

Abstract
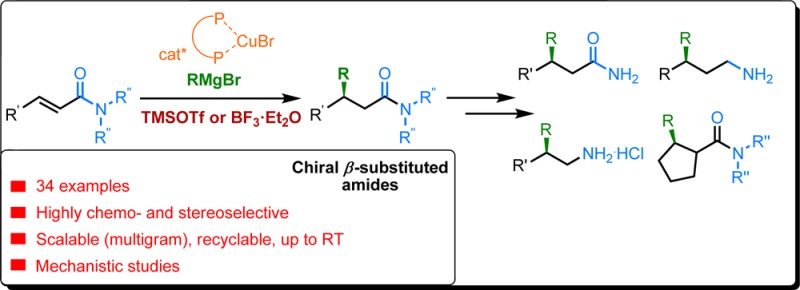
Here we report that readily available silyl- and boron-based Lewis acids in combination with chiral copper catalysts are able to overcome the reactivity issues of unactivated enamides, known as the least reactive carboxylic acid derivatives, toward alkylation with organomagnesium reagents. Allowing unequaled chemo-reactivity and stereocontrol in catalytic asymmetric conjugate addition to enamides, the method is distinguished by its unprecedented reaction scope, allowing even the most challenging and synthetically important methylations to be accomplished with good yields and excellent enantioselectivities. This catalytic protocol tolerates a broad temperature range (−78 °C to ambient) and scale up (10 g), while the chiral catalyst can be reused without affecting overall efficiency. Mechanistic studies revealed the fate of the Lewis acid in each elementary step of the copper-catalyzed conjugate addition of Grignard reagents to enamides, allowing us to identify the most likely catalytic cycle of the reaction.
Introduction
Conjugate addition (CA) reactions of hard carbon nucleophiles to α,β-unsaturated carbonyl derivatives that forge carbon–carbon (C–C) bonds rank among the most fundamental reactions in chemical synthesis.1,2 Chiral copper-based catalysts have proven to permit asymmetric conjugate additions (ACA) to various Michael acceptors (Scheme 1a).2 Deemed one of the most important structural motifs in organic chemistry, amides are found in a plethora of natural products and bioactive compounds, such as proteins and pharmaceuticals.3 However, despite almost 80 years of intensive research in the field of copper promoted CA reactions, a general solution for catalytic ACA to simple α,β-unsaturated amides (enamides) has not been found.2,4 The challenges associated with catalytic ACA to α,β-unsaturated amides are due to the sluggish resonance activation of the olefin moiety via the adjacent carboxamide group (Scheme 1b). The high degree of nitrogen lone-pair delocalization, resulting from the orbital overlap with the antibonding orbital of the carbonyl group, makes carboxamide the least electron-deficient carboxylic acid derivative.3a Thus, contrary to aldehydes, ketones, and esters, the lowest unoccupied molecular orbital (LUMO) of the corresponding enamide is not sufficiently enhanced toward nucleophilic addition at the β-position. As a result of this low reactivity, addition of hard organometallics was only possible at temperatures above −78 °C, at which noncatalyzed blank reactions outcompete the catalytic enantioselective pathway. Therefore, the only reported examples of catalytic ACA to simple enamides are confined to Rh-catalyzed arylations that do not suffer from noncatalyzed additions at high temperatures.5 The challenge faced in the development of efficient and stereoselective alkylations of simple enamides has led to the development of several alternative approaches, with the most common ones based on specific enamide substrates activated by placing an electron-withdrawing group at the N-atoms (Scheme 1c) to allow electronic activation and/or bidentate coordination with the chiral catalyst.5−7 Another nondirect method to β-substituted chiral amides is based on 1,4-addition to α,β-unsaturated esters, followed by quenching of the reaction mixture with the corresponding amines.8 Intriguingly, the only reported direct addition to simple enamides makes use of Grignard reagents, but the limited scope of the resulting chiral β-alkyl substituted amides and the modest enantioselectivities led the authors to switch to a chiral auxiliary strategy.9
Scheme 1. Catalytic ACA of Hard Carbon Nucleophiles to α,β-Unsaturated Carbonyl Compounds: State-of-the-Art.

(a) Cu-catalyzed ACA has been investigated for over the last 70 years. (b) Progress in the development of ACA depending on the reactivity of various conjugated carbonyls is contrasted to the lack of examples for direct ACA to the less reactive conjugated amides (enamides) which could lead to an array of valuable chiral molecules. (c) ACA has been developed only for activated amides or imides. (d) Strategy that was initially aimed at overcoming the intrinsically low reactivity of the enamide through enhancement of its LUMO by coordination with a Lewis acid.
Thus, despite the advances realized, the conjugate alkylation of unactivated enamides still constitutes a daunting, so far unsolved, challenge.
