

Abstract
The aim of this study is to identify genetic variants that harbour signatures of recent positive selection and may facilitate physiological adaptations to hypobaric hypoxia. To achieve this, we conducted whole genome sequencing and lung function tests in 19 Argentinean highlanders (>3500 m) comparing them to 16 Native American lowlanders. We developed a new statistical procedure using a combination of population branch statistics (PBS) and number of segregating sites by length (nSL) to detect beneficial alleles that arose since the settlement of the Andes and are currently present in 15–50% of the population. We identified two missense variants as significant targets of selection. One of these variants, located within the GPR126 gene, has been previously associated with the forced expiratory volume/forced vital capacity ratio. The other novel missense variant mapped to the EPAS1 gene encoding the hypoxia inducible factor 2α. EPAS1 is known to be the major selection candidate gene in Tibetans. The derived allele of GPR126 is associated with lung function in our sample of highlanders (p < 0.05). These variants may contribute to the physiological adaptations to hypobaric hypoxia, possibly by altering lung function. The new statistical approach might be a useful tool to detect selected variants in population studies.
Introduction
High altitude represents an extreme environment characterised by low concentrations of atmospheric oxygen (hypoxia), arid climate, high solar radiation and other environmental stressors. Populations have resided at high elevations in Ethiopia, the Himalayas and the Andes for several millennia1. Each of these populations, faced with ongoing environmental pressure, has developed their unique array of physiological adaptations. In Andeans, these include an enlarged chest, increased lung capacities2, only slightly increased ventilation rate and an elevated haematocrit3.
In this study we focus on the Colla population living in the Northwest Argentinean highlands. Collas also inhabit Southern Bolivia and Northern Chile and are considered to be related to other Andean groups such as Quechua, Aymara and Atacameño4. These groups could trace back to the beginning of human settlement in the Andes, which archaeological evidence places between 12,000 and 9,000 years before present5–7.
The genetic component of high altitude adaptation in Andeans has been the subject of a number of recent studies8–10. Using genotype data genes such as VEGFB, PRKAA1, NOS2A and FAM213A have been suggested to be under selection in Andeans8–10. These genes play a role in a vast array of processes such as cardiac function, oxygen sensing, vasodilation and oxidative stress reduction. In Tibetans, a particular gene involved in the response to hypoxia, the hypoxia inducible factor 2α (HIF-2α) or EPAS1, has been repeatedly shown to have a signature of selection11–14 and to be associated with the almost absent increase of haemoglobin concentration in native Himalayan populations11,14. A gene involved in the oxygen sensitive expression of HIF itself, EGLN1, was selected in both Tibetans and Andeans12. A first subset of Andean whole genome sequences was published comparing 10 healthy Peruvian highlanders with highlanders suffering from chronic mountain sickness15. The authors detected variants in an erythropoiesis regulator gene SENP1, and an oncogene, ANP32D in association with cross-population tests of selection and a higher transcriptional response to hypoxia in individuals with chronic mountain sickness relative to those without.
Compared to other high altitude populations, such as Tibetans, Andeans have been living for less time at elevations above 3500 m. Given that there has been shorter duration for selection to act, rather than being close to fixation, it is likely that proportionally more advantageous gene variants exist at intermediate frequencies. To this end, we complement the scans for hard sweep variants with scans for signatures of incomplete selective processes. These tests are applied on high coverage whole genome sequence data for healthy Andean highlanders from Northwest Argentina together with sequence data from Native American lowlanders and pinpoint to variants advantageous in the adaptation to high altitude.
Results
The sequencing of the whole genome of 19 Collas living above 3500 m resulted in an average genome-wide call rate of 97.4% (min. 97.1%). Coverage of 30x was reached for 95% of the genome with a minimum of 89% in one individual, while the minimum exonic coverage was 93%. On average 280 Gb of sequence data could be mapped for each genome with a minimum of 205 Gb in a single individual. Around 3.3 million SNPs could be identified in each highlander genome. Of these 1.7% were novel SNPs. Within the whole exome only 20,900 SNPs were identified with 2% representing newly discovered SNPs (on average for each individual per genome compared to a reference genome). About 10,200 synonymous SNPs had no effect on the amino acid sequence while 9,350 were non-synonymous. These were further classified as: mainly missense mutations (9,250, average value of all individuals), about 75 nonsense, 10 nonstop, 18 misstart and 70 disruptive mutations.
To investigate population structure, effective population size and split times of highlanders and control populations in the last 300,000 years we conducted multiple sequentially Markovian coalescent analyses (MSMC, Fig. 1). All non-African populations showed a similar pattern 30,000–300,000 years ago dominated by the out of Africa exit. Split dates of non-African populations were around 12,000–30,000 years. Population divergence was consistent with a single Eurasian origin for all analysed populations. The neighbouring populations Collas and Calchaquíes only separated in the last 5,000 years (Fig. 1). Calchaquíes showed high genetic similarity but also population specific differences to the Collas2. The divergence time of Collas and lowland Wichí was estimated to be 5,000 years possibly indicating the onset of permanent high altitude settlement (Fig. 1). The genetic effective population size of Collas declined to 1500 around 9000 years ago and then rose up to 30,000 at 3,000 years ago. The total census size in 2005 of the Collas was estimated to be around 70,500 including Collas residing at lower altitudes16.
Figure 1.

