

ABSTRACT
In addition to the canonical role in protein homeostasis, autophagy has recently been found to be involved in axonal dystrophy and neurodegeneration. Whether autophagy may also be involved in neural development remains largely unclear. Here we report that Mir505–3p is a crucial regulator for axonal elongation and branching in vitro and in vivo, through modulating autophagy in neurons. We identify that the key target gene of Mir505–3p in neurons is Atg12, encoding ATG12 (autophagy-related 12) which is an essential component of the autophagy machinery during the initiation and expansion steps of autophagosome formation. Importantly, axonal development is compromised in brains of mir505 knockout mice, in which autophagy signaling and formation of autophagosomes are consistently enhanced. These results define Mir505–3p-ATG12 as a vital signaling cascade for axonal development via the autophagy pathway, further suggesting the critical role of autophagy in neural development.
KEYWORDS: ATG12, autophagy, axonal development, microRNA, Mir505–3p
Introduction
Macroautophagy (hereafter indicated as autophagy) is a conserved bulk lysosomal-degradation pathway which is involved in protein turnover, cell survival, cellular homeostasis, and stress response.1 Abnormalities in autophagy lead to defective protein degradation, resulting in pathologies including neurodegenerative diseases.2 Mice with specific deletion of Atg5 or Atg7, which are essential genes for autophagosome formation, show neurodegenerative phenotypes.3,4 Specific deletion of Sqstm1/p62 in cerebellar Purkinje neurons causes axon swelling, suggesting the role of autophagy in axon homeostasis.5 Moreover, it is also reported that autophagy activity modulates axon morphological plasticity by regulating axon extension during naive neurite growth in culture neurons.6 However, it is still uncertain whether autophagy is involved in axonal development in vivo and how autophagy is regulated during axonal development.
ATG12 is a key component of the ATG12–ATG5-ATG16L1 complex, involved in membrane initiation and the elongation of the phagophore during autophagosome formation.7,8 After a reaction involving the E1-like enzyme ATG7 and the E2-like enzyme ATG10, ATG12 is covalently conjugated to ATG5 in a ubiquitin-like manner.9 Then, ATG5 interacts with ATG16L1, and ATG12–ATG5-ATG16L1 forms a tetrameric complex through the homo-oligomerization of ATG16L1.10 Apart from the cellular function of membrane scaffold assembly of autophagosomes, ATG12 conjugates to ATG3, then interacts with PDCD6IP/Alix (programmed cell death 6 interacting protein) to promote basal autophagic flux and late endosome function, as well as mitochondrial homeostasis and cell death.11,12 Interestingly, ATG12 directly regulates the apoptotic pathway by binding and inactivating BCL2 (B cell leukemia/lymphoma 2) and MCL1 (myeloid cell leukemia sequence 1), independent of either ATG5 or ATG3.13 Additionally, free ATG12 can be directly ubiquitinated and promotes proteasomal degradation of itself.14 In this study, we showed ATG12 as a novel regulator of axon development. Inhibition of autophagy by Mir505–3p via targeting Atg12 leads to promotion of axon specification, elongation and branching.
In a previous work, we have identified that a quantitative trait locus (QTL) on the X chromosome is involved in the regulation of female mouse puberty onset.15 This locus contains a microRNA gene—Mir505 (Fig. S1A).16 Duplications of the genomic segment containing Mir505 are reported in human males with a neural developmental disorder, X-linked hypopituitarism (Fig. S1B).16-20 As puberty onset is a key event during neural development, including maturation of the hypothalamus–pituitary–gonadal axis (HPG axis), we reasoned that Mir505 may contribute to neural development.21 There are 2 mature microRNA products, Mir505–5p and Mir505–3p, generated from Mir505. In this work, we found that Mir505–3p was required for axonal development in a cultured mouse cortical-neuron model and in mouse in utero electroporation (IUE) experiments. We identified Atg12 as the direct target gene of Mir505–3p in mouse neurons. We showed that ATG12 specifically inhibited polarity establishment, axon elongation and branching in vitro and in vivo. We provided further evidence by generating mir505 knockout mice, in which defects in axonal development, elevated expression of ATG12 and upregulated autophagy signaling were identified. Taken together, these results indicated a novel role of Mir505–3p-ATG12 signaling cascade in regulating axonal development through autophagy.
Results
Mir505–3p regulates axon development in vitro
To investigate the role of Mir505 in neural development, we first examined the mRNA expression pattern of pre-Mir505, and 2 mature versions of Mir505, Mir505–3p and Mir505–5p, in developing mouse cortices.22 We found that pre-Mir505 was highly expressed during embryonic stage and then showed a gradual decrease from postnatal d 0 (P0) (Fig. S2A). Normalized with Mir16–1–5p, a stably expressed miRNA, expression of both Mir505–3p and Mir505–5p increased from P0 then reached to the peak around P7, and decreased around P21 (Fig. S2B, C). These observations imply a potential role of Mir505 in early brain development, as its expression pattern correlates to cortical development and maturation stages, starting from early embryonic stage to the end of the third postnatal wk.23
To determine whether Mir505 may exhibit functional roles in neural development, we designed miRNA mimics and complement siRNA inhibitors for Mir505–3p, as well as a Mir505–3p expression vector (173 base pairs (bps) containing the processing sequences). To confirm the efficacy of genetic modulations for Mir505–3p, we first examined the level of Mir505–3p expression by overexpressing mimics and inhibitors. We found a drastic increase after overexpressing Mir505–3p mimics and a significant decrease in the inhibitor group (Fig. S2D). Also, we found a significant increase of Mir505–3p after transfection of the Mir505–3p expression vector (Fig. S2E). Thus, we showed that in vitro genetic manipulations of Mir505–3p were efficient.