Whereas the resonance stabilization impedes the reactivity of enamides, it also gives rise to a pronounced Lewis basicity of the amide carbonyl oxygen atom. We hypothesized that coordination of a strong LA to the oxygen atom should significantly enhance the electrophilicity of the adjacent olefinic moiety toward nucleophilic addition10,11 thus activating the enamides in situ (Scheme 1d). This in turn could allow direct additions of hard alkyl nucleophiles, namely Grignard reagents, to simple unactivated enamides without the need for specific substrates, while rendering the reaction enantioselective by using chiral catalysts.
Results and Discussion
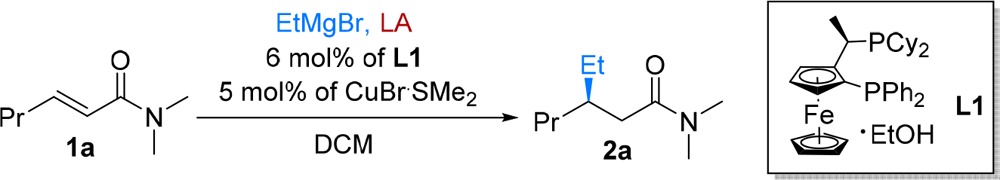
We started exploring this concept by evaluating the reactivity of simple transN,N-dimethyl enamides 1a toward addition of EtMgBr in different reaction conditions. The initial experiments confirmed the inherently poor reactivity of acyclic α,β-unsaturated amides relative to typical Michael acceptors. No addition of the highly reactive EtMgBr to enamide 1a was observed when performing the reaction in CH2Cl2 at −78 °C, regardless of whether copper salt or chiral ligand L1 were present (Table 1, entries 1–3). Raising the temperature to 0 °C resulted in substrate conversion, but unfortunately the reaction with chiral ligand yielded racemic product, and the noncatalyzed reaction was faster than the one promoted by the copper catalyst (entries 4–6). At –50 °C the catalyzed reaction rate started to surpass that of the noncatalytic reaction, but still racemic product was obtained (entries 7 and 8).
Table 1. Selected Optimization Data for the Cu-Catalyzed Alkylation of Enamide 1a with EtMgBra.
| Entry | L1/Cu(I) | LA | T [°C ] | Conv. [%]b | ee [%]c | Entry | L1/Cu(I) | LA | T [°C] | Conv. [%]b | ee [%]c | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | – | – | –78 | 0 | – | 7 | – | – | –50 | 12 | 0 | |
| 2 | Cu(I) | – | –78 | 0 | – | 8 | L1/Cu(I) | – | –50 | 20 | 5 | |
| 3 | L1/Cu(I) | – | –78 | 0 | – | 9 | – | BF3·Et2O | –78 | 0 | – | |
| 4 | – | – | 0 | 97 | – | 10 | – | TMSOTf | –78 | 50 | – | |
| 5 | Cu(I) | – | 0 | 42 | 0 | 11 | L1/Cu(I) | BF3·Et2O | –78 | 94 | 97 | |
| 6 | L1/Cu(I) | – | 0 | 79 | 0 | 12 | L1/Cu(I) | TMSOTf | –78 | 92 | 92 |
Reaction conditions: 0.1 M of 1a in CH2Cl2, LA (2.0 equiv), EtMgBr (2.0 equiv). For details see SI.
Conversion was determined by NMR of reaction crude.
Enantiomeric excess was determined by HPLC on a chiral stationary phase. Absolute configuration was assigned by analogy with literature data (see SI).
These results indicate that the chiral copper catalyst L1/Cu(I) is not capable of either outcompeting the noncatalyzed racemic addition to simple enamides, or of providing CA with enantiodiscrimination. This is a striking difference from the overwhelming literature precedence on Cu-catalyzed asymmetric additions of organometallics to enones and enoates.1,2 At this point we introduced LA to explore the activation of enamides toward additions at low temperature (−78 °C).
In the absence of copper salt, no significant product was formed when using BF3·Et2O. Instead, we observed transmetalation (by NMR spectroscopy, see mechanistic studies below) of the latter with the Grignard reagent, thus effectively destroying the nucleophile in the reaction (entry 9). With the more reactive trimethylsilyl trifluoromethanesulfonate (TMSOTf), 50% of product was formed (entry 10), but also some transmetalation of TMSOTf with EtMgBr was observed. However, combining either BF3·Et2O or TMSOTf with chiral copper catalyst led to an immense acceleration of the ACA reaction. Importantly, apart from outcompeting the noncatalyzed addition of EtMgBr, the catalytic pathway provided the ACA product for the first time with excellent enantioselectivity (entries 11 and 12). Further LA, solvent, chiral ligand, and copper salt screening (see SI) failed to improve these already excellent results, thus establishing the following optimized conditions: 2.0–3.0 equiv of either of these LAs and 2.0 equiv of Grignard reagents in the presence of 6 mol % of chiral ligand L1 and 5 mol % of CuBr·SMe2, with CH2Cl2 as solvent and in a temperature range from −50 to −78 °C.12
With the optimized set of conditions in hand, we investigated the generality of this methodology (Scheme 2), testing both BF3·Et2O and TMSOTf as LA. Although both LA’s enable ACA to almost all tested enamides, TMSOTf generally works best for relatively unreactive and unhindered enamides, while BF3·Et2O is the LA of choice for relatively reactive, both hindered or unhindered, enamides.
