia in humans and Tibetan mastiffs (Beall et al. 2010; Gou et al. 2014; Wang et al. 2014). More specifically, these studies revealed that several HIF-pathway genes experienced positive selection in Tibetan dog and discovered several nonsynonymous mutations in the EPAS1 gene, of which G305S occurred in a well-defined protein domain (PAS domain) and was deleterious (Gou et al. 2014; Wang et al. 2014). Likewise, positive selection and nonsynonymous mutation analyses of the HIF alpha paralogs in the present study also demonstrated that HIF-1A and HIF-2B were subject to positive selection and had several lineage-specific amino acid variants. Positively selected sites were detected in HIF-1A, including sites S329N, S710P, L712I, and L746Y. We tentatively infer that the nonsynonymous mutation in HIF-1A, S329N, might play an important role in the high-altitude hypoxic adaptation of Tibetan loaches, as this PSS is not only conserved in all representative vertebrates but also located in the PAC domain, which was proposed to contribute to the PAS domain fold. However, the remaining S710P, L712I, and L746Y mutations in HIF-1A were variants in the other representative vertebrates and were located in the C-TAD domain that conferred oxygen-dependent regulation through the hydroxylation of a conserved asparagine residue. Additionally, the key S329N, S710P, L712I, and L746Y amino acid mutations that we identified in HIF-1A were predicted to be damaging, which was also likely to cause functional change in HIF-1A. Moreover, several experimental studies revealed that amino acid variants in the alpha- and/or beta-globin genes can undoubtedly change Hb-O2 affinity in high-altitude species, including deer mice (Natarajan et al. 2013; Storz, Runck, et al. 2010; Storz et al. 2009) and Andean hummingbirds (Projecto-Garcia et al. 2013). Therefore, a deep understanding of the contribution of Triplophysa lineage-specific nonsynonymous mutations to adaptation to the Tibetan Plateau in Tibetan loaches can only be achieved by experimental and functional genomics in future.
Conclusions
To the best of our knowledge, this study is the first report to investigate the genetic mechanisms of adaptation to the conditions on Tibetan Plateau in the Triplophysa fishes at the scale of the genome. In particular, the detection of accelerated evolution, the identified candidate genes, and the Triplophysa-specific variations related to energy metabolism and hypoxia response provide evidence for adaptations to the high-altitude environment in the three Triplophysa fishes. These results provide a meaningful contribution to our understanding of the molecular mechanisms underlying the adaptations to high altitudes and provide additional resources for future studies on adaptive evolution in Triplophysa fishes.
Supplementary Material
Acknowledgments
The authors thank two anonymous referees. This work was supported by the Pilot projects (Grant No.XDB13020100).
Literature Cited
- Altschul SF, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ao J, et al. 2015. Genome sequencing of the perciform fish Larimichthys crocea provides insights into molecular and genetic mechanisms of stress adaptation. PLoS Genet. 11:e1005118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awwad K, et al. 2015. Role of secreted modular calcium-binding protein 1 (SMOC1) in transforming growth factor beta signalling and angiogenesis. Cardiovasc Res. 106:284–294. [DOI] [PubMed] [Google Scholar]
- Bakewell MA, Shi P, Zhang J. 2007. More genes underwent positive selection in chimpanzee evolution than in human evolution. Proc Natl Acad Sci U S A. 104:7489–7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall CM, et al. 2010. Natural selection on EPAS1 (HIF2alpha) associated with low hemoglobin concentration in Tibetan highlanders. Proc Natl Acad Sci U S A. 107:11459–11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 57:289–300. [Google Scholar]
- Bickler PE, Buck LT. 2007. Hypoxia tolerance in reptiles, amphibians, and fishes: life with variable oxygen availability. Annu Rev Physiol. 69:145–170. [DOI] [PubMed] [Google Scholar]
- Bigham A, et al. 2010. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet. 6:e1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomgren K, et al. 1999. Calpastatin is up-regulated in response to hypoxia and is a suicide substrate to calpain after neonatal cerebral hypoxia-ischemia. J Biol Chem. 274:14046–14052. [DOI] [PubMed] [Google Scholar]
- Cai Q, et al. 2013. Genome sequence of ground tit Pseudopodoces humilis and its adaptation to high altitude. Genome Biol. 14:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17:540–552. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhu S. 1984. Phylogenetic relationships of the subfamilies in the loach family cobitidae (Pisces). Acta Zootaxonmica Sin. 9:201–207. [Google Scholar]
- Chen K, Cole RB, Rees BB. 2013. Hypoxia-induced changes in the zebrafish (Danio rerio) skeletal muscle proteome. J Proteomics. 78:477–485. [DOI] [PubMed] [Google Scholar]