After electroporation of scrambled miRNAs, Mir505–3p and Mir505–5p mimics into cultured mouse cortical neurons via using the AMAXA nucleofector device, we examined morphological development of transfected neurons in vitro. At 3 d in vitro (DIV), we fixed neuronal cultures and performed immunofluorescence staining with anti-SMI312 antibody, an axonal specific marker, to distinguish the axon from the minor neurite branches including dendrites. Interestingly, we found that only Mir505–3p specifically augmented axon branching (Fig. 1A, B), without a significant effect on minor neurite branching (Fig. S3A). To confirm this data, we transfected the same vectors with Lipofectamine 2000. Consistent with previous observations, neural polarity establishment (Fig. 1C, D) and total axonal length (TAL) (Fig. 1C, E) of 5DIV cultured neurons were greatly promoted by Mir505–3p expression, but not total minor neurite length (TMNL) (Fig. S3B).24
Figure 1.
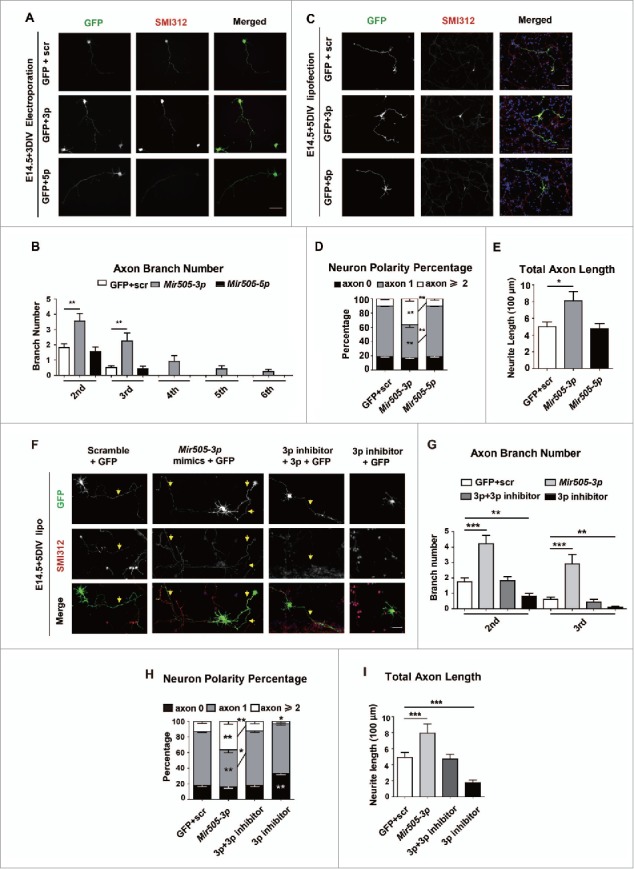
Mir505–3p is required for axonal development in vitro. (A) Example pictures of mouse primary cortical neurons transfected with the AMAXA nucleofector at 0 DIV with GFP and scrambled siRNA or GFP with constructs as indicated. Neurons were fixed and stained for immunofluorescence with anti-SMI312 and GFP antibodies at 3 DIV for measurement of numbers of axon and minor neurites. Scale bar: 50 μm. (B) Measurements of axon numbers in each condition of (A). (C) Example pictures of mouse primary cortical neurons transfected with Lipofectamine 2000 at 1 DIV with GFP and scrambled siRNA or GFP with constructs as indicated. Neurons were fixed at 5 DIV for measurement of neuron polarity and total axon length. Scale bar: 90 μm. (D) Measurements of neuron polarity of each condition in (C). (E) Measurements of total axon length of each condition in (C). (F) Example pictures of mouse primary cortical neurons transfected with Lipofectamine 2000 at 1 DIV with GFP and scrambled siRNA or GFP with constructs as indicated. Scale bar: 45 μm. ((G)to I) Measurements of axon numbers, neuron polarity and total axon length of each condition, respectively. A total 30 to 33 neurons from each condition were randomly selected and measured. Error bars: SEM. *P < 0.05, **P < 0.01, *** P < 0.001 (Student t test).
To further validate the role of Mir505–3p in axonal development, we next transfected scrambled miRNAs, Mir505–3p mimics, Mir505–3p inhibitors or a mixture of Mir505–3p mimics and inhibitors in cultured cortical neurons (Fig. 1F).25 We found that Mir505–3p inhibitors, in contrast to Mir505–3p mimics, exhibited a strong attenuation effect of axon branching, neural polarity establishment and axonal extension, which was rescued by Mir505–3p coexpression (Fig. 1F, G, H and I).26,27 However, no significant difference was observed in minor neurite branching and TMNL after Mir505–3p manipulations (Fig. S3C, D). These results suggest that Mir505–3p is a positive regulator for neural polarity establishment, axonal growth and branching in cultured mouse cortical neurons.
