

Abstract
Skeletal muscle ageing is characterised by atrophy, a deficit in specific force generation, increased susceptibility to injury, and incomplete recovery after severe damage. The hypothesis that increased generation of reactive oxygen species (ROS) in vivo plays a key role in the ageing process has been extensively studied, but remains controversial. Skeletal muscle generates ROS at rest and during exercise. ROS can cause oxidative damage particularly to proteins. Indeed, products of oxidative damage accumulate in skeletal muscle during ageing and the ability of muscle cells to respond to increased ROS becomes defective. The aim of this review is to examine the evidence that ROS manipulation in peripheral nerves and/or muscle modifies mechanisms of proteostasis in skeletal muscle and plays a key role in initiating sarcopenia.
Keywords: Cu/Zn superoxide dismutase, frailty, neuromuscular homeostasis, oxidative stress, sarcopenia
Abbreviations
- CMA
chaperone‐mediated autophagy
- Cu/ZnSOD
copper, zinc superoxide dismutase
- EMG
electromyography
- HSP
heat shock protein
- LAMP‐2A
lysosome‐associated membrane protein type 2A
- MA
macroautophagy
- MnSOD
manganese superoxide dismutase
- mtROS
mitochondrial ROS
- NFκB
nuclear transcription factor kappa B
- NMJs
neuromuscular junctions
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- Sod1
copper, zinc superoxide dismutase
- WT
wild‐type
Introduction
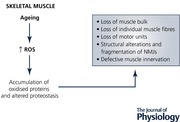
A 30–50% loss of muscle mass occurs between the ages of 50 and 80 years (Fried et al. 2001; Bortz, 2002; Espinoza & Walston, 2005; Cesari et al. 2006) that impacts profoundly on the quality of life of older people resulting in a reduced ability to carry out everyday tasks and increased likelihood of falling. All individuals lose muscle mass and develop age‐related muscle weakness (termed sarcopenia when it reaches clinically relevant severity), although some individuals are more prone to sarcopenia. The underlying mechanisms are unclear, but an individual's initial muscle mass appears to be a critical factor influencing the risk of developing sarcopenia. For example, veteran athletes lose muscle mass at the same rate as sedentary individuals, but have a high peak muscle mass in earlier life and reach the threshold for poor function at a later age than sedentary individuals (Pearson et al. 2002). Muscle mass is dictated by the number of muscle fibres and the size of the fibres. The decline in muscle mass and strength in people after the age of ∼50 appears primarily due to loss of muscle fibres with weakening of the remaining fibres (Marzetti et al. 2009). For example, in humans, Lexell et al. (1988) reported 40% fewer fibres in the vastus lateralis quadriceps muscles of older individuals (Lexell et al. 1988).
Data clearly indicate that in ageing man and rodents, loss of motor neurons accompanies the loss of muscle fibres (Campbell et al. 1973; Brown et al. 1988; Einsiedel & Luff, 1992; Larsson & Ansved, 1995), but whether motor neuron loss is a cause or a consequence of muscle fibre deficits has not been definitively established due largely to the lack of available techniques to directly count motor units (a motor unit consists of a single α‐motor neuron and all of the muscle fibres it innervates) in humans. Measurements have been limited to post mortem anatomical estimates or estimates based on electromyography (EMG). Post mortem anatomical studies have shown that people aged 75 years have 30% fewer motor neurons supplying the muscles of the lower limbs compared with young adults (Kawamura et al. 1977; Tomlinson & Irving, 1977; Mittal & Logmani, 1987). One of the first studies to demonstrate motor unit loss during healthy ageing using EMG was by Campbell et al. (1973), who recorded evoked potentials and compared these with maximum M‐waves to estimate that there were 50% fewer motor units in the extensor digitorum brevis in people above 60 years. Brown et al (1988) also demonstrated that subjects over 60 years of age have approximately half the numbers of motor units of subjects less than 60 years of age (Brown et al. 1988) and more recently, using intramuscular and surface EMG signals from the vastus lateralis during voluntary contractions, Piasecki et al. demonstrated that the total number of motor units in muscles from older individuals (above 65 years) was between 50% and 60% lower compared to those in young (Piasecki et al. 2016b). The range of techniques available to estimate motor unit numbers in humans and their limitations have been reviewed elsewhere (Piasecki et al. 2016a).