- Cheviron ZA, Brumfield RT. 2012. Genomic insights into adaptation to high-altitude environments. Heredity 108:354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F, et al. 2015. Ensembl 2015. Nucleic Acids Res. 43:D662–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Jr, et al. 2003. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4:P3. [PubMed] [Google Scholar]
- Ebersberger I, Strauss S, von Haeseler A. 2009. HaMStR: profile hidden Markov model based search for orthologs in ESTs. BMC Evol Biol. 9:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote AD, et al. 2015. Convergent evolution of the genomes of marine mammals. Nat Genet. 47:272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frickhinger KA. 1991. Fossilien atlas fishes [English translation 1995]. Melle (Germany): Mergus; 1088 pp. [Google Scholar]
- Froese R, Pauly D. 2014. FishBase. World Wide Web electronic publication. Available from: www.fishbase.org, version (11/2014). [Google Scholar]
- Gansser AE. 1964. Geology of the Himalayas. New York: Wiley Interscience. [Google Scholar]
- Ge RL, et al. 2013. Draft genome sequence of the Tibetan antelope. Nat Commun. 4:1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman M, et al. 2009. Phylogenomic analyses reveal convergent patterns of adaptive evolution in elephant and human ancestries. Proc Natl Acad Sci U S A. 106:20824–20829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou X, et al. 2014. Whole-genome sequencing of six dog breeds from continuous altitudes reveals adaptation to high-altitude hypoxia. Genome Res. 24:1308–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracey AY, Troll JV, Somero GN. 2001. Hypoxia-induced gene expression profiling in the euryoxic fish Gillichthys mirabilis. Proc Natl Acad Sci U S A. 98:1993–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He DK, Chen YX, Chen YF. 2006. Molecular phylogeny and biogeography of the genus Triplophysa (Osteichthyes:Nemacheilinae) in the Tibetan Plateau inferred from cytochrome b DNA sequences. Prog Nat Sci. 16:1395–1404. [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4:44–57. [DOI] [PubMed] [Google Scholar]
- Khurana P, Sugadev R, Jain J, Singh SB. 2013. HypoxiaDB: a database of hypoxia-regulated proteins. Database (Oxford) 2013:bat074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 4:1073–1081. [DOI] [PubMed] [Google Scholar]
- Lanfear R, Calcott B, Ho SY, Guindon S. 2012. Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 29:1695–1701. [DOI] [PubMed] [Google Scholar]
- Li C, Ling Q, Ge C, Ye Z, Han X. 2015. Transcriptome characterization and SSR discovery in large-scale loach Paramisgurnus dabryanus (Cobitidae, Cypriniformes). Gene 557:201–208. [DOI] [PubMed] [Google Scholar]
- Li W, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659. [DOI] [PubMed] [Google Scholar]
- Li Y, et al. 2014. Population variation revealed high-altitude adaptation of Tibetan mastiffs. Mol Biol Evol. 31:1200–1205. [DOI] [PubMed] [Google Scholar]
- Liao X, et al. 2013. Transcriptome analysis of crucian carp (Carassius auratus), an important aquaculture and hypoxia-tolerant species. PLoS One 8:e62308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, He X, Li B, Wang Z. 2009. Triplophysa bleekeri pituitary microstructure. Sichuan J Zool. 28:810–812. [Google Scholar]
- Loytynoja A, Goldman N. 2005. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A. 102:10557–10562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Rendon E, et al. 2007. Transcriptional profiling of human cord blood CD133+ and cultured bone marrow mesenchymal stem cells in response to hypoxia. Stem Cells 25:1003–1012. [DOI] [PubMed] [Google Scholar]
- Natarajan C, et al. 2013. Epistasis among adaptive mutations in deer mouse hemoglobin. Science 340:1324–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JS. 2006. Fishes of the world. New York: John Wiley & Sons. [Google Scholar]
- Ohno S. 1970. Evolution by gene duplication. New York: Springer Verlag. [Google Scholar]
- Orr HA. 2005. The probability of parallel evolution. Evolution 59:216–220. [PubMed] [Google Scholar]
- Projecto-Garcia J, et al. 2013. Repeated elevational transitions in hemoglobin function during the evolution of Andean hummingbirds. Proc Natl Acad Sci U S A. 110:20669–20674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Q, et al. 2012. The yak genome and adaptation to life at high altitude. Nat Genet. 44:946–949. [DOI] [PubMed] [Google Scholar]
- Qu Y, et al. 2013. Ground tit genome reveals avian adaptation to living at high altitudes in the Tibetan Plateau. Nat Commun. 4:2071. [DOI] [PubMed] [Google Scholar]
- Ren S, Yan Z, Ma H, Wang Z. 2011. Observation of gill structure of Triplophysa bleekeri. Guizhou Agric Sci. 39:128–130. [Google Scholar]
- Richards JG. 2011. Physiological, behavioral and biochemical adaptations of intertidal fishes to hypoxia. J Exp Biol. 214:191–199. [DOI] [PubMed] [Google Scholar]
- Rose PW, et al. 2013. The RCSB Protein Data Bank: new resources for research and education. Nucleic Acids Res. 41:D475–D482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5:725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki AM, et al. 2015. Polyploidy can drive rapid adaptation in yeast. Nature 519:349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. 2000. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol (1985). 88:1474–1480. [DOI] [PubMed] [Google Scholar]