Scheme 2. Product Scope of Lewis Acid Promoted Copper-Catalyzed ACA Methodology.

Isolated yields for all the products are shown. Absolute configuration was assigned by analogy with literature data (see SI). Reaction conditions (for details see SI). 2.0–3.0 equiv of BF3·Et2O were used as LA at (−78) °C.
In this case 10 mol % of CuBr·SMe2 and 12 mol % of L1 were used.
Weinreb amide was used in this case, and it underwent demethoxylation.
Without LA at (−50) °C.
2.0–3.0 equiv of TMSOTf was used as LA at (−50) °C.
First, we evaluated various substituents at the nitrogen atom and found that a wide variety can be used, allowing efficient transformation to the corresponding β-chiral amides. ACA to N-diallyl, N-dibenzyl, and N-di(p-methoxylbenzyl) groups, with possible subsequent deprotection in mind, are well-tolerated and give the corresponding CA products (2c–2e) with good yields and excellent (98%) enantiomeric excess (ee). Addition of EtMgBr to enamide with a N-phenyl-N-methyl group led to CA product 2f with 77% ee. Notably, CA to highly activated enamide with N-tosyl-N-methyl groups, a substrate that provides the CA product with a dramatic 36% of ee in the absence of a LA (see SI), now yielded product 2g with a high ee of 86%. Addition to Weinreb-type enamide proceeded with excellent chemo- and enantioselectivity and led to the secondary amide product 2h, resulting from demethoxylation. Gratifyingly, CA to morpholine-substituted enamide leading to product 2i, amenable to further synthetic transformations, proceeded with 75% of isolated yield and 96% of ee. Finally, even addition of EtMgBr to the six-membered α,β-unsaturated lactam bearing an endocyclic double bond, resulting in product 2j, succeeded. Interestingly, no Lewis acid was required in this case, most likely due to the higher reactivity of cyclic Michael acceptors toward nucleophiles compared to linear analogues. Carrying out the reaction in the presence of BF3·Et2O or TMSOTf led to side reactions, and the CA product 2j was obtained with an ee of 79% due to the competing background reaction. Low conversions and racemic products were obtained when primary or secondary amides were used as Michael acceptors.
Having established that our catalytic system tolerates a broad scope of variations at the N-atom, we subsequently explored α,β-unsaturated amides with different substitution patterns at the β-position. We were delighted to find that excellent results are obtained with substrates featuring linear as well as branched carbon chains (2a, b, k, l), aromatic rings (2m–2r), heteroaromatic substituents (2s–2v), and functional groups such as halogen and unprotected hydroxyl (2w, x).
It should be noted that the reactivity of the chiral copper catalyst was not affected by the presence of heteroatoms. The consistently first-rate enantioselectivities and good to excellent yields observed during these experiments highlight the prominent role of the catalyst and the LA in the CA to unreactive α,β-unsaturated enamides.
Next, the scope of the reaction in terms of Grignard reagents was examined.13 It is remarkable that most of the assessed Grignard reagents were suitable partners for this catalytic system, with the exception of PhMgBr, which provided low conversion and racemic product.14 It was particularly gratifying that, where previous reports on additions to conjugated enamides were restricted to arylations, our catalytic system enabled the addition of a wide variety of alkyl Grignard reagents (linear as well as α-, β- and γ-substituted and functionalized) with excellent regio- and enatioselectivities (Scheme 2, products 2a, 3a–3g). Because of the utmost synthetic relevance of methylated chiral centers in pharmaceuticals, the addition of MeMgBr deserves a special note. Despite the formidable advances realized in copper-catalyzed additions of organometallics, the methylation of the more reactive α,β-unsaturated esters is still considered a notoriously difficult transformation.1a,2,15 Therefore, we anticipated that the addition of MeMgBr, the least reactive among all alkyl Grignard reagents, to enamides, a substrate far less reactive than ester, would be very challenging.
However, to our delight, the addition of this reagent was successful, providing the β-substituted amide 3h in 50% yield and nearly absolute stereocontrol (99% ee, see SI). Remarkably, the yield was greatly improved to 93% by using TMSOTf as LA, while retaining the enantioselectivity of 99% (Scheme 2, product 3h).
Tests on the temperature tolerance provided a final testament to the robustness and power of our methodology. High levels of selectivity in the ACA of hard organometallics to Michael acceptors are typically possible at temperatures below 0 °C.2,15 From an industrial perspective, this requirement is a major restriction for large-scale applications.1a To challenge our catalytic system further, we carried out the CA reactions to enamide 1a at higher temperatures, using both EtMgBr and the relatively less reactive MeMgBr (Figure 1a). We were pleased that these experiments produced high levels of regio- and enantioselectivity, unprecedented for hard organometallics under these conditions. The corresponding CA products were obtained with good yields and ee’s above 90% at 0 °C in case of the addition of EtMgBr and at both 0 and 25 °C for the addition of MeMgBr (Figure 1a, entries 1–3).