MSMC plot, effective population sizes and split times of Collas, Native American lowlanders, Siberians and Africans. Effective population size (Ne) and split time estimates are based on 1, 2 or 4 genomes for the Native Americans and on 2 and 4 genomes for the other populations. Coloured dots show where the MSMC curves based on 1, 2 or 4 genomes were joined together to provide a comprehensive representation of the changes in Ne over time for each of the analysed population. Around 100,000 years the out of Africa exit starts to reduce population size in non-Africans. Yoruba show a limited decline as they remain in Africa. Ne of Collas rises up to 30,000 units 3,000 years ago. Andean highlanders: Collas; Calchaquíes (Cachi): intermediate altitude population in Argentina (2300 m); American lowlanders: Mexican, Wichí; Siberians: Eskimo, Koryaks, Chukchi.
Signals for positive selection in Collas
We first searched for alleles with high-frequency differences between Collas and other populations that could be driving late-stage selective sweeps. We retrieved all SNPs located in top 1% Tajima’s D17 and number of segregating sites by length (nSL)18 200 kb windows, and then filtered these for variants in the top 1% of population branch statistic (PBS)14 scores. PBS is a statistic that detects variants that have changed substantially in frequency in a target population, using a related population and an outgroup to determine which population the allele frequency change occurred in. The nSL test is a haplotype based statistic identifying haplotypes that are at high frequency but haven’t yet been broken up by recombination, suggesting that the increase in frequency was recent. Both statistics seek to identify a recent change in the frequency of an allele or haplotype, which can reflect the impact of selection in a genomic region. PBS was calculated for 19 Collas in comparison to 19 Siberians19,20 and 16 lowlanders from Central and South America2. The lowland group represented a conservative grouping since it included Calchaquíes, who live at an altitude of 2300 m in Argentina and bear overall genomic similarities to the nearby Collas2. Hence, any genomic signatures shared by Calchaquíes and Collas would be missed by our approach. These filters yielded 72 SNPs. After excluding SNPs with frequency >10% in the 1000 Genomes database, those without gene annotation, and with combined annotation dependent depletion (CADD) scores ≤5 only 9 SNPs remained (Table 1).
Table 1.
Top 1% PBS alleles located within top 1% nSL windows.
| Chr: position | Identifier | Allele frequency Collas | Allele frequency Lowlanders | Allele frequency Siberians | 1000 Genomes DAF | Impact | Associated gene | PBS p-value |
|---|---|---|---|---|---|---|---|---|
| 11:67209515 | rs111451405 | 74% | 44% | 3% | 4% | syn | CORO1B | 0.001 |
| 11:67072382 | rs61731786 | 71% | 44% | 3% | 7% | syn | SSH3 | 0.002 |
| 11:67076748 | rs12417770 | 71% | 44% | 3% | 7% | intronic | SSH3 | 0.002 |
| 11:67159257 | rs17880138 | 71% | 44% | 3% | 7% | promoter | RAD9A | 0.002 |
| 11:67165015 | rs2066494 | 71% | 44% | 3% | 6% | syn | RAD9A | 0.002 |
| 11:67057599 | rs1574103 | 71% | 47% | 3% | 4% | syn | ANKRD13D | 0.005 |
| 6:159062992 | rs882735 | 63% | 38% | 0% | 2% | intronic | DYNLT1 | 0.006 |
| 20:2840929 | rs2297048 | 58% | 25% | 18% | 4% | syn | VPS16 | 0.008 |
| 20:2844911 | rs2274671 | 58% | 25% | 18% | 5% | syn | PTPRA; VPS16 | 0.008 |
Chr = chromosome, DAF = derived allele frequency, syn = synonymous; CADD score for all SNPs > 5.
All of these SNPs were highlighted by the haplotype test nSL and none by Tajima’s D. Notably, six of the nine remaining SNPs were found in the same haplotype context located on chromosome 11 at 67 Mb. Interestingly, this region is 3 Mb upstream of the highest ranking region highlighted previously by the integrated haplotype score (iHS) test in Collas9. The SNP with the highest CADD score of 19.11 was part of the ANKRD13D gene which codes for an ubiquitin binding protein. All hits were in non-coding regions and also showed substantial allele frequency in the lowland population (Table 1).
Given the short time-frame of human occupation in the Andes21, we hypothesize that ongoing selective sweeps acting on intermediate frequency selected variants may contribute a substantial fraction of adaptive genetic changes among the Collas. However, detecting a selective sweep in its early stages poses particular challenges. When selection has not yet driven an allele to high frequency, the impact on linked genetic variation is also low. Consequently, most statistics designed to detect positive selection only have high power towards the final stages of a selective sweep22–24. Using simulations to inform our targeted approach, we chose to combine several aspects of the population genetic data and genome annotation information. To search for these signals, we focused on derived allele frequency (DAF) ≥ 15% and ≤50% to identify alleles at the beginning of selection which are overlooked in the traditional positive selection tests. To this end, we obtained population branch statistic (PBS) and number of segregating sites by length (nSL) scores for exonic variants with a DAF ≥ 15% and ≤50% in Collas, corresponding to 17147 SNPs of which 2831 (16.5%) were classified as missense variants in dbSNP build 141. Calculating nSL + PBS scores as described in the methods (Fig. 2a) yielded a similar score distribution as retrieved from simulations (Fig. 2b); these simulations further suggested that combining the two methods results in a greater power to identify intermediate frequency sweeps than each method on its own (Fig. 2c). This approach yielded 42 missense hits (Supplementary Table S1), as compared to the 28 expected missense SNPs (significant enrichment, corrected Chi Square test, one-sided p = 0.0023). PBS, nSL, and p(PBS|DAFColla) statistics used alone yielded 25, 31 and 31 hits respectively (one-sided p-value not significant). Assuming missense variants are more likely to have an adaptive phenotypic effect, the data supports simulations in suggesting that combining p(PBS|DAFColla) and nSL scores increases power to detect early-stage selection. Based on the degree of enrichment, we expect approximately 14/42 top missense hits to be caused by non-neutral processes.
