

Atomic force microscopy is used to establish that low-affinity integrins bind in cis to P2Y2R. Integrin activation is initiated by a membrane-normal switchblade motion triggered by integrin priming after the virus binds to the integrin PSI domain. Tensile force between the P2Y2R and unbending integrin stimulates outside-in signaling.
Abstract
Pathogenic hantaviruses bind to the plexin-semaphorin-integrin (PSI) domain of inactive, β3 integrins. Previous studies have implicated a cognate cis interaction between the bent conformation β5/β3 integrins and an arginine-glycine-aspartic acid (RGD) sequence in the first extracellular loop of P2Y2R. With single-molecule atomic force microscopy, we show a specific interaction between an atomic force microscopy tip decorated with recombinant αIIbβ3 integrins and (RGD)P2Y2R expressed on cell membranes. Mutation of the RGD sequence to RGE in the P2Y2R removes this interaction. Binding of inactivated and fluorescently labeled Sin Nombre virus (SNV) to the integrin PSI domain stimulates higher affinity for (RGD)P2Y2R on cells, as measured by an increase in the unbinding force. In CHO cells, stably expressing αIIbβ3 integrins, virus engagement at the integrin PSI domain, recapitulates physiologic activation of the integrin as indicated by staining with the activation-specific mAB PAC1. The data also show that blocking of the Gα13 protein from binding to the cytoplasmic domain of the β3 integrin prevents outside-in signaling and infection. We propose that the cis interaction with P2Y2R provides allosteric resistance to the membrane-normal motion associated with the switchblade model of integrin activation, where the development of tensile force yields physiological integrin activation.
INTRODUCTION
Pathogenic hantaviruses cause hemorrhagic fever with renal syndrome (HFRS) and hantavirus cardiopulmonary syndrome (HCPS). Sin Nombre virus (SNV) is a Category A pathogen that causes the most severe form of HCPS with case fatality ratios of 30–50% (Vaheri et al., 2013; Hjelle, 2014). Inhalation of excreta from hantavirus rodent hosts is the primary route of infection. From the lung parenchyma, they are taken up in phagocytes and transported to draining lymph nodes, where they establish infection and then disseminate to various organs, where they replicate principally in vascular endothelial cells (Hjelle, 2014). Tissue tropism involves the vascular endothelium of the heart, kidney, lung, and lymphoid organs. HCPS causes dysregulation of the endothelial barrier function edema and focal hyaline membranes (Zaki et al., 1995; Koster and Mackow, 2012; Mackow et al., 2014). Death results from low-output cardiogenic shock and multi-organ failure (Jonsson et al., 2008, 2010; Vaheri et al., 2013). Currently there is no approved, effective therapy. Treatment of severe cases is supportive and typically includes the use of extracorporeal membrane oxygenation (ECMO) (Wernly et al., 2011).
Previous studies have identified β3 and β1 integrins as the primary receptors for pathogenic and nonpathogenic hantaviruses, respectively (Gavrilovskaya et al., 1998; Mackow and Gavrilovskaya, 2009). Pathogenic hantaviruses bind to the PSI domain of the bent/inactive form (Figure 1A) of the human β3 integrin subunit of αvβ3 and αIIbβ3 integrins (Raymond et al., 2005). However, the mechanism of integrin activation postoccupancy of the PSI domain is unknown.
FIGURE 1:

Switchblade model for integrin activation (adapted from Schurpf and Springer [2011]) and schematic presenting the hypothesis that cis interaction of αIIbβ3 integrin and RGDP2Y2R mediates integrin activation initiated by binding of SNV to the PSI domain. (A) 1) Structure of an inactive integrin. 2–3) Intracellular signaling (inside-out) induces integrin activation mediated by binding of adaptor proteins (such as talin) to the extended conformation with open head-piece bound to soluble and immobilized ligands (see the text for details). 4) Development of mechanochemical force selectively transduced through the β subunit. Integrin binding to immobilized ligand resists lateral translation and causes an increase in force (indicated by arrows) and promotes separation of the α− and β-subunit transmembrane domains. (B) 5) P2Y2R interacts in cis with αIIbβ3 integrin. 6) SNV occupancy of the PSI domain induces an increase in integrin affinity for cis-RGDP2Y2R (based on AFM measurements). Integrin extension exerts a membrane normal/lateral pulling force, due to height differences between the membrane proximal RGDP2Y2R, and the unbending integrin (Takagi et al., 2002; Chigaev et al., 2003, 2015); Outside-in signaling. 7) Recruitment of talin and other adhesion molecules increases force transduction, which is terminated by rupture of RGD interaction. Full extension of αIIbβ3 integrin causes rupture of cis interaction, indicated by PAC1 staining of cells. 8) Cessation of tensile force, and loss of intracellular link to actin, leads to exchange of adhesion proteins, which are replaced by adaptor proteins (e.g., clathrin and Dab2) for integrin endocytosis (Yu et al., 2015).
Integrins are heterodimers of noncovalently associated α- and β-subunits that regulate cell-surface adhesion through changes in the conformation in their ectodomains (Luo and Springer, 2006). Inactive, low-affinity integrins assume a bent structure. Activated integrins adopt a higher affinity for extracellular ligands and convert to an extended or open conformation (Calderwood, 2004). Integrins bind to short linear peptide sequences on adhesion proteins, the most common being arginine-glycine-aspartic acid (RGD), expressed in counter structures and several components of the extracellular matrix proteins (ECM), such as fibronectin, collagens, and vitronectin (Plow et al., 2000). Integrins respond to intracellular or extracellular cues by signaling bidirectionally; this is termed “inside-out” and “outside-in” signaling, respectively (Banno and Ginsberg, 2008; Kim et al., 2011; Schurpf and Springer, 2011; Shen et al., 2012; Ye et al., 2012; Zhu et al., 2013). Inside-out signaling initiates integrin activation by causing the binding of adaptor molecules such talins, kindlins, 14-3-3 proteins, or filamins to the integrin β-subunit (Gahmberg et al., 2009). The engagement of adaptor proteins is necessary for coupling to the underlying actin cytoskeleton, which enables the induction of full integrin activation (Gahmberg et al., 2009). High-affinity integrin binding to the ECM initiates outside-in signaling, which leads to cell spreading, retraction, migration, and proliferation (Takagi et al., 2002; Chen et al., 2006; Zhu et al., 2007; Shen et al., 2012). More recent studies have shown that outside-in signaling requires the binding of a G protein subunit, Gα13 to the cytoplasmic domain of the β3 integrin, which overlaps with the talin binding domain (Gong et al., 2010; Shen et al., 2013).
The linkage between the cytoskeleton and integrins facilitates transduction of force across the membrane (Hu et al., 2007; Kanchanawong et al., 2010). In this way, integrins guide forces generated by the cytoskeleton onto the ECM to produce the traction force necessary for cell adhesion and migration (Volk et al., 1990; Welch et al., 1990; Vogel and Sheetz, 2006; Yu et al., 2015). Recent studies have shown that integrin conformational state is correlated with force exertion (Zhu et al., 2008; Nordenfelt et al., 2016). Force application is optimal when integrins engage immobilized ligands that resist traction force applied to the ligands through the integrin, which results in increased signaling output (Schurpf and Springer, 2011; Nordenfelt et al., 2016). Conversely, soluble ligands cannot transduce tensile force, which is based on allosteric resistance to translation (Schurpf and Springer, 2011; Nordenfelt et al., 2016) (Figure 1).
The PSI domain is believed to be a locus of integrin activation, based either on activating amino acid substitutions (Zang and Springer, 2001; Arnaout, 2002; Sun et al., 2002; Xiong et al., 2004) or epitope location of activation-sensitive monoclonal antibodies such as AP5. AP5 antibodies recognize the hexapeptide sequence GPNICT (residues 1–6) at the amino terminus of β3 integrins of extended conformation (Honda et al., 1995; Smagghe et al., 2010). Nevertheless, studies have shown that polarized epithelial and endothelial cells are susceptible to hantavirus infection exclusively from the apical surface but not the basolateral side (Krautkramer and Zeier, 2008; Buranda et al., 2014), despite integrins being sorted to the latter domain (Drubin and Nelson, 1996). A glycosylphosphatidylinositol-linked protein, CD55/decay-accelerating factor (DAF), which is sorted to the apical surface of polarized cells (Brown et al., 1989), was identified as a cognate cell entry coreceptor for pathogenic hantaviruses (Krautkramer and Zeier, 2008). However, studies of the functional mechanisms of integrin activation associated with these proteins are not yet available. It is known that pathogenic hantaviruses bind to DAF with high-affinity (Buranda et al., 2014) and that H319 anti-DAF antibodies can be used to block infection in polarized cells but not in isotropic cells (Krautkramer and Zeier, 2008; Buranda et al., 2014).
In this study, we sought to investigate an alternative effector for integrin activation during hantavirus infection. We targeted the P2Y2 receptor (P2Y2R), a member of the purinergic family of G protein–coupled receptors (GPCR), which has been shown by biochemical colocalization to interact with β3 and β5 integrins through an integrin-binding RGD motif in its first extracellular loop (Erb et al., 2001). In polarized cells, P2Y2R is sorted to the apical surface (Qi et al., 2005) together with DAF (Brown et al., 1989) and is segregated from integrins which are sorted to the basolateral membrane domains (Drubin and Nelson, 1996). Here we set out to determine whether the presumed cis interaction (Erb et al., 2001) between P2Y2R and β3 was relevant to integrin activation.
Previous studies have shown that monoclonal antibodies (mAbs) N29 and 8E3 that target epitopes in the PSI domain of the α5β1 integrin increased its ligand binding affinity and induced an extended conformation in the integrin (Mould et al., 2005). We speculated that binding of SNV to the PSI domain of a β3 integrin bound in cis to RGDP2Y2R, caused the integrin to transition from an SNV-induced higher affinity state (Calderwood, 2004), to a fully activated state due to tensile forces pulling on the fixed RGDP2Y2R as illustrated in Figure 1B. To test the hypothesis that β3 integrin engages the RGDP2Y2R in cis, we used atomic force microscopy (AFM) to show that an increase in P2Y2R expression (via transfection) in target cells resulted in a concomitant increase in the frequency of adhesive events between the AFM tip and cell surfaces. Conversely, the adhesion rate decreased with RNA interference (RNAi) knockdown of P2Y2R in target cells. In other experiments, we showed that SNV binds to integrins immobilized on the AFM tip and increases the affinity of the integrins when bound to cell surface receptors. We also show a significant increase in cellular staining by the PAC1 antibody, which selectively binds to fully activated αIIbβ3 integrins (Shattil et al., 1985), post SNV binding to αIIbβ3 (Figure 1B). We propose that the cis interaction with P2Y2R provides resistance to the membrane-normal motion associated with the switchblade model of integrin activation, which in turn enhances the force and gives rise to the “PAC1-positive” high-affinity state (Schurpf and Springer, 2011; Nordenfelt et al., 2016).
RESULTS
AFM detection of RGD-specific interactions in astrocytoma cells expressing exogenous P2Y2R
To explore the binding specificity between β3 integrin and P2Y2R we used single-molecule AFM to probe for changes in adhesive interactions between an AFM cantilever functionalized with recombinant αIIbβ3 integrins and astrocytoma 1321N1cells and CHO-K1 cells. The 1321N1 astrocytoma cells that were transfected with hemagglutinin- (HA) tagged human P2Y2R or HA-tagged low-affinity RGE mutant P2Y2R were tested for functional P2Y2 receptor activity and for their ability to be immunoprecipitated with HA antibody, as demonstrated in Liao et al. (2007). Figure 2A shows the expression levels of P2ry2 mRNA in the cell lines that we used in this study. Figure 2B indicates that small interfering RNA (siRNA) transfection of CHO-K1 cells achieved 60% knockdown efficiency of P2ry2 mRNA in CHO-K1.
FIGURE 2:

P2Y2R expression in various cell lines. (A) Plot of P2ry2 mRNA expression in cell lines used in this study, namely P2Y2R-null wild type astrocytoma cells (WT1321N1), 1321N1 cells stably expressing an Arg95-Gly96-Glu97 (RGE) mutation of the Arg95-Gly96-Asp97 (RGD) sequence in the P2Y2R (RGEP2Y2R) and 1321N1 cells expressing wild-type P2Y2R (RGDP2Y2R), CHO-K1 and telomerase-immortalized human microvascular endothelium cell line (TIME). RNA was extracted from 150,000 cells in duplicate wells with RNeasy Qiagen kit. Quantitative RT–PCR was performed in triplicate for each well by Taqman assay as described under Materials and Methods. (B) Plot of P2ry2 mRNA knockdown in CHO-K1 stably transfected with αIIbβ3–integrin measured 24 h after siRNA transfection, **p < 0.05.
The experimental setup for single-molecule interactions between an AFM tip decorated with recombinant αIIbβ3 integrins and cells expressing P2Y2R is shown in Figure 3. To enable measurement of single-molecule interactions between the AFM tip and cellular membranes, we optimized compression force and contact duration to attain an adhesion frequency of ∼33% in the measurements. Figure 3B shows a typical force displacement scan recording the interaction between an AFM tip and substrate in our experimental setting. Single-molecule adhesion between the tip and cell membrane appears as a hysteresis between the engagement and retraction events of the trace. The force jump that accompanies the unbinding of the adhesion is indicated as Fu in Figure 3B.
