

SUMMARY
The cellular abundance of mature microRNAs (miRNAs) is dictated by the efficiency of nuclear processing of primary miRNA transcripts (pri-miRNAs) into pre-miRNA intermediates. The Microprocessor complex of Drosha and DGCR8 carries this out, but it has been unclear what controls Microprocessor’s differential processing of various pri-miRNAs. Here, we show that Drosophila DGCR8 (Pasha) directly associates with the C terminal domain of the RNA polymerase II elongation complex when it is phosphorylated by the Cdk9 kinase (pTEFb). When association is blocked by loss of Cdk9 activity, a global change in pri-miRNA processing is detected. Processing of pri-miRNAs with a UGU sequence motif in their apical junction domain increases, while processing of pri-miRNAs lacking this motif decreases. Therefore, phosphorylation of RNA polymerase II recruits Microprocessor for co-transcriptional processing of non-UGU pri-miRNAs that would otherwise be poorly processed. In contrast, UGU-positive pri-miRNAs are robustly processed by Microprocessor independent of RNA polymerase association.
Keywords: Drosophila, microRNA, RNA polymerase II, DGCR8
eTOC
Nuclear processing of microRNAs is a major determinant of cellular abundance of these RNAs. Church et al find that the DGCR8 subunit of Microprocessor binds to RNA polymerase II. This couples microRNA processing to transcription. If microRNAs lack a sequence motif, co-transcriptional processing plays a more important role in determining abundance.
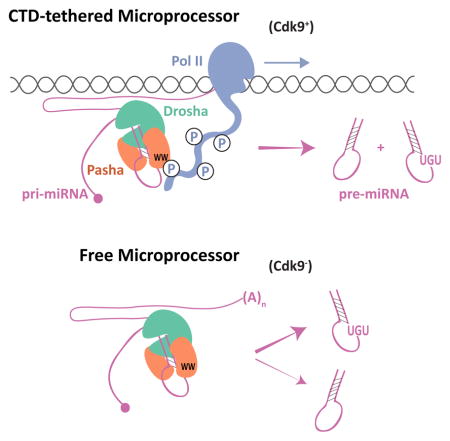
INTRODUCTION
MicroRNAs (miRNAs) are a class of small non-coding RNAs that negatively regulate expression of most protein-coding genes (Bartel, 2009). They play integral roles in a variety of cellular, developmental, and physiological processes in animals (Bushati and Cohen, 2007). It is therefore important to understand how the expression levels of miRNAs are determined within cells.
Biogenesis of miRNAs begins in the nucleus with the synthesis of a capped and polyadenylated primary miRNA (pri-miRNA) transcript by RNA polymerase II (Pol II) (Cai et al., 2004; Lee et al., 2004). Embedded within a single pri-miRNA transcript are one or more hairpins of a defined structure. Each hairpin stem is 33 to 35 bp in length with a terminal loop of variable size, and the hairpin is flanked by unstructured single-stranded RNA (Han et al., 2006; Lee et al., 2003; Zeng and Cullen, 2003). The Microprocessor complex, composed of the RNAse III enzyme Drosha and its cofactor DGCR8, binds to these hairpins and cleaves them from pri-miRNA transcripts (Denli et al., 2004; Gregory et al., 2004; Han et al., 2004; Landthaler et al., 2004; Lee et al., 2003; Nguyen et al., 2015).
It has been found that pri-miRNA processing is more important than pri-miRNA transcription in determining the steady-state abundance of mature miRNAs within HeLa cells (Conrad et al., 2014). Moreover, pri-miRNAs often undergo differential processing. This can be inferred by the differential abundance of discrete miRNAs that are processed from a common pri-miRNA (Chaulk et al., 2011). Often, such polycistronic miRNAs show expression polarity, with the 5′-most miRNA being more abundant than downstream 3′ miRNAs, even though all of the miRNAs originate from a common transcript (Conrad et al., 2014; Pfeffer et al., 2004; Yu et al., 2006). The specific ratios of these polycistronic miRNAs are often functionally important, and perturbing them results in misregulation of the cellular processes that the cluster regulates. This is associated with diseases such as cancer (Olive et al., 2013).
The mechanisms underlying differential processing of pri-miRNAs are not thoroughly understood. Processing can be controlled by RNA-binding proteins that selectively interact with certain pri-miRNAs and/or with Microprocessor (reviewed in (Ha and Kim, 2014). Another mechanism controls Microprocessor processing based on intrinsic structural features of the pri-miRNA itself. For example, in vitro processing activity can be enhanced or repressed by local structural features of the pri-miRNA hairpin and flanking sequences (Alarcon et al., 2015; Auyeung et al., 2013; Fang and Bartel, 2015; Han et al., 2006; Ma et al., 2013; Nguyen et al., 2015). Another potential mechanism for differential processing is that differences in miRNA levels are attributable to co-transcriptional processing of hairpins on nascent transcripts. Indeed, evidence suggests that co-transcriptional pri-miRNA processing can occur (Morlando et al., 2008; Nojima et al., 2015; Pawlicki and Steitz, 2008; Suzuki et al., 2017; Van Wynsberghe et al., 2011; Yin et al., 2015).
The domain organization of Drosha and DGCR8 proteins are strongly conserved throughout the animal kingdom (Fig. 1A). Drosha has two RNAse III domains, which carry out cleavage of the pri-miRNA hairpin (Han et al., 2004). The RNAse domains also bind to the C-terminal tails of two DGCR8 polypeptides (Kwon et al., 2016). This generates a heterotrimeric Microprocessor complex composed of one Drosha subunit and two DGCR8 subunits (Fig. 1B). The complex is further stabilized by DGCR8 homo-dimerization mediated by its Rhed domain (Quick-Cleveland et al., 2014; Weitz et al., 2014). The dimerized Rhed domain also binds to a molecule of ferric heme. Structural modeling predicts that the Microprocessor complex extends the entire length of a pri-miRNA hairpin, with Drosha covering the basal part of the hairpin and the two DGCR8 subunits covering the apical part (Fig. 1B) (Kwon et al., 2016). Based on biochemical experiments, it is likely that the dimerized Rhed domains bind to apical junction and terminal loop RNA (Nguyen et al., 2015).
Figure 1. The Pasha subunit of Microprocessor associates with phosphorylated Pol II.