To further confirm these observations, we applied anti-MAPT/Tau, the marker of microtubule-associated proteins that stabilizes microtubule assembly in axons, to label matured axons. We transfected scrambled miRNAs, Mir505–3p mimics, Mir505–3p inhibitors or mixture of Mir505–3p mimics and inhibitors, with Lipofectamine 2000 in cultured cortical neurons (Fig. S4A). Consistent with SMI312 labeling, we observed that Mir505–3p inhibitors exhibited attenuation effects of neural polarity establishment (Fig. S4B), axonal extension (Fig. S4C) and axon branching (Fig. S4E), which were rescued by coexpression of Mir505–3p (Fig. S4A, B, C and E). Also, no effects of Mir505–3p inhibitors were observed for TMNL (Fig. S4D) and minor neurite branching (Fig. S4F). These results further indicate that Mir505–3p plays a critical role in regulating axonal growth and branching in cultured mouse cortical neurons.
Mir505–3p regulates axonal branching and extension in vivo
Axonal development in newborn cortical neurons can be separated to 3 steps. First, neurons migrate toward to cortical plate with a leading and trailing process, and the trailing process evolves to become the axon (from E11 to E18). Second, the axon extends to the contralateral side across the corpus callosum (at P3) to reach the target region (at P7). Third, the axon experiences a rapid branching progress to find synaptic partners and to form functional synapses (from P8 to P21).28
To explore whether Mir505–3p regulates axonal development in vivo, we performed in utero electroporation to overexpress Mir505–3p in the cortex of E14.5 mice. At P3, projections of GFP labeled axons from the ipsilateral side to the contralateral side across the corpus callosum were measured (Fig. 2A).29 We found that that neurons overexpressing Mir505–3p, showed significantly increased length of projecting axons on the contralateral side (Fig. 2B and D), indicating promotion of axonal extension by Mir505–3p. Intriguingly, significantly increased length of projecting axons of Mir505–3p-overexpressing neurons on the ipsilateral side was also observed (Fig. 2C and E). Typically, neurons possessed monopolar morphologies with a long axon projecting to the contralateral side, thus this aberrant axon bundle in the ipsilateral side of cortex may be generated by multipolarized cortical neurons, further supporting the role of Mir505–3p in neural polarization (Fig. 2K and L).28
Figure 2.

Mir505–3p is sufficient for axonal extension and branching in vivo. (A) Low-magnification images of coronal brain sections of P3 mice in utero electroporated at E 14.5 with the indicated plasmids. Scale bar: 1000 μm. (B) Magnification images of contralateral brain sections in (A). (C) Magnification images of ipsilateral brain sections in (A). (D) Measurements of axon length (from middle line to terminus of axon bundle, indicated by yellow line) in (B). (E) Measurements of axon length (from proximal region to terminus of axon bundle, indicated by yellow line) in (C). (F) Low-magnification images of coronal brain sections of P8 mice in utero electroporated at E 14.5 with plasmids as indicated. Scale bar: 1000 μm. (G) Magnification images of ipsilateral brain sections in (F). Scale bar: 100 μm. (H) Magnification images of contralateral brain sections in (F). Scale bar: 100 μm. (I) Measurements of axonal signal intensity in contralateral cortical plate from layer 1 to white matter of each condition in (H). (J) Measurements of axonal signal intensity in ipsilateral cortical plate layer 5 of each condition in (G). (K) Schematic diagram of coronal brain sections of P8 mice. Boxes with the indicated regions were further illustrated in (L). (L) Schematic diagram of axonal branching morphology in IUE model of each condition. At least 4 litters for each condition were measured. Error bars: SEM. **P < 0.01, *** P < 0.001 (Student t test).
To further investigate the role of Mir505–3p in regulating axonal development in vivo, we harvested IUE mice at P8 to examine terminal axonal branching in barrel field of primary somatosensory cortex on contralateral side (Fig. 2F). At P8, axons of cortical neurons are semiradiated toward deep cortical layers (Fig. 2F and G), and then extended further branches in the contralateral side (Fig. 2F, H, K and L).30 We found that neurons expressing Mir505–3p exhibited enriched axonal branches on both ipsilateral side and contralateral side (Fig. 2G, H and L). We measured the axon branching signal of layer 5 on the ipsilateral side and performed a consecutive measurement from layer 1 to white matter on the contralateral side.31 We showed that Mir505–3p remarkably promoted axon branching on every layer of the contralateral side (Fig. 2H and I) and also promoted axon branching in layer 5 on the ipsilateral side (Fig. 2G and J). Taken together, these data indicate that Mir505–3p plays a critical role in promoting axonal extension and branching in vivo.
Genetic deletion of Mir505–3p inhibits axonal development in vivo
To investigate whether Mir505–3p is required for axonal development genetically, we designed 2 small guide RNAs (sgRNAs) targeting to the Mir505 gene (Fig. 3A) and applied the CRISPR/Cas9 system to generate mir505 knockout mice with a 24-bp deletion (Fig. 3B). To ensure these 2 sgRNAs did not lead to off-target effects on offspring of mir505 knockout mice, we predicted potential off-target genes and designed specific primers for testing and sequencing. We found that all the Mir505 deletion founder mice were without off-target effects (Fig. S5A, B and Table S2, see also CRISPR/Cas9-based mir505 knockout mice in Materials and Methods). Consistently, we confirmed that levels of Pre-Mir505, Mir505–3p and Mir505–5p decreased remarkably in KO mice, compared with WT mice (Fig. 3C).
Figure 3.