FIGURE 3:

Experimental setup for single-molecule force microscopy of αIIbβ3–RGD interaction. (A) Schematic and bottom view microgragh of AFM microcantilever functionalized with αIIbβ3 integrin above RGDP2Y2R binding sites on cell membranes. For the 1321N1 astrocytes, experiments were performed in DMEM containing 5% FBS with addition of 10 mM HEPES (pH 7.4). For CHO-K1, we used Tyrode’s buffer (Sigma-Aldrich) containing 1 mM CaCl2, 1 mM MgCl2, 0.1% glucose, and 0.1% BSA. For Mn2+ activation, 1 mM CaCl2 and 1 mM MgCl2 were replaced with 2 mM MnCl2. (B) A typical cantilever-RGDP2Y2R 1321N1 force scan. Gray and black traces are engagement and retract traces, receptively. Fu is the unbinding force on the retract trace. The lower panel shows the four stages of stretching and rupturing a single ligand-receptor complex using the AFM: 1) AFM-cell surface engagement, 2) retraction, 3) extension, and 4) rupture.
During a force-displacement scan, pulling an integrin across the cell surface is expected to encounter nonspecific adhesions with cell-surface proteins. With 1321N1 cells, we first sought to determine whether expression of exogenous RGDP2Y2R would lead to specific adhesions connected to P2Y2R expression. Substitution in the RGD sequence to RGE reduces affinity by three orders of magnitude (Hautanen et al., 1989). Thus, by comparing AFM measurements using cells transfected with RGEP2Y2R to RGDP2Y2R, we expected to demonstrate specificity for (RGD)–P2Y2R. To assess the strength of single-molecule interactions, we limited the contact between the AFM cantilever and 1321N1 cells expressing RGDP2Y2R, by minimizing contact force (150 pN) and contact time (0.08 s). Under these conditions, an adhesion frequency of ∼33% in the force measurements emerged for RGDP2Y2R cells. In this way, the probabilities of forming single, double, and triple adhesion bonds between AFM tip and surface were 81%, 16%, and 2%, respectively (Chesla et al., 1998). These settings were used to record adhesion events between the AFM tip and the three cell types (P2Y2R-null WT 1321N1, RGEP2Y2R, and RGDP2Y2R expressing cells). For each cell type, a minimum of 400 repeated force scans was conducted using multiple cell-probe pairs. A large number of scans ensured robust statistics. As shown in Figure 4A under the same contact force, duration, and scan rates, the adhesion frequencies associated with WT and RGEP2Y2R were comparable (∼10%), whereas the AFM probe adhered to the RGDP2Y2R cells with higher frequency (∼33%). We plotted samples of 10 consecutive force-distance scans for the three cell types. The WT (Figure 4B) and RGEP2Y2R (Figure 4C) cells displayed adhesions of comparable magnitudes appearing as hysteresis on their respective fourth and sixth traces. Among the 10 consecutive traces associated with RGDP2Y2R expressing cells, four adhesion events were apparent in traces 2, 4, 6, and 9 (Figure 4D). The magnitude of the unbinding force in the fourth trace was comparable to the WT adhesion events.
FIGURE 4:

The frequency of adhesive interactions between an αIIbβ3-functionalized AFM tip and cell surfaces increase with exogenous RGDP2Y2R and not RGEP2Y2R. (A) Adhesion frequency of αIIbβ3-functionalized AFM cell membranes of P2Y2R-null WT, RGEP2Y2R, and RGDP2Y2R 1321N1 cells under similar AFM scan conditions. Values are means ± SD of force scans of greater than or equal to five different cell measurements. Statistical significance was determined by Student’s t test ****p < 0.0001. (B–D) Representative single force distance (retraction) curves between αIIbβ3-functionalized AFM tip and the surface of parent WT 1321N1 cells devoid of P2Y2R, and 1321N1 cells expressing exogenous RGEP2Y2R and RGDP2Y2R. The fractions shown represent the number of positive force scans in the numerator and the denominator, the total number of scans for each cell type.
Sample force histograms are shown in Figure 5. For each cell type, we probed a minimum number of five cells. The ratio of the number of adhesions and the total number of scans is shown in each cell panel. The data from the five cells were used to form a composite histogram. The most probable unbinding forces were determined from the modes of the histograms (Bell, 1978; Evans and Ritchie, 1997). The WT and RGE cells showed similar modes (most frequent unbinding force) at ∼20 pN. The RGD cells histograms showed two peaks. The first peak constituted 30% of the total adhesion events of the RGD cells corresponded to the nonspecific adhesions associated with the WT; the second peak, 60% of the total adhesion events appeared at ∼60 pN. In summary, the 30% adhesion frequency result from probing RGDP2Y2R cells was a composite of 10% nonspecific and 20% αIIbβ3/RGD interactions validated the specificity of the cognate interaction between β3 integrins and RGDP2Y2R.
FIGURE 5:

Distributions of single molecule unbinding force measured under the same conditions of contact force retraction time and pulling speed. Force histograms show nonspecific interactions at P2Y2R-null WT and RGEP2Y2R 1321N1 cell membranes and RGD specific interactions at RGDP2Y2R 1321N1 cell membranes. (A) Force histograms of unitary nonspecific interactions between αIIbβ3 and WT 1321N1 cell membranes (the loading rate, rf, was 880 pN/s). Nonspecific adhesions on five separate cells, and the composite histogram of all force scans are shown. The fractions shown in each panel represent adhesions (numerator) and total number of force distance measurements (denominator). (B) Force histograms of RGEP2Y2R 1321N1 cell membranes measured under similar conditions to WT (rf was 897 pN/s). (C) Force histograms of RGDP2Y2R 1321N1 cell membranes (rf was 930 pN/s). The composite histogram shows a distribution of nonspecific (33% of total events) and RGD specific (66% of total events). The mode of the nonspecific adhesions is similar to WT and RGE.
Measurements of αIIbβ3/RGD interactions in siRNA P2Y2R knockdown and Mn2+- and SNV-treated CHO-K1 cells
The preceding experiments demonstrated that the αIIbβ3-functionalized AFM specifically recognizes exogenous RGDP2Y2R, expressed in astrocytoma cells. To further explore the specificity of the integrin/RGDP2Y2R interaction, we turned to CHO-K1 cells that express endogenous RGDP2Y2R but are devoid of β3 integrins. We optimized contact force (200 pN), contact time (0.15 s), and retraction speed (3.7 µm/s) to achieve an adhesion frequency of ∼33% between the AFM tip and CHO-K1 cells. Figure 6A shows a series of 10 consecutive force–distance curves indicating the interaction of the integrin functionalized tip with WT CHO-K1 cells. The three of 10 unbinding events found in the fourth, sixth, and ninth traces correspond to specific interactions between the AFM tip and RGDP2Y2R. A much weaker unbinding event in the seventh trace was attributed to nonspecific adhesions that we observed with the polyethylene glycol (PEG) linker only (PEG vs. CHO-K1 in Figure 6B). In this environment, we measured adhesion interactions between the AFM tip and siRNA P2Y2R knockdown CHO-K1. We found a >50% drop in adhesion frequency events on the surfaces of P2Y2R knockdown cells compared with the WT CHO-K1 cells (Integrin vs. siRNA vs. CHO-K1 in Figure 6B). This result was consistent with the knockdown efficiency of P2Y2R shown in Figure 2B. We also performed single-molecule force displacement scans in the presence of integrin-activating cation Mn2+ (Dransfield et al., 1992) (Integrin w/Mn2+ vs. CHO-K1 in Figure 6B). In this experiment, 2 mM Mn2+ was added to the cell sample chamber before AFM engagement. In a separate set of experiments, the AFM probe was incubated with UV inactivated and fluorescently labeled SNV (Buranda et al., 2010, 2014) to allow the virus to attach to the immobilized integrins. The probe was washed once and used in force retraction measurements (Integrin w/SNV vs. CHO-K1 in Figure 6B). A peptide derived from the β3 integrin PSI domain is a competitive inhibitor of SNV binding to β3 integrins (Buranda et al., 2010). We inhibited fluorescently labeled SNV from binding to the integrin-decorated cantilever by preincubating the SNV with 6 µM PSI peptide (Integrin w/SNV & PSI vs. CHO-K1 in Figure 6B). The force-distance scans were performed under comparable conditions for probing unitary adhesion events. CHO-K1 cells are devoid of αv and β3 integrins and known to express α5β1 endogenously. We tested whether SNV interacted with CHO-K1 membranes. Therefore SNV particles were directly conjugated to the AFM tip using the same method for immobilizing the integrin. Pulling an SNV functionalized cantilever on the surface of CHO-K1 found weak nonspecific adhesions (SNV vs. CHO-K1 in Figure 6B). This result was expected because SNV does not interact with endogenous β1 integrins (Gavrilovskaya et al., 1999). We therefore used CHO-A24 cells, which are stably transfected with recombinant αIIbβ3 (Zhu et al., 2007) (SNV vs. CHO-A24 in Figure 6B). Sample histograms of unbinding forces measured under the conditions described above are shown in Figure 7 and discussed below.
FIGURE 6:

AFM measurements of unitary αIIbβ3-RGDP2Y2R interactions performed under various conditions in WT CHO-K1 cells that are devoid of β3 integrins. Measurements of SNV-integrin (PSI) interaction measured in CHO-A24 cells stably expressing αIIbβ3 are included. (A) The measurements were performed with an adhesion frequency of ∼33%. Shown are 10 representative consecutive force-distance (retraction) traces between a cantilever tip functionalized with αIIbβ3-integrin and a CHO-K1 cell. The 4th, 6th and 9th force curves reveal unitary RGD-specific adhesive interactions. The 7th force curve shows typical nonspecific interactions, which were present with the heterobifunctional acetal-PEG27-NHS linker (PEG) only. These weak adhesions were ∼6% of the adhesion frequency events as shown in B. (B) Adhesion frequency measurements under various conditions: PEG was used to measure nonspecific interactions. siRNA refers to cells treated with P2Y2R siRNA 24 h prior to the experiment. Experiments were performed in Tyrode’s buffer (Sigma-Aldrich) containing 1 mM CaCl2, 1 mM MgCl2, 0.1% glucose, and 0.1% BSA. For Mn2+ activation, CaCl2 and MgCl2 were replaced with 2 mM MnCl2. For SNV assays, the cantilever was preincubated with fluorescently labeled neat SNVR18 or a mixture of SNVR18 and 25 µM PSI domain polypeptide to competitively block the interaction between the integrin functionalized cantilever and SNVR18. The AFM cantilever was washed before immersion into the sample chamber. Association of SNV and the integrin-functionalized AFM tip was confirmed by imaging SNV fluorescence on the AFM tip. Error bars represent SEM for greater than or equal to five separate measures such as shown in Figure 5. *p < 0.05.
FIGURE 7:

Binding of SNV to integrin PSI induces integrin activation. (A) Force histogram of low affinity αIIbβ3-RGDP2Y2R interactions; (the loading rate, rf, was 720 pN/s; N refers to the total number of adhesion events for all histograms). (B) Force histogram of αIIbβ3-RGDP2Y2R interactions in the presence of 2 mM Mn2+ (rf = 700 pN/s). (C) Fluorescence micrographs (bottom view) of an integrin-functionalized cantilever preincubated with fluorescently labeled SNVR18. (D) Force histogram of αIIbβ3-RGDP2Y2R interactions with SNV bound to the integrin PSI domain (rf = 739 pN/s). (E) Fluorescence micrographs (bottom view) of an integrin-functionalized cantilever preincubated with fluorescently labeled SNVR18 and PSI domain polypeptide for blocking SNV binding. (F) Force histogram of integrin after probe was exposed to SNVR18 and PSI domain peptide. (G) SNV functionalized cantilever. (H) Force histogram of SNV–αIIbβ3 interactions in CHO-A24 cells.
Binding of SNV to integrin PSI induces integrin activation
Using force histogram data, we next explored the effect of SNV binding to the PSI domain of αIIbβ3 on the αIIbβ3/RGDP2Y2R interaction. The most frequent unbinding force of the low-affinity control (Figure 7A) was 55.0 pN. The mode of the unbinding force increased to 65 pN in the presence integrin-activating Mn2+ (Dransfield et al., 1992; Ye et al., 2012). The binding of SNV to the integrin PSI domain increases integrin affinity for the RGD ligand as indicated by the increase in the most probable unbinding force (Garcia et al., 1998; Li et al., 2003; Zhang et al., 2004; Wang and Ha, 2013). We used confocal microscopy imaging to verify decoration of the AFM cantilever with fluorescent SNV (Figure 7C). The specificity of the SNV/PSIαIIbβ3 interactions was confirmed by reduction in SNV decoration of the cantilever by incubating the SNV particles with a PSI domain polypeptide (Buranda et al., 2010). The histogram of SNVR18-treated samples had a peak at 65.0 pN comparable to Mn2+ (Figure 7D). The PSI domain peptide partially blocked SNV binding to the AFM tip (Figure 7E) and yielded a bimodal force histogram with two peaks implying two bound states representing low affinity, 55.0 pN and high-affinity, 65.0 pN (Figure 7F). As noted above, SNV-functionalized cantilever had negligible adhesive interactions with β3 integrin null CHO-K1 cells (SNV vs. CHO-K1 in Figure 6B). Using cloned CHO cell lines, CHO-A24 cells that stably express recombinant αIIbβ3 integrin (Zhu et al., 2007), the unitary unbinding force of SNV/PSIαIIbβ3 (CHO-A24) was determined to be 110.0 pN (Figure 7H), which was significantly higher than the integrin-specific measure shown in Figure 7D. This last result confirmed that SNV binds to the PSI domain with high-affinity and provides a basis for a future study in which we will characterize the affinity of SNV-PSI interactions during infection.