See also Figure S1. (A) Schematic structures of the Drosophila Microprocessor subunits, Drosha and Pasha. Both proteins have nuclear localization sequences (NLS). Drosha has two RNase III domains, RIIIDa and RIIIDb, and a double-strand RNA binding domain (dsRBD). Pasha has two dsRBDs, and a Rhed domain, which drives homotypic dimerization and also binds heme and pri-miRNA hairpins. The WW domain is located within a region (orange) of Rhed that is sufficient for heme binding and dimerization. (B) Model of Microprocessor structure when bound to a pri-miRNA hairpin. The cleavage site in the hairpin is marked by blue arrowheads. The model is adapted from Kim and colleagues (Kwon et al., 2016; Nguyen et al., 2015). (C) Phylogenetic tree of a subset of eukaryotic WW domains. The sub-lineage containing DGCR8 and Pin1 is highlighted. (D) Immunoprecipitation from S2 cell lysate using 4H8 antibody, which recognizes all CTD isoforms of Pol II (Brodsky et al., 2005; Schroder et al., 2013). Molecular weights of standards are shown on the right. 0.3% input was loaded for Pasha and 10% input for Pol II. (E) Immunoprecipitation from S2 cell lysate using anti-GFP to purify GFP-Pasha or GFP. Molecular weights of standards are shown on the left. 2% input was loaded for Pol II and 8% input was loaded for GFP and GFP-Pasha. (F) Immunoprecipitation from S2 cell lysate using an antibody recognizing all Pol II isoforms (4H8), in which some samples were treated with a mixture of RNases. Precipitates were probed for Pasha as shown. 0.3% input was loaded.
Embedded within the Rhed domain is a conserved WW domain whose function is currently unknown (Fig. 1A and Fig. S1A). WW domains are 30 – 40 amino acids in length, and they mediate interactions with short proline-rich motifs in other proteins (Macias et al., 2002). To uncover a role for the DGCR8 WW domain, we performed phylogenetic analysis of the eukaryotic WW domain family. Intriguingly, DGCR8’s WW domain is most highly related to a WW domain that mediates specific binding of the Pin1 protein to the phosphorylated C-terminal domain (CTD) of the Pol II subunit Rpb1 (Fig. 1C) (Morris et al., 1999; Verdecia et al., 2000). The CTD is composed of a heptad sequence YSPTSPS that is repeated 52 times in human Pol II (Jeronimo et al., 2016). It is extensively modified by phosphorylation of specific residues within each repeat. Serine-2 (S2), serine-5 (S5), and serine-7 (S7) are phosphorylated by nuclear kinases to generate a modified Pol II enzyme whose phosphorylation state varies during the transcription cycle (Buratowski, 2009). S5P/S7P-modified Pol II is correlated with early transcription initiation while S2P-modified Pol II is correlated with elongation and termination.
Here, we describe a mechanism by which the Microprocessor complex differentially processes pri-miRNAs by association with elongating Pol II complexes. The Drosophila DGCR8 subunit (called Pasha) of Microprocessor binds to the CTD of Pol II subunit Rpb1 through its highly conserved WW domain. When the nuclear kinase that modifies CTD and marks elongating Pol II is inhibited, this impairs the interaction of Pasha with Pol II. Kinase inhibition causes a major shift in the differential processing of miRNAs. This effect has functional consequences, since miRNA-mediated gene silencing is impaired in vivo.
RESULTS
Pasha interacts with the elongation isoforms of Pol II
In light of the phylogenetic data, we wondered if the Drosophila DGCR8 homolog’s WW domain mediates an interaction between Microprocessor and phosphorylated Pol II. We immunoprecipitated all isoforms of Pol II from Drosophila S2 cells, and observed that Pasha co-immunoprecipitated with S2P- and S5P-modified isoforms of Pol II (Fig. 1D). To perform the reverse immunoprecipitation, we created S2 cell lines stably expressing GFP-Pasha or GFP, and used an antibody specific to GFP to immunoprecipitate these proteins from lysates. Both S2P- and S5P-modified Pol II specifically co-immunoprecipitated with GFP-Pasha (Fig. 1E). Co-immunprecipitation of total Pol II with Pasha was insensitive to RNase treatment, indicating that it occurs via a protein-protein interaction (Fig. 1F). Thus, Pol II and Pasha physically associate in Drosophila S2 cells.
We wondered if the association between Pol II and Pasha was mediated by Pasha’s WW domain and the phosphorylated CTD domain of Pol II. We generated two GST fusion proteins: GST-WW containing only Pasha’s WW domain, and GST-WWD, which contains the WW domain plus the adjacent heme-binding segment of the Rhed-domain (Fig. S1A). We first asked if the WWD domain is sufficient to interact with endogenous phosphorylated Pol II. We incubated Drosophila S2 cell nuclear extract with GST-WWD protein, and pulled down resulting complexes with glutathione beads (Fig. 2A). S2P-modified Pol II was specifically pulled down with GST-WWD (Fig. 2B). We next asked if the WW and WWD proteins directly interact with the phosphorylated CTD heptad repeats. GST fusion proteins were incubated with biotinylated peptides containing four repeats of the canonical CTD heptad sequence, and complexes were pulled down using streptavidin beads. Neither GST-WW nor GST-WWD formed stable complexes with peptides that were unmodified by phosphorylation (Fig. 2C,D). In contrast, both proteins formed stable complexes with peptides that were either singly or doubly phosphorylated at the S2 and S5 positions. The Kd for binding was estimated to be approximately 4 μM (see Methods). To ensure that binding was not simply due to electrostatic interactions with phosphate-laden peptide, we performed the binding reaction in phosphate buffer, which competes for such electrostatic interactions (Kim et al., 2004). Specific binding to peptide with S2P was still detected (Fig. S1B). Altogether, these results indicate that Pasha directly binds to the phosphorylated CTD of Pol II, and that Pasha’s WW domain is sufficient for the interaction to occur.
Figure 2. The WW domain of Pasha associates with the phosphorylated CTD of Pol II.

See also Figure S1. (A) Scheme of assay for binding between GST proteins and phospho-Pol II present in nuclear extracts. (B) Nuclear extract was incubated with GST proteins, as indicated, and pulled down material was probed for S2-phosphorylated Pol II. Input represents 4% of the total pulldown reaction. (C) Binding between GST proteins and biotinylated peptides containing four CTD heptad repeats. Peptide-associated GST proteins were visualized by Western blot. Inputs shown represent 5% of total binding reactions. (D) Biotinylated peptides with different phospho-modifications were assayed for binding to GST-WWD. Input is 5% of the binding reactions.
Interaction between Pasha and Pol II requires the Cdk9 kinase
Pol II CTD phosphorylation depends upon several serine-threonine kinases (Buratowski, 2009; Jeronimo et al., 2016). Cdk7 is a subunit of TFIIH, and it phosphorylates S5 and S7 in the CTD repeat. This occurs in the pre-initiation complex, and is thought to aid in promoter escape and pausing by Pol II. The marks are erased in two phases; first, shortly after initiation and second, when Pol II reaches the 3′ end of the gene. In Drosophila, the S2 residue is phosphorylated by two kinases. Cdk9 is a subunit of pTEFb, and it phosphorylates S2 residues in Pol II situated at the 5′ end of genes (Price, 2000). Cdk12 phosphorylates S2 residues in Pol II situated in the middle and at the 3′ end of genes (Bartkowiak et al., 2010).
