

Abstract
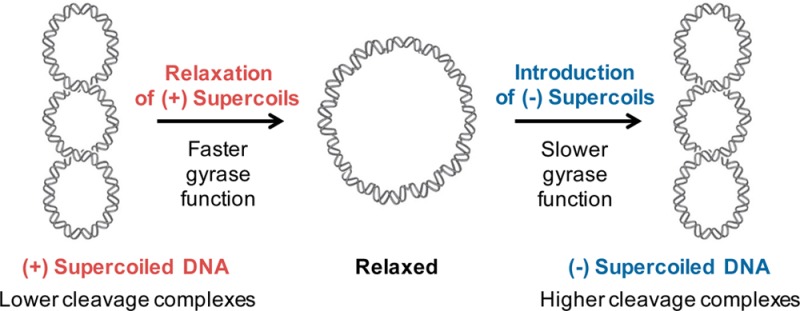
Mycobacterium tuberculosis encodes only a single type II topoisomerase, gyrase. As a result, this enzyme likely carries out the cellular functions normally performed by canonical gyrase and topoisomerase IV, both in front of and behind the replication fork. In addition, it is the sole target for quinolone antibacterials in this species. Because quinolone-induced DNA strand breaks generated on positively supercoiled DNA ahead of replication forks and transcription complexes are most likely to result in permanent genomic damage, the actions of M. tuberculosis gyrase on positively supercoiled DNA were investigated. Results indicate that the enzyme acts rapidly on overwound DNA and removes positive supercoils much faster than it introduces negative supercoils into relaxed DNA. Canonical gyrase and topoisomerase IV distinguish supercoil handedness differently during the DNA cleavage reaction: while gyrase maintains lower levels of cleavage complexes on overwound DNA, topoisomerase IV maintains similar levels of cleavage complexes on both over- and underwound substrates. M. tuberculosis gyrase maintained lower levels of cleavage complexes on positively supercoiled DNA in the absence and presence of quinolone-based drugs. By retaining this important feature of canonical gyrase, the dual function M. tuberculosis type II enzyme remains a safe enzyme to act in front of replication forks and transcription complexes. Finally, the N-terminal gate region of the enzyme appears to be necessary to distinguish supercoil handedness during DNA cleavage, suggesting that the capture of the transport segment may influence how gyrase maintains cleavage complexes on substrates with different topological states.
The vast majority of bacterial species encode two type II topoisomerases, topoisomerase IV and gyrase.1−4 These enzymes function as heterotetramers (GrlA2GrlB2 and ParC2ParE2 for topoisomerase IV in Gram-positive and Gram-negative species, respectively, and GyrA2GyrB2 for gyrase) and display sequence homology.1−4 Both enzymes regulate the topological state of DNA by creating a transient double-stranded break in one segment of DNA (the gate- or G-segment) and passing a second intact segment (the transport- or T-segment) through the break.2,5−7 To maintain genomic integrity during this process, topoisomerase IV and gyrase covalently attach to the 5′-terminus of each DNA strand.2,5−7 This “cleavage complex” is a hallmark of enzyme activity.8
Despite this shared catalytic mechanism, differences in the C-terminal domains of GrlA/ParC and GyrA confer topoisomerase IV and gyrase with a unique array of catalytic activities.3 The C-terminal domain of GrlA/ParC allows topoisomerase IV to interact with distal DNA segments, which permits it to capture existing intra- or intermolecular DNA crossovers.1,4 This property confers the enzyme with the ability to relax (i.e., remove) positive or negative DNA supercoils. It also allows the enzyme to remove DNA knots and tangles in a highly efficient manner.1,3,4
In contrast, the C-terminal domain of GyrA wraps DNA, inducing a positive supercoil between the G- and T-segments.1,4,9−14 Because these captured DNA segments are proximal to one another,1,4,15 gyrase greatly favors the catalysis of intramolecular strand passage reactions. As a result, the enzyme can efficiently alter superhelical density but is very poor at removing knots and tangles.1,4,15,16 Furthermore, because gyrase acts on the induced positive crossover, it carries out a unidirectional reaction in the presence of ATP in which it always decreases the DNA linking number.1,4,17,18 This allows the enzyme to remove positive, but not negative, supercoils and to reduce the linking number beyond that of relaxed DNA. Thus, among all known topoisomerases, gyrase is the only enzyme able to negatively supercoil DNA.3,5
As a result of the differences described above, gyrase and topoisomerase IV play distinct roles during DNA replication and transcription (Figure 1).1,3,19−25 Gyrase removes the positive supercoils that accumulate ahead of replication forks and goes on to restore the negative superhelicity of the genome. Although topoisomerase IV can also relax positive supercoils, its primary function is to remove the precatenanes/catenanes that are formed behind replication forks, which allows chromosomes to be segregated appropriately.
Figure 1.
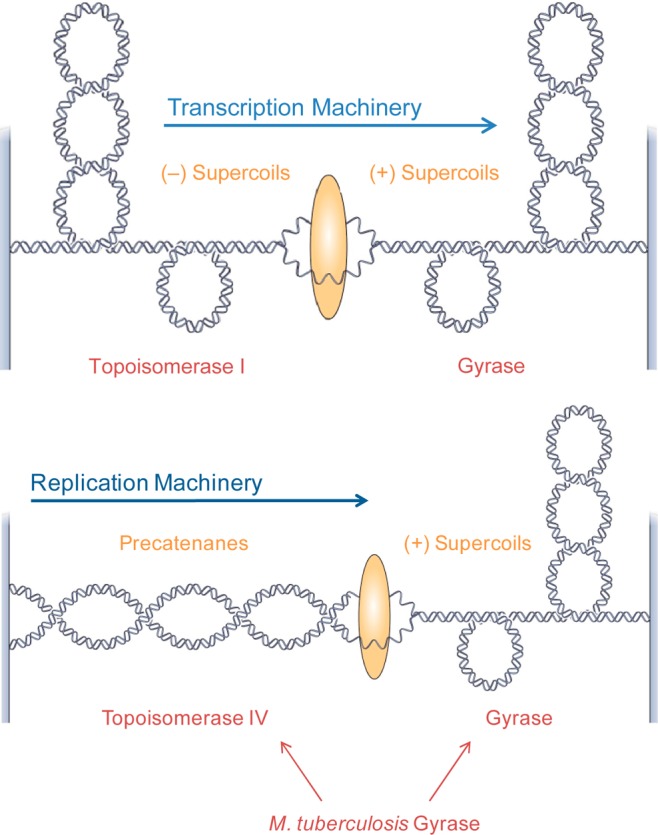
Cellular functions of bacterial topoisomerases. Gyrase removes positively supercoiled DNA ahead of transcription (top) and replication (bottom) complexes and in conjunction with topoisomerase I, also maintains the negative superhelicity of the genome.1,3,19−25 Topoisomerase IV can remove positive supercoils, but primarily acts behind the replication fork to resolve precatenanes and unlink daughter chromosomes. Topoisomerase I acts to remove negative supercoils that are generated behind transcription complexes. In species that encode gyrase as the only type II topoisomerase, such as M. tuberculosis, the enzyme likely carries out the functions of both gyrase and topoisomerase IV and acts ahead of transcription complexes and ahead of and behind replication forks.25
Beyond their essential cellular functions, both bacterial type II topoisomerases are targets for quinolone antibacterials, which kill cells by stabilizing cleavage complexes.2,7,26,27 As replication forks, transcription complexes, or other DNA tracking systems encounter these protein-bound DNA roadblocks, transient cleavage complexes are converted to nonligatable DNA breaks that induce DNA damage response pathways. When these pathways are overwhelmed, the DNA breaks result in cell death. Because quinolone-stabilized cleavage complexes formed ahead of moving forks and transcription complexes are most likely to be converted to nonligatable strand breaks, cleavage complexes stabilized on positively supercoiled DNA are the most dangerous for the cell. A previous study on type II topoisomerases from Bacillus anthracis and Escherichia coli found that gyrase maintains lower levels of cleavage complexes with positively (as compared to negatively) supercoiled DNA,28 making it a safer enzyme to function ahead of replication forks and transcription complexes.25 Conversely, topoisomerase IV, which acts behind replication forks, reportedly maintains levels of cleavage complexes on positively supercoiled DNA that are similar to28 or higher than29 those generated on negatively supercoiled molecules.