Figure 2.

Distribution of nSL + PBS and power calculations. (a) Observed joint distribution of nSL and p(PBS|DAF) of all exonic SNPs (blue), with top-1% outliers coloured teal (not missense), orange (missense) or red with black border (missense; the 11 highly damaging variants). (b) Simulated joint distribution of nSL and p(PBS|DAF), with X- and Y-axes as in (a). Results from neutral scenarios are shown in blue, with those from selected scenarios (s = 0.02) in red. Note the clustering of selected SNPs in the low-p(PBS|DAF), low-nSL regime, corresponding to that detected by our PBS + nSL selection statistic; also note the especial clustering of most damaging missense variants in (a) in this region of the plot. (c) Power from the simulations of the three statistics to detect selection, with selected variants with a p-value ≤ 0.0062 classified as successful detections. X- and Y-axes are derived allele frequency and power, respectively.
We found that 23/42 (54.8%) missense hits had evidence of a deleterious effect (as indicated by a deleterious SIFT or PolyPhen2 categorisation), compared to 960/2733 (35.1%) over all scored missense variants. This enrichment (corrected Chi-Square, one-sided p-value = 0.0066) suggests that PBS + nSL outliers are likely to be damaging. Given that we would expect 42 * 0.344 ≈ 14.7 damaging hits in 42 random missense variants, finding 23 suggests an excess 8–9 damaging missense variants that may be non-neutral.
The enrichment of selection signatures among deleterious missense variants with outlier PBS + nSL scores prompted us to define a set of variants with especially high-probability of being damaging. We supplemented SIFT and PolyPhen2 scores with CADD scores for our 42 missense hits and defined a high-probability damaging variants as (SIFT: “damaging”, PolyPhen2: “possibly damaging” or “probably damaging”, and CADD Phred >5). This identified 11 SNPs (Table 2).
Table 2.
Top PBS + nSL missense hits based on intermediate derived allele frequencies between 15–50%.
| Chr: Position | Identifiera | Ref | Alt | Allele frequency Collas | Allele frequency Lowlanders | Allele frequency Siberians | 1000 Genomes DAF | Gene | Amino acid change | PBS + nSL score |
|---|---|---|---|---|---|---|---|---|---|---|
| 8:24349417 | rs3736281 | T | C | 47% | 6% | 11% | 2.8% | ADAM7 | I453T | 0.000 |
| 6:142688969 | rs17280293 | A | G | 50% | 3% | 8% | 2.8% | GPR126 | S123G | 0.000 |
| 21:33689199 | rs762225 | G | C | 34% | 3% | 5% | 1.4% | URB1 | P2071R | 0.001 |
| 1:40766943 | rs140041506 | G | T | 37% | 6% | 5% | 0.7% | COL9A2 | P661T | 0.001 |
| 14:58958923 | rs3783697 | A | G | 29% | 6% | 32% | 2.4% | KIAA0586 | D1263G | 0.001 |
| 14:33165230 | rs543290190 | T | C | 16% | 0% | 0% | 0.1%b | AKAP6 | W972R | 0.002 |
| 3:77629200 | rs188582283 | C | T | 24% | 0% | 0% | 0.9% | ROBO2 | R811W | 0.002 |
| 2:46588031 | rs570553380 | A | G | 32% | 6% | 0% | 0.2%c | EPAS1 | H194R | 0.004 |
| 6:76631823 | rs76604824 | C | T | 24% | 3% | 3% | 4.6% | IMPG1 | D793N | 0.004 |
| 22:30888494 | rs17738527 | C | T | 50% | 16% | 0% | 14.4% | SEC. 14L4 | E211K | 0.005 |
| 16:3708170 | rs2791 | C | T | 24% | 0% | 0% | 1.7% | TRAP1 | R692H | 0.006 |
aAll variants were classed as possibly/probably damaging in PolyPhen 2 and damaging in SIFT and had a CADD score > 17, for values see Supplementary Table S1
bOnly found in 4 heterozygote Peruvians from Lima out of 85 individuals. cOnly found in 12 heterozygote Peruvians from Lima out of 85 individuals; lowland allele frequency strongly influenced by 20% allele frequency in Calchaquíes (n = 2). Chr = chromosome, Ref = reference allele, Alt = alternative allele, DAF = derived allele frequency.