To test if the interaction between Pasha and Pol II requires CTD phosphorylation in vivo, we treated Drosophila S2 cells with flavopiridol, a small molecule inhibitor of Cdk activity. At the dose applied to cells, Cdk9 was predicted to be strongly inhibited by flavopiridol while other Cdks were less affected (Ni et al., 2004). Although 20 hours of flavopiridol treatment strongly inhibited Pol II phosphorylation, it had no effect on total Pol II and Pasha protein levels (Fig. 3A). We then immunoprecipitated all Pol II isoforms and determined that co-immunoprecipitation of Pasha was greatly reduced in the flavopiridol-treated samples (Fig. 3B). As expected, little or no S2P- or S5P-modified Pol II was immunoprecipitated from these samples. We next prepared nuclear extracts from cells treated for 2 hours with flavopiridol. Since Pol II must be completely dephosphorylated before flavopiridol can have an effect on its phosphorylation state, 2 hours was not long enough for many Pol II molecules to go through a dephosphorylation-phosphorylation cycle. Extracts were incubated with GST-WWD protein, which was used to pull down newly associated complexes. A physical interaction between GST-WWD and S2P-modified Pol II was strongly inhibited in extracts from flavopiridol-treated cells (Figs. 3C and S1C). Thus, de novo binding of added GST-WWD protein appeared to be highly specific for newly phosphorylated Pol II. The newly phosphorylated molecules might not be masked by interacting nuclear factors and thus are more free to associate with the exogenous bait protein. In summary, dynamic phosphorylation of Pol II by Cdk9, and possibly other Cdks, is necessary for the in vitro and in vivo association of Pasha and Pol II.
Figure 3. Pasha binding to Pol II requires Cdk9 activity.

See also Figure S1. (A) S2 cells were treated with flavopiridol for 20 hours. Whole cell extracts prepared from cells were blotted for Pasha, total Pol II, S2P-modified Pol II, S5P-modified Pol II, and tubulin. (B) Extracts analyzed in (A) were subjected to immunoprecipitation with antibody specific for all isoforms of Pol II. Immunoprecipitates were probed for Pasha, total Pol II, S2P-modified Pol II and S5P-modified Pol II. (C) S2 cells were treated with flavopiridol for 2 hours. Nuclear extracts were subjected to pulldown reactions with GST proteins, as indicated, and pulled down material was probed for S2P-modified Pol II. Inputs represent 5% of total pulldown reactions.
Cdk9 activity regulates miRNA levels in vivo
We asked whether the Pasha-Pol II interaction has a functional impact on Microprocessor activity. Point mutations in the WW domain of DGCR8 that potentially would disrupt Pol II binding result in impaired dimerization or heme binding, both of which lead to insoluble protein (Quick-Cleveland et al., 2014). Homologous mutations in Pasha also caused protein insolubility (data not shown). Therefore, we could not generate cis-mutations that would specifically block Pol II binding. Instead, we reasoned that loss of Cdk9 would affect Microprocessor activity if there was such a functional connection, since loss of Cdk9 activity impairs the physical association between the proteins. We isolated a mutant strain of Drosophila that had a point mutation in the cdk9 gene. The point mutation results in a single amino acid substitution of the first invariant glycine in the ATP-binding motif GXGXXG located in the kinase domain (Fig. S2A). The G57S mutant allele is homozygous lethal, and mutant larvae arrest their development at the L1-L2 transition before dying (Fig. S2B,C). To demonstrate that the mutation was responsible for the lethal phenotype, we created a Cdk9 transgene (Fig. S2D). The transgene fully rescued the lethal mutant phenotype, and a single transgene copy was sufficient to rescue the developmental defects (Fig. S2E) and produce fertile adult flies (data not shown). Although the mutant is organismal lethal, it is not cell-lethal (Fig. S2F).
We tested the mutant for its effect on S2 and S5 phosphorylation of Pol II. Western analysis of Pol II from mutant animals showed that Pol II protein levels were normal but S2P- and S5P-modified Pol II levels were strongly inhibited (Figs. 4A and S3A). Thus, S2 and S5 phosphorylation is strongly dependent upon Cdk9 activity in vivo. Although Cdk7 and Cdk12 have been found to contribute to CTD phosphorylation in Drosophila (Bartkowiak et al., 2010; Schwartz et al., 2003), Cdk9 may provide phospho-marks on elongating Pol II, with the other kinases transiently marking initiating and terminating Pol II complexes. We attempted to perform Pasha-Pol II co-immunoprecipitations in extracts derived from the mutant larvae, but non-specific proteolysis in these extracts precluded their completion (data not shown).
Figure 4. Cdk9 is required for differential miRNA processing.

See also Figure S2. (A) Western blot of total Pol II and specific phospho-isoforms of Pol II from cdk9 mutant tissue. (B) Schematic structures of five polycistronic genes that were analyzed for miRNA expression. (C) Levels of mature miRNAs expressed from polycistronic genes. Shown are normalized RNA-seq read levels (1000s) from animals that are wildtype and cdk9 mutant. (D) Differential expression of mature miRNAs between mutant and wildtype animals as determined by RNA-seq. (*, p ≤ .01; **, p ≤ .001; ***, p ≤ .0001; edgeR exact test). (E) Differential expression of pri-miRNAs between mutant and wildtype animals as determined by RT-qPCR. Shown below are positions of the various RT-qPCR products being assayed. Error bars are standard deviations. (*, p ≤ .01; t-test)
If Cdk9 regulates Microprocessor activity, we reasoned that the cdk9 mutant would have abnormal levels of processed miRNAs. Therefore, we performed small-RNA sequencing on homozygous wildtype and mutant larvae collected at the L1-L2 transition (Table S1). This time point was chosen because it was close to the lethal phase but the mutant animals were still developmentally synchronized with the wildtype animals. Thus, any differences in miRNA levels between mutant and wildtype would not be due to asynchronous development. As a further effort to exclude mutant effects unrelated to processing, we first considered pri-miRNA transcripts that contain more than one hairpin, and individually generate multiple miRNAs. If Cdk9 had differential effects on polycistronic miRNAs originating from the same pri-miRNA transcript, this would suggest that processing was affected.