Figure 1.

Practical aspects and application of the methodology. (a) Temperature dependence and 10 g scale reaction. (b) Scale-up reaction procedure. (c) ACA product transformations. (d) ACA to trifluoromethylated enamide 1y for further applications in the synthesis of a drug candidate. (e) Effect of the nature of the LA on the structure of the final ACA product.
These results convincingly demonstrate the synergistic power of the chiral copper catalyst and the LA, allowing them to outcompete the noncatalyzed reaction at relatively high temperatures for this chemistry. The nature of the LA is critical to the success of these reactions at high temperatures, with TMSOTf found to be superior in terms of yield and ee.
This catalytic protocol is scalable and operationally simple, as we corroborated by performing the addition of MeMgBr to enamide 1a at 0 °C on a preparative scale (10 g, 71 mmol), using 5 mol % of chiral catalyst (L1-CuBr). Full conversion was reached once the addition of the last reaction component, MeMgBr, to the reaction mixture was completed (within few minutes, Figure 1b). The CA product 3h was obtained with excellent yield and enantioselectivity (entry 3) with no need for special equipment. The catalyst was recovered with 80% yield and reused for another ACA reaction with similar performance (see SI).
β-Alkyl-substituted chiral secondary amides as well as β-alkyl substituted chiral amines are interesting synthetic targets as these structures are present in various pharmaceutically active ingredients16−18 including Cyclotheonamide E5 and Orbiculamide A, both known for their cytotoxic activities.17 Similarly, β-alkyl-substituted chiral amines, and in particular trifluoromethylated ones, are known precursors in the synthesis of leukotriene receptor antagonists used, for instance, to treat asthma.18 To showcase the utility of our catalytic protocol, we demonstrated that chiral β-substituted amide 2e can easily be transformed into a number of corresponding valuable molecules (Figure 1c). Deprotection19 of 2e afforded chiral β-ethyl amide 4, which in turn can be used for the synthesis of the chiral γ-ethyl chiral amine 5 via reduction of the carbonyl moiety or to β-ethyl chiral amine 6 through Hoffman rearrangement.20 We have also applied our ACA methodology for the methylation of trifluoromethylated enamide 1y, leading to β-methyl-substituted amide product 3j with 99% ee (Figure 1d). When subjected to deprotection and Hoffman rearrangement, this product could lead to a direct precursor of the drug candidate ZENECA ZD 3523.18 Another synthetically important transformation in which this catalytic system can be engaged is the trapping of the product enolate (Figure 1e). To demonstrate this, we performed the CA reaction to Br-substituted enamide 1w. When BF3·Et2O is used as LA, conjugate addition product 2w is obtained. However, switching to TMSOTf as LA allows the CA reaction to be followed by intramolecular trapping of the intermediate silyl enolate, providing cyclic product trans-7 with contiguous stereocenters and as a single diastereoisomer.
To gain more detailed insight into this catalytic system, and particularly clarify the role of the LA, we carried out mechanistic studies (Figure 2, more details are in SI). It is generally assumed that the mechanism of the Cu-catalyzed enantioselective CA of organometallic compounds follows similar principles as proposed for the noncatalytic organocuprate addition, involving an oxidative addition-reductive elimination pathway.2,21−24 However, the necessary presence of LA to accomplish enantioselective CA to enamides adds another level of mechanistic complexity. LA additives have been known for decades to accelerate the CA of organometallics to various α,β-unsaturated carbonyl derivatives.22 In particular, the use of very weak TMSCl became common practice in CA of various hard organometallics.21a,23 In contrast, relatively strong LAs, such as BF3·Et2O, have been used only in CA of stoichiometric organocopper reagents.21a,24
Figure 2.

Mechanistic studies. (a) Undesired reaction pathways in the CA of Grignard reagents to enamides in the presence of LA followed by NMR spectroscopy in CD2Cl2 at −80 °C. (b) Reaction scheme for the sections c, d. Reaction conditions: 0.1 M 1a in CH2Cl2, LA (2.0 equiv), RMgBr (2.0 equiv) at −78 °C, 18 h. (c) Effect of different LAs in the Cu-catalyzed CA of EtMgBr to 1a. (d) Cu-catalyzed CA of MeMgBr to (E)- or (Z)-1a. (e) 31P NMR spectra of: L1-CuBr (red), L1-CuBr and 2.0 equiv of MeMgBr (species 10, orange), L1-CuBr and 10 equiv of TMSOTf (green) or BF3·Et2O (purple), addition of MeMgBr to L1-CuBr prior mixed with TMSOTf (blue). (f) 1H NMR spectra of LA-enamide complexes: free enamide (red), with TMSOTf (orange), BF3·Et2O (purple), with MeMgBr (blue), the reaction media before completion (green). (g) Types of enolates formed as end product of the CA of MeMgBr to enamide (for detailed discussion see SI) in the reaction using TMSOTf (blue), BF3·Et2O (green), and in the absence of LA (red) determined by TOCSYs experiments. (h) Proposed catalytic cycle.