SNV-activated αIIbβ3-RGDP2Y2R bonds are more resistant to tensile force compared with Mn2+ activation
The preceding data show that the presence of Mn2+ and SNV caused the formation of higher affinity αIIbβ3-RGDP2Y2R interactions characterized by higher modal rupture forces compared resting integrins. To further explore the relative strengths of Mn2+ and SNV-activated αIIbβ3-RGDP2Y2R bonds, we determined the modal forces for breaking single integrin-RGDP2Y2R over a wide range of loading rates (300–7000 pN/s). The resulting dynamic force spectra (DFS), represented by a plot of most frequent unbinding forces of αIIBβ3-integrin/RGD interactions versus the logarithm of loading rate is shown in Figure 8. As predicted by the Bell-Evans model, the most frequent unbinding force, (f* in Eq. 2) is linearly dependent on the logarithm of the loading rate (rf in Eq. 2) (Bell, 1978; Evans and Ritchie, 1997). The two-dimensional dissociation rates in the absence of force (k0 in Eq. 2), as well as the barrier widths (γ in Eq. 2), were determined from a linear regression fit to Eq. 2 (Figure 8A). The best-fit parameters for ko were 0.39 s–1, 0.097 s–1, and 0.13 s–1 for resting, Mn2+ and SNV treated integrins, respectively. These force-free dissociation rates indicated that the activation of αIIBβ3-integrin by Mn2+ and SNV resulted in comparable increases (four- and threefold, respectively) in strengths of the αIIbβ3-RGDP2Y2R bonds relative to the resting state. The scales of the barrier widths were: control = 0.39 nm; Mn2+ = 0.40 nm; SNV = 0.35 nm. We next used Eq. 1 to simulate the relative effects of increasing tensile force (in the range of 0–100 pN), on the lifetimes of αIIBβ3-integrin interactions in the presence of SNV and Mn2+. The semi-logarithmic plot of bond lifetimes versus applied tensile force shows an exponential decrease of bond lifetimes with increasing tensile force (Figure 8C). The graph shows that the force-dependent decreases in the bond lifetimes of SNV and Mn2+-activated integrins diverge with increasing tensile force. The SNV activated bond appears to be more resistant to force compared with the resting and Mn2+ activated bonds. This behavior is rationalized in Figure 8D. According to the Bell-Evans model, a pulling force (f, in Eq. 1) distorts the intermolecular potential of a ligand-receptor complex, leading to a lowering of the activation energy and a shortening of the bond lifetime. As illustrated in Figure 8D, the net effect of tensile force on bond dissociation is attenuated by f•γ. In this way, the applied tensile force is less attenuated during SNV activation compared Mn2+ activation because of the smaller value of γ associated with the former. In summary, the DFS data suggest that, in a nonphysiological setting, SNV and Mn2+ induce stabilization of the αIIbβ3–RGDP2Y2R bond to a comparable extent in the absence of a tensile force (i.e., k0(SNV) ∼ k0(Mn2+)). For our purposes, this finding is important because it allows us to make estimates on the scale of SNV-induced activity based on what is known about the effect of Mn2+ on induced integrin activity (reviewed in Ye et al., 2012).
FIGURE 8:

Analysis of potential barrier energy landscapes and bond lifetimes by dynamic force spectroscopy (A) Dynamic force spectrum: Plot of most probable unbinding force (f* in Eq. 2) of αIIBβ3–integrin/RGD interactions vs. logarithm of loading rate, experimental data are shown as follows: red square (SNV activated integrin), blue circle (Mn2+-activated integrin), and open circle (low affinity integrin). Lines represent fits to Eq. 2. γ, the distance of the potential barrier position from the bottom of the potential well, is derived from the slope. The force-free dissociation rate, k0, is derived from the intercept of the fit. (B) Potential energy landscape of the αIIBβ3–RGDP2Y2R bond qualitatively showing the relative magnitudes of energy barrier widths of SNV (0.35 nm) and Mn2+- (0.40 nm) activated interactions derived from the DFS. k0 values for SNV and Mn2+-associated interactions were 0.13 s–1 and 0.097 s–1, respectively. (C) Effect of tensile force (f in Eq. 1) on the lifetime of the αIIBβ3-RGDP2Y2R interactions, for control and SNV- and Mn2+-treated samples. (D) Effect of tensile force on the potential energy barrier landscape of SNV- and Mn2+-treated integrin–RGDP2Y2R interactions (see the text for details). The mechanical energy for each applied force lowers the transition state energy barrier by -f•γ as shown.
Occupancy of the PSI domain induces physiological integrin activation in living cells
The canonical switch-blade model for integrin activation postulates that integrins undergo conformational changes involving integrin extension and an opening of the headpiece leading to high affinity (Shimaoka et al., 2002; Schurpf and Springer, 2011; Zhu et al., 2013). We tested whether SNV binding to the PSI domain caused conformational changes in αIIbβ3 similar to physiologically activated αIIbβ3 (cf. Figure 1). The IgM mAb PAC1 with an RYD integrin-targeting site (Abrams et al., 1992) selectively binds to the open conformation fibrinogen-binding site of physiologically active αIlbβ3 integrins, with ∼1 nM affinity (Hantgan and Stahle, 2009). It is used as a surrogate marker for physiologic integrin activation (O’Toole et al., 1990; Du et al., 1991; Ye et al., 2012). We used CHO-A24 cells stably expressing wild-type αIIbβ3 integrin and CHO cells expressing the αIIbW968C/β3I693C mutant (C3) for this assay (Zhu et al., 2007). In the C3 cells, the αIIb 968 tryptophan (W) and the 693 isoleucine (I) residues were replaced with cysteines to prevent the separation of the cytoplasmic domains through constitutive disulfide bond formation (Luo et al., 2004) (Figure 9). Exposure of CHO-A24 and CHO-C3 cells to SNV for 5 min produced a threefold increase in PAC1 binding to A24 cells compared with the resting state (Figure 9A), with no significant change in PAC1 staining of C3 cells in the same setting (Figure 9B). These results indicated that transmembrane domain separation is required for the high-affinity integrin that is induced by SNV binding.
FIGURE 9:

SNV binding to integrin PSI domain recapitulates physiological activation of αIlbβ3. (A, B) Model of SNV induced extended conformation associated with a high-affinity state with separated α and β transmembrane and cytoplasmic domains, which allows the ligand mimetic monoclonal anti-αIlbβ3 antibody PAC1 to bind to CHO-A24 cells stably expressing wild-type αIIbβ3. Flow cytometry histogram inserts show fluorescence associated with (a) PAC-1 staining of 50,000 cells activated with SNV particles, (b) staining of resting cells, and (c) autofluorescence of unstained cells. The graph is a plot of PAC-1 staining (1:20 dilution) of CHO-A24 corrected for autofluorescence. Cells were exposed to SNV for 5 min at 37°C and then quenched on ice, fixed with 3% paraformaldehyde for 10 min, and stained with PAC1 at 1:20 dil. for 30 min on shaker at 37°C. Poor PAC1 staining of CHO-C3 cells expressing integrin mutant with an inner membrane clasp at the αIIbW968C/β3I693, which prevents subunit separation. Experiments were performed in parallel with CHO-A24. Error bar is SE for duplicate measurements, *p < 0.05. (C) Plot of fold increase over resting cells in PAC-1 staining of CHO-A24 cells in suspension after 30 min exposure to killed SNV. 10,000 CHO-A24 cells suspended in 20 µl media containing 1:10 dilution PAC-1 antibody, coadministered with 1 µM GRGDSP. (D) Titration of soluble GRGDSP peptide, which abrogates cis-interaction of the integrin and RGDP2Y2R, inhibits infection of suspension CHO-24 cells. CHO-A24 cells were infected in suspension with 0.1 moi SNV for 30 min. Cells were then washed in low pH media three times and then incubated in normal media for 24 h and then assayed for viral N-protein.
Integrin activation to full extension is dependent on the transmission of mechanical force through the integrin β subunit (Figure 1) (Gahmberg et al., 2009; Nordenfelt et al., 2016). To test the idea that integrin tension was required for stabilizing the fully active state (Zhu et al., 2008), in our experiments, we used the soluble fibronectin hexapeptide GRGDSP to disrupt the presumed cis interaction of αIIbβ3 with RGDP2Y2R in suspension cells. The affinity constant, Kd, for GRGDSP binding to a low-affinity purified αIIbβ3 headgroup is ∼0.4 ± 0.26 µM (Lin et al., 2016). Elsewhere GRGDSP was shown to inhibit fibrinogen binding to purified αIIbβ3 with an IC50 of 2.0 µM (Suehiro et al., 1996). PAC1 (1:10 dilution) and 1 µM GRGDSP were coadministered to 20,000 CHO-A24 cells in suspension, before exposure of the cells to SNV. Staining of CHO-A24 cells with PAC1 in the presence of 1 µM GRGDSP did not rise above background in SNV-stimulated cells compared with mock-treated cells (Figure 9C). This result suggests that competitive inhibition of the relatively low-affinity αIIbβ3-RGDP2Y2R interaction by GRGDSP prevented the integrin from achieving full activation post SNV binding. In this way, the cognate binding site for PAC1 (Kd ∼ 1 nM) (Hantgan and Stahle, 2009) remained obstructed. In a second set of experiments, CHO-A24 cells were infected in the presence of various titrations (0.3–10 µM) of GRGDSP. Cell entry and replication by SNV was analyzed by Western blot analysis of SNV N-protein expression as described in the methods. Loading controls of the gels were established by using a typical standard housekeeping gene, calnexin, on the same membrane. Quantitative analysis of the gel was performed using a BioRad Molecular Imager. The data indicate that GRGDSP inhibited infection (Figure 9D). GRGDSP was also shown to inhibit infection in telomerase-immortalized human microvascular endothelium cell line (TIME) cells (Supplemental Figure S1B). Our results therefore demonstrate that SNV-induced global conformational changes in integrin conformation that are necessary for PAC1 binding and infection require the integrin to engage an immobilized ligand (Schurpf and Springer, 2011).
Gα13 binding to β3 integrin mediates infection
Integrin binding to immobilized ligands mediates outside-in signaling. Recent studies have shown that the heterotrimeric Gα13 protein binds to a fully activated ligand-occupied integrin (Gong et al., 2010). We therefore asked whether a myristoylated peptide, mP6, (Myr-FEEERA-OH) (Shen et al., 2013), which blocks the binding of Gα13 to the cytoplasmic domain of β3 integrin, prevents outside-in signaling and infection of cells by SNV. Outside-in signaling stimulates cytoskeletal remodeling, receptor clustering (Qin et al., 2004), internalization, and trafficking. GTP loading of several GTPases was used as a surrogate measure of SNV-induced integrin activation as previously shown (Buranda et al., 2013). We used a multiplex G-trap effector binding assay which employs a quantitative flow cytometric read-out (Buranda et al., 2013) (Figure 10). We measured GTP loading of (Rap1) associated integrin activation (Bos, 2005), cytoskeletal remodeling (RhoA, Rac1) (Burridge et al., 2004) and cargo trafficking (Rab7) (Feng et al., 1995) within 5 min after exposure to SNV to capture early events of integrin activity. mP6 blocked the GTP loading of Rac1, Rap1, and Rab7 (Figure 10, B–D). GTP loading of RhoA was minimal, which was consistent with an expected down-regulation of Rho signaling during the initial wave of Gα13-mediated outside-in signaling (Figure 4F in rGong et al. [2010] and Figure 3G in Shen et al. [2013]).
FIGURE 10:

Gα13 is an effector of SNV-induced GTPase activation and infection of CHO-A24 cells. (A) Histograms of a mixture of five bead populations with graded levels of red fluorescence to form a bead array. Each bead is functionalized with an effector molecule for Rac1 (Pak1), H-Ras (Raf), Rho A (Rhotekin), Rap1(Ral), and Rab 7 (Rilp). The beads act as sensors for GTP-GTPases in a cell lysate solution. Gates were used to select beads associated with known effector proteins labeled with green fluorescent antibodies. (B) Top panels show typical histograms of beads incubated in CHO-A24 cell lysates derived from cells treated with SNV and vehicle (0.1 DMSO), 250 µM DMSO solubilized mP6 and SNV, and resting cells. Bottom panels represent beads incubated in cell lysis buffer to determine nonspecific binding of multiplex antibodies. In 48-well plates 50,000 CHO-A24 cells in each well were exposed to purified and fluorescently labeled UV inactivated virus for 5 min. The reaction was quenched on ice, after which cells were lysed with radio immunoprecipitation assay buffer (RIPA) buffer. Subsequent incubations with effector beads and antibodies were performed at 4°C to limit GTP hydrolysis. The assay took 4 h to complete. (C) Respective plots of site occupancy/bead of active GTPases, determined from median channel fluorescence (MCF) of CHO-A24 cell-lysate histograms after correction for nonspecific binding. The error bars represent triplicate measurements for each target. (D) CHO-A24 cells were plated at confluence into a 96-well plate and then loaded with Fura 2AM and 10 µM BAPTA-AM following standard protocols and analyzed on a Biotek H2 Synergy Plate Reader. Thapsigargin (Tg, 10 µM) was used to stimulate the discharge intracellular Ca2+ stores in the Fura 2 AM-loaded CHO-A24 cells. The traces show fluorescence intensity of the Ca2+ sensitive Fura 2 AM in the presence of Ca2+ (Tg) or absence (Tg + BAPTA) after [Ca2+]i. Each trace was background-corrected using fluorescence of nonstimulated cells. The traces are an average of three separate wells. (E) Cells were treated with 250 µM mP6 for 30 min. The 10 µM Tg was added to cells 5 min before infection with 0.1 moi SNV inocula and incubated for 30 min and washed in triplicate at low pH to remove unbound SNV and resuspended in fresh media and analyzed after 24 h. Data show comparison of SNV N-protein in CHO-A24 cells infected with SNV inocula among samples treated with mock treated (Veh.), mP6 (mP6), 10 µM Tg (Tg), and mP6 and 10 µM Tg (mP6+Tg). Error bars are SEs for the four data points shown in the graph, *p < 0.05.
mP6 is known to inhibit Gα13- β3 interaction without disrupting talin-dependent integrin functions (Shen et al., 2013), and therefore we investigated how mP6 affects chemically induced inside-out signaling under our experimental conditions. Intracellular free Ca2+ ([Ca2+]i) and Rap1GTP have been implicated in the activation of inside-out signaling for integrins (Crittenden et al., 2004). We therefore exploited the fact that thapsigargin (Tg) (Thastrup et al., 1990) stimulates the discharge of intracellular Ca2+ stores to gain the [Ca2+]i-mediated inside-out signaling function in CHO-A24 cells (Figure 10D). We tested the effect of mP6 in an infection assay measuring the production of viral N-protein. mP6 inhibited infection of CHO-A24 cells (Figure 10E). Thus the results confirmed that SNV-induced integrin outside-in signaling and infectivity required Gα13 binding to the β3 cytoplasmic domain. The infection of Tg-treated cells, replete with a sustained bolus of elevated [Ca2+]i, was enhanced by a factor of 2 (Figure 10E), and mP6 did not block SNV infection of Tg-treated cells. This result suggested that inside-out signaling not associated with SNV engagement dispenses the requirement of Gα13 binding to the β3 integrin tail for infection (Figure 10E).