There were five loci predicted to encode polycistronic miRNAs that were detected in the sequenced libraries (Fig. 4B) (Ryazansky et al., 2011; Sun et al., 2015; Truscott et al., 2011). Using nested RT-PCR and 3′RACE, we validated that all five loci generate pri-miRNAs encompassing the relevant miRNAs, i.e. the pri-miRNAs are polycistronic in L1 larvae (Fig. S3B–D). Examination of the sequencing data found that three of the five clusters exhibited an expression polarity such that the 5′-most mature miRNA was more abundant than the 3′ mature miRNAs derived from the same cluster (Fig. 4C). Strikingly, this expression polarity was impaired in the cdk9 mutant for all three clusters (Fig. 4C,D). Abundance of the 5′-most miRNA was reduced, while 3′ miRNAs either increased in abundance or remained constant. We also observed differences with the other two clusters. The miR-279/996 cluster exhibited the opposite polarity in wildtype animals, with the 3′-most miR-996 being more abundant than miR-279. However, approximately equal levels of miR-279 and miR-996 were detected in the cdk9 mutant (Fig. 4C). The fifth miRNA cluster was unique in that the two hairpins encoding miR-275 and miR-305 are spaced only 80 basepairs apart, significantly less than the 500 or more basepairs that exist between hairpins in the other clusters (Fig. 4B). Interestingly, miR-275 and miR-305 are expressed at equivalent levels in wildtype, but the 3′ miR-305 is almost exclusively expressed in the mutant (Fig. 4C). We validated the results of RNA-seq analysis by independently measuring levels of the more abundant miRNAs in wildtype and mutant, using a splinted-ligation assay (Maroney et al., 2007) (Fig. S4A). Thus, Cdk9 affects differential abundance of miRNAs originating from common precursor RNAs.
One possible reason for the differential effect is that Cdk9 affects pri-miRNA synthesis, and therefore pri-miRNA levels change in the mutant in concordance with altered mature miRNA levels. To test this possibility, we performed RT-qPCR on pri-miRNA transcripts. To ensure that our RT-qPCR analysis was measuring bona fide pri-miRNAs, we measured RNA levels in a drosha mutant (Fig. S4B). Since Drosha is essential for pri-miRNA processing, its loss should increase abundance of the pri-miRNA precursors. Indeed, most probes measured higher levels of mutant RNA, consistent with their detection of pri-miRNAs. We then compared pri-miRNA levels between cdk9 mutant and wildtype animals (Fig. 4E). The miR-279/-996 pri-miRNA was increased in the cdk9 mutant, suggesting an effect on processing. Three of five clusters showed no significant change in pri-miRNA levels in the cdk9 mutant. The reason why the mutant did not elicit a significant change in these pri-miRNA levels is likely because the mature miRNA levels are only reduced 1.5 to 3 fold in the mutant relative to wildtype, in contrast to the drosha mutant which had a greater effect since processing was abolished. The miR-275/-305 pri-miRNA was reduced in the mutant, whereas the level of mature miR-305 was increased, suggesting an effect on processing and not synthesis. Overall, these results indicate that altered miRNA levels cannot be explained by an effect of Cdk9 on pri-miRNA synthesis.
An apical junction motif in pri-miRNA hairpins is related to differential processing
Sequence motifs in the pri-miRNA hairpin ensure accurate and efficient pri-miRNA processing in vitro (Auyeung et al., 2013; Fang and Bartel, 2015; Ma et al., 2013; Nguyen et al., 2015). These determinants include an apical junction motif composed of UGU, and two basal junction motifs: UG and CNNC (Fig. 5A). They enhance in vitro processing but are not essential. We wondered whether these motifs or some unknown sequence motif might be related to the differential processing regulated by Cdk9. Therefore, we analyzed polycistronic miRNA hairpin sequences that were affected by the cdk9 mutant. The only motif that showed hints of a correlation was the apical junction UGU. MicroRNAs that did not decrease in cdk9 mutants typically contained a UGU in their apical junctions (Fig. 5B), whereas those that decreased in cdk9 mutants did not have a UGU (Fig. 5C). Although this might suggest that presence of a UGU motif in hairpins is related to differential processing events mediated by Cdk9, the number of hairpins was too small to make any definite conclusion. However, the polycistronic miRNAs we examined represent only a small fraction of the miRNAs that were affected by the cdk9 mutant. In total, out of 77 miRNAs detected (Table S1), 30 miRNAs were significantly over-expressed in cdk9 mutants, while 26 miRNAs were under-expressed (Fig. 5D). Thus, many monocistronic miRNAs were also affected by cdk9. To confirm that monocistronic miRNA levels were affected by cdk9 in the same manner as polycistronic miRNAs, we examined two of the monocistronic miRNAs: miR-7 and miR-14. By RNA-seq, miR-7 was reduced and miR-14 was increased in the cdk9 mutant (Fig. S4C). These effects were also observed when mature miR-7 and miR-14 levels were assayed by Northern blot (Fig. S4D,E). However, the level of pri-miR-7 RNA was not reduced in the cdk9 mutant (Fig. S4D,F), consistent with what we had observed with polycistronic miRNAs. Therefore, it appears that processing of monocistronic and polycistronic miRNAs is regulated by Cdk9.
Figure 5. An apical junction sequence is related to Cdk9-dependent processing.

See also Figures S3 and S4. (A) Generalized structural and sequence features of pri-miRNA hairpin. Sequence motifs that affect in vitro processing specificity and efficiency are highlighted. Cleavage sites for Drosha and Dicer are shown with arrowheads. (B) Predicted pre-miRNA structures for those miRNAs whose abundance did not decrease in cdk9 mutants. Highlighted are the UGU motif (yellow) and mature miRNA (blue). (C) Predicted pre-miRNA structures for those miRNAs whose abundance decreased in cdk9 mutants. For B and C, mFold and RNAfold were independently used to predict the pre-miRNA structures. (D) Differential expression of all miRNAs detected by RNA-seq between mutant and wildtype samples. miRNAs are ranked by fold-change, and those whose fold-change is considered significant (a FDR below 5%) are colored. Those miRNAs that are derived from polycistronic genes are noted. (E,F) Logo graphs of nucleotide sequence bias within the apical junctions of pri-miRNA hairpins. Contrasted are the 30 miRNAs whose expression is greater in cdk9 mutants (E) versus the 26 miRNAs whose expression is reduced in cdk9 mutants (F). The y-axes correspond to the binomial probability of residue frequencies, with respect to the background of all Drosophila pre-miRNA sequences. Threshold values of p < 0.05 significance (2.75) are shown in red and marked with red horizontal lines.
Polycistronic miRNAs showed a bias for having or not having the apical UGU motif dependent on their response to Cdk9 activity. We looked to see if all miRNAs responsive to Cdk9 showed a similar bias. MicroRNAs that were over-expressed in cdk9 mutants had apical junctions that were strongly enriched for a UGU motif (Fig. 5E). There was also a small enrichment for C and U at positions 6 and 7 downstream of the U at position 1. MicroRNAs that were under-expressed in cdk9 mutants had apical junctions that were strongly depleted for the UGU motif (Fig. 5F). In particular, position 1 was typically lacking a U, and there was no enrichment for C at positions 6 and 7. These data suggest that Cdk9 is required for regulating differential processing events related to the apical UGU motif in pri-miRNA hairpins.