In contrast to the great majority of bacteria, a few species encode only a single type II topoisomerase II, gyrase. Among these are several disease-causing organisms, including Treponema pallidum (syphilis),30Helicobacter pylori (stomach and intestinal ulcers),31Campylobacter jejuni (gastroenteritis),32Mycobacterium leprae (leprosy),33 and Mycobacterium tuberculosis (tuberculosis).34 Worldwide, tuberculosis is one of the top 10 causes of death and ranks above HIV/AIDS as the leading cause of death due to an infectious disease.35 Because gyrase is the only type II topoisomerase in M. tuberculosis, it must carry out the activities of both gyrase and topoisomerase IV.25 Therefore, it is functionally distinct from the “canonical” gyrases found in most species (i.e., species that encode both gyrase and topoisomerase IV).36,37 Furthermore, M. tuberculosis gyrase is the sole target for quinolones in this species. Although the standard treatment regimen for tuberculosis does not typically include quinolones, the fourth-generation quinolone antibacterials moxifloxacin and gatifloxacin are critical drugs for treating patients who have multidrug-resistant tuberculosis or are intolerant of first-line therapies.35
Because M. tuberculosis gyrase, a “dual function” gyrase/topoisomerase IV enzyme,36,37 is the sole type II topoisomerase and quinolone target in the cell and because quinolone-induced cleavage complexes formed on positively supercoiled DNA ahead of replication forks and transcription complexes are the most dangerous for the cell, we investigated the actions of this enzyme on overwound DNA. Results indicate that M. tuberculosis gyrase removes positive supercoils much more rapidly than it introduces negative supercoils into relaxed DNA. Like “canonical” gyrase, and in contrast to topoisomerase IV, M. tuberculosis gyrase maintains lower levels of cleavage complexes on positively supercoiled DNA in the absence and presence of quinolone-based drugs. By retaining this important feature of canonical gyrase, the dual function M. tuberculosis type II enzyme remains a safe enzyme to act in front of replication forks and transcription complexes. Finally, the ability of gyrase to distinguish supercoil handedness during DNA cleavage is not dependent on elements in the C-terminal domain of GyrA but appears to require the N-terminal portion of GyrB.
Materials and Methods
Enzymes and Materials
Full-length M. tuberculosis gyrase subunits (GyrA and GyrB) were expressed and purified as described previously.38 A C-terminally deleted GyrA mutant (GyrAΔCTD, residues 2–500 of GyrA) was cloned into a pET28b derivative that added an N-terminal tobacco etch virus protease (TEV)-cleavable 6xHis tag. The catalytic core (a fusion of residues 426–675 of GyrB with residues 2–500 of GyrA) was cloned into a pET28b derivative that added N-terminal TEV-cleavable 6xHis and maltose binding protein tags, as described previously.39 Each construct was expressed and purified as described for the wild-type subunits,38 except that cells carrying the catalytic core construct were grown in medium containing 100 μg/mL ampicillin.
Negatively supercoiled pBR322 DNA was prepared from E. coli using a Plasmid Mega kit (Qiagen) as described by the manufacturer. Positively supercoiled pBR322 DNA was prepared by treating negatively supercoiled molecules with recombinant Archaeoglobus fulgidus reverse gyrase.40,41 The number of positive supercoils induced by this process was comparable to the number of negative supercoils in the original pBR322 preparations.40 In all experiments that compared negatively with positively supercoiled DNA, the negatively supercoiled plasmid preparations were processed identically to the positively supercoiled molecules except that reaction mixtures did not contain reverse gyrase. Relaxed pBR322 plasmid DNA was generated by treating negatively supercoiled pBR322 with calf thymus topoisomerase I (Invitrogen) and purified as described previously.42
Ciprofloxacin was obtained from LKT Laboratories, stored at 4 °C as a 40 mM stock solution in 0.1 N NaOH, and diluted 5-fold with 10 mM Tris-HCl (pH 7.9) immediately prior to use. Moxifloxacin was obtained from LKT Laboratories, and levofloxacin was obtained from Sigma-Aldrich. Both drugs were stored at 4 °C as 20 mM stock solutions in 100% DMSO. 8-Methyl-moxifloxacin (1-cyclopropyl-6-fluoro-1,4-dihydro-8-methyl-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-3-quinolinecarboxylic acid) and 3′-(AM)P-dione (3-amino-7-[(3S)-3-(aminomethyl)-1-pyrrolidinyl]-1-cyclopropyl-6-fluoro-8-methyl-2,4(1H,3H)-quinazolinedione) were generous gifts from Dr. Robert J. Kerns (University of Iowa). These compounds were synthesized as reported previously43,44 and were stored at 4 °C as 20 mM stock solutions in DMSO. CP-115,953 was the generous gift of Thomas D. Gootz and Paul R. McGuirk (Pfizer Global Research), was stored at −20 °C as a 40 mM solution in 0.1 N NaOH, and was diluted 5-fold with 10 mM Tris-HCl (pH 7.9) immediately prior to use. All other chemicals were analytical reagent grade.
DNA Supercoiling
DNA supercoiling assays were based on previously published protocols.38,43 Assays contained 50 nM gyrase (2:1 GyrA/GyrB ratio), 5 nM positively supercoiled or relaxed pBR322, and 1.5 mM ATP in 20 μL of 10 mM Tris-HCl (pH 7.5), 40 mM KCl, 6 mM MgCl2, 2 mM DTT, 0.1 mg/mL BSA, and 10% glycerol. Reactions were incubated at 37 °C for various times and stopped by the addition of 3 μL of a mixture of 0.77% SDS and 77.5 mM Na2EDTA. Samples were mixed with 2 μL of agarose loading dye [60% sucrose in 10 mM Tris-HCl (pH 7.9), 0.5% bromophenol blue, and 0.5% xylene cyanol FF] and subjected to electrophoresis in 1% agarose gels in TBE [100 mM Tris-borate (pH 8.3) and 2 mM EDTA]. Gels were stained with 1 μg/mL ethidium bromide for 30 min. DNA bands were visualized with medium-range ultraviolet light on an Alpha Innotech digital imaging system.