We explored in several assays the functional significance of RGDP2Y2R-mediated integrin activation post-SNV binding. First, we measured SNV infectivity in P2Y2R-knockdown CHO-A24 cells and in mP6-treated and GRGDSP-treated TIME endothelial cells. siRNA knockdown of P2Y2R expression in CHO-A24 cells decreased SNV infectivity by 60% (Supplemental Figure S1A). mP6 and GRGDSP equally inhibited infection of TIME cells (Supplemental Figure S1B). These results further suggested cis binding to P2Y2R and binding of Gα13 to β3 were both required for SNV infection. Second, the P2Y2R is a Gαq coupled receptor. We speculated that inside-out signaling initiated by P2Y2R agonists, ATP, UTP, and ATPγs, a nonhydrolyzable form of ATP, could enhance SNV infectivity in CHO-A24, TIME, and Vero E6 cells. We initially compared SNV infectivity of CHO-A24 cells in regular media and media containing P2Y2R agonists or apyrase, a nucleotidase that acts extracellularly to hydrolyze ATP and ADP (Corriden et al., 2010). The exogenous P2Y2R agonists and nucleotidase had no significant effect on SNV infectivity of CHO-A24 (Supplemental Figure S1C). This result was not surprising as αIIbβ3 integrins expressed in CHO cells are known to be refractory to agonist stimulation (Watanabe et al., 2008). However, the CHO-A24 data suggested that binding of Gα13 to the RGDP2Y2R-occupied integrin was independent of inside-out signaling as expected (Shen et al., 2013). On the contrary, UTP-stimulated a nominal two-fold increase in viral N-protein expressed in TIME cells (Supplemental Figure S1D). To obtain a more quantitative measure of this effect, we turned to Vero E6 cells, a modified kidney epithelial cell line from the African green monkey that lacks the interferon gene, which suppresses viral replication in normal Vero cells (Emeny and Morgan, 1979). SNV focus forming units (FFU) were assayed using serial supernatant samples of infected Vero E6 cells cultured in ATPγs replete media. The results show that at 7 d postinfection (pi), cells infected in ATPγs-replete media yielded a 10-fold increase in progeny SNV compared with cells infected without ATPγs (Supplemental Figure S1E). Thus, collectively, the data suggest that integrin bidirectional signaling synergistically stimulates SNV infectivity even if SNV induced outside-in integrin activation is sufficient for productive infection.
Cell polarity disrupts the cis interaction between P2Y2R and β3 integrin and infectivity
Adhesion of cells to the extracellular matrix and neighboring cells provides external cues that establish structurally and functionally distinct apical and basolateral membrane domains (Drubin and Nelson, 1996). As mediators of cell adhesion, integrins are sorted to the basolateral surface, (Drubin and Nelson, 1996) while the P2Y2R is sorted to the apical surface (Qi et al., 2005) (Figure 11A). We hypothesized that SNV infectivity of isotropic suspension cells was more efficient than polarized cells because nonpolarized cells allow for optimal cis interactions, where polarization segregated β3 integrins and RGDP2Y2R. To test this idea, we plated CHO-A24, TIME, and Vero cells in a 48-well plate, at confluence overnight in vitronectin-coated plates. Cells in selected wells were detached with Accutase cell detachment solution, washed, and resuspended in media and infected under the same conditions as the plated cells. Suspension TIME and CHO –A24 cells yielded four times more progeny virions than plated cells (Figure 11B). However, the infection rates for plated and suspension Vero E6 were similar. DAF/CD55, a cognate apical coreceptor for pathogenic hantaviruses (Krautkramer and Zeier, 2008), is highly expressed on Vero E6 cells (250,000 DAF/CD55 per cell) (Buranda et al., 2014). Thus the segregation of integrins and P2Y2R does not affect infection of Vero E6 cells as DAF/CD55 provides an alternate pathway for integrin activation. Conversely, CHO cells are devoid of DAF, while TIME cells (derived from microvascular cells of the foreskin) express low levels of DAF. These data provide additional evidence that cis interaction between RGDP2Y2R and β3 integrin is necessary for SNV induced integrin activation, which leads to infection. Given the above result on receptor distribution in polarized cells, it is important to note that under normal physiological conditions integrins are expressed on the apical surface of vascular endothelial cells (Conforti et al., 1991, 1992). The luminal expression of integrins in vivo allows the potential for cis interaction between β3 integrins and P2Y2R in vivo to be exploited as a mechanism for cell entry by pathogenic hantaviruses.
FIGURE 11:
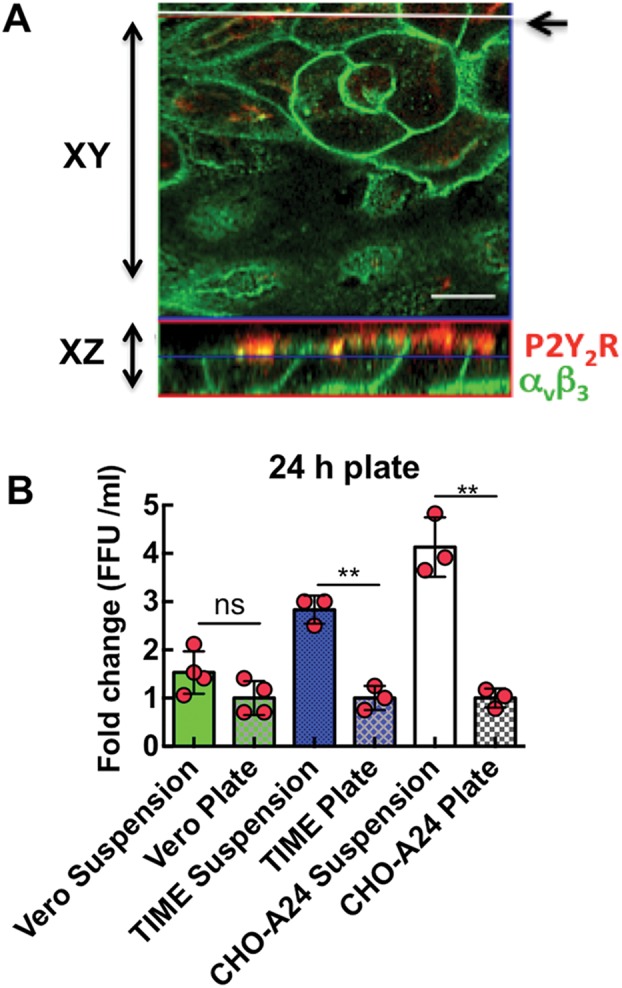
Cell polarity disrupts the cis interaction between P2Y2R and β3 integrin and infectivity. (A) Confocal image of polarized Vero E6 cells expressing antibody-stained basolateral αvβ3 and apical P2Y2R. The top panel is a confocal image in which the focus plane was parallel to the monolayer (XY scan), whereas the bottom panel shows the focus plane as a vertical cross section of the monolayer (XZ scan). The white line indicated by the arrow in the XY scan indicates the path of the XZ scan. To enable visualization, Vero E6 cells were transiently transfected with P2Y2R and plated at confluence in eight-well Nunc Lab-Tek chambers and allowed to propagate for 1–2 d. At room temperature, cells were fixed with 3% paraformaldehyde for 20 min and subsequently permeabilized with 0.2% Triton X-100 (in PBS) for 15 min. Cells were blocked for nonspecific staining with 1% BSA in PBS for 30 min and then incubated with mouse monoclonal anti-integrin αvβ3 (1:40, Millipore MsxHu, MAB#1976) and rabbit polyclonal anti-P2Y2 (1:100, Abcam, ab10270) were used with Alexa 488 and Alexa 580, respectively. (B) Plot of SNV FFU/ml vs. cells in suspension and on plates. CHO-A24, TIME, and Vero E6 cells were plated at confluence in vitronectin treated 48-well plates for 24 h. Confluent cells in triplicate wells were inoculated with 0.1 moi SNV for 1 h, washed in low pH media, and then incubated in normal media for 24 h. Media from infected cells was collected after 24 h diluted and subjected to a 7-day focus assay in Vero cells (see Supplemental Figure S1E for typical SNV FFU). **p < 0.001.
DISCUSSION
In the present study, we investigate the role of P2Y2R-β3 integrin cis interaction in integrin activation during the first step of viral infection, the binding of SNV to low-affinity β3 integrins. We draw the following conclusions from the data. SNV binds to the PSI domain of the integrin and activates the integrin by stimulating an increase in its ligand binding affinity coupled with changes in conformation. Unbending of the integrin generates a tensile force between the integrin and the purinergic receptor in the plane of the membrane, which induces full integrin activation. For these studies, we used single-molecule force spectroscopy and multiple cell lines that were chosen because of targeted expression of receptor proteins relevant to the study. Wild-type astrocytoma cells are devoid of P2Y2R expression (Parr et al., 1994). Therefore, using wild-type 1321N1 cells and well-characterized stable transfectants of wild-type RGDP2Y2R (positive control) and a nonfunctional RGEP2Y2R (negative control) enabled us to demonstrate the specificity of the cognate interaction between RGDP2Y2R and β3 integrin (Figures 4 and 5). The astrocytoma cells have been well characterized previously in biochemical experiments showing the possible cis integrin-RGDP2Y2R interactions mediated Gα12- and Go-dependent signaling in αvβ5 integrin-RGDP2Y2R expressing cells but not αvβ5 integrin-RGEP2Y2R cells (Erb et al., 2001; Bagchi et al., 2005; Liao et al., 2007; Erb and Weisman, 2012). 1321N1 cells are devoid of β3 integrin subunits and are not permissive to productive SNV infection in our hands. We therefore used CHO-K1 cells that are devoid of DAF/CD55 (a coreceptor for SNV cell entry) to establish, by AFM, that interaction of SNV with the integrin PSI domain causes integrin activation. The AFM provides a unique capability of measuring the cell metabolism independent activation of integrins on the binding of SNV to the integrin PSI domain (Figure 7, C and D).
Under conditions established for unitary adhesive interactions, the frequency of adhesion events is governed by Poisson statistics (Chesla et al., 1998). A large number of scans ensured robust statistics. The frequency of adhesive interactions between the AFM tip and cell membranes depends on several factors. Surface decoration of the AFM tip with αIIbβ3 was minimized to make sure that the preponderance of the observed rupture events was the result of single αIIbβ3-RGDP2Y2R interactions. The topography of the cell membrane is also a contributing factor. Unlike the relatively smooth surface of CHO cells, the surface of astrocytes is known to have many projected structures such as dendrites. During the course of our studies, we found that differential design of the AFM cantilever was a critical determinant for establishing unitary binding events in astrocytes and CHO cells. Because of projections located on the surface of astrocytes, a relatively sharp probe such as the MLCT AFM (with an open angle of ∼40°) presents a narrow radius (∼50 nm) that is comparable to the cellular projections. This probe was found to adhere nonspecifically to the protrusions, which manifested as nonspecific forces. However, a “dull” probe tip (open angle of 70°) was effective at limiting the probability of nonspecific interactions. Thus the frequency of binding events comprising ≤33% of total force displacement scans and >95% of unbinding events occurring in a single step (cf. step 4 in Figure 3B) indicated unitary adhesive interactions (Zhang et al., 2002; Rankl et al., 2008). At our established set-point for unitary adhesive interactions, the probability for weaker nonspecific interactions was ∼10% in 1321N1 cells (Figure 5) and ∼6% in CHO-K1 cells (Figure 6). The nonspecific events were solely present in WT and RGE 1321N1 cells, whereas both nonspecific and RGD-specific interactions were present in RGDP2Y2R expressing 1321N1 and CHO-K1 cells. We found that the presence of Mn2+ and SNV induced an increase in the population of higher affinity interactions represented in the force histograms relative to quiescent samples (Figure 7). In the resting integrins, the histogram data show that only 35% of unbinding interactions are defined by Fu values >55 pN (the most frequent unbinding force of quiescent samples measured in CHO-K1 cells) (Figure 7A). For stimulated integrins, the population of adhesions above this threshold increases to 67% in the presence of Mn2+ (Figure 7B) and 70% in the presence of SNV (Figure 7D).