Cdk9 is required for proper miRNA-mediated mRNA silencing in vivo
Microprocessor appears to associate with Pol II, which leads to differential processing of certain pri-miRNA hairpins. Is this of functional importance? To explore the question, we looked at the effects of the cdk9 mutant on miRNA-mediated gene regulation. We had previously generated a transgenic reporter for miRNA regulation of the Bearded (Brd) gene (Pressman et al., 2012). This reporter is regulated by miR-7, which is reduced in the cdk9 mutant (Fig. S5A). Therefore, we assayed the reporter in a developing eye that contained clones of cdk9 mutant cells (Fig. 6A). Mutant cells showed a dramatic upregulation of reporter expression. If cdk9 mutant eye clones also contained the Cdk9 rescue transgene, then reporter expression was not upregulated to the same degree (Fig. 6B). Reporter mRNA abundance and structure were unchanged in the mutant, as demonstrated by 3′-RACE (Fig. S5B). These results indicate that miR-7-mediated Brd gene regulation is impaired in cdk9 mutant cells.
Figure 6. Differential processing has a significant effect on gene silencing in vivo.

See also Figure S5. (A,B) Expression of the Brd reporter gene in the developing eye. Each image contains approximately 800 cells, some of which are genetically wildtype and some of which are mutant for the cdk9 gene. Cell genotypes have been marked by the presence or absence of an RFP marker, as indicated. RFP and GFP channels are shown separately along with a merged image. Since cdk9 mutant cells in (B) also expressed the Cdk9 rescue transgene, silencing of the Brd reporter is restored within these cells. (C) When rbf mutant eye discs are stained for activated caspase protein, they show a zone of prevalent cell apoptosis within the morphogenetic furrow. (D) This zone is absent if eye cells overexpress miR-998 via the GAL4/UAS system. (E) When rbf mutant cells are also mutant for cdk9, then there is greatly reduced apoptosis. (F) A summary of the genetic experiments showing the pathways of gene regulation downstream of Cdk9. Scale bars for A–E, 10 μm.
Other miRNAs are overexpressed in the cdk9 mutant, including miR-998. This miRNA has an anti-apoptotic function in the developing eye, where it enhances survival signaling via EGFR (Truscott et al., 2014). MiR-998 does so by directly repressing Cbl, a negative regulator of EGFR signaling. Anti-apoptotic function of miR-998 was studied in the context of an rbf mutation, which induces a high level of apoptosis (Fig. 6C). If miR-998 is overexpressed, apoptosis is completely blocked (Fig. 6D). We reasoned that since cdk9 mutant cells overexpressed miR-998, this might also be sufficient to block apoptosis in the eye. Therefore, we generated clones of cdk9 mutant cells in the rbf eye and found that these cells, like cells overexpressing miR-998, were completely protected from apoptosis (Fig. 6E). In summary, Cdk9 is required for generating levels of miRNAs appropriate for regulating normal cellular functions (Fig. 6F).
DISCUSSION
We provide evidence for a direct interaction between a component of the Microprocessor complex and Pol II. Our results are consistent with previous studies suggesting that select miRNAs are co-transcriptionally processed, and that co-transcriptional processing is specifically associated with CTD-phosphorylated Pol II (Morlando et al., 2008; Nojima et al., 2015; Pawlicki and Steitz, 2008). Our results are also consistent with Drosha association with Pol II and ChIP detection of DGCR8 at pri-miRNA loci in a variety of cells (Gromak et al., 2013; Suzuki et al., 2017). We found that Cdk9 stimulates processing of miRNAs that lack a UGU apical junction motif, whereas miRNAs that contain a UGU motif are not stimulated by Cdk9. From in vitro studies it was found that a pri-miRNA is processed by purified Microprocessor more efficiently if the pri-miRNA has a UGU motif (Nguyen et al., 2015). However, if DGCR8 lacks its Rhed domain, then the UGU motif has no effect on processing efficiency. This indicates that the Rhed domain is essential for freely diffusing Microprocessor to functionally interact with the UGU motif. It is also consistent with binding studies that find the Rhed domain physically associates with the apical junction (Nguyen et al., 2015; Quick-Cleveland et al., 2014).
How then does Pol II association stimulate the processing of UGU-negative miRNAs but not UGU-positive miRNAs? One model is that when bound to Pol II, Microprocessor alters its intrinsic preference for RNA processing based on the UGU motif. Another model is that tethering of Microprocessor to Pol II does not affect its intrinsic processing activity. Instead, tethering increases the effective concentration of Microprocessor at the site of hairpin RNA synthesis, consequently enhancing the rate of co-transcriptional processing. The latter model is favored by in vitro processing studies. When phosphorylated peptide is added to in vitro Microprocessor reactions, the peptide has no effect on pri-miRNA processing efficiency, regardless of whether the substrate RNA contains a UGU motif or not (Narry Kim and Tuan Ahn Nguyen, personal communication). Although the model would predict that tethering stimulates UGU and non-UGU RNA substrates equally, this is not seen. Possibly when tethering is lost, UGU-containing RNA substrates are still efficiently processed in the nucleoplasm. However, because non-UGU substrates are processed so inefficiently as free entities, the bulk of their processing would instead be achieved by tethering Microprocessor to Pol II and localizing it near nascent pri-mRNA transcripts. This would make non-UGU substrates more dependent upon tethering. To summarize, pri-miRNA processing is accelerated by either the presence of a UGU motif in the RNA substrate or by tethering of Microprocessor. In diverse animal species, only one-third of pri-miRNA hairpins contain a UGU motif, suggesting that these two mechanisms might be an important and conserved feature of differential pri-miRNA processing.
The most parsimonious mechanism to account for the results of our study and other studies is that the Pasha-Pol II interaction directly mediates differential processing. If so, then a cis-mutation in Pasha’s WW domain that blocks Pol II interaction would phenocopy the processing defects of the cdk9 mutant. However, a systematic mutation of the Pasha WWD domain was unsuccessful since proteins were insoluble (data not shown). Therefore, it remains a formal possibility that the Cdk9 effect on processing is indirect. However, we found that Cdk9-dependent processing is strongly related to the UGU motif in the apical junction of the pre-miRNA precursor, which directly associates with the Rhed domain. If the Cdk9 effects on differential processing were indirect, then it becomes difficult to parse how the presence or absence of a UGU motif relates to it.
Why are there distinct mechanisms for nuclear processing of pri-miRNAs? We do not think that these mechanisms act on miRNAs according to their expression level. Analysis of our data did not find a significant correlation between the abundance of miRNAs and either cdk9 responsiveness or the UGU motif. Rather, we think that these mechanisms enable efficient processing of polycistronic transcripts. If only one or two Microprocessor molecules are tethered to each elongating Pol II, then tethered Microprocessor would encounter the 5′ hairpins first, dissociate from Pol II, and process them. Processing of more distal miRNAs would necessitate a non-tethered mechanism, which is more efficient if the hairpins contain a UGU motif. The net result would be differential expression of miRNAs from a polycistronic transcript, which is frequently observed. There might be other reasons for processing to occur by distinct mechanisms. For hairpins embedded within introns, they would be more efficiently excised by tethered Microprocessor from nascent transcripts before splicing occurs (Pawlicki and Steitz, 2008). Indeed, intronic hairpins can be processed from unspliced introns (Kim and Kim, 2007). For hairpins that reside in exons, tethered processing might not be essential for efficient excision, and so free Microprocessor could act on them. Finally, tethered processing could be susceptible to regulatory processes distinct from processing in the free nucleoplasm. Local chromatin organization, histone modification, and the proximity of cis-acting RNAs and proteins might regulate Microprocessor activity when tethered to Pol II. Regulated RNA splicing is controlled by such mechanisms (Naftelberg et al., 2015), and so it is plausible for pri-miRNA processing to be as well.