Alternatively, reaction products were analyzed by two-dimensional gel electrophoresis as described previously.40 The first dimension was run for 2 h as described in the preceding paragraph. The gel was then soaked in TBE containing 4.5 μg/mL chloroquine for 2 h with gentle shaking followed by electrophoresis in the orthogonal direction (90° clockwise) for 2 h in fresh TBE containing 4.5 μg/mL chloroquine. Gels were stained and DNA bands were visualized as described above.
Gyrase-DNA Binding
DNA binding by M. tuberculosis gyrase was monitored by an electrophoretic mobility shift assay.45 Reactions contained 0–1000 nM gyrase (3:2 GyrA/GyrB ratio) and 10 nM positively or negatively supercoiled pBR322 in a total volume of 20 μL of 10 mM Tris-HCl (pH 7.5), 40 mM KCl, 0.1 mg/mL BSA, and 10% glycerol. Reactions were incubated at 37 °C for 10 min. Samples were mixed with 2 μL of agarose loading dye and subjected to electrophoresis in 1% agarose gels in TAE [40 mM Tris-acetate (pH 8.3) and 2 mM Na2EDTA]. Gels were stained and DNA bands were visualized as described above.
DNA Cleavage
DNA cleavage reactions were based on the procedure of Aldred et al.43 Reactions contained 100 nM wild-type or GyrAΔCTD gyrase (3:2 GyrA/GyrB ratio) or 100 nM catalytic core and 10 nM positively supercoiled, negatively supercoiled, or relaxed pBR322 in a total volume of 20 μL of 10 mM Tris-HCl (pH 7.5), 40 mM KCl, 6 mM MgCl2, 0.1 mg/mL BSA, and 10% glycerol. Reactions were incubated at 37 °C for 10 min. Enzyme–DNA cleavage complexes were trapped by adding 2 μL of 5% SDS followed by 2 μL of 250 mM Na2EDTA and 2 μL of 0.8 mg/mL Proteinase K. Reaction mixtures were incubated at 45 °C for 30 min to digest the enzyme. Samples were mixed with 2 μL of agarose loading dye and incubated at 45 °C for 2 min before loading on gels. Reaction products were subjected to electrophoresis in 1% agarose gels in TAE containing 0.5 μg/mL ethidium bromide (except for reactions containing relaxed DNA, which were run in gels without EtBr and stained with 1 μg/mL EtBr after electrophoresis) and visualized as described above. DNA cleavage was monitored by the conversion of supercoiled plasmid to linear molecules and quantified by comparison to a control reaction in which an equal mass of DNA was digested by EcoRI (New England BioLabs).
Results and Discussion
M. tuberculosis Gyrase Relaxes Positive DNA Supercoils More Rapidly than It Introduces Negative Supercoils
The sole type II topoisomerase in M. tuberculosis is defined as a gyrase based on its ability to introduce negative supercoils into relaxed DNA.36 However, the enzyme displays a higher decatenation activity than observed in a canonical gyrase and is therefore a dual function gyrase/topoisomerase IV enzyme.36,37 The ability of this enzyme to supercoil relaxed DNA is well documented,36−38,46 but no studies have investigated its actions on positively supercoiled DNA. To determine how this dual function gyrase/topoisomerase IV type II enzyme interacts with overwound DNA, we assessed the ability of M. tuberculosis gyrase to relax a positively supercoiled plasmid and subsequently convert it to a negatively supercoiled molecule. Enzyme activity was monitored over 45 min in order to observe both the removal of positive supercoils (the relaxation reaction) and the introduction of negative supercoils (the supercoiling reaction) within the same assay.
As shown in Figure 2 (top), gyrase rapidly relaxed the overwound DNA and subsequently introduced negative supercoils much more slowly. To further investigate the removal of positive supercoils, we monitored relaxation over a shorter time course (Figure 2, middle). All of the positive supercoils appear to be removed by ∼2 min. Conversely, the conversion of relaxed plasmid to negatively supercoiled DNA took 30–45 min. A similar time course for DNA supercoiling was observed whether the initial substrate was positively supercoiled (Figure 2, top) or relaxed (Figure 2, bottom), highlighting the distinction between rates of relaxation and supercoiling. This pattern of activity is similar to that observed for B. anthracis and E. coli gyrase.28 Therefore, like these canonical gyrases, the dual function type II enzyme from M. tuberculosis functions differently on positively supercoiled and relaxed DNA.
Figure 2.

Gyrase removes positive supercoils more rapidly than it introduces negative supercoils into relaxed DNA. Top: Gyrase activity on positively supercoiled DNA. A time course is shown for the relaxation of positive supercoils followed by the introduction of negative supercoils. Positively supercoiled [(+)SC] and negatively supercoiled [(−)SC] standards are shown. Middle: Expanded time course for the relaxation of (+)SC DNA by gyrase. Bottom: Time course for the introduction of negative supercoils into relaxed DNA (Rel) by gyrase. Gel images are representative of at least three independent experiments.
To determine the handedness of the topoisomers in the later stages of the relaxation reaction (Figure 2, middle), reaction products were analyzed by two-dimensional gel electrophoresis. As shown in Figure 3, all positive supercoils were removed by 2 min, and negatively supercoiled DNA started to appear between 2 and 3 min. This result confirms the conclusion that the relaxation of positive supercoils occurs at least 20-fold faster than the introduction of negative supercoils. However, under the conditions employed, both of these processes appear to take place in a distributive manner with the bulk of the DNA moving synchronously through all of the intermediate topoisomers between the positively supercoiled substrate and the fully negatively supercoiled product.
Figure 3.

Two-dimensional gel analysis of gyrase activity on positively supercoiled DNA. The mobility of nicked, positively supercoiled [(+)SC], relaxed (Rel), and negatively supercoiled [(−)SC] DNA are indicated in a control gel (top left). DNA products generated after 1 min (top right), 2 min (bottom left), and 3 min (bottom right) reactions are shown. Gel images are representative of at least three independent experiments.