Mn2+ is a widely used positive control for integrin activation. However, the extent to which Mn2+ induces integrin activation is variable and depends on context and isotype of the target integrin (reviewed in Ye et al., 2012). While stimulation of αvβ3 integrin by Mn2+ is known to induce both integrin extension and change in affinity, Mn2+ stimulation produces only a change in affinity without the extension in αIIbβ3 integrin (Kamata et al., 2005). Elsewhere Cluzel et al. (2005) observed Mn2+-induced clustering of αvβ3 integrins in focal complexes when attached to immobilized ligands. To extend the comparison between SNV- and Mn2+-induced states of integrin activation, we analyzed the dynamic force spectrum (DFS) of resting Mn2+- and SNV-treated samples. From the DFS analysis, we showed that Mn2+ and SNV exposure confers a ∼3.5-fold decrease in the two-dimensional dissociation rate of the αIIbβ3-RGDP2Y2R bond in the absence of force. Using the Bell-Evans formalism, we found that the bond lifetimes of resting Mn2+- and SNV-treated samples decreased monotonically with increasing tensile force, suggesting slip-bond dissociation (Dembo et al., 1988; Marshall et al., 2003) in all cases. However, the difference in the potential energy barrier widths for the αIIbβ3-RGDP2Y2R interaction, under SNV (0.35 nm) or Mn2+ (0.40 nm) activation, are small but consequential in that the narrower barrier width associated with SNV activity confers a nominally longer bond lifetime under tensile force (Figure 8C). In recent studies, the threshold limit of tensile force required to activate outside-in signaling has been set to 56 pN (Wang and Ha, 2013; Wang et al., 2015). Because SNV-activated αIIbβ3-RGDP2Y2R bonds are shown here to be more resistant to tensile force compared with Mn2+, we suggest that SNV activated αIIbβ3-RGDP2Y2R bonds are sufficiently stable to allow outside-in mediated adhesion signaling, like Mn2+ (Ye et al., 2012). In summary, these single molecule two-dimensional events could be extended to biologically relevant bulk experiments induced by other stimuli.
Biochemical evidence for cis interaction between integrins and P2Y2R is extensive. Studies to establish this association used human1321N1 astrocytoma cells that are devoid of P2Y2R (Parr et al., 1994; Erb et al., 2001). A set of cells were stably transfected with the wild-type RGDP2Y2R designed to test whether integrin interaction with the P2Y2R was an essential noncanonical effector for the GqPCR to couple to Gα12 and Gαo proteins. Nonfunctional RGE mutant P2Y2R served as negative controls. Evidence for cis interaction between αv integrins and the RGD domain of the P2Y2R was demonstrated in a series of papers that showed that the αv integrin coimmunoprecipitates with the RGDP2Y2R but not with the nonfunctional RGEP2Y2R in 1321N1 astrocytoma cells (Erb et al., 2001; Bagchi et al., 2005; Liao et al., 2007; Erb and Weisman, 2012). The authors then showed that the presumptive RGDP2Y2R- integrin engagement conferred to the GqPCR the ability to activate the heterotrimeric G-protein Gα12, which stimulated RhoA signaling (Liao et al., 2007). Under these circumstances Gα12 associated with the wild-type P2Y2R and with αvβ5 integrins. Also, function-blocking antibodies directed against αvβ5 inhibited UTP-induced Rho activation and stress fiber formation. Conversely, UTP treatment of the RGE mutant neither caused Gα12 activation nor conferred RhoA activity (Liao et al., 2007).
SNV-permissive CHO-A24 cells stably expressing αIIbβ3 were used to establish the requirement of cis interaction for infectivity as follows. We first showed that SNV binding to the PSI domain recapitulates physiological integrin activation. SNV binding to the PSI domain in CHO-A24 cells induces full extension of the integrin ectodomain and opening of the headpiece as indicated by binding of the fibrinogen-mimetic PAC-1 mAb. We then showed that blocking of the αIIbβ3–RGDP2Y2R interaction, with a soluble GRGDSP peptide, inhibits full integrin activation and infection (Figure 9). siRNA knockdown of P2Y2R in CHO-A24 cells inhibits infectivity (Supplemental Figure S1). Cell polarization limits infection of CHO-A24 and physiologically relevant TIME cells by segregating β3 integrins from the P2Y2R by differential sorting of the two receptors to the basolateral and apical domains, respectively (Figure 11 and Supplemental Figure S1). Integrin activation to an extended open structure is associated with a >100 Å vertical shift of the integrin’s ligand recognition domain away from the plasma membrane (Takagi et al., 2002; Chigaev et al., 2003, 2015). Thus extension displaces the head of the integrin above the RGDP2Y2R (Figure 1). In this setting, one might imagine the development of tensile force from the integrin head pulling away from a membrane-anchored P2Y2R. Zhu et al. have described a mechano-mechanical model for bidirectional integrin activation that involves engagement to immobilized extracellular matrix ligands (Zhu et al., 2008). Thus, for this study, one might expect a combination of vertical and lateral (cis) tensile forces to be exerted during the change in conformation while being resisted by the membrane fixed RGDP2Y2R (Figure 1B [5 and 6]) (Schurpf and Springer, 2011; Nordenfelt et al., 2016). Importantly, our study shows that SNV infectivity is inhibited when soluble GRGDSP abrogates possible cis interaction of the integrin with P2Y2R.
Integrin signaling is bidirectional and involves, inside-out and outside-in signaling processes. This process involves dynamic interactions of the integrins with cyto- and ectoplasmic proteins (Shattil et al., 2010). External forces transmitted through integrins’ connection to the cellular cytoskeleton contribute to outside-in signaling (Iskratsch et al., 2014). Prototypical inside-out signaling, is initiated by receptor signaling (commonly a GPCR), where a downstream intracellular integrin activator, such as talin, binds to the cytoplasmic β-integrin domain (Calderwood et al., 1999). This leads to conformational changes that result in increased affinity for extracellular ligands. Integrin binding to extracellular ligands and subsequent changes in conformation initiates outside-in signaling, which in turn can generate signals that cause inside-out signaling. Recent studies have shown that inside-out and outside-in signaling phases are regulated by a “timeshare occupancy” of a common domain on the β-integrin cytoplasmic domain by talin and Gα13. These proteins are principle effectors for inside-out and outside-in signaling, respectively (Gong et al., 2010; Shen et al., 2013).
Our results indicate that the binding of Gα13 to the integrin β3 subunit is a critical determinant of SNV infectivity. We demonstrate that mP6, a membrane permeable peptide inhibitor of integrin–Gα13 binding inhibits cell signaling and infection. Of importance, our results indicate that the implicit binding of Gα13 to β3 integrin does not require upstream activity by a GPCR. In previous studies, Gα13 activation by a ligand-activated GPCR was viewed as an important upstream signaling component for Gα13–integrin binding (Gong et al., 2010; Shen et al., 2013). Ultimately, physiologic integrin activation in the presence of macromolecular ligands such as fibrinogen (but not small soluble peptides) are indispensable for Gα13 coupling to the integrin (Shen et al., 2013). Our data recapitulate the model shown in Figure 1, where the ligand-binding pocket of bent integrins faces toward the plasma membrane, which favors physical interaction with membrane-proximal RGDP2Y2R. After SNV binds to the integrin PSI domain, activation of the integrin initiates switchblade-like conformational change and development of tensile force. The increase in PAC1 staining of SNV-activated cells indicates that the rupture of P2Y2R–integrin interactions occurs after the integrin is fully activated, which favors Gα13 binding to β3 integrin with higher affinity (Shen et al., 2013). It is now established that outside-in signaling is localized at focal adhesions in a process that includes the recruitment of adhesion molecules such as talin and vinculin (Askari et al., 2010; Yu et al., 2015). Termination of the tensile force results in integrin turnover with the exchange of adhesion related molecules by endocytic adaptor proteins (Puklin-Faucher and Sheetz, 2009; Yu et al., 2015; Nordenfelt et al., 2016). It is therefore likely that the rupture of the integrin–P2Y2R junction results in endocytosis. Studies of the signaling steps that lead to internalization are ongoing. In summary, our study reports a new mechanism for integrin activation post binding of SNV to the PSI domain of the integrin. The findings have potential impact for the discovery of novel therapeutic targets of hantavirus infection, due to a high case mortality ratio.
MATERIALS AND METHODS
Materials
Octadecyl rhodamine B chloride (R18), 5-octadecanoyl amino fluorescein (F18), BAPTA-AM, amine-reactive Alexa Fluor 488 carboxylic acid, and succinimidyl ester probe were purchased from Life Technologies (Carlsbad, CA). Recombinant DAF was purchased from R&D Systems. Rabbit polyclonal H319 anti-DAF antibody was purchased from Santa Cruz Biotechnology. Primary antibodies used for immunostaining were mouse monoclonal anti-integrin αvβ3 (1:40, Millipore MsxHu, #MAB 1976), rabbit polyclonal anti-P2Y2 (1:100, Abcam, ab10270), and fluorescein isothiocyanate mouse anti-human PAC-1 (BD Biosciences). Secondary antibodies used were anti-mouse immunoglobulin G (IgG) Alexa488, anti-mouse IgG Alexa647, anti-mouse IgG Cy5, and anti-rabbit Alexa647, all from Molecular Probes. Phosphate-buffered saline (PBS) was purchased from Life Technologies. Tyrode’s buffer, dimethyl sulfoxide (DMSO), and Sephadex G-50 were purchased from Sigma. TRIS (10 mM or 25 mM Tris, 150 mM NaCl, pH 7.5) and HHB (30 mM HEPES, 110 mM NaCl, 10 mM KCl, 1 mM MgCl26H2O, and 10 mM glucose, pH 7.4) buffer, and Hanks balanced saline solution (HBSS) (0.35 g NaH2CO3, 0.049 g MgSO4, and 1 mM CaCl2 or 1 mM MnCl2) were prepared under sterile conditions and stored in 50-ml tubes at -20°C. Chemical reagents sources and conditions of use are summarized in Table 1.
TABLE 1:
Signaling modulation assays.
| Reagent (source) | Target; concentration | Incubation time | Solvent |
|---|---|---|---|
| ATPγs, ATP, UTP (Sigma) | P2Y2R, Gaq; 100 µM | 0 | H2O |
| BAPTA-AM (Life Technologies) | [Ca2+]i; 10 µM | 30 min | DMSO |
| H319 (Santa Cruz) | DAF/CD55; 40 µg/ml | 1 h | H2O |
| mP6 (New England Peptide) | G13; 250 µM | 30 min | DMSO |
| Tg (Sigma) | [Ca2+]i stores; 10 µM | ±2 min | DMSO |
A24 cells were incubated with inhibitors or activators for the indicated time and concentration. Control samples were incubated with the solvent used to solubilize specific drugs.
Effector proteins
The GST-effector chimeras consisting of the minimal GTPase binding domains (RBD) used for the studies were as follows. PAK-1 RBD, a Rac1 effector, and Raf-1 RBD, a RAS effector protein, were obtained from Millipore. Rhotekin-RBD, a Rho effector protein, was purchased from Cytoskeleton. RalGDS-RBD, a RAP1 effector protein, was expressed and purified from a plasmid kindly provided by Keith Burridge (UNC Chapel Hill) (Wittchen and Burridge, 2008). GST-RILP RBD was purified as described in Buranda et al. (2013).
Cell culture
Vero E6 were purchased from the American Type Culture Collection and maintained in DMEM (Life Technologies) at 37°C in a water-jacketed 5% CO2 incubator. The medium contains 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin/streptomycin, 2 mM l-glutamine.
CHO cells lines: Zhu et al. (2007) have generated stable CHO-K1 cell lines expressing wild-type recombinant αIIbβ3 integrins (A24) or disulfide mutant αIIbW968C/β3I693C (C3). In the C3, the αIIb 968 tryptophan (W) and the 693 isoleucine (I) residues were replaced with cysteines to prevent the separation of the cytoplasmic domains through constitutive disulfide bond formation (Luo et al., 2004). They showed that the mutant integrin was defective in adhesion-induced outside-in signaling (Zhu et al., 2007). CHO A24 were grown in DMEM/F12 HAM mixture (Life Technologies) with 2 mM l-glutamine and 1 mg/ml G418 (Sigma). CHO C3 cells were cultured in F-12K media (Cellgro, Mediatech), 100 U/ml penicillinsStreptomycin, and 2 mM l-glutamine.
Human lung microvascular endothelial cell (HLMVEC) and TIME were maintained in EBM-2 Basal Medium with EGM-2 SingleQuot Kit supplements and growth factors (Lonza, Walkersville, MD).
Human 1321N1 astrocytoma cells were transfected with HA-tagged human P2Y2R, or HA-tagged low-affinity RGE mutant P2Y2R. The cells were tested for functional P2Y2 receptor activity and for their ability to be immunoprecipitated with HA antibody, as demonstrated in Liao et al. (2007). Wild-type (WT) human 1321N1 astrocytoma cells (lacking detectable expression of any subtype of P2Y receptor), human 1321N1 astrocytoma cells stably transfected with the human P2Y2 receptor (pLXSN vector with an mino-terminal HA tag for detection with HA antibodies), and human 1321N1 astrocytoma cells stably transfected with the RGE mutant human P2Y2 receptor (pLXSN vector with an amino-terminal HA tag) were cultured in DMEM containing 5% FBS, 100 U/ml penicillin, 100 µg/ml streptomycin at 37°C in a humidified atmosphere of 5% CO2, and 95% air (Bagchi et al., 2005). The amount of 500 µg/ml Geneticin (G418, Life Technologies) was added to maintain the expression of pLXSN-P2Y2 receptor constructs in the transfected cells.
All cell lines are tested for the presence of mycoplasma at every cell passage using commercial kits such as the MycoAlert mycoplasma detection kit (www.lonza.com).