Genetic deletion of Mir505–3p impairs axon development in vivo. (A) Schematic diagram of 2 sgRNAs targeting pre-Mir505. (B) Sequencing results show a total deletion of Mir505–3p induced by CRISPR/Cas9 system (upper). DNA gel electrophoresis show genotyping results of WT, heterozygous and KO littermates (lower). A 24 bp deletion mutation was generated by the CRISPR/Cas9 system. (C) Examination of mRNA levels of Pre-Mir505, Mir505–3p and Mir505–5p in mir505 KO mice comparing to WT littermates by Q-PCR. (D and E) Coronal brain sections of mature mice with hematoxylin-eosin (D) and toluidine blue staining (E). Alteration of axon morphology is indicated by dotted lines. Corpus callosum (cc) and cingulum (cg) regions were amplified to represent loss of axon bundles in KO mice. Scale bar: 100 μm. (F) Magnification images of the indicated regions of each genotype. Immunostaining was performed with anti-SMI312 and RBFOX3 antibodies. Scale bar: 50 μm. Het, heterozygous. (G to I) Measurements of axon signal intensity in cc (G), fi (H) and cg (I) regions of mice of each genotype, respectively. (J) Measurements of RBFOX3-positive cells in cingulum of mice of each genotype. For immunostaining on brain slices, at least 4 litters for each condition were measured. Error bars are SEM. ** P < 0.05, *** P < 0.001 (t test).
Next, we performed hematoxylin-eosin and toluidine blue staining to examine brain anatomy of WT and mir505 KO mice.32 We found that, in axon-concentrated regions such as in corpus callosums (cc) and cingulums (cg), axonal density was clearly decreased in KO mice, in comparison with WT mice (Fig. 3D and E). To further confirm this observation, we performed immunofluorescence staining on SMI312 to monitor axon bundles and on RBFOX3/NeuN to label mature neurons. We found that density of axon bundles significantly decreased in cc, cg, and fimbria (fi) regions of KO mice comparing to WT littermates, with no significant differences between heterozygous and WT mice (Fig. 3F, G, H and I). Intriguingly, no difference in cell number of RBFOX3-positive neuron was observed, indicating that attenuation of axons resulted from defects in axonal development, rather than neuronal proliferation (Fig. 3F and J). Thus, this evidence demonstrate that genetic ablation of Mir505–3p has a strong impact on axonal density of brain in vivo.
Atg12 is a target gene of Mir505–3p
To identify target genes of Mir505–3p in mouse neurons, we first performed a bioinformatics screen. We found 172 potential target genes in Targetscan 6.2 database, which predicted target genes based on conservation and position of seed-match region in the 3′ untranslated region (UTR),33,34 and found that 57 genes emerged more than twice from 7 other databases. Then we performed seed-match predictions based on RNA sequencing results of cortical neurons, and found 799 candidate genes, which are expressed in relatively high levels in neurons.35 Comparing these gene lists, we screened out 8 predicted candidate target genes, which have potentially important functions in neurons. We also selected Srsf1, which has previously been reported as a target gene in mouse embryonic fibroblasts (MEFs). Together, these 9 candidate genes were selected for further investigation (Fig. 4A and Table S1).36
Figure 4.
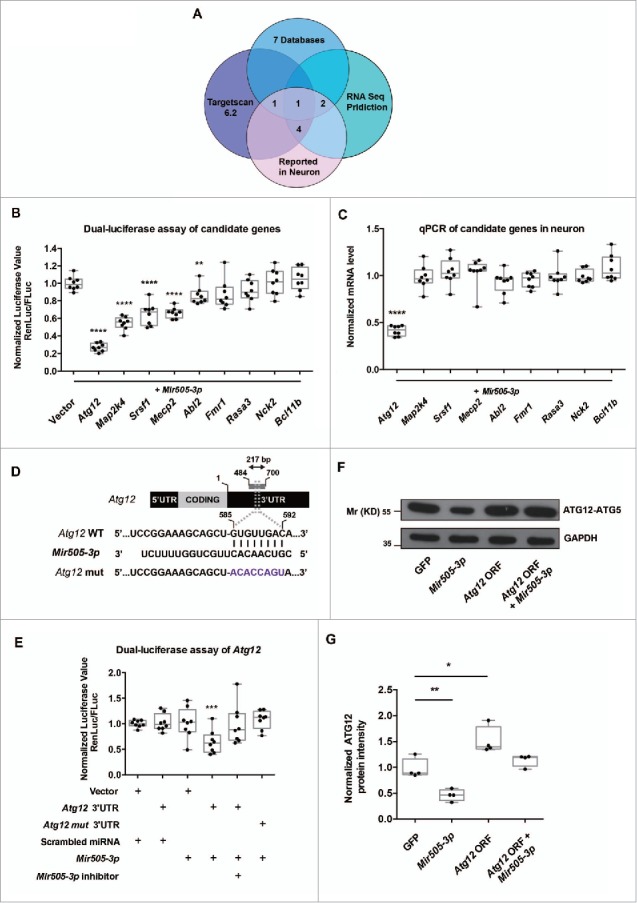
Atg12 is a direct target gene of Mir505–3p in mouse cortical neuron. (A) Schematic diagram of bioinformation screen of candidate target genes of Mir505–3p in mouse cortical neurons. See also Table S1. (B) Normalized luciferase (Renilla:firefly) values of all candidate targets. The seed match region of 3′UTR of candidate targets were cloned into psi-CHECK-2 dual-luciferase vector and cotransfected with Mir505–3p plasmid in the HEK 293 cell line. (C) Q-PCR examination of mRNA levels of all candidate targets in cultured cortical neuron with lentivirus infection to overexpress Mir505–3p. (D) Schematic diagram of the exact position which Mir505–3p targets on the mouse Atg12 3′UTR. Seed match region was replaced in a site-directed mutation experiment. (E) Normalized luciferase (Renilla:firefly) values are shown. The Atg12 3′UTR psi-CHECK-2 vector was cotransfected with the constructs as indicated. Mir505–3p exhibited a specific inhibition on Atg12 WT 3′UTR, rescued by Mir505–3p inhibitors. (F) ATG12 was regulated by Mir505–3p in cultured cortical neurons. Immunoblottings were performed using ATG12 and GAPDH antibodies of 3DIV neurons transfected by AMAXA nucleofector with vectors as indicated. (G) Measurements of immunoblotting in (F). At least 3 independent assays were measured. Error bars: SEM. *P < 0.05, ** P < 0.01, *** P < 0.001 (Student t test).