Gyrase Has a Higher Affinity for Positively Supercoiled than Negatively Supercoiled DNA
A previous study found that E. coli gyrase displays an ∼10-fold higher affinity for relaxed over negatively supercoiled DNA.47 However, binding interactions with positively supercoiled molecules have yet to be assessed for gyrase. Because M. tuberculosis gyrase carries out distributive reactions for both the removal of positive supercoils and the introduction of negative supercoils, we determined whether the differences in reaction rates reflect differences in enzyme affinity for positively and negatively supercoiled DNA. An electrophoretic mobility shift assay was employed to assess the relative affinities of gyrase for positively or negatively supercoiled molecules. As shown in Figure 4, gyrase induced much greater shifts with positively supercoiled DNA than negatively supercoiled molecules and did so at lower enzyme concentrations. The nicked DNA in these gels served as an internal control for molecules that were not under torsional stress. In the presence of positively supercoiled DNA, gyrase did not substantially shift the nicked/relaxed molecules, suggesting that the enzyme has a significantly higher affinity for the positively supercoiled plasmid. In contrast, in incubations with negatively supercoiled DNA, gyrase preferentially shifted the nicked plasmid and altered its mobility to a much greater extent than it did with the negatively supercoiled molecules. Thus, gyrase appears to have a higher affinity for positively supercoiled DNA than negatively supercoiled DNA and has an intermediate affinity for DNA molecules that lack torsional stress. This pattern is consistent with positively supercoiled DNA being the preferred substrate for gyrase-catalyzed DNA strand passage, with relaxed DNA as an intermediary product and negatively supercoiled DNA as the final reaction product.
Figure 4.

Gyrase preferentially binds positively supercoiled DNA. The electrophoretic mobility shifts of positively supercoiled [(+)SC), top] and negatively supercoiled [(−)SC, bottom] DNA induced by increasing concentrations of gyrase are shown. Original positions of nicked, (+)SC, and (−)SC DNA are indicated. Gel images are representative of at least three independent experiments.
Gyrase Maintains Lower Levels of Cleavage Complexes with Positively Supercoiled DNA
The cleavage complexes that are most dangerous for the cell are those that are formed on overwound DNA ahead of moving tracking systems. Previous studies indicate that gyrase and topoisomerase IV differ in their ability to recognize DNA geometry while they are cleaving the double helix.28,29 This specificity may be related to the position of each enzyme relative to replication forks and transcription complexes.1,3,19−25 Canonical gyrase, which functions ahead of these tracking systems, often works on overwound DNA and has the potential to create dangerous cleavage complexes. Consistent with this function, gyrase maintains lower levels of cleavage complexes on positively supercoiled DNA.28 In contrast, topoisomerase IV works primarily behind replication forks and is less likely to encounter moving DNA tracking systems. Thus, it does not distinguish between overwound and underwound DNA during cleavage (or actually maintains higher levels of cleavage complexes with positively supercoiled substrates).28,29
As a dual function enzyme, M. tuberculosis gyrase must be able to work both ahead of and behind the replication fork and other DNA tracking systems and could conceivably distinguish supercoil handedness during DNA cleavage as a gyrase or as a topoisomerase IV. To address this issue, the ability of M. tuberculosis gyrase to cleave positively and negatively supercoiled DNA was determined. Like canonical gyrase,28M. tuberculosis gyrase maintained ∼3-fold lower levels of cleavage complexes on overwound compared to underwound DNA in the absence of drugs (Figure 5). This discrimination is despite the fact that the enzyme displays a higher affinity for positively supercoiled molecules and suggests that M. tuberculosis gyrase cycles through the DNA cleavage reaction more rapidly on positively supercoiled substrates.
Figure 5.
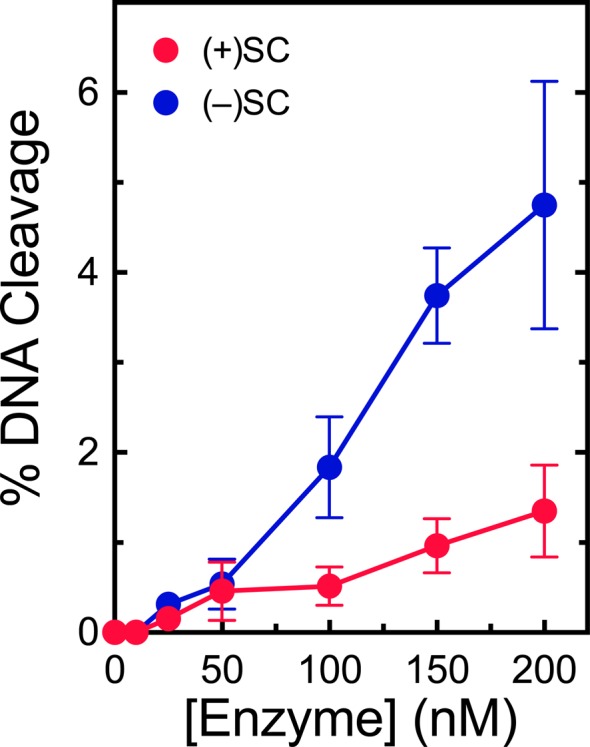
Gyrase maintains lower levels of cleavage complexes on positively supercoiled DNA in the absence of drugs. Levels of cleavage complexes generated by varying concentrations of gyrase on positively supercoiled [(+)SC DNA, red] or negatively supercoiled [(−)SC DNA, blue] are shown. Error bars represent the standard deviations for at least three independent experiments.
Because quinolones stabilize cleavage complexes and are clinically important drugs for the treatment of tuberculosis, we also monitored cleavage complexes generated by gyrase in the presence of moxifloxacin (Figure 6). Similar to the results seen in the absence of drugs, gyrase maintained ∼3-fold lower levels of cleavage complexes on positively supercoiled molecules. Levels of cleavage complexes generated by the enzyme on relaxed DNA were between those generated on positively and negatively supercoiled plasmid. Gyrase also maintained the distinction between over and underwound DNA in the presence of ciprofloxacin or levofloxacin, two other clinically relevant quinolones; CP-115,953, a quinolone that displays high activity against both bacterial and eukaryotic type II topoisomerases;48−51 and two quinolone-derived drugs, 8-methyl-moxifloxacin and 3′-(AM)P-dione, which overcome quinolone resistance caused by mutations in M. tuberculosis gyrase38,39 (Figure 7). These results suggest that, although gyrase in M. tuberculosis has adapted to perform some of the functions of topoisomerase IV, it still retains the characteristics that make gyrase a safe enzyme to function ahead of replication forks and other DNA tracking systems such as RNA polymerase.
Figure 6.
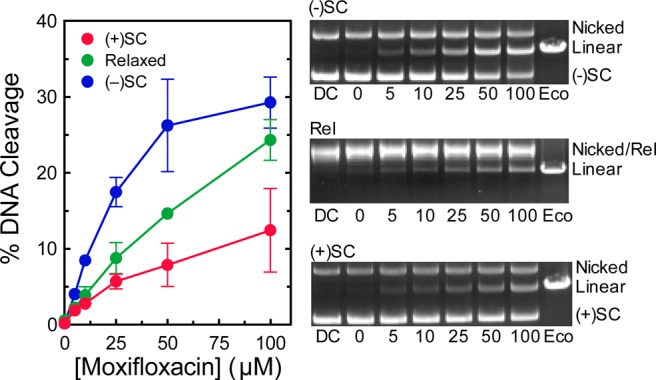
Gyrase maintains lower levels of cleavage complexes on positively supercoiled DNA in the presence of moxifloxacin. Left: Levels of cleavage complexes generated by gyrase on positively supercoiled [(+)SC DNA, red], relaxed (Rel, green), or negatively supercoiled [(−)SC DNA, blue] in the presence of the quinolone moxifloxacin. Error bars represent the standard deviations for at least three independent experiments. Right: Representative gels of DNA cleavage induced by gyrase on negatively supercoiled [(−)SC, top], relaxed (Rel, middle), or positively supercoiled [(+)SC, bottom] DNA in the presence of moxifloxacin. DNA controls in the absence of enzyme (DC) and DNA digested by EcoRI (EcoRI) are shown. The positions of nicked, relaxed, linear, and supercoiled DNA are indicated.