Cell imaging
Tight junction markers occludin or ZO1 were imaged in cell monolayers with Zeiss META or LSM 510 systems using 63 × 1.4 oil immersion objectives as previously described. One hundred fifty thousand cells were plated at confluency in eight-well Nunc Lab-Tek chambers (thermoscientific.com) or in inverted transwell filter supports and allowed to propagate for 2 d. At room temperature, cells were fixed with 3% paraformaldehyde for 20 min and subsequently permeabilized with 0.2% Triton X-100 (in PBS) for 15 min. Cells were blocked for nonspecific staining with 1% bovine serum albumin (BSA) in PBS for 30 min and then incubated with either anti-ZO1 (rabbit polyclonal from Abcam, 1:100 dilution in blocking buffer) or occludin (mouse mAb from Invitrogen, 1:200 dilution in blocking buffer) overnight at 4°C. Cells were then incubated with a suitable Alexa488 tagged secondary antibody (Millipore; 1:100 dilution in blocking buffer for 1.5 h at room temperature). Samples were washed and imaged on the confocal microscope. For receptor imaging, mouse monoclonal anti-integrin αvβ3 (1:40, Millipore MsxHu, MAB#1976), and rabbit polyclonal anti-P2Y2 (1:100, Abcam, ab10270) were used with Alexa 488 and Alexa 580, respectively.
Knockdown of P2Y2R expression by siRNA
RNAi knockdown of P2Y2R in TIME was performed using P2Y2R siRNA (h): sc-42579 purchased from Santa Cruz Biotechnologies. siRNA for CHO cells was custom designed from a partial sequence of Cricetulus griseus and synthesized by Dharmacon. P2Y2R siRNA ribonucleotides (40 nM) were pooled from four target-specific duplex components (sense: 5′ U.C.U.G.C.A.A.A.C.U.G.G.U.G.C.G.A.U.U.U.U 3′ and antisense: 5′ A.A.U.C.G.C.A.C.C.A.G.U.U.U.G.C.A.G.A.U.U 3′; sense: 5′ C.A.G.C.U.G.G.G.A.G.C.G.A.-G.A.C.U.A.A.U.U 3′ and antisense: 5′ U.U.A.G.U.C.U.C.-G.C.U.C.C.C.A.G.C.U.G.U.U 3′; sense: 5′ G.G.G.A.U.-G.A.A.C.U.G.G.G.C.U.A.C.A.A.U.U 3′ and antisense: 5′ U.U.G.U.A.G.C.C.C.A.G.U.U.C.A.U.C.C.C.U.U 3′; sense: 5′ C.A.G.U.C.A.U.C.C.U.G.G.U.C.U.G.U.U.A.U.U 3′ and antisense: 5′ U.A.A.C.A.G.A.C.C.A.G.G.-A.U.G.A.C.U.G.U.U 3′) and was used to knock down P2Y2R in CHO-A24 cells expressing wild-type integrin (Zhu et al., 2007). Transfection was performed with Lipofectamine 2000 (Thermofisher) following the manufacturer’s protocol. Knockdown efficiency was determined by quantitative PCR.
Quantitative PCR
Total RNA was isolated from CHO A24 and TIME cells, and TaqMan gene expression assays were performed to determine the expression of P2Y2R in wild-type and siRNA knockdown cells. RNA was extracted using a Qiagen RNeasy kit (cat. no. 74104), quantified with a Thermo Fisher Scientific Nanodrop. PCR and TaqMan reactions were carried out using GeneAmp PCR System 9700 from ABI and Bio-Rad CFX96 Real-Time PCR Systems, respectively. Quantitative reverse transcription PCR (RT-PCR) was performed using primers for P2Y2R (Hs04176264_s1 FG from Life Technologies) and a standard curve derived from serial dilutions of a WT P2Y2R plasmid.
Production of sin nombre virus
We propagated and titered SNV in Vero E6 cells inside biosafety level 3 (BSL3) facilities and practices (CDC registration number C20041018-0267) (Bharadwaj et al., 1999). To inactivate SNV with UV radiation, we placed 100 μl of virus stock (isolate SN77734, typically (1.5–2) × 106 FFU/ml). In each well of a 96-well plate we subjected the virus to UV irradiation at 254 nm for various time intervals (∼5 mW/cm2) as described elsewhere (Buranda et al., 2010).
Fluorescence labeling of SNV
The envelope membrane of hantavirus particles was stained with the lipophilic lipid probe octadecyl rhodamine (R18) and purified as previously described (Buranda et al., 2010). The typical yield of viral preparation was 1 ± 0.5 × 108 particles/µl in 300 µl tagged with ∼10,000 R18 probes/particle. Small aliquots for single use were stored at –80°C.
Infection assays
Virus inocula (0.1 moi) were added to cells in a serum-free medium and incubated for 30 min at 37°C. Various cell signal-inhibiting or -activating drugs were used at the concentrations shown in Table 1. Unbound virions were removed by triple washing, and cells were incubated for 24 h at 37°C. The infection was analyzed by a standard Western blot analysis of N-protein expression where cell lysates were boiled in SDS buffer and separated by 8% SDS–PAGE and transferred to a nitrocellulose membrane. SNV N protein was detected with αSNV/N (hyperimmune rabbit anti-SN virus N protein) (Elgh et al., 1997) used at a 1:1000 dilution, with overnight incubation, washed, and then stained for an hour incubation with a secondary antibody (Peroxidase AffiniPure Goat Anti-Rabbit IgG; used at 1:1000 dilution) from Jackson Immuno Research Laboratories (cat. no. 111-035-003, lot no. 104668). The nitrocellulose membrane was then treated with an HRP substrate from Pierce, SuperSignal West Pico (product no. 0034077) for 5 min before imaging. Quantitative analysis of the gel was performed using a BioRad Molecular Imager, ChemiDoc XRS+ equipped with Image Lab Software 4.1. Equal loading was verified by the detection of β-actin or calnexin on the same membrane with the anti-β-actin (clone AC-74 used at 1:2000 from Sigma) or anti-calnexin (clone H-70 used at 1:500 dilution; from Santa Cruz) monoclonal antibodies.
Focus assay
These experiments were performed under a protocol that was approved by the University of New Mexico Biosafety Committee. This protocol is typically applied to grow the virus in our lab (Buranda et al., 2010, 2013, 2014) The medium for propagating virus is typically limited in nutrients (2% FBS) to minimize cell proliferation. We inoculated Vero E6 monkey kidney cells with a low multiplicity of infection (moi) of virus (<0.01) and then incubated infected cells, serially collecting supernatants of the culture into aliquots for 1–9 d postinoculation (pi) and freezing those supernatants down at -80°C as 1-ml aliquots. The virus titers were determined via a Focus Reduction Neutralization Test (FRNT) (Bharadwaj et al., 1999). A 7-d FRNT assay was carried out to quantify the viral progeny in each serial sample using 1:10 and 1:1000 dilutions for samples collected on day 1 and day 7 samples, respectively. Viral antigen was visualized by addition of rabbit anti-SN virus nucleocapsid protein serum followed by peroxidase-conjugated goat anti-rabbit IgG and DAB/metal concentrate as substrate.
GTPase effector trap flow cytometry assay (G-trap)
This has been previously described (Buranda et al., 2013). CHO A24 were plated in 48-well plates and starved overnight in serum-free medium followed by stimulation with activator or inhibitor at concentrations and times shown in Table 1. After incubating cells with various inhibitors, activators, and SNVR18 for desired times, cells were lysed with 100 µl ice-cold RIPA buffer. For standardization, a fraction of the supernatant was collected to measure protein concentrations. Lysates were kept cold on ice at all times to limit hydrolysis of active GTPases. For each effector assay, 10,000 beads were added to 50 µg of protein in 100 µl of cell lysate in RIPA buffer. Optimizing the assay for each new lot of antibody is critical.
Cytometry experiments
Equilibrium binding interactions of fluorescent antibodies with cells or beads were analyzed by flow cytometry as described previously (Buranda et al., 2010, 2011, 2014).
Atomic force microscopy measurements of individual integrin–P2Y2R interactions
Recombinant human αIIbβ3 integrin (R&D Systems, Minneapolis, MN) was attached to an AFM cantilever (MLCT or MLCT-BIO-DC, Bruker Nano, Camarillo, CA) by covalently crosslinking the two using a heterobifunctional acetal-PEG27-NHS linker (Ebner et al., 2007; Wildling et al., 2011). MN). Recombinant heterodimeric soluble human αIIbβ3 integrin was from R&D Systems (Minneapolis, MN). The protein contains the ectodomains of αIIb (Leu32-Arg993) and β3 (Gly27Asp718) followed by an ACID peptide and a His(6) tag at the carboxyl terminus of αIIb, and by a BASE peptide at β3. The ACID/BASE coiled-coil peptides can stabilize the heterodimer and have been widely used in expressing soluble integrins (Takagi et al., 2002, 2003; Xiao et al., 2004; Bennett, 2005; Jeong et al., 2013; Ozawa et al., 2016). All single-molecule force measurements were conducted using a custom-designed apparatus designed for operation in the force spectroscopy mode (Zhang et al., 2002, 2004). The cantilever (320-µm-long by 22-µm-wide triangle) was calibrated by characterizing the spring constant via thermally induced fluctuations. The spring constants (13 ± 3 pN/nm) of the calibrated cantilevers agreed with the values specified by the manufacturer. The objectives of these experiments were twofold: First, to directly measure detachment force associated with specific binding interactions between the adhesion sites at cell membranes and AFM cantilever decorated with human αIIbβ3 integrin, we probed P2Y2R-null human astrocytoma 1321N1 cells, as well as 1321N1 cells, stably expressing wild-type P2Y2R or the RGE mutant human P2Y2 receptor. After modification with PEG, the MLCT cantilever showed very low nonspecific adhesion (i.e., 6–8%) to CHO-K1 cells. However, high nonspecific adhesion (∼30%) was found between the PEG-modified MLCT and astrocytoma cells. This is likely due to the specific surface structure of astrocytoma, such as dendrites. To address this issue, a special type of MLCT probe, designated MLCT-BIO-DC, was used. Whereas the regular MLCT has a relatively sharp tip with an open angle of 35–40°, MLCT-BIO-DC offers a dull tip with an open angle of ∼70°. The blunt tip produces gentler measurement on the cell surfaces and is less likely to slide against cell surface structures such as dendrites. Moreover, the MLCT-BIO-DC is compensated for thermal drifts. Each cantilever on the MLCT-BIO-DC is coated with reflective metal at the tip region, thus reducing its thermal drift and improving its detection stability. Also, 5% FBS was included in the experimental buffer for astrocytoma studies to reduce nonspecific interactions further. With the MLCT-BIO-DC and additional 5% FBS, we were able to lower the nonspecific adhesions on astrocytoma to ∼10%. We next determined whether SNV binding to the PSI domain causes a change in affinity at the RGD binding site in CHO-K1 cells. The positive control for integrin activation was 2 mM Mn2+ added to the media (Zhang et al., 2002; Li et al., 2003; Trache et al., 2010; Ligezowska et al., 2011). For SNV assays, the cantilever was preincubated with fluorescently labeled neat SNVR18, or a mixture of SNVR18 and 25 µM PSI domain polypeptide (Buranda et al., 2010) to competitively block the interaction between the integrin functionalized cantilever and SNVR18. Association of SNV and the integrin functionalized AFM tip was confirmed by imaging SNV fluorescence on the AFM tip. The binding interaction between the AFM tip and binding sites was then determined from the deflection of the cantilever via a position-sensitive two-segment photodiode (Figure 3C). Experimental conditions allowing for measurement of single-molecule unbinding forces (Fu) of the αIIbβ3-RGD site complex were established following a method described by Evans (2001).
Typically, 5–10 separate cells were probed for each setting. For each pulling speed, 200–600 force curves were recorded, which yielded ≥50 to ≥150 unbinding forces. Curve fitting was performed using IGOR Pro or Origin software by minimizing the chi-square statistic for the optimal fit. The unbinding force (Fu) of the αIIbβ3–integrin/P2Y2R complex was derived from the force jump that accompanies the unbinding of the complex (Figure 3B) (Hinterdorfer and Dufrene, 2006; Rankl et al., 2008).
Dynamic force spectroscopy
The Bell-Evans model, a theory to determine energy landscape properties, describes the influence of an external force on the rate of bond dissociation (Bell, 1978; Evans and Ritchie, 1997). According to this model, a pulling force, f, distorts the intermolecular potential of a ligand-receptor complex, which leads to a lowering of the activation energy and an increase in the dissociation rate k(f) as follows:
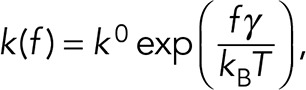 (1) (1)
|
where k0 is the dissociation rate constant in the absence of a pulling force, γ is the linear distance (nm) of the transition state from the bottom of the potential well, T is the absolute temperature, and kB is the Boltzmann constant. For a constant loading rate rf, the most probable, or most frequent unbinding force f* is given by
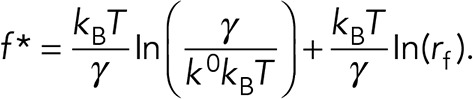 (2) (2)
|
Hence the Bell model predicts that f* is a linear function of the logarithm of the loading rate. Experimentally, f* was determined from the mode of the unbinding force histograms. The Bell model parameters were determined by fitting Eq. 2 to the plot of f* versus ln(rf).
Statistical analysis
Statistical analysis was performed using GraphPad Prism. Differences between the data were tested for statistical significance using Dunnett’s multiple comparisons or Student’s t test; p < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported by the University of New Mexico School of Medicine, Department of Pathology and Cardiovascular and Metabolic Diseases Signature Program. We thank Larry Sklar, Nancy Kanagy, and Eric Prossnitz for helpful comments on integrin and GPCR biology.