To validate the direct targets of Mir505–3p, we first used dual-luciferase assays in human embryonic kidney 293 cells. We cotransfected Mir505–3p with a dual-luciferase vector containing a ∼200-bp sequence including seed regions of the 3′UTR of candidate genes and detected signals of both Renilla luciferase and firefly luciferase. Among all the candidate genes, Atg12 showed the sharpest decrease in ratio of normalized Renilla:firefly signals (Fig. 4B). To further confirm whether they are target genes in neurons, we then transfected Mir505–3p in E14.5 cultured cortical neurons with lentivirus and performed q-PCR to monitor the mRNA level of candidate genes. Consistent with dual-luciferase assays in human embryonic kidney 293 cells, only Atg12 showed a clear downregulation in mRNA level (Fig. 4C). Thus, Atg12 may be a direct target gene under regulation of Mir505–3p.
Furthermore, we performed dual luciferase assays with a vector containing the site-directed mutation in the seed match region of Atg12 3′UTR (Fig. 4D). The site-directed mutation experiment suggested a direct interaction between the Mir505–3p and the 3′UTR of Atg12 (Fig. 4D and E). Moreover, we examined the level of ATG12 protein after electroporation of Mir505–3p by an AMAXA nucleofector in cultured cortical neurons at 3DIV. We found that the protein level of ATG12 was indeed regulated by Mir505–3p in neurons (Fig. 4F and G). Thus, we showed that Atg12 is a direct target gene of Mir505–3p in mouse cortical neurons.
ATG12 is required in axonal development in utero and in vivo
To determine whether ATG12 plays a role in regulating axonal development, we constructed a plasmid expressing the Atg12 open reading frame (ORF) without 5′- or 3′-UTR regions. We first electroporated the plasmid encoding the Atg12 ORF into cortical neurons with an AMAXA nucleofector and performed immunofluorescence staining at 3DIV (Fig. 5A). We found that overexpression of the Atg12 ORF significantly decreased axon branching (Fig. 5A and C). However, no significant influence was observed on minor neurite branching (Fig. S3E), suggesting that ATG12 is critical for axonal development. Moreover, we found that overexpression of the Atg12 ORF by Lipofectamine 2000 transfection delayed establishment of neural polarity at 5DIV (Fig. 5B and D). Consistently, the TAL was decreased by the Atg12 ORF (Fig. 5B and E) whereas no significant changes occurred in TMNL (Fig. S3F). Furthermore, we found that axonal extension and branching caused by expression of ATG12 were rescued by Mir505–3p coexpression, indicating that Mir505–3p was indeed an upstream signal of ATG12 for establishment of neuron polarity (Fig. 5A, B, C, D and E). Therefore, we showed that ATG12 is a critical regulator of axonal development in cultured neurons.
Figure 5.
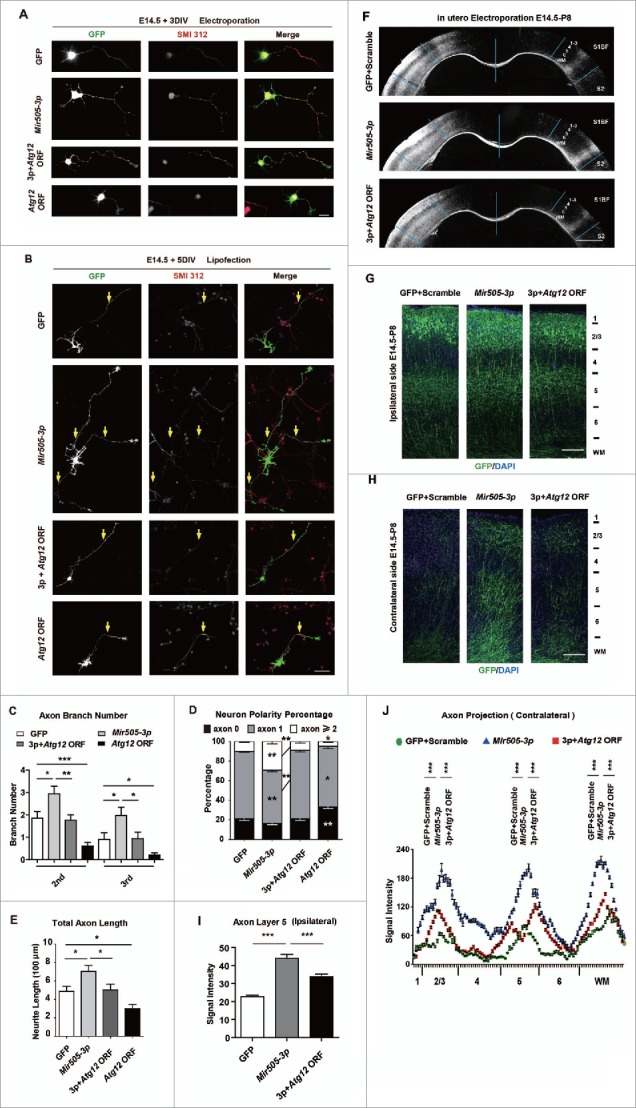
ATG12 is a negative regulator of axonal development. (A) Example pictures of mouse primary cortical neurons transfected with AMAXA nucleofector with GFP or constructs indicated. Neurons were fixed and stained for immunofluorescence with SMI312 and GFP antibodies at 3 DIV for measuring axon numbers. Scale bar: 20 μm. (B) Example pictures of mouse primary cortical neurons transfected with Lipofectamine 2000 at 1 DIV with GFP or constructs as indicated. Scale bar: 20 μm. (C) Measurements of axon numbers of each condition in (A). (D) Measurements of neuron polarity of each condition in (B). (E) Measurements of total axon length of each