Figure 7.
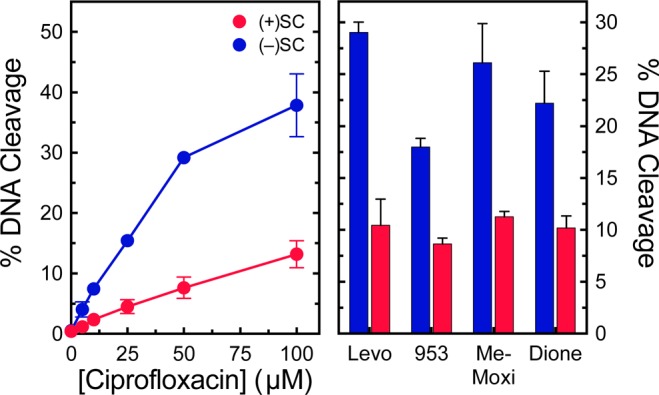
Gyrase maintains lower levels of cleavage complexes on positively supercoiled DNA in the presence of ciprofloxacin, levofloxacin, and other quinolone-derived compounds. Left: Levels of cleavage complexes generated by gyrase on positively supercoiled [(+)SC DNA, red] or negatively supercoiled [(−)SC DNA, blue] in the presence of ciprofloxacin. Right: Levels of cleavage complexes generated by gyrase on positively supercoiled [(+)SC DNA, red] or negatively supercoiled [(−)SC DNA, blue] in the presence of levofloxacin (Levo, 100 μM), CP-115,953 (953, 100 μM), 8-methyl-moxifloxacin (Me-Moxi, 20 μM), and 3′-(AM)P-dione (dione, 20 μM). Error bars represent the standard deviations for at least three independent experiments.
Recognition of Supercoil Geometry During Cleavage Is Not Dependent on the C-Terminal Domain of GyrA but Does Require the N-Terminal Portion of GyrB
A previous study with E. coli gyrase demonstrated that removal of the entire C-terminal domain (CTD) of GyrA converted gyrase to a “conventional” type II topoisomerase.12 The enzyme was no longer able to introduce negative supercoils into relaxed DNA but, like topoisomerase IV, gained the ability to relax negatively supercoiled DNA in the presence of ATP and exhibited a higher decatenation activity than the parent enzyme. However, the effects of this alteration on DNA cleavage have not been examined. For human topoisomerase IIα, removal of the CTD does not change the ability of the enzyme to discern supercoil handedness during cleavage.52 To determine the role of the CTD in DNA geometry recognition by a bacterial enzyme during cleavage, we monitored the ability of M. tuberculosis gyrase that contained wild-type GyrB and a mutant GyrA in which the CTD was deleted (GyrAΔCTD gyrase, which contains residues 2–500 of GyrA) to cleave over- and underwound DNA substrates (Figure 8). GyrAΔCTD gyrase still maintained lower levels of cleavage complexes with positively supercoiled DNA. Therefore, like the human enzyme, the ability of M. tuberculosis gyrase to distinguish supercoil handedness during the cleavage reaction does not rely on the CTD.
Figure 8.
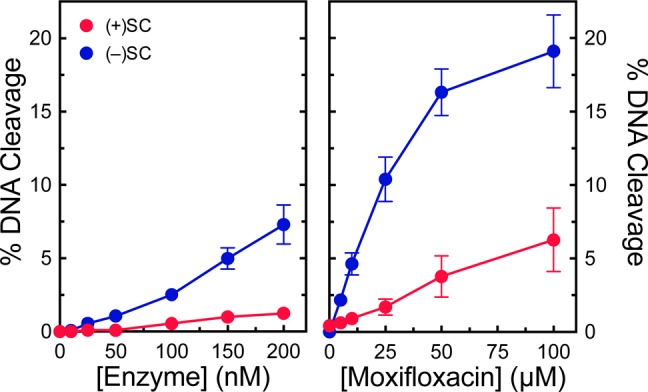
GyrAΔCTD gyrase maintains lower levels of cleavage complexes on positively supercoiled DNA. Left: Levels of cleavage complexes generated by varying concentrations of GyrAΔCTD gyrase on positively supercoiled [(+)SC, red] or negatively supercoiled [(−)SC, blue] DNA in the absence of quinolones. Right: Levels of cleavage complexes generated by GyrAΔCTD gyrase on positively supercoiled [(+)SC, red] or negatively supercoiled [(−)SC, blue] DNA in the presence of moxifloxacin. Error bars represent the standard deviations for at least three independent experiments.
It is not obvious which portion of M. tuberculosis gyrase is responsible for the recognition of DNA geometry during cleavage. Although DNA scission occurs within the catalytic core of the enzyme (defined as residues 426–675 of GyrB and residues 2–500 of GyrA), the N-terminal gate region (residues 2–425 of GyrB) is crucial for capturing the T-segment.53−55 The N-terminal gate in eukaryotic type II topoisomerases is also important for capturing the T-segment56 and changes within this region have been shown to modulate rates of DNA cleavage.57,58 Therefore, this region of M. tuberculosis gyrase could have an important role in distinguishing supercoil handedness. Previous work with the catalytic core of human topoisomerase IIα showed that the N-terminal gate was not required for the recognition of supercoil geometry during cleavage.56 To determine if this was also the case for the bacterial type II topoisomerase, we evaluated the ability of the catalytic core of M. tuberculosis to cleave positively and negatively supercoiled DNA (Figure 9). Unlike full-length or GyrAΔCTD gyrase, the catalytic core did not distinguish between overwound and underwound DNA and maintained similar levels of cleavage complexes on both substrates in the absence or presence of moxifloxacin. Therefore, the ability of M. tuberculosis gyrase to recognize DNA geometry during cleavage appears to be embedded, at least in part, in the N-terminal region of the enzyme.
Figure 9.