EXPERIMENTAL PROCEDURES
Genetics
A list of all Drosophila genotypes used is presented in Table S2. An EMS-mutagenesis screen previously described was used to generate the cdk9G57S mutant strain (Pressman et al., 2012). The mutation was meiotically mapped to the cdk9 locus, and a base substitution of G to A in the gene’s coding sequence was detected. To prove that the cdk9 mutation was responsible for the phenotypes, a 2.5 kb fragment of genomic DNA encompassing the cdk9 gene (Fig. S2) was cloned into the pYES vector (Patton et al., 1992). Transgenic lines were created using P-element mediated transformation.
Cell Culture
Drosophila S2* cells were cultured in Schneider’s media supplemented with 10% FBS (Invitrogen). To make stable cell lines, cells were transfected with either GFP or GFP-Pasha expressing pMK33 plasmid vectors, and selected for 4–5 weeks on Hygromycin B until >90% cells expressed GFP or GFP-Pasha when induced with 250 μM CuSO4 for 12–16 hours. For flavopiridol treatment, cells were plated at 2 x 106 cells/ml and treated with 0.5 μM flavopiridol (Sigma) for 2 or 12 hours. Longer treatments were necessary to allow sufficient induction of GFP or GFP-Pasha after addition of CuSO4.
Analysis of cdk9 mutant larvae
Balanced cdk9G57S animals were crossed and progeny were collected. Balancer chromosomes carried a twist> GFP transgene, and so after 48 hours, non-fluorescent larval progeny were isolated. They were gently washed in deionized water and either frozen or immediately extracted for RNA. RNA extraction was performed using Trizol. For Western blotting, larvae were lysed in RIPA buffer (50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) plus protease and phosphatase inhibitors and spun 16,000xg for 15 minutes 4°C before performing SDS-PAGE.
RNA-seq
Small-RNA seq libraries were prepared and sequenced on an Illumina platform as described (Gu et al., 2009; Shirayama et al., 2012). Since Drosophila expresses a 2S rRNA that is 30 nts in length, we size-selected RNAs between 15 and 29 nts by gel purification before library preparation. Reads were mapped to the Drosophila genome build dm3 using Bowtie. Annotation was performed using Avadis NGS software. Read counts were normalized by library size in EdgeR/DESEQ and the data were filtered so that only miRNAs that had 500 or more reads across samples were analyzed.
Northern Blot, Splinted-Ligation, RT-qPCR and 3′RACE
For all RNA analysis, total RNA purified by Trizol was assayed. Oligos used for RNA analysis are listed in Table S3. For Northern blots, 20 μg of total RNA was electrophoresed on a 15% acrylamide/8M urea gel. Membranes were chemically cross-linked with EDC (0.15 M 1-methylimidazole, 0.16 M N-Ethyl-N′-(3-dimethylaminopropyl)carbodiimide, pH 8.0) for 2 hours at 60°C. The membrane was then pre-hybridized in ULTRAhyb (Ambion) for 30 minutes at 60°C. Oligonucleotide complimenta ry to 2S rRNA (IDT), and locked nucleic acid oligos complementary to mature miR-7 and miR-14 (Exiqon) were hybridized overnight at 37°C. Blots were washed at 37°C in 2X SSC, 0.1% SDS and then in 0.1X SSC, 0.1% SDS.
For splinted ligation assays of mature miRNAs, reactions were carried out as described (Lee et al., 2009; Maroney et al., 2007). A ligation-oligo was 5′-end labeled with [γ-32P]-ATP. The labeled product was phenol-cholorform extracted and diluted in 100 μL RNase free H2O. Bridge-oligos were designed based off of the corresponding miRbase miRNA sequences. Bridge-oligos were synthesized with a 3 carbon spacer at the 3′end. 5–20μg total RNA from 48 hr cdk9G57S and wildtype larvae was used for each ligation reaction including T4 DNA ligase (NEB). For each assay, three negative controls were performed, leaving out either: ligase, RNA, or bridge-oligo. Reaction products were resolved on 12% urea-polyacrylamide gels. Gels were imaged overnight on a PhosphorImager. Data was quantified in ImageQuant and signals for each miRNA product were corrected for control backgrounds, were normalized to 2S rRNA, and then reported as the log2(Fold Change) for cdk9G57S relative to wildtype.
For RT-qPCR, 2.5 μg of total RNA was primed with a 1:2 molar mix of oligo-dT: random 9mers for the RT reaction. qPCR was performed on a Biorad ICycler and the delta-delta Ct method was used to measure output. RpL32 was used to normalize RNA levels.
3′ RACE and nested RT-PCR were performed using gene specific RT primers (listed in Table S3) and Qo, QI, QT primers (Scotto-Lavino et al., 2006). Briefly, total RNA was purified from 48 hour wildtype or droshaQ884X larvae using Trizol. Reverse transcription was performed with Superscript III using 5 μg total RNA per reaction and QT or Ro primers (for 3′RACE or nested PCR, respectively). cDNA reactions were treated with 0.75 μL RNase H (Promega) for 20 minutes at 37°C and diluted in 1 m L of TE (Tris-EDTA buffer pH 8.0). The first PCR round was performed with 1 μL of cDNA per 50 μL PCR reaction and the following primers: Qo and GSP1 primers for 3′RACE or the “outer” gene specific primers for nested PCR. The second PCR round was performed with 1 μL of a 1:20 dilution of round one PCR, using QI and GSP2 primers for 3′RACE or the “inner” gene specific primers for nested PCR. All PCRs were performed with GoTaq DNA polymerase following the supplied protocol (Promega).
Apoptosis assay
Eye imaginal discs were dissected from third instar larvae in Schneider’s insect medium (Sigma) and then fixed in phosphate-buffered saline (PBS) buffer with 4% formaldehyde. Discs were then blocked in PBS + 0.1% Triton X-100 (PBST) + 10% normal donkey serum followed by overnight incubation with rabbit anti-C3 (Cleaved Caspase 3), lot 26, 1:50 (Cell Signaling). Discs were washed in PBST and incubated in blocking solution containing the appropriate secondary antibodies: Cy3-, or Cy5- conjugated anti-rabbit antibodies, 1:300 (Jackson Immunoresearch). After washing in TBST, discs were placed into glycerol containing 0.5% propyl gallate in preparation for slide mounting. Imaging was performed using a Zeiss LSM Observer.Z1.