Several studies have also reported swollen, segmental demyelinated and remyelinated axons in peripheral nerve of old animals and humans (Sharma et al. 1980; Grover‐Johnson & Spencer, 1981; Adinolfi et al. 1991; Verdu et al. 2000) and such neuronal changes have been proposed to play a major role in the age‐related loss of muscle mass and function (Delbono, 2003). Neuromuscular junctions (NMJs) in muscle fibres of old mice show a variety of alterations, including axonal swellings, sprouting, synaptic detachment, withdrawal of axons from postsynaptic sites and fragmentation of the acetylcholine receptors (AChRs) (Valdez et al. 2010; Chai et al. 2011; Vasilaki et al. 2016).
Reactive oxygen species and their role in the loss of skeletal muscle mass during ageing
Regulated changes in reactive oxygen species (ROS) formation are important in signalling to maintain normal physiological processes in all cells including muscles and neurons. The main physiological mechanism by which cells regulate ROS activities (and hence protect against oxidative damage) is by modification of the expression and activities of regulatory enzymes such as manganese superoxide dismutase (MnSOD), copper, zinc superoxide dismutase (Cu/ZnSOD), catalase, glutathione peroxidases and haem oxygenase‐1 (Powers & Jackson, 2008). Acute increases in ROS generation in skeletal muscle lead to activation of a number of redox‐sensitive transcription factors, including nuclear transcription factor kappa B (NFκB) and activator protein‐1 (Vasilaki et al. 2006b; Gomez‐Cabrera et al. 2008) with the subsequent increased expression of antioxidant defence enzymes such as superoxide dismutase (SOD) and catalase and cytoprotective proteins such as heat shock proteins (HSPs) (McArdle et al. 2001; Vasilaki et al. 2006b). Motor neurons also have the capacity to upregulate some of these cytoprotective proteins in response to exogenous reactive oxygen and nitrogen species (Bishop et al. 1999).
ROS activities in many tissues increase with age and there is evidence that increased ROS generation may be involved in several age‐related pathologies. In skeletal muscle, in vitro and in vivo studies have provided evidence of an age‐related increase in ROS production (Vasilaki et al. 2006a; Palomero et al. 2013). Increased ROS production can lead to changes in the redox state of muscle cells with potentially serious effects on muscle such as cumulative damage to cellular macromolecules including lipids in cell membranes, DNA, and subcellular membranes and structures. A major effect of aberrant ROS generation is oxidative damage to proteins (Levine & Stadtman, 2001; Dalle‐Donne et al. 2003; Ghezzi & Bonetto, 2003). There is a large body of evidence supporting the accumulation of ROS‐induced damaged proteins in senescent cells and tissues from old animals and our previous work has shown that muscles of old wild‐type (WT) mice have an elevated content of a marker of oxidative damage, 3‐nitrotyrosine residues, in the major cytosolic protein carbonic anhydrase III (Vasilaki et al. 2007) and increased protein carbonyl content (Broome et al. 2006) in comparison with muscles from adult WT mice. Accumulation of oxidised proteins can lead to formation of insoluble protein aggregates, and therefore carbonylated proteins and other irreversibly modified proteins must be degraded in order to prevent them from forming aggregates.
The main players in proteostasis maintenance influencing skeletal muscle are the chaperones, the calcium‐dependent calpains, and the ubiquitin–proteasome and the lysosome–autophagy systems. These components are responsible for the fate of unfolded, misfolded or oxidised proteins, i.e. whether they will refold into their original conformation or whether they will be removed from the cell (Kaushik & Cuervo, 2015; Anthony, 2016; Hohn et al. 2017). When a protein is targeted for degradation, chaperones often dictate which proteolytic pathway these unfolded, misfolded or oxidised proteins will follow. Many chaperones are HSPs, which are named according to their molecular mass, e.g. the HSP70 family (which consists of the constitutively expressed HSC70 and highly inducible HSP70) and HSP90, and interventions that maintain overexpression of HSPs, such as HSP70, prevent the accumulation of oxidative damage and preserve some aspects of age‐related muscle dysfunction (McArdle et al. 2004; Broome et al. 2006).