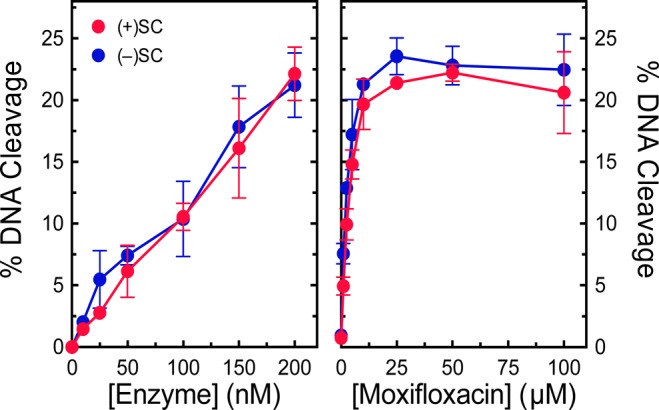
Catalytic core of gyrase maintains similar levels of cleavage complexes on positively and negatively supercoiled DNA. Left: Levels of cleavage complexes generated by varying concentrations of the catalytic core on positively supercoiled [(+)SC, red] or negatively supercoiled [(−)SC, blue] DNA in the absence of quinolones. Right: Levels of cleavage complexes generated by the catalytic core on positively supercoiled [(+)SC, red] or negatively supercoiled [(−)SC, blue] DNA in the presence of moxifloxacin. Error bars represent the standard deviations for at least three independent experiments.
Conclusions
Gyrase from M. tuberculosis and other species encoding a single type II topoisomerase must carry out the functions usually associated with topoisomerase IV. Thus, gyrase from these species has a mix of characteristics from canonical gyrases (ability to induce negative supercoils into DNA) and topoisomerase IV (ability to decatenate DNA).36,37 Our results indicate that the dual function M. tuberculosis gyrase retains features of canonical gyrase that make it a safer enzyme to function ahead of moving DNA tracking systems. In particular, it is able to rapidly remove positive supercoils, while its introduction of negative supercoils occurs more slowly. Furthermore, it is able to distinguish between positively and negatively supercoiled DNA during the cleavage reaction and maintains significantly lower levels of cleavage complexes on overwound DNA. The N-terminal gate region of the enzyme is required to distinguish supercoil handedness, suggesting that the capture of the transport segment may influence how gyrase maintains cleavage complexes on substrates with different topological states. The involvement of the N-terminal region in geometry recognition also points to a mechanistic difference between M. tuberculosis gyrase and human topoisomerase IIα,56 implying that bacterial and eukaryotic topoisomerases may utilize distinct methods for sensing the supercoil geometry of their substrates.
Acknowledgments
The authors would like to thank Ethan Tyler of NIH Medical Arts for design work on Figure 1. We are grateful to Elizabeth G. Gibson for critical reading of the manuscript.
Author Present Address
# Department of Biosciences and Department of Chemistry, Durham University, Durham DH1 3LE, United Kingdom.
This work was supported by the US Veterans Administration (Merit Review Award I01 Bx002198 to N.O.) and the National Institutes of Health (R01 GM033944 to N.O. and R01 CA077373 to J.M.B.). R.E.A. was supported by a predoctoral fellowship from the National Science Foundation (DGE-0909667) and T.R.B. was supported by a European Molecular Biology Organization Long-Term Fellowship.
The authors declare no competing financial interest.
References
- Levine C.; Hiasa H.; Marians K. J. (1998) DNA gyrase and topoisomerase IV: biochemical activities, physiological roles during chromosome replication, and drug sensitivities. Biochim. Biophys. Acta, Gene Struct. Expression 1400, 29–43. 10.1016/S0167-4781(98)00126-2. [DOI] [PubMed] [Google Scholar]
- Anderson V. E.; Osheroff N. (2001) Type II topoisomerases as targets for quinolone antibacterials: turning Dr. Jekyll into Mr. Hyde. Curr. Pharm. Des. 7, 337–353. 10.2174/1381612013398013. [DOI] [PubMed] [Google Scholar]
- Sissi C.; Palumbo M. (2010) In front of and behind the replication fork: bacterial type IIA topoisomerases. Cell. Mol. Life Sci. 67, 2001–2024. 10.1007/s00018-010-0299-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush N. G., Evans-Roberts K., and Maxwell A.. DNA topoisomerases. EcoSal Plus, 2015, 6 10.1128/ecosalplus.ESP-0010-2014. [DOI] [PubMed] [Google Scholar]
- Champoux J. J. (2001) DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 70, 369–413. 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- Vos S. M.; Tretter E. M.; Schmidt B. H.; Berger J. M. (2011) All tangled up: how cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 12, 827–841. 10.1038/nrm3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; Kerns R. J.; Osheroff N. (2014) Mechanism of quinolone action and resistance. Biochemistry 53, 1565–1574. 10.1021/bi5000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deweese J. E.; Osheroff N. (2009) The DNA cleavage reaction of topoisomerase II: wolf in sheep’s clothing. Nucleic Acids Res. 37, 738–749. 10.1093/nar/gkn937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L. F.; Wang J. C. (1978) Micrococcus luteus DNA gyrase: active components and a model for its supercoiling of DNA. Proc. Natl. Acad. Sci. U. S. A. 75, 2098–2102. 10.1073/pnas.75.5.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L. F.; Wang J. C. (1978) DNA-DNA gyrase complex: the wrapping of the DNA duplex outside the enzyme. Cell 15, 979–984. 10.1016/0092-8674(78)90281-7. [DOI] [PubMed] [Google Scholar]
- Reece R. J.; Maxwell A. (1991) The C-terminal domain of the Escherichia coli DNA gyrase A subunit is a DNA-binding protein. Nucleic Acids Res. 19, 1399–1405. 10.1093/nar/19.7.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampranis S. C.; Maxwell A. (1996) Conversion of DNA gyrase into a conventional type II topoisomerase. Proc. Natl. Acad. Sci. U. S. A. 93, 14416–14421. 10.1073/pnas.93.25.14416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramlinger V. M.; Hiasa H. (2006) The ″GyrA-box″ is required for the ability of DNA gyrase to wrap DNA and catalyze the supercoiling reaction. J. Biol. Chem. 281, 3738–3742. 10.1074/jbc.M511160200. [DOI] [PubMed] [Google Scholar]
- Basu A.; Parente A. C.; Bryant Z. (2016) Structural dynamics and mechanochemical coupling in DNA gyrase. J. Mol. Biol. 428, 1833–1845. 10.1016/j.jmb.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullsperger C.; Cozzarelli N. R. (1996) Contrasting enzymatic acitivites of topoisomerase IV and DNA gyrase from Escherichia coli. J. Biol. Chem. 271, 31549–31555. 10.1074/jbc.271.49.31549. [DOI] [PubMed] [Google Scholar]
- Marians K. J. (1987) DNA gyrase-catalyzed decatenation of multiply linked DNA dimers. J. Biol. Chem. 262, 10362–10368. [PubMed] [Google Scholar]