Abbreviations used:
- AFM
atomic force microscopy
- BSA
bovine serum albumin
- DAF
decay-accelerating factor
- DFS
dynamic force spectroscopy
- ECM
extracellular matrix proteins
- ECMO
extracorporeal membrane oxygenation
- FFU
focus-forming units
- FITC
fluorescein isothiocyanate
- GPCR
G protein-coupled receptors
- HA
hemagglutinin
- HCPS
hantavirus cardiopulmonary syndrome
- HFRS
hemorrhagic fever with renal syndrome
- HLMVEC
human lung microvascular endothelial cell
- mAB
monoclonal antibody
- PEG
polyethylene glycol
- PSI
plexin-semaphorin-integrin
- RGD
arginine-glycine-aspartic acid
- RGE
arginine-glycine-glutamic acid
- RIPA
radio immunoprecipitation assay buffer
- RT-PCR
reverse transcription PCR
- SNV
sin nombre virus
- Tg
thapsigargin
- TIME
telomerase-immortalized human microvascular endothelium cell line
- WT
wild-type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-01-0082) on August 23, 2017.
REFERENCES
- Abrams CS, Ruggeri ZM, Taub R, Hoxie JA, Nagaswami C, Weisel JW, Shattil SJ. Anti-idiotypic antibodies against an antibody to the platelet glycoprotein (GP) IIb-IIIa complex mimic GP IIb-IIIa by recognizing fibrinogen. J Biol Chem. 1992;267:2775–2785. [PubMed] [Google Scholar]
- Arnaout MA. Integrin structure: new twists and turns in dynamic cell adhesion. Immunol Rev. 2002;186:125–140. doi: 10.1034/j.1600-065x.2002.18612.x. [DOI] [PubMed] [Google Scholar]
- Askari JA, Tynan CJ, Webb SE, Martin-Fernandez ML, Ballestrem C, Humphries MJ. Focal adhesions are sites of integrin extension. J Cell Biol. 2010;188:891–903. doi: 10.1083/jcb.200907174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi S, Liao Z, Gonzalez FA, Chorna NE, Seye CI, Weisman GA, Erb L. The P2Y2 nucleotide receptor interacts with alphav integrins to activate Go and induce cell migration. J Biol Chem. 2005;280:39050–39057. doi: 10.1074/jbc.M504819200. [DOI] [PubMed] [Google Scholar]
- Banno A, Ginsberg MH. Integrin activation. Biochem Soc Trans. 2008;36:229–234. doi: 10.1042/BST0360229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell GI. Models for the specific adhesion of cells to cells. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- Bennett JS. Structure and function of the platelet integrin alphaIIbbeta3. J Clin Invest. 2005;115:3363–3369. doi: 10.1172/JCI26989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj M, Lyons CR, Wortman IA, Hjelle B. Intramuscular inoculation of Sin Nombre hantavirus cDNAs induces cellular and humoral immune responses in BALB/c mice. Vaccine. 1999;17:2836–2843. doi: 10.1016/s0264-410x(99)00096-1. [DOI] [PubMed] [Google Scholar]
- Brown DA, Crise B, Rose JK. Mechanism of membrane anchoring affects polarized expression of two proteins in MDCK cells. Science. 1989;245:1499–1501. doi: 10.1126/science.2571189. [DOI] [PubMed] [Google Scholar]
- Buranda T, Basuray S, Swanson S, Bondu-Hawkins V, Agola J, Wandinger-Ness A. Rapid parallel flow cytometry assays of active GTPases using effector beads. Anal Biochem. 2013;144:149–157. doi: 10.1016/j.ab.2013.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buranda T, Swanson S, Bondu V, Schaefer L, Maclean J, Mo ZZ, Wycoff K, Belle A, Hjelle B. Equilibrium and kinetics of sin nombre hantavirus binding at DAF/CD55 functionalized bead surfaces. Viruses-Basel. 2014;6:1091–1111. doi: 10.3390/v6031091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buranda T, Wu Y, Perez D, Jett SD, BonduHawkins V, Ye C, Edwards B, Hall P, Larson RS, Lopez GP, et al. Recognition of decay accelerating factor and alpha(v)beta(3) by inactivated hantaviruses: toward the development of high-throughput screening flow cytometry assays. Anal Biochem. 2010;402:151–160. doi: 10.1016/j.ab.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buranda T, Wu Y, Sklar LA. Quantum dots for quantitative flow cytometry. Methods Mol Biol. 2011;699:67–84. doi: 10.1007/978-1-61737-950-5_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood DA. Integrin activation. J Cell Sci. 2004;117:657–666. doi: 10.1242/jcs.01014. [DOI] [PubMed] [Google Scholar]
- Calderwood DA, Zent R, Grant R, Rees DJ, Hynes RO, Ginsberg MH. The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J Biol Chem. 1999;274:28071–28074. doi: 10.1074/jbc.274.40.28071. [DOI] [PubMed] [Google Scholar]
- Chen J, Yang W, Kim M, Carman CV, Springer TA. Regulation of outside-in signaling and affinity by the beta2 I domain of integrin alphaLbeta2. Proc Natl Acad Sci USA. 2006;103:13062–13067. doi: 10.1073/pnas.0605666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesla SE, Selvaraj P, Zhu C. Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophys J. 1998;75:1553–1572. doi: 10.1016/S0006-3495(98)74074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluzel C, Saltel F, Lussi J, Paulhe F, Imhof BA, Wehrle-Haller B. The mechanisms and dynamics of (alpha)v(beta)3 integrin clustering in living cells. J Cell Biol. 2005;171:383–392. doi: 10.1083/jcb.200503017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti G, Dominguezjimenez C, Zanetti A, Gimbrone MA, Cremona O, Marchisio PC, Dejana E. Human endothelial-cells express integrin receptors on the luminal aspect of their membrane. Blood. 1992;80:437–446. [PubMed] [Google Scholar]
- Conforti G, Zanetti A, Jimenez CD, Cremona O, Marchisio PC, Dejana E. Human endothelial-cells express an inactive alpha-V-beta-3 receptor on their apical surface whose activity can be modulated. Thromb Haemost. 1991;65:962–962. [Google Scholar]
- Crittenden JR, Bergmeier W, Zhang Y, Piffath CL, Liang Y, Wagner DD, Housman DE, Graybiel AM. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat Med. 2004;10:982–986. doi: 10.1038/nm1098. [DOI] [PubMed] [Google Scholar]
- Dembo M, Torney DC, Saxman K, Hammer D. The reaction-limited kinetics of membrane-to-surface adhesion and detachment. Proc Roy Soc Lond B. 1988;234:55–83. doi: 10.1098/rspb.1988.0038. [DOI] [PubMed] [Google Scholar]
- Dransfield I, Cabanas C, Craig A, Hogg N. Divalent cation regulation of the function of the leukocyte integrin LFA-1. J Cell Biol. 1992;116:219–226. doi: 10.1083/jcb.116.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin DG, Nelson WJ. Origins of cell polarity. Cell. 1996;84:335–344. doi: 10.1016/s0092-8674(00)81278-7. [DOI] [PubMed] [Google Scholar]
- Du XP, Plow EF, Frelinger AL, 3rd, O’Toole TE, Loftus JC, Ginsberg MH. Ligands “activate” integrin alpha IIb beta 3 (platelet GPIIb-IIIa) Cell. 1991;65:409–416. doi: 10.1016/0092-8674(91)90458-b. [DOI] [PubMed] [Google Scholar]
- Ebner A, Wildling L, Kamruzzahan AS, Rankl C, Wruss J, Hahn CD, Holzl M, Zhu R, Kienberger F, Blaas D, et al. A new, simple method for linking of antibodies to atomic force microscopy tips. Bioconjug Chem. 2007;18:1176–1184. doi: 10.1021/bc070030s. [DOI] [PubMed] [Google Scholar]
- Elgh F, Lundkvist A, Alexeyev OA, Stenlund H, Avsic-Zupanc T, Hjelle B, Lee HW, Smith KJ, Vainionpaa R, Wiger D, et al. Serological diagnosis of hantavirus infections by an enzyme-linked immunosorbent assay based on detection of immunoglobulin G and M responses to recombinant nucleocapsid proteins of five viral serotypes. J Clin Microbiol. 1997;35:1122–1130. doi: 10.1128/jcm.35.5.1122-1130.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emeny JM, Morgan MJ. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. JGenVirol. 1979;43:247–252. doi: 10.1099/0022-1317-43-1-247. [DOI] [PubMed] [Google Scholar]
- Erb L, Liu J, Ockerhausen J, Kong Q, Garrad RC, Griffin K, Neal C, Krugh B, Santiago-Perez LI, Gonzalez FA, et al. An RGD sequence in the P2Y(2) receptor interacts with alpha(V)beta(3) integrins and is required for G(o)-mediated signal transduction. J Cell Biol. 2001;153:491–501. doi: 10.1083/jcb.153.3.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb L, Weisman GA. Coupling of P2Y receptors to G proteins and other signaling pathways. Wiley Interdiscip Rev Membr Transp Signal. 2012;1:789–803. doi: 10.1002/wmts.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E. Probing the relation between force–lifetime–and chemistry in single molecular bonds. Annu Rev Biophys Biomol Struct. 2001;30:105–128. doi: 10.1146/annurev.biophys.30.1.105. [DOI] [PubMed] [Google Scholar]
- Evans E, Ritchie K. Dynamic strength of molecular adhesion bonds. Biophys J. 1997;72:1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahmberg CG, Fagerholm SC, Nurmi SM, Chavakis T, Marchesan S, Gronholm M. Regulation of integrin activity and signalling. Biochim Biophys Acta. 2009;1790:431–444. doi: 10.1016/j.bbagen.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AJ, Huber F, Boettiger D. Force required to break alpha5beta1 integrin-fibronectin bonds in intact adherent cells is sensitive to integrin activation state. J Biol Chem. 1998;273:10988–10993. doi: 10.1074/jbc.273.18.10988. [DOI] [PubMed] [Google Scholar]
- Gavrilovskaya I, LaMonica R, Fay ME, Hjelle B, Schmaljohn C, Shaw R, Mackow ER. New York 1 and Sin Nombre viruses are serotypically distinct viruses associated with hantavirus pulmonary syndrome. J Clin Microbiol. 1999;37:122–126. doi: 10.1128/jcm.37.1.122-126.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilovskaya IN, Shepley M, Shaw R, Ginsberg MH, Mackow ER. beta3 Integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc Natl Acad Sci USA. 1998;95:7074–7079. doi: 10.1073/pnas.95.12.7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong H, Shen B, Flevaris P, Chow C, Lam SC, Voyno-Yasenetskaya TA, Kozasa T, Du X. G protein subunit Galpha13 binds to integrin alphaIIbbeta3 and mediates integrin “outside-in” signaling. Science. 2010;327:340–343. doi: 10.1126/science.1174779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantgan RR, Stahle MC. Integrin priming dynamics: mechanisms of integrin antagonist-promoted alphaIIbbeta3:PAC-1 molecular recognition. Biochemistry. 2009;48:8355–8365. doi: 10.1021/bi900475k. [DOI] [PubMed] [Google Scholar]
- Hautanen A, Gailit J, Mann DM, Ruoslahti E. Effects of modifications of the RGD sequence and its context on recognition by the fibronectin receptor. J Biol Chem. 1989;264:1437–1442. [PubMed] [Google Scholar]
- Hinterdorfer P, Dufrene YF. Detection and localization of single molecular recognition events using atomic force microscopy. Nat Methods. 2006;3:347–355. doi: 10.1038/nmeth871. [DOI] [PubMed] [Google Scholar]
- Hjelle B. Epidemiology and diagnosis of hantavirus infections. In: Rose BD, editor. UpToDate. Wellesley, MA: Wolters Kluwer; 2014. [Google Scholar]
- Honda S, Tomiyama Y, Pelletier AJ, Annis D, Honda Y, Orchekowski R, Ruggeri Z, Kunicki TJ. Topography of ligand-induced binding sites, including a novel cation-sensitive epitope (AP5) at the amino terminus, of the human integrin beta 3 subunit. J Biol Chem. 1995;270:11947–11954. doi: 10.1074/jbc.270.20.11947. [DOI] [PubMed] [Google Scholar]
- Hu K, Ji L, Applegate KT, Danuser G, Waterman-Storer CM. Differential transmission of actin motion within focal adhesions. Science. 2007;315:111–115. doi: 10.1126/science.1135085. [DOI] [PubMed] [Google Scholar]
- Iskratsch T, Wolfenson H, Sheetz MP. Appreciating force and shape-the rise of mechanotransduction in cell biology. Nat Rev Mol Cell Biol. 2014;15:825–833. doi: 10.1038/nrm3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong SJ, Luo R, Singer K, Giera S, Kreidberg J, Kiyozumi D, Shimono C, Sekiguchi K, Piao X. GPR56 functions together with alpha3beta1 integrin in regulating cerebral cortical development. PLoS One. 2013;8:e68781. doi: 10.1371/journal.pone.0068781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson CB, Figueiredo LTM, Vapalahti O. A global perspective on hantavirus ecology, epidemiology, and disease. Clin Microbiol Rev. 2010;23:412–441. doi: 10.1128/CMR.00062-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson CB, Hooper J, Mertz G. Treatment of hantavirus pulmonary syndrome. Antiviral Res. 2008;78:162–169. doi: 10.1016/j.antiviral.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata T, Handa M, Sato Y, Ikeda Y, Aiso S. Membrane-proximal α/β stalk interactions differentially regulate integrin activation. J Biol Chem. 2005;280:24775–24783. doi: 10.1074/jbc.M409548200. [DOI] [PubMed] [Google Scholar]
- Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, Waterman CM. Nanoscale architecture of integrin-based cell adhesions. Nature. 2010;468:580–584. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Ye F, Ginsberg MH. Regulation of integrin activation. Annu Rev Cell Dev Biol. 2011;27:321–345. doi: 10.1146/annurev-cellbio-100109-104104. [DOI] [PubMed] [Google Scholar]