condition in (B). A total 30 to 33 neurons from each condition were randomly selected and measured. Error bars are SEM. *P < 0.05, **P < 0.01, *** P < 0.001 (t test). (F) Images of coronal brain sections of P8 mice electroporated at E14.5 with plasmids indicated. Scale bar: 1000 μm. (G) Magnification images of ipsilateral brain sections in (F). Scale bar: 100 μm. (H) Magnification images of contralateral brain sections in (F). Scale bar: 100 μm. (I) Measurements of axonal signal intensity in ipsilateral cortical plate layer 5 of each condition in (G). (J) Measurements of axonal signal intensity in the contralateral cortical plate from layer 1 to white matter of each condition in (H). At least 4 litters for each condition were measured. Error bars are SEM. *** P < 0.001 (Student t test).
To further determine whether ATG12 is required for Mir505–3p regulating axonal development in vivo, we performed IUE to express Mir505–3p or a mixture of Mir505–3p with the Atg12 ORF in brains of embryonic mice, then examined projections of GFP-labeled axons from the ipsilateral side to the contralateral side at P8. We found that the Atg12 ORF was able to fully rescue the overgrowth of axonal branching caused by expression of Mir505–3p on each layer of contralateral side, from measurements of signal intensity of axonal branching on both the ipsilateral and the contralateral side, (Fig. 5F, H and J) and axon branching on layer 5 of the ipsilateral side (Fig. 5G and I). Taken together, these results indicate that ATG12 is required for Mir505–3p regulating axon development in vivo.
Genetic ablation of Mir505–3p activates autophagic flux in brain
To investigate the dynamics of the autophagy pathway are altered in the brain of mir505 knockout mice, we dissected cortical neurons and transfected GFP-LC3B to trace autophagic flux.37-39 We performed immunostaining and labeled subcellular GFP-LC3B on the membrane of autophagic vesicles. However, due to the rapid turnover of autophagy in neurons we could not monitor local GFP-LC3B in mouse cortices.40
Thus, we used a transmission electron microscopy (TEM) strategy to examine subcellular autophagosomes in cultured cortical neuron or cortices from mir505 knockout mice.41 Surprisingly, we observed a significant promotion of autophagic flux correlate to loss of Mir505–3p (Fig. 6A and E). In 3 DIV cultured neurons, more and larger autophagosomes were accumulated in both the soma and the axonal part of KO mice, comparing to WT mice (Fig. 6A, E and C). To confirm these observations, we killed P0 mice and dissected cortices followed by TEM capture. Consistently, autophagosome formation greatly increased in terms of number and sizes, in cortices of mir505 KO mice (Fig. 6B, D and F).
Figure 6.
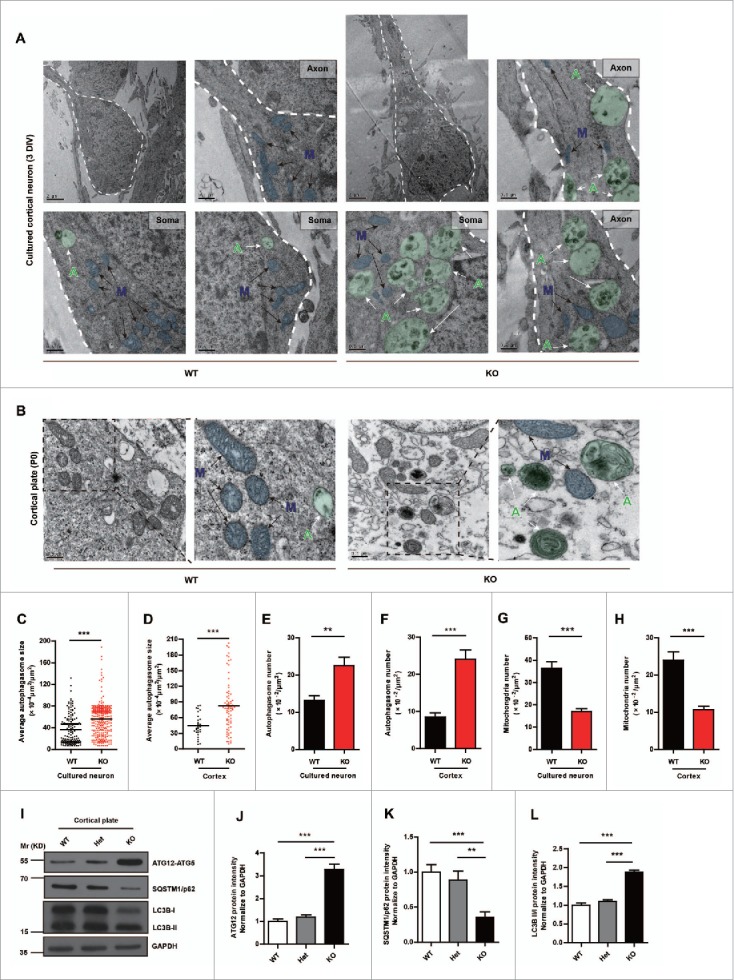
Genetic deletion of Mir505–3p activates autophagy in the brain. (A) Example pictures and enlarged pictures of cortical neurons of each genotype. (B) Example pictures and enlarged pictures of cortices of each genotype. Pseudo colors were applied to represent individual autophagosome (green circle, indicated by green capital letter “A” with white arrow) and mitochondria (blue circle, indicated by blue capital letter “M” with black arrow). (C and D) Measurements of autophagosome sizes in the unit area of cytoplasm in cortical neurons and cortex, respectively. (E and F) Measurements of autophagosome numbers in the unit area of cytoplasm in cortical neurons and cortex, respectively. (G and H) Measurements of mitochondria numbers in the unit area of cytoplasm in cortical neurons and cortex, respectively. At least 4 litters were measured for each condition. (I) Increase of ATG12, LC3B-II/-I and decrease of SQSTM1/p62 in mir505 KO mice. Immunoblotting was performed with the antibodies as indicated. (J to L) Measurements of protein levels of ATG12, SQSTM1/p62 and LC3B-II/-I in (I). Error bars are SEM. **P < 0.01, *** P < 0.001 (Student t test).