Of these 11 SNPs, two (rs570553380 and rs543290190) are not found in 1000 Genome Phase 1 data and are polymorphic only in Peruvians from Lima in the extended 1000 Genome Phase 3 data set; this was only true for one other variant in our list of 42 missense hits. Tellingly, one of these two variants (rs570553380) is located in the EPAS1 gene, which is a major oxygen sensor and has been identified multiple times as subject to high altitude selection in Tibetans11–14. The other SNP (rs543290190) is located in AKAP6, which is involved in cardiac myocyte reaction to stresses such as hypoxia, hypertrophic remodelling of the heart25 and stabilises the hypoxia master regulator HIF-1α during hypoxia while enhancing its degradation at normoxia26. Furthermore, of the 11 shortlisted SNPs, rs17280293, located in GPR126, is the variant with the second most extreme nSL + PBS score. During the course of our data analysis, a study of a European cohort was published identifying a strong association between lung function and SNP rs148274477 (largest effect size and most extreme p-value reported in that study, p = 9.58e−26)27. In that cohort, the GPR126 SNP rs17280293 that we identify here is mentioned as being in strong LD with rs148274477 (r2 = 0.85). While the same level of LD is observed in our Colla population (r2 = 0.848) the intriguing possibility exists that it, rather than rs148274477, is driving a strong lung function association in a population unrelated to the Colla. The derived alleles at both SNPs in GPR126 rs148274477 and rs17280293 are associated with increased forced expiratory volume in the first second/forced vital capacity (FEV1/FVC). As we have FEV1/FVC measurements for our 19 Colla individuals2, we were able to speculatively test for a similar association in our small Colla sample. We could identify a positive association (Fig. 3; one-tailed p = 0.029) with each additional derived allele at rs17280293 associated with an increase in FEV1/FVC of 0.075 (standard error = 0.037). While this association does not persist when phenotypic and genetic data from Wichí (a lowland population) and Calchaquíes (an intermediate altitude population) are incorporated into the linear regression (one-tailed p = 0.158, data not shown) there could be several confounding factors such as population structure or an effect on FEV1/FVC associated with the altitude at which the lung function measurements were taken.
Figure 3.
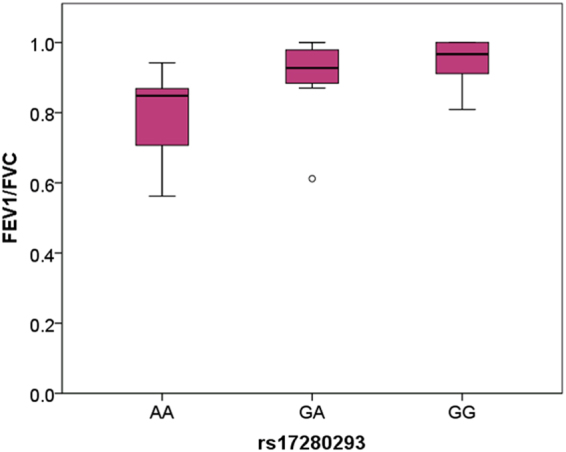
FEV1/FVC in Collas sorted by derived allele frequency. The ratio increases with number of derived alleles indicating an improvement of lung function in Collas. One-tailed p-value = 0.029, r2 = 0.195; FEV1: forced expiratory volume in the first second, FVC: forced vital capacity.
Based on a wide range of evidence - low worldwide frequency DAF (1000 Genome) and local population differentiation (PBS), relatively extended derived-allele haplotype homozygosity (nSL) and evidence for disruption of the protein (SIFT, PolyPhen2, CADD) – as well as evidence based on prior selection signals and gene function, we consider the two missense variants in EPAS1 and GPR126 as high-probability selection candidates.
Discussion
In this study, we have detected missense variants as novel candidates for local positive selection in the genomes of Andean highlanders with possible functional consequences. By combining different selection statistics, along with genome annotation information, we were able to identify intermediate frequency variants that may contribute to high altitude adaptation in two very prominent candidate genes, GPR126 and EPAS1. The first gene has been previously associated with lung function measurements; the second, with the main adaptive phenotype in Tibetans. In contrast, no functional variant could be identified in Collas by scans of late-stage selective sweeps.
Our first main finding is the second strongest hit in the intermediate frequency analysis originated from a missense SNP (rs17280293) in GPR126 (or ADGRG6, Adhesion G Protein-Coupled Receptor G6) found at 2.8% in 1000 Genomes Project data. This mutation causes an amino acid change (p.S123G) classed as damaging by SIFT and possibly damaging by PolyPhen2 within the seven transmembrane domain of the protein. Several previous studies have shown a strong association of this gene with lung function27,28. Furthermore, this SNP has been identified as in strong linkage disequilibrium with a variant (rs148274477) associated with FEV1/FVC. We found rs17280293 to be associated with FEV1/FVC in our own phenotype data, suggesting that Andeans with a derived allele may have better equipped lungs for life at high altitude. This is the first time a phenotype in Andeans could be correlated with genomic data. Taken together, the extreme PBS + nSL score, evidence of damaging effect, and association with lung function situate rs17280293 as an interesting selection candidate.