During chaperone‐mediated autophagy (CMA), substrate proteins are first recognised and bind to heat shock cognate, HSC70. The resulting complex is then targeted to the lysosomes by binding to the lysosomal CMA receptor known as lysosome‐associated membrane protein type 2A (LAMP‐2A), at which point the target protein is unfolded and translocated into the lysosomal lumen for degradation (Cuervo & Wong, 2014; Zhou et al. 2017). CMA operates at basal conditions in most mammalian cells, but it is mostly activated in response to stressors, such as oxidative stress (Xilouri & Stefanis, 2016). The activity of CMA has been shown to decline with age in some tissues such as the central nervous system and this decline, which associates with the accumulation of damaged/oxidised/aggregated proteins has been proposed to contribute to tissue dysfunction and possibly to some common age‐related human disorders, such as Parkinson's and Alzheimer's disease (Xilouri & Stefanis, 2016). However, little is known about CMA in other tissues such as skeletal muscle during ageing. Our previous work has shown that oxidative damage to proteins in muscle during ageing is associated with an increase in HSC70 content in quiescent muscles of old mice (Vasilaki et al. 2006b) suggesting that muscles of old mice are trying to adapt in order to prevent the accumulation of oxidised proteins. This is in contrast to the findings from a recent study by Zhou and colleagues who demonstrated decreased protein levels of HSC70 and LAMP‐2A in muscle from old mice but an increase in ubiquitinated proteins (Zhou et al. 2017). Currently, the function of CMA in skeletal muscle is not well understood and since we did not measure LAMP‐2A or ubiquitinated proteins in our study, we can only speculate that the increased content of HSC70 in muscles of old mice does not result in functional CMA as it may be the case that the formation of damaged proteins overwhelms their degradation and instead contributes to protein aggregation and subsequent loss in proteostasis mechanisms. It is also worth noting that once misfolded proteins organise into oligomers or insoluble aggregates, the only option for their elimination is by degradation in lysosomes via macroautophagy (MA) or expulsion outside the cell by exosomes (Kaushik & Cuervo, 2015). Therefore further studies are required in order to identify the exact mechanisms involved.
A novel mouse model of frailty: the Cu/ZnSOD knockout mouse
A clear link between age‐related muscle loss and increased ROS production has been indicated by studies of mice lacking Cu/ZnSOD (Sod1−/− mice). These mice have a significantly shortened lifespan (∼30%) compared to WT mice and have an accelerated decline in muscle mass and function (Muller et al. 2006). In addition, adult Sod1−/− mice show loss of motor function and contractility, declines in nerve conduction, decline in the number of motor units, partial denervation, degeneration of NMJs and increased muscle mitochondrial ROS (mtROS) generation and mitochondrial dysfunction (Flood et al. 1999; Shefner et al. 1999; Jang et al. 2010; Vasilaki et al. 2010; Larkin et al. 2011; Sims‐Robinson et al. 2013; Deepa et al. 2017). In contrast to adult WT mice, but in common with old WT mice, adult mice lacking Cu/ZnSOD have an elevated content of 3‐nitrotyrosine residues (Vasilaki et al. 2007) and also demonstrate a constitutive activation of NFκB and a constitutive increase in the content of HSPs in muscle at rest (Vasilaki et al. 2010).