- Koster F, Mackow E. Pathogenesis of the hantavirus pulmonary syndrome. Future Virol. 2012;7:41–51. [Google Scholar]
- Krautkramer E, Zeier M. Hantavirus causing hemorrhagic fever with renal syndrome enters from the apical surface and requires decay-accelerating factor (DAF/CD55) J Virol. 2008;82:4257–4264. doi: 10.1128/JVI.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Redick SD, Erickson HP, Moy VT. Force measurements of the alpha5beta1 integrin-fibronectin interaction. Biophys J. 2003;84:1252–1262. doi: 10.1016/S0006-3495(03)74940-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Z, Seye CI, Weisman GA, Erb L. The P2Y2 nucleotide receptor requires interaction with alpha v integrins to access and activate G12. J Cell Sci. 2007;120:1654–1662. doi: 10.1242/jcs.03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligezowska A, Boye K, Eble JA, Hoffmann B, Klosgen B, Merkel R. Mechanically enforced bond dissociation reports synergistic influence of Mn2+ and Mg2+ on the interaction between integrin alpha7beta1 and invasin. J Mol Recognit. 2011;24:715–723. doi: 10.1002/jmr.1108. [DOI] [PubMed] [Google Scholar]
- Lin FY, Zhu J, Eng ET, Hudson NE, Springer TA. beta-dubunit binding is sufficient for ligands to open the integrin alphaIIbbeta3 headpiece. J Biol Chem. 2016;291:4537–4546. doi: 10.1074/jbc.M115.705624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo BH, Springer TA. Integrin structures and conformational signaling. Curr Opin Cell Biol. 2006;18:579–586. doi: 10.1016/j.ceb.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo BH, Springer TA, Takagi J. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biol. 2004;2:e153. doi: 10.1371/journal.pbio.0020153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackow ER, Gavrilovskaya IN. Hantavirus regulation of endothelial cell functions. Thromb Haemost. 2009;102:1030–1041. doi: 10.1160/TH09-09-0640. [DOI] [PubMed] [Google Scholar]
- Mackow ER, Gorbunova EE, Gavrilovskaya IN. Endothelial cell dysfunction in viral hemorrhage and edema. Front Microbiol. 2014;5:733. doi: 10.3389/fmicb.2014.00733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall BT, Long M, Piper JW, Yago T, McEver RP, Zhu C. Direct observation of catch bonds involving cell-adhesion molecules. Nature. 2003;423:190–193. doi: 10.1038/nature01605. [DOI] [PubMed] [Google Scholar]
- Mould AP, Travis MA, Barton SJ, Hamilton JA, Askari JA, Craig SE, Macdonald PR, Kammerer RA, Buckley PA, Humphries MJ. Evidence that monoclonal antibodies directed against the integrin beta subunit plexin/semaphorin/integrin domain stimulate function by inducing receptor extension. J Biol Chem. 2005;280:4238–4246. doi: 10.1074/jbc.M412240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordenfelt P, Elliott HL, Springer TA. Coordinated integrin activation by actin-dependent force during T-cell migration. Nat Commun. 2016;7:13119. doi: 10.1038/ncomms13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole TE, Loftus JC, Du XP, Glass AA, Ruggeri ZM, Shattil SJ, Plow EF, Ginsberg MH. Affinity modulation of the alpha IIb beta 3 integrin (platelet GPIIb-IIIa) is an intrinsic property of the receptor. Cell Regul. 1990;1:883–893. doi: 10.1091/mbc.1.12.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa A, Sato Y, Imabayashi T, Uemura T, Takagi J, Sekiguchi K. Molecular Basis of the Ligand Binding Specificity of alphavbeta8 Integrin. J Biol Chem. 2016;291:11551–11565. doi: 10.1074/jbc.M116.719138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr CE, Sullivan DM, Paradiso AM, Lazarowski ER, Burch LH, Olsen JC, Erb L, Weisman GA, Boucher RC, Turner JT. Cloning and expression of a human P2U nucleotide receptor, a target for cystic fibrosis pharmacotherapy. Proc Natl Acad Sci USA. 1994;91:3275–3279. doi: 10.1073/pnas.91.8.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plow EF, Haas TA, Zhang L, Loftus J, Smith JW. Ligand binding to integrins. J Biol Chem. 2000;275:21785–21788. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- Puklin-Faucher E, Sheetz MP. The mechanical integrin cycle. J Cell Sci. 2009;122:179–186. doi: 10.1242/jcs.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi AD, Wolff SC, Nicholas RA. The apical targeting signal of the P2Y2 receptor is located in its first extracellular loop. J Biol Chem. 2005;280:29169–29175. doi: 10.1074/jbc.M501301200. [DOI] [PubMed] [Google Scholar]
- Qin J, Vinogradova O, Plow EF. Integrin bidirectional signaling: a molecular view. PLoS Biol. 2004;2:e169. doi: 10.1371/journal.pbio.0020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankl C, Kienberger F, Wildling L, Wruss J, Gruber HJ, Blaas D, Hinterdorfer P. Multiple receptors involved in human rhinovirus attachment to live cells. Proc Natl Acad Sci USA. 2008;105:17778–17783. doi: 10.1073/pnas.0806451105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond T, Gorbunova E, Gavrilovskaya IN, Mackow ER. Pathogenic hantaviruses bind plexin-semaphorin-integrin domains present at the apex of inactive, bent alphavbeta3 integrin conformers. Proc Natl Acad Sci USA. 2005;102:1163–1168. doi: 10.1073/pnas.0406743102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurpf T, Springer TA. Regulation of integrin affinity on cell surfaces. EMBO J. 2011;30:4712–4727. doi: 10.1038/emboj.2011.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shattil SJ, Hoxie JA, Cunningham M, Brass LF. Changes in the platelet membrane glycoprotein IIb.IIIa complex during platelet activation. J Biol Chem. 1985;260:11107–11114. [PubMed] [Google Scholar]
- Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Delaney MK, Du X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr Opin Cell Biol. 2012;24:600–606. doi: 10.1016/j.ceb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Zhao X, O’Brien KA, Stojanovic-Terpo A, Delaney MK, Kim K, Cho J, Lam SC, Du X. A directional switch of integrin signalling and a new anti-thrombotic strategy. Nature. 2013;503:131–135. doi: 10.1038/nature12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimaoka M, Takagi J, Springer TA. Conformational regulation of integrin structure and function. Annu Rev Biophys Biomol Struct. 2002;31:485–516. doi: 10.1146/annurev.biophys.31.101101.140922. [DOI] [PubMed] [Google Scholar]
- Smagghe BJ, Huang PS, Ban YE, Baker D, Springer TA. Modulation of integrin activation by an entropic spring in the β-knee. J Biol Chem. 2010;285:32954–32966. doi: 10.1074/jbc.M110.145177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suehiro K, Smith JW, Plow EF. The ligand recognition specificity of beta3 integrins. J Biol Chem. 1996;271:10365–10371. doi: 10.1074/jbc.271.17.10365. [DOI] [PubMed] [Google Scholar]
- Sun QH, Liu CY, Wang R, Paddock C, Newman PJ. Disruption of the long-range GPIIIa Cys(5)-Cys(435) disulfide bond results in the production of constitutively active GPIIb-IIIa (alpha(IIb)beta(3)) integrin complexes. Blood. 2002;100:2094–2101. doi: 10.1182/blood-2002-02-0418. [DOI] [PubMed] [Google Scholar]
- Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002a;110:599–611. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- Takagi J, Strokovich K, Springer TA, Walz T. Structure of integrin a5b1 in complex with fibronectin. EMBO J. 2003;22:4607–4615. doi: 10.1093/emboj/cdg445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trache A, Trzeciakowski JP, Meininger GA. Mg2+ modulates integrin-extracellular matrix interaction in vascular smooth muscle cells studied by atomic force microscopy. J Mol Recognit. 2010;23:316–321. doi: 10.1002/jmr.985. [DOI] [PubMed] [Google Scholar]
- Vaheri A, Strandin T, Hepojoki J, Sironen T, Henttonen H, Makela S, Mustonen J. Uncovering the mysteries of hantavirus infections. Nat Rev Microbiol. 2013;11:539–550. doi: 10.1038/nrmicro3066. [DOI] [PubMed] [Google Scholar]
- Vogel V, Sheetz M. Local force and geometry sensing regulate cell functions. Nat Rev Mol Cell Biol. 2006;7:265–275. doi: 10.1038/nrm1890. [DOI] [PubMed] [Google Scholar]
- Volk T, Fessler LI, Fessler JH. A role for integrin in the formation of sarcomeric cytoarchitecture. Cell. 1990;63:525–536. doi: 10.1016/0092-8674(90)90449-o. [DOI] [PubMed] [Google Scholar]
- Wang X, Ha T. Defining single molecular forces required to activate integrin and notch signaling. Science. 2013;340:991–994. doi: 10.1126/science.1231041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Sun J, Xu Q, Chowdhury F, Roein-Peikar M, Wang Y, Ha T. Integrin molecular tension within motile focal adhesions. Biophys J. 2015;109:2259–2267. doi: 10.1016/j.bpj.2015.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Bodin L, Pandey M, Krause M, Coughlin S, Boussiotis VA, Ginsberg MH, Shattil SJ. Mechanisms and consequences of agonist-induced talin recruitment to platelet integrin alphaIIbbeta3. J Cell Biol. 2008;181:1211–1222. doi: 10.1083/jcb.200803094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch MP, Odland GF, Clark RA. Temporal relationships of F-actin bundle formation, collagen and fibronectin matrix assembly, and fibronectin receptor expression to wound contraction. J Cell Biol. 1990;110:133–145. doi: 10.1083/jcb.110.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernly JA, Dietl CA, Tabe CE, Pett SB, Crandall C, Milligan K, Crowley MR. Extracorporeal membrane oxygenation support improves survival of patients with hantavirus cardiopulmonary syndrome refractory to medical treatment. Eur J Cardio-Thorac. 2011;40:1334–1340. doi: 10.1016/j.ejcts.2011.01.089. [DOI] [PubMed] [Google Scholar]
- Wildling L, Unterauer B, Zhu R, Rupprecht A, Haselgrubler T, Rankl C, Ebner A, Vater D, Pollheimer P, Pohl EE, et al. Linking of sensor molecules with amino groups to amino-functionalized AFM tips. Bioconjug Chem. 2011;22:1239–1248. doi: 10.1021/bc200099t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittchen ES, Burridge K. Analysis of low molecular weight GTPase activity in endothelial cell cultures. Methods Enzymol. 2008;443:285–298. doi: 10.1016/S0076-6879(08)02014-4. [DOI] [PubMed] [Google Scholar]
- Xiao T, Takagi J, Coller BS, Wang JH, Springer TA. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432:59–67. doi: 10.1038/nature02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong JP, Stehle T, Goodman SL, Arnaout MA. A novel adaptation of the integrin PSI domain revealed from its crystal structure. J Biol Chem. 2004;279:40252–40254. doi: 10.1074/jbc.C400362200. [DOI] [PubMed] [Google Scholar]
- Ye F, Kim C, Ginsberg MH. Reconstruction of integrin activation. Blood. 2012;119:26–33. doi: 10.1182/blood-2011-04-292128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CH, Rafiq NB, Cao F, Zhou Y, Krishnasamy A, Biswas KH, Ravasio A, Chen Z, Wang YH, Kawauchi K, et al. Integrin-beta3 clusters recruit clathrin-mediated endocytic machinery in the absence of traction force. Nat Commun. 2015;6:8672. doi: 10.1038/ncomms9672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki SR, Greer PW, Coffield LM, Goldsmith CS, Nolte KB, Foucar K, Feddersen RM, Zumwalt RE, Miller GL, Khan AS, et al. Hantavirus pulmonary syndrome. Pathogenesis of an emerging infectious disease. Am J Pathol. 1995;146:552–579. [PMC free article] [PubMed] [Google Scholar]
- Zang Q, Springer TA. Amino acid residues in the PSI domain and cysteine-rich repeats of the integrin beta2 subunit that restrain activation of the integrin alpha(X)beta(2) J Biol Chem. 2001;276:6922–6929. doi: 10.1074/jbc.M005868200. [DOI] [PubMed] [Google Scholar]
- Zhang X, Craig SE, Kirby H, Humphries MJ, Moy VT. Molecular basis for the dynamic strength of the integrin alpha4beta1/VCAM-1 interaction. Biophys J. 2004;87:3470–3478. doi: 10.1529/biophysj.104.045690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wojcikiewicz E, Moy VT. Force spectroscopy of the leukocyte function-associated antigen-1/intercellular adhesion molecule-1 interaction. Biophys J. 2002;83:2270–2279. doi: 10.1016/S0006-3495(02)73987-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Carman CV, Kim M, Shimaoka M, Springer TA, Luo BH. Requirement of alpha and beta subunit transmembrane helix separation for integrin outside-in signaling. Blood. 2007;110:2475–2483. doi: 10.1182/blood-2007-03-080077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Luo BH, Xiao T, Zhang C, Nishida N, Springer TA. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol Cell. 2008;32:849–861. doi: 10.1016/j.molcel.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhu J, Springer TA. Complete integrin headpiece opening in eight steps. J Cell Biol. 2013;201:1053–1068. doi: 10.1083/jcb.201212037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.