Our second main finding highlighted a missense SNP in the hypoxia inducible factor 2 α (HIF-2α, EPAS1) at intermediate frequencies in the Collas. While the HIF-1α is activated during short-term hypoxia, HIF-2α is rather expressed as a chronic hypoxia response29. Both transcription factors regulate a plethora of genes involved in energy metabolism and differential oxygen use during hypoxic conditions. EPAS1 has already been repeatedly shown to have a signature of selection in Tibetan populations11–14,30, but this is the first study to indicate selection also in Andeans. Since this gene was highlighted with a test investigating variants at intermediate levels it may indicate that a potentially advantageous allele of EPAS1 is on its way to higher frequencies also in Andeans but has not yet reached levels equivalent to Tibetans. Interestingly, the selected haplotype in Sherpas and Tibetans was suggested to be an introgression from the Denisovan genome31 and its frequency increased with altitude of populations in the Himalayas32. The Tibetan genotype has been associated with an inhibition of haemoglobin concentrations rise at high altitude11,14. However, the specific functional mechanism has not yet been identified in Tibetans. This might be due to the fact that all highlighted SNPs so far fall within non-coding regions of the gene33. In contrast, the highlighted variant in Andeans (rs570553380) encodes a missense mutation predicted to have a damaging effect on protein function. Knock-out studies in mice underlined the essential role of HIF-2α for normal erythropoiesis. Complete knock-out leads to a drastically reduced haematocrit34. Heterozygous knock-out mice had a similar haematocrit compared to wild type mice but showed a resistance to hypoxia induced pulmonary hypertension and thus no subsequent right ventricular hypertrophy35. This functional advantage under hypoxic conditions suggests a protective role of at least some loss of function mutations in EPAS1 for life at high altitude. In contrast, human patients with gain of function mutations were characterised by pulmonary hypertension and erythrocytosis showing a continuous activation of the HIF response36. Hypoxia induced pulmonary hypertension is a universal physiological response to lowlanders going to high altitude37 and is constantly present in Andeans38. While in healthy highlanders pressures are only moderately increased39 they are exaggerated in individuals suffering from chronic mountain sickness, which is more common in the Andean population than in other high altitude dwellers40. Pulmonary hypertension leads to a remodelling of the pulmonary vasculature, inducing changes which may result in right heart failure. Thus, a protective allele in EPAS1 might not only reduce the red blood cell count as seen in Tibetans but also protect from extensive pulmonary hypertension in the Andean population.
We based our results on 19 whole genome sequences from the Argentinean Collas. The results of this study should be confirmed in a larger sample possibly including further Andean high altitude populations. Additionally, further work to clarify the effect of the EPAS1 variant, for example by assessing any association with haematocrit in these groups, would be valuable.
In conclusion, we introduce a new approach for detecting early-stage selective sweeps which enables us to identify moderate frequency (0.15–0.50) derived variants in Andean high altitude populations as potential targets of recent selection. Among exonic variants with functional annotation we identify two missense variants in the GPR126 and EPAS1 genes. GPR126 is known to be involved in lung function and the selected EPAS1 variant might have a potential protective effect on hypoxia induced erythrocytosis and pulmonary hypertension. In case of the EPAS1 gene, our results are the first to point to a plausible target of selection at variant level. Considering this variant is restricted to South America these results provide strong evidence of convergent evolution in Andeans and Tibetans living with the same environmental pressures on opposite sides of the world. The approach of detecting early-stage selective sweeps has the potential to be applied to other cases of recent local selection.
Methods
Subjects and ethical approval
All participants provided their written informed consent. The study was approved by the by the Ethics Committee at the University of East Anglia and the Ministry of Health of the Province of Salta (Ministerio de Salud Pública, Salta, Argentina). Ethical approval for the whole genome sequencing of the samples was obtained from University of Cambridge Human Biology Research Ethics Committee (HBREC.2011.01). All methods were performed in accordance with the relevant guidelines and regulations. High altitude samples were collected from an Andean population, the Colla, living in Northern Argentina at an altitude >3500 m9. Native American lowland control samples for selection analyses originated from 5 Calchaquí and 4 Wichí from neighbouring regions in Argentina2, plus publicly available sequences from the Complete Genomics database including 5 Mexican (NA19648-200-37-ASM, NA19669-200-37-ASM, NA19735-200-37-ASM, NA19649-200-37-ASM, NA19670-200-37-ASM) and 2 Puerto Rican (HG00732-200-37-ASM, GS000015711-ASM) samples20. Additional North-East Siberian samples served as controls for threesided population statistics including 2 Chukchi, 4 Eskimo and 13 Koryak individuals19,20. Two additional Chukchi individuals were added in the demographic analyses.
Whole genome sequences
Whole genome sequences were obtained with the Complete Genomics platform and quality filtered as described elsewhere20. The newly sequenced Colla genomes were deposited in the European Nucleotide Archive: PRJEB14828. Data are also freely available through the Estonian Biocentre website. All genomes were annotated against the Ensembl GRCh37 database and compared to dbSNP Human Build 141 and Phase 1 of the 1000 Genomes Project dataset41 as described previously20. For 1000 Genomes variant frequency Phase 3 data42 was used. We focused on variants annotated as “exonic” in Ensembl occurring in Hardy-Weinberg equilibrium in our sample resulting in 402,227 autosomal variants. Variants with a combined annotation dependent depletion (CADD) score >5 were investigated further. The CADD score was calculated to estimate impact of synonymous and non-synonymous variable alike43. Missense variants were assessed with the in silico prediction programs SIFT44 and Polyphen245.
Tests of positive selection
Two statistics were calculated for each SNP – the population branch statistic (PBS)14 and the number of segregating sites by length (nSL)18. The PBS score was based on pairwise F ST values for the three populations using a weighted analysis of variance46 with the program GENEPOP’00747. PBS was estimated as in Yi and colleagues14. The nSL scores were calculated only on the Colla sample.
Two statistics were window based. We calculated Tajima’s D on the Colla sample using non-overlapping 200 kb windows17. A window-based nSL score was also calculated as the average proportion of common (minor allele frequency < 95%) SNPs with |nSL| > 2.0 in a 200 kb window.
In scans for late-stage hard sweeps we first identified the 200 kb windows that showed genome-wide the lowest 1% of Tajima’s D or nSL scores. The top 1% PBS scoring variants in these windows were further filtered for SNPs with less than 10% frequency in the 1000 Genomes Phase 3 data. We only retained those with a CADD score higher than 5.