- Brown P. O.; Cozzarelli N. R. (1979) A sign inversion mechanism for enzymatic supercoiling of DNA. Science 206, 1081–1083. 10.1126/science.227059. [DOI] [PubMed] [Google Scholar]
- Bates A. D., and Maxwell A. (2005) DNA Topology, Oxford University Press, New York, USA. [Google Scholar]
- Khodursky A. B.; Peter B. J.; Schmid M. B.; DeRisi J.; Botstein D.; Brown P. O.; Cozzarelli N. R. (2000) Analysis of topoisomerase function in bacterial replication fork movement: use of DNA microarrays. Proc. Natl. Acad. Sci. U. S. A. 97, 9419–9424. 10.1073/pnas.97.17.9419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadesse S.; Graumann P. L. (2006) Differential and dynamic localization of topoisomerases in Bacillus subtilis. J. Bacteriol. 188, 3002–3011. 10.1128/JB.188.8.3002-3011.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu Y. H.; Chung M. W.; Li T. K. (2006) Distribution of gyrase and topoisomerase IV on bacterial nucleoid: implications for nucleoid organization. Nucleic Acids Res. 34, 3128–3138. 10.1093/nar/gkl392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmott C. J.; Critchlow S. E.; Eperon I. C.; Maxwell A. (1994) The complex of DNA gyrase and quinolone drugs with DNA forms a barrier to transcription by RNA polymerase. J. Mol. Biol. 242, 351–363. 10.1006/jmbi.1994.1586. [DOI] [PubMed] [Google Scholar]
- Rovinskiy N.; Agbleke A. A.; Chesnokova O.; Pang Z.; Higgins N. P. (2012) Rates of gyrase supercoiling and transcription elongation control supercoil density in a bacterial chromosome. PLoS Genet. 8, e1002845. 10.1371/journal.pgen.1002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawadzki P.; Stracy M.; Ginda K.; Zawadzka K.; Lesterlin C.; Kapanidis A. N.; Sherratt D. J. (2015) The localization and action of topoisomerase IV in Escherichia coli chromosome segregation is coordinated by the SMC complex, MukBEF. Cell Rep. 13, 2587–2596. 10.1016/j.celrep.2015.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed W.; Sala C.; Hegde S. R.; Jha R. K.; Cole S. T.; Nagaraja V. (2017) Transcription facilitated genome-wide recruitment of topoisomerase I and DNA gyrase. PLoS Genet. 13, e1006754. 10.1371/journal.pgen.1006754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drlica K.; Hiasa H.; Kerns R.; Malik M.; Mustaev A.; Zhao X. (2009) Quinolones: action and resistance updated. Curr. Top. Med. Chem. 9, 981–998. 10.2174/156802609789630947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper D. C.; Jacoby G. A. (2015) Mechanisms of drug resistance: quinolone resistance. Ann. N. Y. Acad. Sci. 1354, 12–31. 10.1111/nyas.12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley R. E.; Dittmore A.; McPherson S. A.; Turnbough C. L.; Neuman K. C.; Osheroff N. (2017) Activities of gyrase and topoisomerase IV on positively supercoiled DNA. Nucleic Acids Res. 45, 9611–9624. 10.1093/nar/gkx649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisona N. J.; Strick T. R.; Bensimon D.; Croquette V.; Cozzarelli N. R. (2000) Preferential relaxation of positively supercoiled DNA by E. coli topoisomerase IV in single-molecule and ensemble measurements. Genes Dev. 14, 2881–2892. 10.1101/gad.838900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser C. M.; Norris S. J.; Weinstock G. M.; White O.; Sutton G. G.; Dodson R.; Gwinn M.; Hickey E. K.; Clayton R.; Ketchum K. A.; Sodergren E.; Hardham J. M.; McLeod M. P.; Salzberg S.; Peterson J.; Khalak H.; Richardson D.; Howell J. K.; Chidambaram M.; Utterback T.; McDonald L.; Artiach P.; Bowman C.; Cotton M. D.; Fujii C.; Garland S.; Hatch B.; Horst K.; Roberts K.; Sandusky M.; Weidman J.; Smith H. O.; Venter J. C. (1998) Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science 281, 375–388. [DOI] [PubMed] [Google Scholar]
- Tomb J. F.; White O.; Kerlavage A. R.; Clayton R. A.; Sutton G. G.; Fleischmann R. D.; Ketchum K. A.; Klenk H. P.; Gill S.; Dougherty B. A.; Nelson K.; Quackenbush J.; Zhou L.; Kirkness E. F.; Peterson S.; Loftus B.; Richardson D.; Dodson R.; Khalak H. G.; Glodek A.; McKenney K.; Fitzegerald L. M.; Lee N.; Adams M. D.; Hickey E. K.; Berg D. E.; Gocayne J. D.; Utterback T. R.; Peterson J. D.; Kelley J. M.; Cotton M. D.; Weidman J. M.; Fujii C.; Bowman C.; Watthey L.; Wallin E.; Hayes W. S.; Borodovsky M.; Karp P. D.; Smith H. O.; Fraser C. M.; Venter J. C. (1997) The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388, 539–547. 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- Parkhill J.; Wren B. W.; Mungall K.; Ketley J. M.; Churcher C.; Basham D.; Chillingworth T.; Davies R. M.; Feltwell T.; Holroyd S.; Jagels K.; Karlyshev A. V.; Moule S.; Pallen M. J.; Penn C. W.; Quail M. A.; Rajandream M. A.; Rutherford K. M.; van Vliet A. H.; Whitehead S.; Barrell B. G. (2000) The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403, 665–668. 10.1038/35001088. [DOI] [PubMed] [Google Scholar]
- Cole S. T.; Eiglmeier K.; Parkhill J.; James K. D.; Thomson N. R.; Wheeler P. R.; Honore N.; Garnier T.; Churcher C.; Harris D.; Mungall K.; Basham D.; Brown D.; Chillingworth T.; Connor R.; Davies R. M.; Devlin K.; Duthoy S.; Feltwell T.; Fraser A.; Hamlin N.; Holroyd S.; Hornsby T.; Jagels K.; Lacroix C.; Maclean J.; Moule S.; Murphy L.; Oliver K.; Quail M. A.; Rajandream M. A.; Rutherford K. M.; Rutter S.; Seeger K.; Simon S.; Simmonds M.; Skelton J.; Squares R.; Squares S.; Stevens K.; Taylor K.; Whitehead S.; Woodward J. R.; Barrell B. G. (2001) Massive gene decay in the leprosy bacillus. Nature 409, 1007–1011. 10.1038/35059006. [DOI] [PubMed] [Google Scholar]
- Cole S. T.; Brosch R.; Parkhill J.; Garnier T.; Churcher C.; Harris D.; Gordon S. V.; Eiglmeier K.; Gas S.; Barry C. E. 3rd; Tekaia F.; Badcock K.; Basham D.; Brown D.; Chillingworth T.; Connor R.; Davies R.; Devlin K.; Feltwell T.; Gentles S.; Hamlin N.; Holroyd S.; Hornsby T.; Jagels K.; Krogh A.; McLean J.; Moule S.; Murphy L.; Oliver K.; Osborne J.; Quail M. A.; Rajandream M. A.; Rogers J.; Rutter S.; Seeger K.; Skelton J.; Squares R.; Squares S.; Sulston J. E.; Taylor K.; Whitehead S.; Barrell B. G. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- WHO. (2016) Global tuberculosis report.