- Sherva R, et al. 2007. A whole genome scan for pulse pressure/stroke volume ratio in African Americans: the HyperGEN study. Am J Hypertens. 20:398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NG, Eyre-Walker A. 2002. Adaptive protein evolution in Drosophila. Nature 415:1022–1024. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. [DOI] [PubMed] [Google Scholar]
- Storz JF, et al. 2009. Evolutionary and functional insights into the mechanism underlying high-altitude adaptation of deer mouse hemoglobin. Proc Natl Acad Sci U S A. 106:14450–14455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz JF, Runck AM, Moriyama H, Weber RE, Fago A. 2010. Genetic differences in hemoglobin function between highland and lowland deer mice. J Exp Biol. 213:2565–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz JF, Scott GR, Cheviron ZA. 2010. Phenotypic plasticity and genetic adaptation to high-altitude hypoxia in vertebrates. J Exp Biol. 213:4125–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 24:1596–1599. [DOI] [PubMed] [Google Scholar]
- Tomaro ML, Frydman J, Frydman RB. 1991. Heme oxygenase induction by CoCl2, Co-protoporphyrin IX, phenylhydrazine, and diamide: evidence for oxidative stress involvement. Arch Biochem Biophys. 286:610–617. [DOI] [PubMed] [Google Scholar]
- Urbina MA, Glover CN. 2012. Should I stay or should I go?: physiological, metabolic and biochemical consequences of voluntary emersion upon aquatic hypoxia in the scaleless fish Galaxias maculatus. J Comp Phys B Biochem Syst Environ Physiol. 182:1057–1067. [DOI] [PubMed] [Google Scholar]
- Val AL, Gomes KRM, de Almeida-Val VMF. 2015. Rapid regulation of blood parameters under acute hypoxia in the Amazonian fish Prochilodus nigricans. Comp Biochem Physiol A Mol Integr Physiol. 184:125–131. [DOI] [PubMed] [Google Scholar]
- Valkovskaya NV. 2008. Hypoxia-dependent expression of ADAM8 in human pancreatic cancer cell lines. Exp Oncol. 30:129–132. [PubMed] [Google Scholar]
- Vilella AJ, et al. 2009. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 19:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Zhong XP, Qiao ZX, Gui JF. 2008. Inductive transcription and protective role of fish heme oxygenase-1 under hypoxic stress. J Exp Biol. 211:2700–2706. [DOI] [PubMed] [Google Scholar]
- Wang GD, et al. 2014. Genetic convergence in the adaptation of dogs and humans to the high-altitude environment of the Tibetan Plateau. Genome Biol Evol. 6:2122–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yang LD, Wu B, Song ZB, He SP. 2015. Transcriptome analysis of the plateau fish (Triplophysa dalaica): implications for adaptation to hypoxia in fishes. Gene 565:211–220. [DOI] [PubMed] [Google Scholar]
- Wu MZ, et al. 2011. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell. 43:811–822. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wu C. 1992. The fishes of the Qinghai–Xizang Plateau. Chengdu (China): Sichuan Science and Technology Press. [Google Scholar]
- Xiao W. 2015. The hypoxia signaling pathway and hypoxic adaptation in fishes. Sci China Life Sci. 58:148–155. [DOI] [PubMed] [Google Scholar]
- Xu SH, et al. 2011. A genome-wide search for signals of high-altitude adaptation in Tibetans. Mol Biol Evol. 28:1003–1011. [DOI] [PubMed] [Google Scholar]
- Yang L, Wang Y, Zhang Z, He S. 2015. Comprehensive transcriptome analysis reveals accelerated genic evolution in a Tibet fish, Gymnodiptychus pachycheilus. Genome Biol Evol. 7:251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Qi Y, Fu J. 2014. Exploring the genetic basis of adaptation to high elevations in reptiles: a comparative transcriptome analysis of two toad-headed agamas (genus Phrynocephalus). PLoS One 9:e112218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WZ, Qi Y, Bi K, Fu JZ. 2012. Toward understanding the genetic basis of adaptation to high-elevation life in poikilothermic species: a comparative transcriptomic analysis of two ranid frogs, Rana chensinensis and R. kukunoris. BMC Genomics 13:588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- Yin S, Ding F, Dokholyan NV. 2007. Eris: an automated estimator of protein stability. Nat Methods. 4:466–467. [DOI] [PubMed] [Google Scholar]
- Yu XY, Li YC, Zhou T. 1990. Karyotype studies of Cyprinid fishes in China-comparative study of the karyotypes of 8 species of schizothoracine fishes. J Wuhan Univ. 2:97–104. [Google Scholar]
- Zan R, Liu W, Song Z. 1985. Tetraploid-hexaploid relationship in schizothoracinae. Acta Genet Sin. 12:137–142. [Google Scholar]
- Zhang JZ, Nielsen R, Yang ZH. 2005. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 22:2472–2479. [DOI] [PubMed] [Google Scholar]
- Zhang W, et al. 2014. Hypoxia adaptations in the grey wolf (Canis lupus chanco) from Qinghai-Tibet Plateau. PLoS Genet. 10:e1004466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu SQ. 1989. The loaches of the subfamily Nemacheilinae in China (Cypriniformes: Cobitidae). Nanjing (China): Jiangsu Scientific Technology Press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.