Intermediate frequency based selection
As the timescale of human occupation of the Andes is relatively short21, we supplemented our selection scans with an attempt to detect positive selection acting on intermediate frequency variants. To this end, we first chose to focus on SNP-by-SNP selection statistics, considering each SNP a potential target of selection. Although this vastly increases the number of tests we performed, early-stage selective sweeps have not yet had a substantial impact on variation far from the selected locus such that they may be difficult to detect using average statistic values or the number of outliers over large genomic windows. We first calculated the two statistics PBS and nSL for each SNP in the Colla sample. For nSL approximate p-values were generated, based on nSL scores that were standardised in derived allele frequency (DAF) bins assuming unstandardised SL follows a roughly normal distribution in frequency bins. PBS values were calculated using lowland Americans as the comparative closely related control population and Siberian populations as outgroup. These were then converted into empirical p-values given DAF in Collas based on 4% DAF bins (e.g. if a PBS value for the 15% ≤ DAFColla < 19% bin is the 990th largest of 1000 values, its p(PBS|DAFColla) would be 0.01). nSL has been shown to have power to detect early-stage selective sweeps18, while PBS has been an effective test-statistic for detecting local selection. Simulations suggest that combining these two statistics using Fisher’s method provides a composite statistic with high power to detect early-stage sweeps (see Fig. 2).
To reduce the number of hypothesis tests and focus on variants for which annotation information exists, we cut our Fisher-combined PBS + nSL score data to SNPs in the exome (Ensemble Biomart B37Rel75), and only considered SNPs with DAF in the Colla of 15% ≤ DAF ≤ 50% to focus on early-stage selective sweeps. We considered the set of top 1% most extreme statistic value SNPs as enriched for SNPs subject to or impacted by selection. We filtered this set for missense variants and obtained CADD, SIFT and Polyphen2 scores for each SNP.
Multiple sequentially Markovian coalescent analyses
Effective population sizes (Ne) and population split times between Collas and other Native Americans were estimated using multiple sequentially Markovian coalescent model (MSMC)48 on up to four phased genomes per population, as described elsewhere20. To provide a comprehensive picture of the changes in effective population size over time of each of the analysed populations, the MSMC curves obtained using 1, 2 or 4 genomes were joined together at points of their overlap. The joining was performed to maximise the MSMC accuracy within a given time interval, as suggested by Schiffels and Durbin48 and implemented by Clemente, Cardona and colleagues49.
Simulations
Simulations were performed using the forward-time population genetic simulator SLiM50. Briefly, we generated a null distribution of nSL, p(PBS|DAF) and the combined nSL + PBS statistic by generating 250 neutral simulations of the Koryak, Wichí and Colla populations, taking population sizes and population split times from our MSMC analysis. Here, the Colla and Wichí split 5478 years before present (ybp), and the Koryak and ancestral Wichí/Colla population 19451 ybp. These split times are inferred based on cross-coalescence, and no migration is included in the model. We assumed a generation time of 25 years, and started the model at 1 million years ago (with a short, 500-generation burn-in). In each simulation, we generated 1 MB of genetic data, using a mutation rate of 1.7e−8/generation and human-realistic variable recombination rate by randomly sampling from non-centromeric regions of the chromosome 2 of the HapMap combined genetic map51. At the end of each simulation we retrieved samples according to our study (38 Colla chromosomes, 32 Wichí and 38 Koryak), and then randomly chose 50 mutations with DAF 0.15–0.75 in the Colla sample from the central region (450 kb-550 kb) of the data. We calculated SL and PBS for each of these mutations.
To approximate the distribution of these statistics under selection, we performed 160 simulations following the neutral model until the time of the Colla/Wichí split. At this point, for 20 times in each replicate, we randomly chose a variant in the central region (450 kb-550 kb) and applied positive selection (heterozygote s = 0.01, homozygote derived s = 0.02) for the next 5478 years in the Colla population. We restarted any of the 20 ‘sub-replicate’ routines if the selected allele did not have 0.15 ≤ DAF ≤ 0.75 at sampling time in the Colla sample. On successful completion of a sub-replicate, SL and PBS were calculated for the selected variant.
For both neutral and selected results, SL was normalised to nSL and PBS to p(PBS|DAF) using 5% frequency bins and the neutral distribution of SL and PBS. The nSL score was converted to an approximate p-value by assuming a standard normal distribution of SL in frequency bins, and this was combined with p(PBS|DAF) using Fisher’s method. Each frequency bin included at least 440 neutral results and 100 selected results. Power was estimated using the Fisher-combined p-values with a p-value cut off of 0.0064 (corresponding to the top-1% combined p-value in our genomic data, with weak correlations between nSL and p(PBS|DAF) driving this from the expected value of 0.01).
Genotype-phenotype assessment
Lung function measurements of forced vital capacity (FVC) and forced expiratory volume 1 second (FEV1) for Collas were reported previously2. A ratio of FEV1/FVC was calculated and linear regression was used to assess effects of the derived allele with a one-sided p-value.