- Aubry A.; Fisher L. M.; Jarlier V.; Cambau E. (2006) First functional characterization of a singly expressed bacterial type II topoisomerase: the enzyme from Mycobacterium tuberculosis. Biochem. Biophys. Res. Commun. 348, 158–165. 10.1016/j.bbrc.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Tretter E. M.; Berger J. M. (2012) Mechanisms for defining supercoiling set point of DNA gyrase orthologs: II. The shape of the GyrA subunit C-terminal domain (CTD) is not a sole determinant for controlling supercoiling efficiency. J. Biol. Chem. 287, 18645–18654. 10.1074/jbc.M112.345736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; Blower T. R.; Kerns R. J.; Berger J. M.; Osheroff N. (2016) Fluoroquinolone interactions with Mycobacterium tuberculosis gyrase: enhancing drug activity against wild-type and resistant gyrase. Proc. Natl. Acad. Sci. U. S. A. 113, E839–E846. 10.1073/pnas.1525055113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower T. R.; Williamson B. H.; Kerns R. J.; Berger J. M. (2016) Crystal structure and stability of gyrase-fluoroquinolone cleaved complexes from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 113, 1706–1713. 10.1073/pnas.1525047113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClendon A. K.; Rodriguez A. C.; Osheroff N. (2005) Human topoisomerase IIα rapidly relaxes positively supercoiled DNA: implications for enzyme action ahead of replication forks. J. Biol. Chem. 280, 39337–39345. 10.1074/jbc.M503320200. [DOI] [PubMed] [Google Scholar]
- Rodriguez A. C. (2002) Studies of a positive supercoiling machine: nucleotide hydrolysis and a multifunctional ″latch″ in the mechanism of reverse gyrase. J. Biol. Chem. 277, 29865–29873. 10.1074/jbc.M202853200. [DOI] [PubMed] [Google Scholar]
- Aldred K. J.; Breland E. J.; Vlčková V.; Strub M. P.; Neuman K. C.; Kerns R. J.; Osheroff N. (2014) Role of the water-metal ion bridge in mediating interactions between quinolones and Escherichia coli topoisomerase IV. Biochemistry 53, 5558–5567. 10.1021/bi500682e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; McPherson S. A.; Wang P.; Kerns R. J.; Graves D. E.; Turnbough C. L.; Osheroff N. (2012) Drug interactions with Bacillus anthracis topoisomerase IV: biochemical basis for quinolone action and resistance. Biochemistry 51, 370–381. 10.1021/bi2013905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred K. J.; Schwanz H. A.; Li G.; McPherson S. A.; Turnbough C. L.; Kerns R. J.; Osheroff N. (2013) Overcoming target-mediated quinolone resistance in topoisomerase IV by introducing metal-ion-independent drug–enzyme interactions. ACS Chem. Biol. 8, 2660–2668. 10.1021/cb400592n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osheroff N. (1986) Eukaryotic topoisomerase II. Characterization of enzyme turnover. J. Biol. Chem. 261, 9944–9950. [PubMed] [Google Scholar]
- Bouige A.; Darmon A.; Piton J.; Roue M.; Petrella S.; Capton E.; Forterre P.; Aubry A.; Mayer C. (2013) Mycobacterium tuberculosis DNA gyrase possesses two functional GyrA-boxes. Biochem. J. 455, 285–294. 10.1042/BJ20130430. [DOI] [PubMed] [Google Scholar]
- Higgins N. P.; Cozzarelli N. R. (1982) The binding of gyrase to DNA: analysis by retention by nitrocellulose filters. Nucleic Acids Res. 10, 6833–6847. 10.1093/nar/10.21.6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. J.; Martin B. A.; Gootz T. D.; McGuirk P. R.; Moynihan M.; Sutcliffe J. A.; Osheroff N. (1991) Effects of quinolone derivatives on eukaryotic topoisomerase II: A novel mechanism for enhancement of enzyme-mediated DNA cleavage. J. Biol. Chem. 266, 14585–14592. [PubMed] [Google Scholar]
- Elsea S. H.; Osheroff N.; Nitiss J. L. (1992) Cytotoxicity of quinolones toward eukaryotic cells. Identification of topoisomerase II as the primary cellular target for the quinolone CP-115,953 in yeast. J. Biol. Chem. 267, 13150–13153. [PubMed] [Google Scholar]
- Anderson V. E.; Zaniewski R. P.; Kaczmarek F. S.; Gootz T. D.; Osheroff N. (1999) Quinolones inhibit DNA religation mediated by Staphylococcus aureus topoisomerase IV: changes in drug mechanism across evolutionary boundaries. J. Biol. Chem. 274, 35927–35932. 10.1074/jbc.274.50.35927. [DOI] [PubMed] [Google Scholar]
- Aldred K. J.; Schwanz H. A.; Li G.; Williamson B. H.; McPherson S. A.; Turnbough C. L. Jr.; Kerns R. J.; Osheroff N. (2015) Activity of quinolone CP-115,955 against bacterial and human type II topoisomerases is mediated by different interactions. Biochemistry 54, 1278–1286. 10.1021/bi501073v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClendon A. K.; Gentry A. C.; Dickey J. S.; Brinch M.; Bendsen S.; Andersen A. H.; Osheroff N. (2008) Bimodal recognition of DNA geometry by human topoisomerase IIα: preferential relaxation of positively supercoiled DNA requires elements in the C-terminal domain. Biochemistry 47, 13169–13178. 10.1021/bi800453h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt B. H.; Osheroff N.; Berger J. M. (2012) Structure of a topoisomerase II-DNA-nucleotide complex reveals a new control mechanism for ATPase activity. Nat. Struct. Mol. Biol. 19, 1147–1154. 10.1038/nsmb.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams N. L.; Howells A. J.; Maxwell A. (2001) Locking the ATP-operated clamp of DNA gyrase: probing the mechanism of strand passage. J. Mol. Biol. 306, 969–984. 10.1006/jmbi.2001.4468. [DOI] [PubMed] [Google Scholar]
- Kumar R.; Riley J. E.; Parry D.; Bates A. D.; Nagaraja V. (2012) Binding of two DNA molecules by type II topoisomerases for decatenation. Nucleic Acids Res. 40, 10904–10915. 10.1093/nar/gks843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey R. H. Jr.; Pendleton M.; Ashley R. E.; Mercer S. L.; Deweese J. E.; Osheroff N. (2014) Catalytic core of human topoisomerase IIα: insights into enzyme-DNA interactions and drug mechanism. Biochemistry 53, 6595–65602. 10.1021/bi5010816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird C. L.; Harkins T. T.; Morris S. K.; Lindsley J. E. (1999) Topoisomerase II drives DNA transport by hydrolyzing one ATP. Proc. Natl. Acad. Sci. U. S. A. 96, 13685–13690. 10.1073/pnas.96.24.13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Jung S. R.; Heo K.; Byl J. A.; Deweese J. E.; Osheroff N.; Hohng S. (2012) DNA cleavage and opening reactions of human topoisomerase IIα are regulated via Mg2+-mediated dynamic bending of gate-DNA. Proc. Natl. Acad. Sci. U. S. A. 109, 2925–2930. 10.1073/pnas.1115704109. [DOI] [PMC free article] [PubMed] [Google Scholar]