The great extent to which changes in Sod1−/− mice mimic normal ageing indicates that Sod1−/− mice provide a model in which to study mechanistic links between oxidative stress and sarcopenia and to gain insight into mechanisms of age‐associated atrophy and weakness. While strong associations exist between degeneration of NMJs and declines in mass and force both in Sod1−/− mice and with normal ageing (Jang & Van Remmen, 2011), knowledge of whether age‐associated muscle wasting and weakness are due to changes proximal or distal to neuromuscular synapses is a major gap in our understanding of sarcopenia. To address questions of the relative importance of pre‐ vs. postsynaptic changes, we developed unique mouse models with tissue‐specific targeting of Cu/ZnSOD. These models have generated several key findings. (1) Partial restoration of Cu/ZnSOD only in neurons of Sod1−/− mice prevented the increases in muscle mtROS production, premature muscle atrophy, and weakness observed in Sod1−/− mice (Sakellariou et al. 2014). In contrast to Sod1−/− mice, the level of protein nitration and the protein content of a peroxynitrite reductase, peroxiredoxin 5 (PRXV), as well as the contents of key HSPs in skeletal muscle from these mice, were not different from WT levels, indicating no change in the overall redox status. (2) Mice with Sod1 deficiency in neurons alone (nSod1KO mice) do not show atrophy in gastrocnemius muscles, show only mild weakness and limited evidence of NMJ disruption, and show no significant changes in either mtROS generation or oxidative damage measured by 3‐nitrotyrosine residues suggesting that Cu/ZnSOD deficit in the motor neuron alone is not sufficient to initiate a full sarcopenic phenotype (Sataranatarajan et al. 2015). (3) Finally, mice lacking Cu/ZnSOD only in muscle fibres do not show NMJ degeneration or muscle atrophy, show no changes in the HSP content, and oxidative damage is not elevated, but they do show weakness and increased susceptibility to injury (Zhang et al. 2013). Collectively, these data suggest that redox homeostasis in motor neurons is a critical factor in initiating sarcopenia, but that the progression of sarcopenia is determined by complex interactions between both pre‐ and postsynaptic factors (Fig. 1).
Figure 1. Cu/ZnSOD deficits in either the motor neuron or muscle alone are not sufficient to initiate a full sarcopenic phenotype as seen in Sod1−/− mice.

Because adult mice lacking Cu/ZnSOD reproduce many of the main features seen in old WT mice, they may indicate important mechanisms that lead to loss of muscle fibres and function that are relevant to the ageing of WT mice. The Sod1−/− mice as well as our other novel mouse models with tissue specific modulation of Cu/ZnSOD have also demonstrated the importance of nerve–muscle interactions in the maintenance of neuromuscular function where ROS homeostasis is altered. Because our data indicate that motor neuron deficits arising from an oxidised redox status are critical in sarcopenia, our future work will focus on determining the impact of oxidative stress in motor neurons on NMJ formation and maintenance and the impact of directly disrupting NMJs on key postsynaptic muscle functions.
In summary, current findings support the hypothesis that increased generation of ROS is an important component of the ageing process, providing a link between accumulation of oxidative damage and muscle dysfunction.
Additional information
Competing interests
None
Author contributions
A.V. produced the manuscript. All authors approved the final version of the manuscript and all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The authors would like to thank National Institutes of Health AG051442 for funding. A.R. and H.V.R. are supported by a Senior Research Career Scientist award from the US Department of Veterans Affairs.
Acknowledgements
The authors would like to thank their present and previous collaborators and colleagues.
Biography
Aphrodite Vasilaki (Liverpool), Arlan Richardson (Oklahoma), Holly Van Remmen (Oklahoma), Susan Brooks (Michigan), Lisa Larkin (Michigan), Anne McArdle (Liverpool) and Malcolm Jackson (Liverpool) (shown left to right) are long term collaborators. This collaborative work was initiated in 2002 by Professor John Faulkner from the Muscle Mechanics Laboratory at the University of Michigan. The major funding for the collaborative work comes from a Program‐project supported by the US National Institute on Aging (NIH). The overall aim of the work is to understand the role of ROS in normal muscle physiology and skeletal muscle ageing.
![]()
This review was presented at the symposium ‘The Modulation of Aging through Altered Proteostasis’ which took place at Experimental Biology 2017, Chicago, USA, 22–26 April 2017.