Electronic supplementary material
Acknowledgements
We would like to thank all participants of this study. This work was supported by the European Research Council with a ERC Starting Investigator grant (grant number FP7 - 261213 to T.K.); the Singapore Ministry of Education (grant number MOE2015-T2-1-127, supporting G.S.J.); the Estonian Research Council with an Estonian Institutional Research grant (grant number IUT24-1 to E.M., G.H., M.Me., M.Mi., R.M., T.K.) and an Estonian Research Infrastructure Roadmap grant for sequencing (grant number 3.2.0304.11-0312); the European Commission with an European Regional Development Fund through the Centre of Excellence in Genomics to the Estonian Biocentre (to E.M., G.H., M.Me., M.Mi., R.M., T.K.); the European Union with a New Zealand Estonian French Research Exchange Scheme grant (People Marie Curie Actions), International Research Staff Exchange Scheme, call FP7-PEOPLE-2012-IRSES-number (grant number 318979 to M.Me., G.H.); the Estonian Science Foundation (grant number 8973 to M.Me.); the National Geographic Society with a Young Explorers Grant (grant number 8900-11 to C.A.E.); the University of East Anglia with a Starting Investigator Grant (grant number RC-158 to M.Mo.).
Author Contributions
T.K., C.A.E., G.S.J. designed and initiated the study. Colla samples were collected by C.A.E., M.Mo. DNA was extracted and prepared for sequencing at Complete Genomics by C.A.E., T.K. and E.M. Quality checks were run and sequence data were prepared for further analyses by A.C., E.M., L.P., M.Mi., R.M., G.H., M.Me., T.A. L.P. carried out the MSMC analysis. A.C., A.M., C.E.I., F.J.C., G.H., G.S.J., L.P., T.A. performed selection tests. G.S.J., T.J.S., T.K. developed the approach for detecting early-stage selective sweeps. C.A.E., G.S.J., M.Mo., T.K. wrote the initial draft of the manuscript. All authors have critically revised the manuscript and approved the final version.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Guy S. Jacobs and Toomas Kivisild jointly supervised this work.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-13382-4.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Christina A. Eichstaedt, Email: Christina.eichstaedt@med.uni-heidelberg.de
Guy S. Jacobs, Email: guysjacobs@ntu.edu.sg
References
- 1.Pleurdeau D. Human Technical Behavior in the African Middle StoneAge: The Lithic Assemblage of Porc-Epic Cave (Dire Dawa, Ethiopia) Afr. Archaeol. Rev. 2005;22:177–197. doi: 10.1007/s10437-006-9000-7. [DOI] [Google Scholar]
- 2.Eichstaedt CA, et al. Genetic and phenotypic differentiation of an Andean intermediate altitude population. Physiol. Rep. 2015;3:e12376. doi: 10.14814/phy2.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rupert JL, Hochachka PW. Genetic approaches to understanding human adaptation to altitude in the Andes. J. Exp. Biol. 2001;204:3151–3160. doi: 10.1242/jeb.204.18.3151. [DOI] [PubMed] [Google Scholar]
- 4.Frank, S. Pueblos Originarios de América. (Ediciones de Sol S.R.L., 2008).
- 5.Rothhammer F, Silva C. Peopling of Andean South America. Am. J. Phys. Anthropol. 1989;78:403–410. doi: 10.1002/ajpa.1330780308. [DOI] [PubMed] [Google Scholar]
- 6.Acreche N, et al. Diversidad biológica humana en la provincia de Salta (Human biological diversity in Salta) Cuadernos FHYCS-UNJu. 2004;22:171–194. [Google Scholar]
- 7.Rothhammer F, Santoro C. Cultural development in the Azapa Valley in the far north of Chile, and its connection with population displacement in the highlands. Latin American Antiquity. 2001;12:59–66. doi: 10.2307/971757. [DOI] [Google Scholar]
- 8.Bigham AW, et al. Identifying positive selection candidate loci for high-altitude adaptation in Andean populations. Hum. Genomics. 2009;4:79–90. doi: 10.1186/1479-7364-4-2-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eichstaedt CA, et al. The Andean Adaptive Toolkit to Counteract High Altitude Maladaptation: Genome-wide and Phenotypic Analysis of the Collas. PLoS One. 2014;9:e93314. doi: 10.1371/journal.pone.0093314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valverde, G. et al. A novel candidate region for genetic adaptation to high altitude in Andean populations. PLoS One10, e0125444 (2015). [DOI] [PMC free article] [PubMed]
- 11.Beall CM, et al. Natural selection on EPAS1 (HIF2α) associated with low hemoglobin concentration in Tibetan highlanders. Proc. Natl. Acad. Sci. USA. 2010;107:11459–11464. doi: 10.1073/pnas.1002443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bigham AW, et al. Andean and Tibetan patterns of adaptation to high altitude. Am. J. Hum. Biol. 2013;25:190–197. doi: 10.1002/ajhb.22358. [DOI] [PubMed] [Google Scholar]
- 13.Simonson TS, McClain DA, Jorde LB, Prchal JT. Genetic determinants of Tibetan high-altitude adaptation. Hum. Genet. 2012;131:527–533. doi: 10.1007/s00439-011-1109-3. [DOI] [PubMed] [Google Scholar]
- 14.Yi X, et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. 2010;329:75–78. doi: 10.1126/science.1190371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou D, et al. Whole-Genome Sequencing Uncovers the Genetic Basis of Chronic Mountain Sickness in Andean Highlanders. Am. J. Hum. Genet. 2013;93:452–462. doi: 10.1016/j.ajhg.2013.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Instituto Nacional de Estadística y Censos de la República Argentina (INDEC). Encuesta Complementaria de Pueblos Indígenas 2004–2005 [Complementary Survey of Indigenous People 2004-2005]. (Buenos Aires, Argentina, 2005).