We then examined protein levels of ATG12, LC3B and SQSTM1/p62 in cortex tissue from WT, heterozygous and homozygous mir505 KO mice, respectively (Fig. 6I). We found that deletion of Mir505 significantly enhanced the protein level of ATG12, indicating release of ATG12 from Mir505–3p inhibition (Fig. 6I and J). Moreover, the ratio of LC3B-II:LC3B-I increased, whereas SQSTM1/p62 decreased (Fig. 6I, K and L). Furthermore, we electroporated complement siRNAs as Mir505–3p inhibitors with an AMAXA nucleofector in cultured neurons and examined mRNA levels with q-PCR. We found a significant increase of Atg12 and a slight decrease of SQSTM1/p62 in conditions of Mir505–3p downregulation (Fig. S6). Therefore, these results indicate that loss of Mir505–3p activates autophagy by upregulating ATG12 in the mouse brain.
Mir505–3p regulates autophagy through ATG12
Autophagy is conserved from yeast to mammal. Therefore, we reasoned that regulation of autophagy by Mir505–3p can be widely observed in different types of cells as well. Thus, we isolated MEF cells for subcellular observations of autophagy. As accumulation of autophagosomes may result from either activation of autophagic flux or blockage of autolysosomes, we set out to determine which pathway Mir505–3p may directly interfere with. We applied rapamycin (RPMC) or chloroquine (CQ) to mimic autophagic activation or blockage respectively, comparing with expression of the Atg12 ORF alone (Fig. 7A). We found that ATG12 led to more and larger early-stage autophagosome formation, containing double or multiple membranes (Fig. 7A, B and C). This procedure was closer to activation by RPMC rather than blockage by CQ, as morphology of autophagosomes in RPMC was represented as early-medium-stage, while in CQ, as late-stage of single-membrane autolysosomes (Fig. 7A, B and C). We observed that Mir505–3p effectively interfered with autophagosome formation by decreasing ATG12. This decrease of ATG12 may result in shortage of scaffold components, which is necessary for constructing autophagosomes with normal size and quality (as shown in the model of Fig. 8H).
Figure 7.
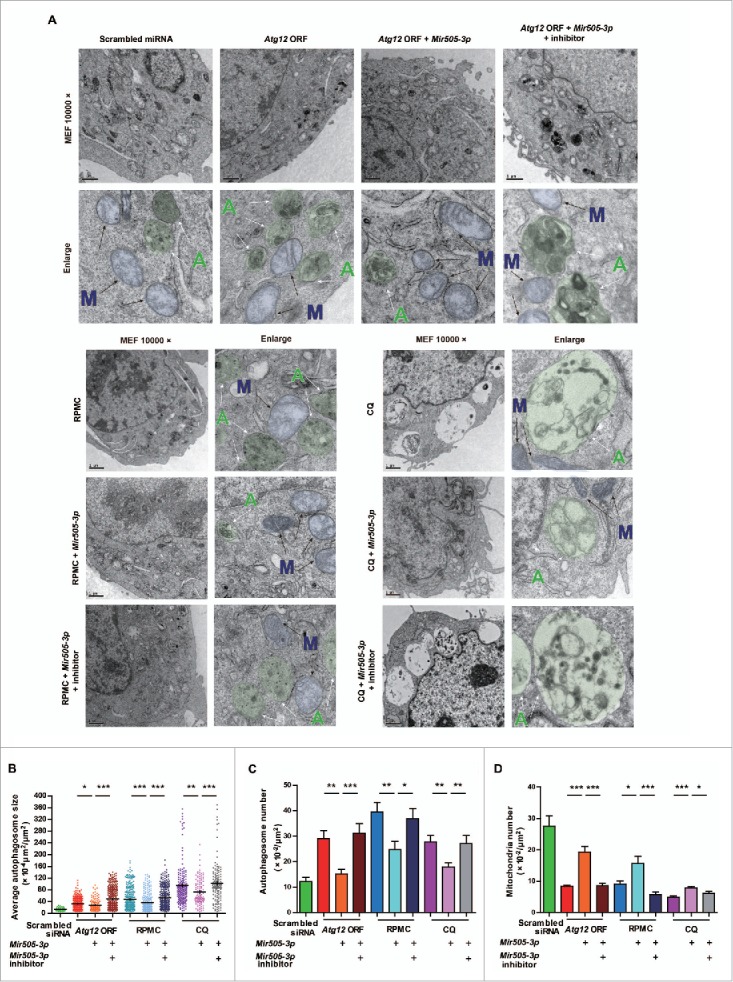
Mir505–3p regulates autophagy by targeting Atg12. (A) Example pictures and enlarged pictures of MEF cells from TEM strategy. Cells were transfected with constructs as indicated. Two d after transfection, MEF cells were fed with various treatments as indicated, for 3 h followed by fixing with 2.5% glutaraldehyde in PBS. Pseudo colors were applied to represent individual autophagosomes (green circle, indicated by green capital letter “A” with white arrow) and mitochondria (blue circle, indicated by blue capital letter “M” with black arrow). (B) Measurements of autophagosome sizes in the unit area of cytoplasm in each condition, respectively. (C and D) Measurements of autophagosome number and mitochondria numbers in the unit area of cytoplasm in each condition, respectively. Error bars are SEM. *P < 0.05, **P < 0.01, *** P < 0.001 (Student t test).