In our case, the presence of a strong LA together with only few percent of chiral Cu(I)-catalyst and highly reactive Grignard reagents makes for a complex system. The outcome of the reaction depends critically not only on the relative rates of the desired catalyzed and the undesired noncatalyzed pathways but also on those of several competing processes, indicated in Figure 2a.
We expect the catalytic cycle (Figure 2h) to be initiated by the formation of species 8 through transmetalation of chiral catalyst L1-CuBr by Grignard reagents. However, the chiral ligand L1, reversibly bound to copper, is Lewis basic, and thus a strong LA competes with copper for binding to L1, potentially destroying the chiral catalyst (Figure 2a, e). This was confirmed by a control experiment that saw the formation of a mixture of unidentified species lacking bidentate coordination to copper (singlets versus doublets in 31P NMR) upon addition of either LA to chiral copper complex L1-CuBr. Remarkably though, addition of MeMgBr to this mixture resulted in an immediate recovery of either the L1-CuBr or the transmetalated copper complex 8, depending on the remaining amount of Grignard reagent in the media. Similarly, adding an excess of LA to the transmetalated copper complex did not affect its structure. Even when adding copper salt and Grignard after combining LA with L1, species 8 is formed, demonstrating its remarkable formation rate and stability. Furthermore, transmetalation of the LA by the Grignard reagent can deplete both components. NMR guided control experiments, in the absence of enamide or L1-CuBr complex, performed in CD2Cl2 at −80 °C confirmed all indicated in Figure 2a pathways occur (see SI). Fortunately, the excellent results observed for our system constitute evidence that these processes are outcompeted by the catalyzed reaction. Following the formation of transmetalated copper complex 8, the next step in the catalytic cycle is π-complexation with the activated enamide to form species 11 or 11′, followed by the formation of σ-complex intermediates 12 or 12′ (silyl enolate in case of TMSOTf and boron enolate in case of BF3·Et2O).
We anticipated the activated enamide to be an LA-enamide complex (9), the formation of which was indeed observed by 1H NMR spectroscopy when using either BF3·Et2O or TMSOTf (Figure 2h, f). However, subsequent addition of a stoichiometric amount of MeMgBr led to the formation of a new species corresponding to MeMgBr-enamide complex 10.
On the one hand it seems reasonable to assume that π-complexation of species 8 with the relatively more stable activated enamide 10 would occur next, forming π-complex 11 in step 1. On the other hand, based on the Curtin–Hammett principle, direct formation of π-complex 11′ in step 1 cannot be excluded.
Thus, the main question is at which stage the LA is involved in the catalytic cycle: before, during, or after oxidative addition step or during or after reductive elimination step.
Based on the E–Z isomerizations of α,β-unsaturated carbonyl substrates often observed in copper-catalyzed CA reactions and specific isotope effects observed, the oxidative addition step is thought to be reversible, with the rate enhancement upon addition of TMSCl attributed to making the oxidative step irreversible.2,21,23
To verify whether this holds true for the role of BF3·Et2O and TMSOTf in our reaction, two more sets of experiments were executed (Figure 2b–d). A number of different LAs were tested varying in strength and sterics (Figure 2b, c). The results reveal clearly that the strength of the LA is crucial for our catalytic cycle, with the very weak TMSCl producing <7% conversion, the relatively stronger TMSBr a poor 44%, and the stronger TMS-, TBS- and TBDPS- substituted triflates as well as BF3·Et2O providing excellent results, both in terms of reactivity and selectivity. Furthermore, the nearly identical ee’s given by the three sterically varying triflates imply that the LA does not affect the enantiodiscrimination step. The differences observed in the ee’s using various LA are explained by different rates between the enantioselective Cu-catalyzed and the noncatalyzed racemic CA reactions.
Double-bond isomerization of the enamide substrate ((Z)-1a to the more stable (E)-1a) was studied as well (Figure 2b, d). Copper-catalyzed CA of MeMgBr to (Z)-1a led to the final products with absolute configuration opposite to that obtained with (E)-1a and with ee’s of 99% and 46% when using BF3·Et2O and TMSOTf, respectively. Analysis of substrate samples obtained during the reaction confirmed that no isomerization to the more stable (E)-1a takes place, consistent with the interpretation that step 2 (or 3′) of the catalytic cycle is not reversible.
Combining the results on the strength of the LA, its lack of effect on the enantiodiscrimination, the lack of isomerization and Cu-catalyzed ACA in the absence of LA, and all the discussed experimental data, we believe that the LA is almost certainly involved in one of the steps preceding reductive elimination (step 4), making the oxidative addition overall irreversible. Strong LA is either required to increase the π-acidity of the π-complex (as in π-complex 11′ formed in step 2′) or to trap the magnesium enolate σ-complex 12 into the more stable silyl or boron enolate σ-complex 12′ (step 3). In both scenarios, that cannot be distinguished with the current data, the resulting more stable silyl or boron enolate σ-complex 12′ is expected to undergo faster reductive elimination than the magnesium enolate σ-complex 12. Further support for the formation of σ-complex 12′ comes from the fact that only Mg-activated enamide 10 and silyl or boron enolates of the CA-addition products (Figure 2g) are observed, with no traces of 9 or Mg-enolate of the CA-product throughout the reaction.