- 17.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrer-Admetlla A, Liang M, Korneliussen T, Nielsen R. On detecting incomplete soft or hard selective sweeps using haplotype structure. Mol. Biol. Evol. 2014;31:1275–1291. doi: 10.1093/molbev/msu077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cardona A, et al. Genome-wide analysis of cold adaptation in indigenous Siberian populations. PLoS One. 2014;9:e98076. doi: 10.1371/journal.pone.0098076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pagani L, et al. Genomic analyses inform on migration events during the peopling of Eurasia. Nature. 2016;538:238–242. doi: 10.1038/nature19792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aldenderfer M. The Pleistocene/Holocene transition in Peru and its effects upon human use of the landscape. Quat. Int. 1999;53/54:11–19. doi: 10.1016/S1040-6182(98)00004-4. [DOI] [Google Scholar]
- 22.Sabeti PC, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng K, Fu YX, Shi S, Wu CI. Statistical tests for detecting positive selection by utilizing high-frequency variants. Genetics. 2006;174:1431–1439. doi: 10.1534/genetics.106.061432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Passariello CL, Li J, Dodge-Kafka K, Kapiloff MS. mAKAP-a master scaffold for cardiac remodeling. J. Cardiovasc. Pharmacol. 2015;65:218–225. doi: 10.1097/FJC.0000000000000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong W, et al. mAKAP compartmentalizes oxygen-dependent control of HIF-1α. Sci. Signal. 2008;1:ra18. doi: 10.1126/scisignal.2000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soler Artigas M, et al. Sixteen new lung function signals identified through 1000 Genomes Project reference panel imputation. Nat. Commun. 2015;6:8658. doi: 10.1038/ncomms9658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hancock DB, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat. Genet. 2010;42:45–52. doi: 10.1038/ng.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uchida T, et al. Prolonged hypoxia differentially regulates hypoxia-inducible factor (HIF)-1α and HIF-2α expression in lung epithelial cells: implication of natural antisense HIF-1α. J. Biol. Chem. 2004;279:14871–14878. doi: 10.1074/jbc.M400461200. [DOI] [PubMed] [Google Scholar]
- 30.Peng Y, et al. Genetic variations in Tibetan populations and high-altitude adaptation at the Himalayas. Mol. Biol. Evol. 2011;28:1075–1081. doi: 10.1093/molbev/msq290. [DOI] [PubMed] [Google Scholar]
- 31.Huerta-Sanchez E, et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature. 2014;512:194–197. doi: 10.1038/nature13408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hackinger S, et al. Wide distribution and altitude correlation of an archaic high-altitude-adaptive EPAS1 haplotype in the Himalayas. Hum. Genet. 2016;135:393–402. doi: 10.1007/s00439-016-1641-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bigham AW, Lee FS. Human high-altitude adaptation: forward genetics meets the HIF pathway. Genes Dev. 2014;28:2189–2204. doi: 10.1101/gad.250167.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scortegagna M, et al. The HIF family member EPAS1/HIF-2alpha is required for normal hematopoiesis in mice. Blood. 2003;102:1634–1640. doi: 10.1182/blood-2003-02-0448. [DOI] [PubMed] [Google Scholar]
- 35.Brusselmans K, et al. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J. Clin. Invest. 2003;111:1519–1527. doi: 10.1172/JCI15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gale DP, et al. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood. 2008;112:919–921. doi: 10.1182/blood-2008-04-153718. [DOI] [PubMed] [Google Scholar]
- 37.West JB. Pulmonary hypertension at high altitude. High. Alt. Med. Biol. 2013;14:89. doi: 10.1089/ham.2013.1421. [DOI] [PubMed] [Google Scholar]
- 38.Penaloza D, Arias-Stella J. The heart and pulmonary circulation at high altitudes: healthy highlanders and chronic mountain sickness. Circulation. 2007;115:1132–1146. doi: 10.1161/CIRCULATIONAHA.106.624544. [DOI] [PubMed] [Google Scholar]
- 39.Soria R, et al. Pulmonary artery pressure and arterial oxygen saturation in people living at high or low altitude: systematic review and meta-analysis. J. Appl. Physiol. 2016;121:1151–1159. doi: 10.1152/japplphysiol.00394.2016. [DOI] [PubMed] [Google Scholar]
- 40.Hainsworth R, Drinkhill MJ. Cardiovascular adjustments for life at high altitude. Respir. Physiol. Neurobiol. 2007;158:204–211. doi: 10.1016/j.resp.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 41.The 1000 Genomes Project Consortium et al. An integrated map of genetic variation from 1,092 human genomes. Nature491, 56–65 (2012). [DOI] [PMC free article] [PubMed]
- 42.The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature526, 68–74 (2015). [DOI] [PMC free article] [PubMed]
- 43.Kircher M, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 45.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 47.Rousset F. Genepop'007: a complete reimplementation of the Genepop software for Windows and Linux. Mol. Ecol. Resour. 2008;8:103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- 48.Schiffels S, Durbin R. Inferring human population size and separation history from multiple genome sequences. Nat. Genet. 2014;46:919–925. doi: 10.1038/ng.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clemente FJ, et al. A Selective Sweep on a Deleterious Mutation in CPT1A in Arctic Populations. Am. J. Hum. Genet. 2014;95:584–589. doi: 10.1016/j.ajhg.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Messer PW. SLiM: simulating evolution with selection and linkage. Genetics. 2013;194:1037–1039. doi: 10.1534/genetics.113.152181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.International HapMap C, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.