References
- Adinolfi AM, Yamuy J, Morales FR & Chase MH (1991). Segmental demyelination in peripheral nerves of old cats. Neurobiol Aging 12, 175–179. [DOI] [PubMed] [Google Scholar]
- Anthony TG (2016). Mechanisms of protein balance in skeletal muscle. Domest Anim Endocrinol 56(Suppl), S23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop A, Marquis JC, Cashman NR & Demple B (1999). Adaptive resistance to nitric oxide in motor neurons. Free Radic Biol Med 26, 978–986. [DOI] [PubMed] [Google Scholar]
- Bortz WM 2nd (2002). A conceptual framework of frailty: a review. J Gerontol A Biol Sci Med Sci 57, M283–M288. [DOI] [PubMed] [Google Scholar]
- Broome CS, Kayani AC, Palomero J, Dillmann WH, Mestril R, Jackson MJ & McArdle A (2006). Effect of lifelong overexpression of HSP70 in skeletal muscle on age‐related oxidative stress and adaptation after nondamaging contractile activity. FASEB J 20, 1549–1551. [DOI] [PubMed] [Google Scholar]
- Brown WF, Strong MJ & Snow R (1988). Methods for estimating numbers of motor units in biceps‐brachialis muscles and losses of motor units with aging. Muscle Nerve 11, 423–432. [DOI] [PubMed] [Google Scholar]
- Campbell MJ, McComas AJ & Petito F (1973). Physiological changes in ageing muscles. J Neurol Neurosurg Psychiatry 36, 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesari M, Leeuwenburgh C, Lauretani F, Onder G, Bandinelli S, Maraldi C, Guralnik JM, Pahor M & Ferrucci L (2006). Frailty syndrome and skeletal muscle: results from the Invecchiare in Chianti study. Am J Clin Nutr 83, 1142–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai RJ, Vukovic J, Dunlop S, Grounds MD & Shavlakadze T (2011). Striking denervation of neuromuscular junctions without lumbar motoneuron loss in geriatric mouse muscle. PLoS One 6, e28090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuervo AM & Wong E (2014). Chaperone‐mediated autophagy: roles in disease and aging. Cell Res 24, 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle‐Donne I, Rossi R, Giustarini D, Milzani A & Colombo R (2003). Protein carbonyl groups as biomarkers of oxidative stress. Clin Chim Acta 329, 23–38. [DOI] [PubMed] [Google Scholar]
- Deepa SS, Bhaskaran S, Espinoza S, Brooks SV, McArdle A, Jackson MJ, Van Remmen H & Richardson A (2017). A new mouse model of frailty: the Cu/Zn superoxide dismutase knockout mouse. Geroscience 39, 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbono O (2003). Neural control of aging skeletal muscle. Aging Cell 2, 21–29. [DOI] [PubMed] [Google Scholar]
- Einsiedel LJ & Luff AR (1992). Alterations in the contractile properties of motor units within the ageing rat medial gastrocnemius. J Neurol Sci 112, 170–177. [DOI] [PubMed] [Google Scholar]
- Espinoza S & Walston JD (2005). Frailty in older adults: insights and interventions. Cleve Clin J Med 72, 1105–1112. [DOI] [PubMed] [Google Scholar]
- Flood DG, Reaume AG, Gruner JA, Hoffman EK, Hirsch JD, Lin YG, Dorfman KS & Scott RW (1999). Hindlimb motor neurons require Cu/Zn superoxide dismutase for maintenance of neuromuscular junctions. Am J Pathol 155, 663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried LP, Tangen CM, Walston J, Newman AB, Hirsch C, Gottdiener J, Seeman T, Tracy R, Kop WJ, Burke G & McBurnie MA (2001). Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci 56, M146–M156. [DOI] [PubMed] [Google Scholar]
- Ghezzi P & Bonetto V (2003). Redox proteomics: identification of oxidatively modified proteins. Proteomics 3, 1145–1153. [DOI] [PubMed] [Google Scholar]
- Gomez‐Cabrera MC, Domenech E & Vina J (2008). Moderate exercise is an antioxidant: upregulation of antioxidant genes by training. Free Radic Biol Med 44, 126–131. [DOI] [PubMed] [Google Scholar]
- Grover‐Johnson N & Spencer PS (1981). Peripheral nerve abnormalities in aging rats. J Neuropathol Exp Neurol 40, 155–165. [DOI] [PubMed] [Google Scholar]