Conclusions
We have presented a versatile approach to ACA reactions of readily available Grignard reagents to α,β-unsaturated amides, aided by LAs and chiral copper catalysts. The broad scope of substrates as well as Grignard reagents allows even the most challenging and synthetically important methylations to be achieved with good yields and excellent enantioselectivities and makes our methodology by far the most general strategy for CA to carbonyl-based Michael acceptors. The demonstrated temperature tolerance, scalability, and possibilities for catalyst recovery add to its attractiveness. Our mechanistic studies and experimental data support the notion that the role of the Lewis acid is in the enhancement of the copper-catalyzed pathway. As a result, LA allows both the ACA to occur as well as to outcompete the blank reactions that occur at higher temperatures. Furthermore, the experimental data point to a very similar mechanistic behavior for both Lewis acids employed in this ACA of Grignard reagents to enamides, namely BF3·Et2O and TMSOTf. Finally, the unexpected compatibility observed in our catalytic system between highly reactive Grignard reagents, Lewis acids, and phosphine ligands was found to be due to the remarkable stability of the active catalyst toward the deleterious effect of Lewis acids.
Acknowledgments
Financial support from The Netherlands Organization for Scientific Research (NWO-ECHO to S.R.H.), the China Scholarship Council (CSC, to X.Y.), and the Ministry of Education, Culture and Science (Gravity program 024.001.035 to S.R.H.) is acknowledged. We thank M. Veenstra for few additional experiments. Solvias is acknowledged for the supply for chiral ferrocenyl diphosphine ligands
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b07344.
Author Contributions
§ These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Howell G. P. Org. Process Res. Dev. 2012, 16, 1258–1272. 10.1021/op200381w. [DOI] [Google Scholar]; b Perlmutter P.Conjugate Addition Reactions in Organic Synthesis; Tetrahedron Organic Chemistry, Series 9; Pergamon: Oxford, 1992. [Google Scholar]; c Rossiter B. E.; Swingle N. M. Chem. Rev. 1992, 92, 771–806. 10.1021/cr00013a002. [DOI] [Google Scholar]; d Comprehensive Asymmetric Catalysis I–III; Jacobsen E. N., Pfaltz A., Yamamoto H., Eds.; Springer-Verlag: Berlin Heidelberg, 1999. [Google Scholar]; e Catalytic Asymmetric Synthesis, 3rd ed.; Ojima I., Ed.; Wiley: Hoboken, NJ, 2010. [Google Scholar]
- a Alexakis A.; Krause N.; Woodward S.. Copper-Catalysed Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]; b Harutyunyan S. R.Progress in Enantioselective Cu(I)-catalyzed Formation of Stereogenic Centers; Springer: Heidelberg, 2015. [Google Scholar]; c Alexakis A.; Bäckvall J. E.; Krause N.; Pàmies O.; Diéguez M. Chem. Rev. 2008, 108, 2796–2823. 10.1021/cr0683515. [DOI] [PubMed] [Google Scholar]; d Harutyunyan S. R.; den Hartog T.; Geurts K.; Minnaard A. J.; Feringa B. L. Chem. Rev. 2008, 108, 2824–2852. 10.1021/cr068424k. [DOI] [PubMed] [Google Scholar]; e Jerphagnon T.; Pizzuti M. G.; Minnaard A. J.; Feringa B. L. Chem. Soc. Rev. 2009, 38, 1039–1075. 10.1039/b816853a. [DOI] [PubMed] [Google Scholar]; f Schmid T. E.; Drissi-Amraoui S.; Crévisy C.; Baslé O.; Mauduit M. Beilstein J. Org. Chem. 2015, 11, 2418–2434. 10.3762/bjoc.11.263. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Fraser P. K.; Woodward S. Chem. - Eur. J. 2003, 9, 776–783. 10.1002/chem.200390087. [DOI] [PubMed] [Google Scholar]
- a Greenberg A.; Breneman C. M.; Liebman J. F.. The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Materials Science; Wiley: New York, 2000. [Google Scholar]; b Ruider S. A.; Maulide N. Angew. Chem., Int. Ed. 2015, 54, 13856–13858. 10.1002/anie.201508536. [DOI] [PubMed] [Google Scholar]; c Bray B. L. Nat. Rev. Drug Discovery 2003, 2, 587–593. 10.1038/nrd1133. [DOI] [PubMed] [Google Scholar]; d Pattabiraman V. R.; Bode J. W. Nature 2011, 480, 471–479. 10.1038/nature10702. [DOI] [PubMed] [Google Scholar]; e Valeur E.; Bradley M. Chem. Soc. Rev. 2009, 38, 606–631. 10.1039/B701677H. [DOI] [PubMed] [Google Scholar]