Protein Association Assays
Immunoprecipitations, GST pulldowns, and peptide binding assays were performed essentially as described (Green and Sambrook, 2012; Kim et al., 2004), with some modifications. See Supplemental Information for precise details of these assays.
Supplementary Material
Highlights.
Differential nuclear processing of microRNAs affects microRNA abundance
Microprocessor subunit DGCR8 binds to the CTD of elongating Pol II
Co-transcriptional processing targets microRNAs lacking a UGU motif
Acknowledgments
We thank Narry Kim and Tuan Ahn Nguyen for kindly sharing their results on peptide interactions with Microprocessor activities in vitro. We thank Matt Schipma of the Northwestern Genomics Core and Weifeng Gu for assistance with RNA-seq adaptor removal and genome alignment. We also thank J. Brickner, G. Hannon and C. Horvath for gifts of reagents. We thank G. Beitel and our lab colleagues for their advice. This work was supported by the NIH (T32GM08061 to V.A.C.; R01GM110018 to M.V.F.; R01GM56663 to S.B.; R01GM077581 and R35GM118144 to R.W.C.).
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, V.C., S.P., and R.C.; Methodology, V.C., S.B., M.F., and R.C.; Investigation, V.C., S.P., M.I., M.T. and N.C.; Formal Analysis, V.C.; Writing–Original Draft, V.C. and R.C.; Writing–Review and Editing, S.P., M.I., M.T., N.C., S.B., and M.F; Visualization, V.C., and R.C.; Supervision, M.F., S.B., and R.C.
ACCESSION NUMBERS
RNA-seq data has been deposited in the Gene Expression Omnibus (GEO) under accession number GSE103234.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alarcon CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519:482–485. doi: 10.1038/nature14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auyeung VC, Ulitsky I, McGeary SE, Bartel DP. Beyond secondary structure: primary-sequence determinants license pri-miRNA hairpins for processing. Cell. 2013;152:844–858. doi: 10.1016/j.cell.2013.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkowiak B, Liu P, Phatnani HP, Fuda NJ, Cooper JJ, Price DH, Adelman K, Lis JT, Greenleaf AL. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010;24:2303–2316. doi: 10.1101/gad.1968210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky AS, Meyer CA, Swinburne IA, Hall G, Keenan BJ, Liu XS, Fox EA, Silver PA. Genomic mapping of RNA polymerase II reveals sites of co-transcriptional regulation in human cells. Genome Biol. 2005;6:R64. doi: 10.1186/gb-2005-6-8-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratowski S. Progression through the RNA polymerase II CTD cycle. Mol Cell. 2009;36:541–546. doi: 10.1016/j.molcel.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004;10:1957–1966. doi: 10.1261/rna.7135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaulk SG, Thede GL, Kent OA, Xu Z, Gesner EM, Veldhoen RA, Khanna SK, Goping IS, MacMillan AM, Mendell JT, et al. Role of pri-miRNA tertiary structure in miR-17~92 miRNA biogenesis. RNA Biol. 2011;8:1105–1114. doi: 10.4161/rna.8.6.17410. [DOI] [PubMed] [Google Scholar]
- Conrad T, Marsico A, Gehre M, Orom UA. Microprocessor activity controls differential miRNA biogenesis In Vivo. Cell Rep. 2014;9:542–554. doi: 10.1016/j.celrep.2014.09.007. [DOI] [PubMed] [Google Scholar]
- Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–235. doi: 10.1038/nature03049. [DOI] [PubMed] [Google Scholar]
- Fang W, Bartel DP. The Menu of Features that Define Primary MicroRNAs and Enable De Novo Design of MicroRNA Genes. Mol Cell. 2015;60:131–145. doi: 10.1016/j.molcel.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M, Sambrook J. A Laboratory Manual. 4. New York: Cold Spring Harbor Laboratory Press; 2012. Molecular Cloning. [Google Scholar]
- Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, Shiekhattar R. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- Gromak N, Dienstbier M, Macias S, Plass M, Eyras E, Caceres JF, Proudfoot NJ. Drosha regulates gene expression independently of RNA cleavage function. Cell Rep. 2013;5:1499–1510. doi: 10.1016/j.celrep.2013.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Shirayama M, Conte D, Jr, Vasale J, Batista PJ, Claycomb JM, Moresco JJ, Youngman EM, Keys J, Stoltz MJ, et al. Distinct argonaute-mediated 22G-RNA pathways direct genome surveillance in the C. elegans germline. Mol Cell. 2009;36:231–244. doi: 10.1016/j.molcel.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509–524. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- Jeronimo C, Collin P, Robert F. The RNA Polymerase II CTD: The Increasing Complexity of a Low-Complexity Protein Domain. J Mol Biol. 2016;428:2607–2622. doi: 10.1016/j.jmb.2016.02.006. [DOI] [PubMed] [Google Scholar]
- Kim M, Krogan NJ, Vasiljeva L, Rando OJ, Nedea E, Greenblatt JF, Buratowski S. The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature. 2004;432:517–522. doi: 10.1038/nature03041. [DOI] [PubMed] [Google Scholar]
- Kim YK, Kim VN. Processing of intronic microRNAs. EMBO J. 2007;26:775–783. doi: 10.1038/sj.emboj.7601512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SC, Nguyen TA, Choi YG, Jo MH, Hohng S, Kim VN, Woo JS. Structure of Human DROSHA. Cell. 2016;164:81–90. doi: 10.1016/j.cell.2015.12.019. [DOI] [PubMed] [Google Scholar]
- Landthaler M, Yalcin A, Tuschl T. The human DiGeorge syndrome critical region gene 8 and Its D. melanogaster homolog are required for miRNA biogenesis. Curr Biol. 2004;14:2162–2167. doi: 10.1016/j.cub.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. Embo J. 2004;23:4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Pressman S, Andress AP, Kim K, White JL, Cassidy JJ, Li X, Lubell K, Lim DH, Cho IS, et al. Silencing by small RNAs is linked to endosomal trafficking. Nat Cell Biol. 2009;11:1150–1156. doi: 10.1038/ncb1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Wu Y, Choi JG, Wu H. Lower and upper stem-single-stranded RNA junctions together determine the Drosha cleavage site. Proc Natl Acad Sci U S A. 2013;110:20687–20692. doi: 10.1073/pnas.1311639110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias MJ, Wiesner S, Sudol M. WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 2002;513:30–37. doi: 10.1016/s0014-5793(01)03290-2. [DOI] [PubMed] [Google Scholar]