- Hohn A, Weber D, Jung T, Ott C, Hugo M, Kochlik B, Kehm R, Konig J, Grune T & Castro JP (2017). Happily (n)ever after: aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol 11, 482–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YC, Lustgarten MS, Liu Y, Muller FL, Bhattacharya A, Liang H, Salmon AB, Brooks SV, Larkin L, Hayworth CR, Richardson A & Van Remmen H (2010). Increased superoxide in vivo accelerates age‐associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J 24, 1376–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YC & Van Remmen H (2011). Age‐associated alterations of the neuromuscular junction. Exp Gerontol 46, 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S & Cuervo AM (2015). Proteostasis and aging. Nat Med 21, 1406–1415. [DOI] [PubMed] [Google Scholar]
- Kawamura Y, Okazaki H, O'Brien PC & Dych PJ (1977). Lumbar motoneurons of man: I) number and diameter histogram of alpha and gamma axons of ventral root. J Neuropathol Exp Neurol 36, 853–860. [DOI] [PubMed] [Google Scholar]
- Larkin LM, Davis CS, Sims‐Robinson C, Kostrominova TY, Remmen HV, Richardson A, Feldman EL & Brooks SV (2011). Skeletal muscle weakness due to deficiency of CuZn‐superoxide dismutase is associated with loss of functional innervation. Am J Physiol Regul Integr Comp Physiol 301, R1400–R1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson L & Ansved T (1995). Effects of ageing on the motor unit. Prog Neurobiol 45, 397–458. [DOI] [PubMed] [Google Scholar]
- Levine RL & Stadtman ER (2001). Oxidative modification of proteins during aging. Exp Gerontol 36, 1495–1502. [DOI] [PubMed] [Google Scholar]
- Lexell J, Taylor CC & Sjostrom M (1988). What is the cause of the ageing atrophy? Total number, size and proportion of different fiber types studied in whole vastus lateralis muscle from 15‐ to 83‐year‐old men. J Neurol Sci 84, 275–294. [DOI] [PubMed] [Google Scholar]
- Marzetti E, Lees HA, Wohlgemuth SE & Leeuwenburgh C (2009). Sarcopenia of aging: underlying cellular mechanisms and protection by calorie restriction. Biofactors 35, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArdle A, Dillmann WH, Mestril R, Faulkner JA & Jackson MJ (2004). Overexpression of HSP70 in mouse skeletal muscle protects against muscle damage and age‐related muscle dysfunction. FASEB J 18, 355–357. [DOI] [PubMed] [Google Scholar]
- McArdle A, Pattwell D, Vasilaki A, Griffiths RD & Jackson MJ (2001). Contractile activity‐induced oxidative stress: cellular origin and adaptive responses. Am J Physiol Cell Physiol 280, C621–C627. [DOI] [PubMed] [Google Scholar]
- Mittal KR & Logmani FH (1987). Age‐related reduction in 8th cervical ventral nerve root myelinated fiber diameters and numbers in man. J Gerontol 42, 8–10. [DOI] [PubMed] [Google Scholar]
- Muller FL, Song W, Liu Y, Chaudhuri A, Pieke‐Dahl S, Strong R, Huang TT, Epstein CJ, Roberts LJ 2nd, Csete M, Faulkner JA & Van Remmen H (2006). Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age‐dependent skeletal muscle atrophy. Free Radic Biol Med 40, 1993–2004. [DOI] [PubMed] [Google Scholar]
- Palomero J, Vasilaki A, Pye D, McArdle A & Jackson MJ (2013). Aging increases the oxidation of dichlorohydrofluorescein in single isolated skeletal muscle fibers at rest, but not during contractions. Am J Physiol Regul Integr Comp Physiol 305, R351–R358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson SJ, Young A, Macaluso A, Devito G, Nimmo MA, Cobbold M & Harridge SD (2002). Muscle function in elite master weightlifters. Med Sci Sports Exerc 34, 1199–1206. [DOI] [PubMed] [Google Scholar]
- Piasecki M, Ireland A, Jones DA & McPhee JS (2016a). Age‐dependent motor unit remodelling in human limb muscles. Biogerontology 17, 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piasecki M, Ireland A, Stashuk D, Hamilton‐Wright A, Jones DA & McPhee JS (2016b). Age‐related neuromuscular changes affecting human vastus lateralis. J Physiol 594, 4525–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK & Jackson MJ (2008). Exercise‐induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev 88, 1243–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakellariou GK, Davis CS, Shi Y, Ivannikov MV, Zhang Y, Vasilaki A, Macleod