- Byrd K. M. Beilstein J. Org. Chem. 2015, 11, 530–562. 10.3762/bjoc.11.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples using rhodium-catalyzed asymmetric addition:; a Yasukawa T.; Saito Y.; Miyamura H.; Kobayashi S. Angew. Chem., Int. Ed. 2016, 55, 8058–8061. 10.1002/anie.201601559. [DOI] [PubMed] [Google Scholar]; b Sakuma S.; Miyaura N. J. Org. Chem. 2001, 66, 8944–8946. 10.1021/jo010747n. [DOI] [PubMed] [Google Scholar]; c Shintani R.; Kimura T.; Hayashi T. Chem. Commun. 2005, 3213–3214. 10.1039/b502921j. [DOI] [PubMed] [Google Scholar]; d Oi S.; Taira A.; Honma Y.; Sato T.; Inoue Y. Tetrahedron: Asymmetry 2006, 17, 598–602. 10.1016/j.tetasy.2005.12.032. [DOI] [Google Scholar]; e Pucheault M.; Michaut V.; Darses S.; Genet J.-P. Tetrahedron Lett. 2004, 45, 4729–4732. 10.1016/j.tetlet.2004.04.075. [DOI] [Google Scholar]; f Defieber C.; Paquin J.-F.; Serna S.; Carreira E. M. Org. Lett. 2004, 6, 3873–3876. 10.1021/ol048240x. [DOI] [PubMed] [Google Scholar]; g Shintani R.; Kimura T.; Hayashi T. Chem. Commun. 2005, 3213–3214. 10.1039/b502921j. [DOI] [PubMed] [Google Scholar]
- For examples using copper-catalyzed ACA:; a Hird A. W.; Hoveyda A. H. Angew. Chem., Int. Ed. 2003, 42, 1276–1279. 10.1002/anie.200390328. [DOI] [PubMed] [Google Scholar]; b Pineschi M.; Del Moro F.; Di Bussolo V.; Macchia F. Adv. Synth. Catal. 2006, 348, 301–304. 10.1002/adsc.200505309. [DOI] [Google Scholar]
- For copper-catalyzed ACA to lactams:; a Pineschi M.; Del Moro F.; Gini F.; Minnaard A. J.; Feringa B. L. Chem. Commun. 2004, 1244–1245. 10.1039/B403793F. [DOI] [PubMed] [Google Scholar]; b Cottet P.; Müller D.; Alexakis A. Org. Lett. 2013, 15, 828–831. 10.1021/ol303505k. [DOI] [PubMed] [Google Scholar]; c Pace V.; Rae J. P.; Procter D. J. Org. Lett. 2014, 16, 476–479. 10.1021/ol4033623. [DOI] [PubMed] [Google Scholar]
- Schoonen A. K.; Fernández-Ibáñez M. Á.; Fañanás-Mastral M.; Teichert J. F.; Feringa B. Org. Biomol. Chem. 2014, 12, 36–41. 10.1039/C3OB41923A. [DOI] [PubMed] [Google Scholar]
- Biswas K.; Woodward S. Tetrahedron: Asymmetry 2008, 19, 1702–1708. 10.1016/j.tetasy.2008.06.017. [DOI] [Google Scholar]
- a Yamamoto H.Lewis Acids in Organic Synthesis; Wiley-VCH: Weinheim, 2000; Vol. 1–2. [Google Scholar]; b Yamamoto Y.; Yamamoto S.; Yatagai H.; Ishihara Y.; Maruyama K. J. Org. Chem. 1982, 47, 119–126. 10.1021/jo00340a026. [DOI] [Google Scholar]; c Pace V.; Holzer W.; Olofsson B. Adv. Synth. Catal. 2014, 356, 3697–3736. 10.1002/adsc.201400630. [DOI] [Google Scholar]
- Jumde R. P.; Lanza F.; Veenstra M. J.; Harutyunyan S. R. Science 2016, 352, 433–437. 10.1126/science.aaf1983. [DOI] [PubMed] [Google Scholar]
- CH2Cl2 was found to be the optimal solvent. The presence of even traces of THF is detrimental for the reaction conversion and enantioselectivity (see SI). On the other hand, copper salts other than CuBr can be used as well, as long as the halide in the Grignard reagent is a bromide (RMgBr).
- Grignard reagents must be used either in Et2O or tBuOMe. THF must be avoided, even in a small quantities. For example, Cu(I)-catalyzed conjugate addition of iPrMgBr in THF led to racemic product, while in Et2O the same reaction afforded the product with 67% ee.
- Racemic product being obtained with PhMgBr is due to slower reductive elimination of Cu from Ar-Cu species as compared to Alk-Cu, resulting in a slower copper-catalyzed reaction, which consequently is outcompeted by the direct, noncatalytic addition of PhMgBr. The low conversion obtained with PhMgBr is the direct consequence of its high reactivity, since this causes the reaction with the LA to be faster than the desired CA.