- Maroney PA, Chamnongpol S, Souret F, Nilsen TW. A rapid, quantitative assay for direct detection of microRNAs and other small RNAs using splinted ligation. RNA. 2007;13:930–936. doi: 10.1261/rna.518107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlando M, Ballarino M, Gromak N, Pagano F, Bozzoni I, Proudfoot NJ. Primary microRNA transcripts are processed co-transcriptionally. Nat Struct Mol Biol. 2008;15:902–909. doi: 10.1038/nsmb.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris DP, Phatnani HP, Greenleaf AL. Phospho-carboxyl-terminal domain binding and the role of a prolyl isomerase in pre-mRNA 3′-End formation. J Biol Chem. 1999;274:31583–31587. doi: 10.1074/jbc.274.44.31583. [DOI] [PubMed] [Google Scholar]
- Naftelberg S, Schor IE, Ast G, Kornblihtt AR. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu Rev Biochem. 2015;84:165–198. doi: 10.1146/annurev-biochem-060614-034242. [DOI] [PubMed] [Google Scholar]
- Nguyen TA, Jo MH, Choi YG, Park J, Kwon SC, Hohng S, Kim VN, Woo JS. Functional Anatomy of the Human Microprocessor. Cell. 2015;161:1374–1387. doi: 10.1016/j.cell.2015.05.010. [DOI] [PubMed] [Google Scholar]
- Ni Z, Schwartz BE, Werner J, Suarez JR, Lis JT. Coordination of transcription, RNA processing, and surveillance by P-TEFb kinase on heat shock genes. Mol Cell. 2004;13:55–65. doi: 10.1016/s1097-2765(03)00526-4. [DOI] [PubMed] [Google Scholar]
- Nojima T, Gomes T, Grosso AR, Kimura H, Dye MJ, Dhir S, Carmo-Fonseca M, Proudfoot NJ. Mammalian NET-Seq Reveals Genome-wide Nascent Transcription Coupled to RNA Processing. Cell. 2015;161:526–540. doi: 10.1016/j.cell.2015.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive V, Sabio E, Bennett MJ, De Jong CS, Biton A, McGann JC, Greaney SK, Sodir NM, Zhou AY, Balakrishnan A, et al. A component of the mir-17–92 polycistronic oncomir promotes oncogene-dependent apoptosis. Elife. 2013;2:e00822. doi: 10.7554/eLife.00822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton JS, Gomes XV, Geyer PK. Position-independent germline transformation in Drosophila using a cuticle pigmentation gene as a selectable marker. Nucleic Acids Res. 1992;20:5859–5860. doi: 10.1093/nar/20.21.5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlicki JM, Steitz JA. Primary microRNA transcript retention at sites of transcription leads to enhanced microRNA production. J Cell Biol. 2008;182:61–76. doi: 10.1083/jcb.200803111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S, Zavolan M, Grasser FA, Chien M, Russo JJ, Ju J, John B, Enright AJ, Marks D, Sander C, et al. Identification of virus-encoded microRNAs. Science. 2004;304:734–736. doi: 10.1126/science.1096781. [DOI] [PubMed] [Google Scholar]
- Pressman S, Reinke CA, Wang X, Carthew RW. A Systematic Genetic Screen to Dissect the MicroRNA Pathway in Drosophila. G3 (Bethesda) 2012;2:437–448. doi: 10.1534/g3.112.002030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DH. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol. 2000;20:2629–2634. doi: 10.1128/mcb.20.8.2629-2634.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick-Cleveland J, Jacob JP, Weitz SH, Shoffner G, Senturia R, Guo F. The DGCR8 RNA-binding heme domain recognizes primary microRNAs by clamping the hairpin. Cell Rep. 2014;7:1994–2005. doi: 10.1016/j.celrep.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryazansky SS, Gvozdev VA, Berezikov E. Evidence for post-transcriptional regulation of clustered microRNAs in Drosophila. BMC Genomics. 2011;12:371. doi: 10.1186/1471-2164-12-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder S, Herker E, Itzen F, He D, Thomas S, Gilchrist DA, Kaehlcke K, Cho S, Pollard KS, Capra JA, et al. Acetylation of RNA polymerase II regulates growth-factor-induced gene transcription in mammalian cells. Mol Cell. 2013;52:314–324. doi: 10.1016/j.molcel.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz BE, Larochelle S, Suter B, Lis JT. Cdk7 is required for full activation of Drosophila heat shock genes and RNA polymerase II phosphorylation in vivo. Mol Cell Biol. 2003;23:6876–6886. doi: 10.1128/MCB.23.19.6876-6886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotto-Lavino E, Du G, Frohman MA. Amplification of 5′ end cDNA with ‘new RACE’. Nat Protoc. 2006;1:3056–3061. doi: 10.1038/nprot.2006.479. [DOI] [PubMed] [Google Scholar]
- Shirayama M, Seth M, Lee HC, Gu W, Ishidate T, Conte D, Jr, Mello CC. piRNAs initiate an epigenetic memory of nonself RNA in the C. elegans germline. Cell. 2012;150:65–77. doi: 10.1016/j.cell.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Jee D, de Navas LF, Duan H, Lai EC. Multiple In Vivo Biological Processes Are Mediated by Functionally Redundant Activities of Drosophila mir-279 and mir-996. PLoS Genet. 2015;11:e1005245. doi: 10.1371/journal.pgen.1005245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki HI, Young RA, Sharp PA. Super-Enhancer-Mediated RNA Processing Revealed by Integrative MicroRNA Network Analysis. Cell. 2017;168:1000–1014. e1015. doi: 10.1016/j.cell.2017.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truscott M, Islam ABMMK, Lightfoot J, Lopez-Bigas N, Frolov MV. An intronic microRNA links Rb/E2F and EGFR signaling. PLoS genetics. 2014;10:e1004493. doi: 10.1371/journal.pgen.1004493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truscott M, Islam ABMMK, Lopez-Bigas N, Frolov MV. mir-11 limits the proapoptotic function of its host gene, dE2f1. Genes & development. 2011;25:1820–1834. doi: 10.1101/gad.16947411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wynsberghe PM, Kai ZS, Massirer KB, Burton VH, Yeo GW, Pasquinelli AE. LIN-28 co-transcriptionally binds primary let-7 to regulate miRNA maturation in Caenorhabditis elegans. Nat Struct Mol Biol. 2011;18:302–308. doi: 10.1038/nsmb.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat Struct Biol. 2000;7:639–643. doi: 10.1038/77929. [DOI] [PubMed] [Google Scholar]
- Weitz SH, Gong M, Barr I, Weiss S, Guo F. Processing of microRNA primary transcripts requires heme in mammalian cells. Proc Natl Acad Sci U S A. 2014;111:1861–1866. doi: 10.1073/pnas.1309915111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S, Yu Y, Reed R. Primary microRNA processing is functionally coupled to RNAP II transcription in vitro. Sci Rep. 2015;5:11992. doi: 10.1038/srep11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Wang F, Yang GH, Wang FL, Ma YN, Du ZW, Zhang JW. Human microRNA clusters: Genomic organization and expression profile in leukemia cell lines. Biochemical and Biophysical Research Communications. 2006;349:59–68. doi: 10.1016/j.bbrc.2006.07.207. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Cullen BR. Sequence requirements for micro RNA processing and function in human cells. RNA. 2003;9:112–123. doi: 10.1261/rna.2780503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.