GT, Richardson A, Van Remmen H, Jackson MJ, McArdle A & Brooks SV (2014). Neuron‐specific expression of CuZnSOD prevents the loss of muscle mass and function that occurs in homozygous CuZnSOD‐knockout mice. FASEB J 28, 1666–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sataranatarajan K, Qaisar R, Davis C, Sakellariou GK, Vasilaki A, Zhang Y, Liu Y, Bhaskaran S, McArdle A, Jackson M, Brooks SV, Richardson A & Van Remmen H (2015). Neuron specific reduction in CuZnSOD is not sufficient to initiate a full sarcopenia phenotype. Redox Biol 5, 140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma AK, Bajada S & Thomas PK (1980). Age changes in the tibial and plantar nerves of the rat. J Anat 130, 417–428. [PMC free article] [PubMed] [Google Scholar]
- Shefner JM, Reaume AG, Flood DG, Scott RW, Kowall NW, Ferrante RJ, Siwek DF, Upton‐Rice M & Brown RH Jr (1999). Mice lacking cytosolic copper/zinc superoxide dismutase display a distinctive motor axonopathy. Neurology 53, 1239–1246. [DOI] [PubMed] [Google Scholar]
- Sims‐Robinson C, Hur J, Hayes JM, Dauch JR, Keller PJ, Brooks SV & Feldman EL (2013). The role of oxidative stress in nervous system aging. PLoS One 8, e68011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson BE & Irving D (1977). The numbers of limb motor neurons in the human lumbosacral cord throughout life. J Neurol Sci 34, 213–219. [DOI] [PubMed] [Google Scholar]
- Valdez G, Tapia JC, Kang H, Clemenson GD Jr, Gage FH, Lichtman JW & Sanes JR (2010). Attenuation of age‐related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc Natl Acad Sci USA 107, 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilaki A, Mansouri A, Remmen H, van der Meulen JH, Larkin L, Richardson AG, McArdle A, Faulkner JA & Jackson MJ (2006a). Free radical generation by skeletal muscle of adult and old mice: effect of contractile activity. Aging Cell 5, 109–117. [DOI] [PubMed] [Google Scholar]
- Vasilaki A, McArdle F, Iwanejko LM & McArdle A (2006b). Adaptive responses of mouse skeletal muscle to contractile activity: the effect of age. Mech Ageing Dev 127, 830–839. [DOI] [PubMed] [Google Scholar]
- Vasilaki A, Pollock N, Giakoumaki I, Goljanek‐Whysall K, Sakellariou GK, Pearson T, Kayani A, Jackson MJ & McArdle A (2016). The effect of lengthening contractions on neuromuscular junction structure in adult and old mice. Age (Dordr) 38, 259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilaki A, Simpson D, McArdle F, McLean L, Beynon RJ, Van Remmen H, Richardson AG, McArdle A, Faulkner JA & Jackson MJ (2007). Formation of 3‐nitrotyrosines in carbonic anhydrase III is a sensitive marker of oxidative stress in skeletal muscle. Proteomics Clin Appl 1, 362–372. [DOI] [PubMed] [Google Scholar]
- Vasilaki A, van der Meulen JH, Larkin L, Harrison DC, Pearson T, Van Remmen H, Richardson A, Brooks SV, Jackson MJ & McArdle A (2010). The age‐related failure of adaptive responses to contractile activity in skeletal muscle is mimicked in young mice by deletion of Cu,Zn superoxide dismutase. Aging Cell 9, 979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdu E, Ceballos D, Vilches JJ & Navarro X (2000). Influence of aging on peripheral nerve function and regeneration. J Peripher Nerv Syst 5, 191–208. [DOI] [PubMed] [Google Scholar]
- Xilouri M & Stefanis L (2016). Chaperone mediated autophagy in aging: starve to prosper. Ageing Res Rev 32, 13–21. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Davis C, Sakellariou GK, Shi Y, Kayani AC, Pulliam D, Bhattacharya A, Richardson A, Jackson MJ, McArdle A, Brooks SV & Van Remmen H (2013). CuZnSOD gene deletion targeted to skeletal muscle leads to loss of contractile force but does not cause muscle atrophy in adult mice. FASEB J 27, 3536–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Chong SY, Lim A, Singh BK, Sinha RA, Salmon AB & Yen PM (2017). Changes in macroautophagy, chaperone‐mediated autophagy, and mitochondrial metabolism in murine skeletal and cardiac muscle during aging. Aging 9, 583–599. [DOI] [PMC free article] [PubMed] [Google Scholar]