- a Des Mazery R.; Pullez M.; López F.; Harutyunyan S. R.; Minnaard A. J.; Feringa B. L. J. Am. Chem. Soc. 2005, 127, 9966–9967. 10.1021/ja053020f. [DOI] [PubMed] [Google Scholar]; b Wang S.-Y.; Ji S.-J.; Loh T.-P. J. Am. Chem. Soc. 2007, 129, 276–277. 10.1021/ja0666046. [DOI] [PubMed] [Google Scholar]
- a Omura S.Macrolide Antibiotics; Academic Press: Orlando, FL, 1984. [Google Scholar]; b Lazarevski G.; Kobrehel G.; Metelko B.; Duddeck H. J. Antibiot. 1996, 49, 1066–1069. 10.7164/antibiotics.49.1066. [DOI] [PubMed] [Google Scholar]; c Nicholas G. M.; Molinski T. F. Tetrahedron 2000, 56, 2921–2927. 10.1016/S0040-4020(00)00189-7. [DOI] [Google Scholar]
- a Fusetani N.; Sugawara T.; Matsunaga S.; Hirota H. J. Am. Chem. Soc. 1991, 113, 7811–7812. 10.1021/ja00020a080. [DOI] [Google Scholar]; b Murakami Y.; Takei M.; Shindo K.; Kitazume C.; Tanaka J.; Higa T.; Fukamachi H. J. Nat. Prod. 2002, 65, 259–261. 10.1021/np010304e. [DOI] [PubMed] [Google Scholar]
- Jacobs R. T.; Bernstein P. R.; Cronk L. A.; Vacek E. P.; Newcomb L. F.; Aharony D.; Buckner C. K.; Kusner E. J. J. Med. Chem. 1994, 37, 1282–1297. 10.1021/jm00035a008. [DOI] [PubMed] [Google Scholar]; b Jacobs R. T.; Yee Y. K.; Bernstein P. R.; Brewster A. G.; Sependa G. J.. (2R)-Methyl-4,4,4- trifluorobutylamine. EP 489548A1, June 10, 1992.
- For the deprotection:Nagaki A.; Takahashi Y.; Yoshida J.-I. Angew. Chem., Int. Ed. 2016, 55, 5327–5331. 10.1002/anie.201601386. [DOI] [PubMed] [Google Scholar]
- Miyamoto K.; Sakai Y.; Goda S.; Ochiai M. Chem. Commun. 2012, 48, 982–984. 10.1039/C2CC16360H. [DOI] [PubMed] [Google Scholar]
- a Yoshikai N.; Nakamura E. Chem. Rev. 2012, 112, 2339–2372. 10.1021/cr200241f. [DOI] [PubMed] [Google Scholar]; b Harutyunyan S. R.; López F.; Browne W. R.; Correa A.; Peña D.; Badorrey R.; Meetsma A.; Minnaard A. J.; Feringa B. L. J. Am. Chem. Soc. 2006, 128, 9103–9118. 10.1021/ja0585634. [DOI] [PubMed] [Google Scholar]
- For comprehensive list of references on use of Lewis acids in copper-catalyzed conjugate additions see ref (21a).
- a Corey E. J.; Boaz N. W. Tetrahedron Lett. 1985, 26, 6019–6022. 10.1016/S0040-4039(00)95114-1. [DOI] [Google Scholar]; b Corey E. J.; Boaz N. W. Tetrahedron Lett. 1985, 26, 6015–6018. 10.1016/S0040-4039(00)95113-X. [DOI] [Google Scholar]; c Alexakis A.; Berlan J.; Besace Y. Tetrahedron Lett. 1986, 27, 1047–1050. 10.1016/S0040-4039(86)80044-2. [DOI] [Google Scholar]; d Matsuzawa S.; Horiguchi Y.; Nakamura E.; Kuwajima I. Tetrahedron 1989, 45, 349–362. 10.1016/0040-4020(89)80064-X. [DOI] [Google Scholar]; e Bertz S. H.; Miao G.; Rossiter B. E.; Snyder J. P. J. Am. Chem. Soc. 1995, 117, 11023–11024. 10.1021/ja00149a032. [DOI] [Google Scholar]; f Frantz D. E.; Singleton D. A. J. Am. Chem. Soc. 2000, 122, 3288–3295. 10.1021/ja993373c. [DOI] [Google Scholar]
- a Yamamoto Y.; Yamamoto S.; Yatagai H.; Ishihara Y.; Maruyama K. J. Org. Chem. 1982, 47, 119–126. 10.1021/jo00340a026. [DOI] [Google Scholar]; b Nakamura E.; Yamanaka M.; Mori S. J. Am. Chem. Soc. 2000, 122, 1826–1827. 10.1021/ja993124o. [DOI] [Google Scholar]; c Lipshutz B. H.; Ellsworth E. L.; Dimock S. H. J. Am. Chem. Soc. 1990, 112, 5869–5871. 10.1021/ja00171a031. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.