Figure 8.
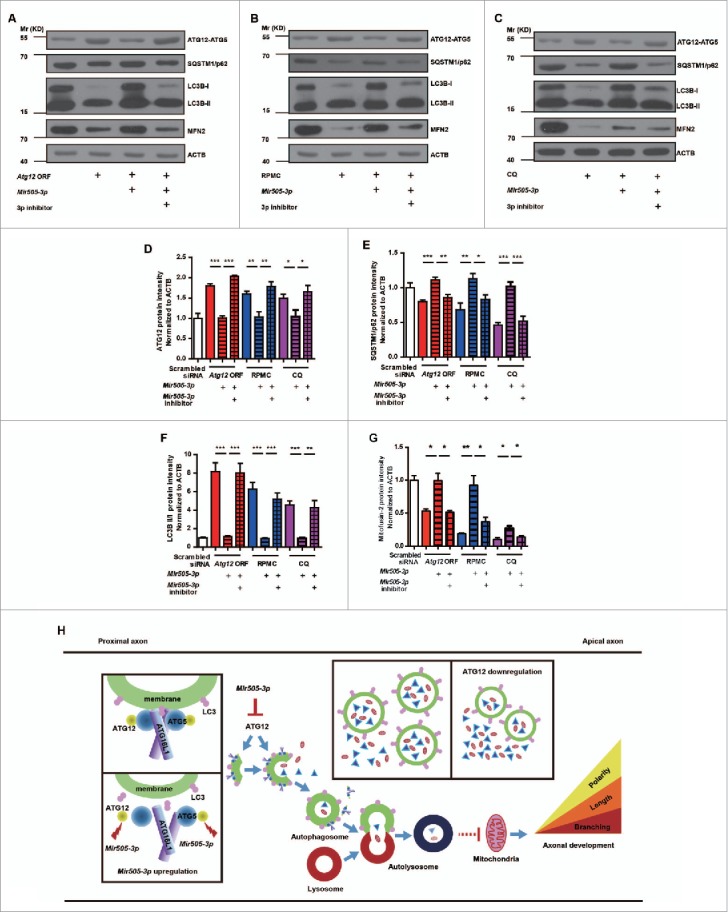
Mir505–3p influences mitochondria numbers by inhibiting ATG12 and the autophagy pathway. (A to C) Immunoblotting was performed in the expression of the Atg12 ORF, RPMC induction and CQ induction conditions with antibodies as indicated. (D to G) Measurements of protein levels of ATG12, SQSTM1/p62, LC3B-II/-I and MFN2 in (A, B, C). (H) Schematic diagram of hypothetical regulatory mechanisms of the Mir505–3p-ATG12 axonal development pathway. Autophagosome formation consists of 3 main steps: nucleation, expansion and maturation. ATG12 participates in the ATG12–ATG5-ATG16L1 complex which is required for autophagosome scaffold construction. In a developing axon, downregulation of ATG12 by Mir505–3p results in decrease of the autophagy signal, which leads to an abundant storage of the local axonal positive regulatory component and a greater supply of energy generated from mitochondria. Thus, the axonal specification, extension and branching are promoted.
At the third generation of primary cultured MEF, we transfected GFP-LC3B to monitor autophagy flux. We co-overexpressed Mir505–3p mimics or a mixture of Mir505–3p mimics and inhibitors, with either RPMC or CQ stimulation. Consistently, the Mir505–3p mimics reduced ATG12 puncta formation (Fig. S7A and B). Taken together, these results suggest that Mir505–3p generally downregulates autophagy by targeting ATG12 and impairing autophagosome formation.
The amount of mitochondria was increased by Mir505–3p, along with autophagy inhibition
In TEM images of cultured cortical neuron or cortices from mir505 KO mice, we found a clear decrease of mitochondria (Fig. 6A, B, G and H). We further found that Mir505–3p also leads to alterations of mitochondria numbers in primary cultured MEF (Fig. 7A and D). Intriguingly, this trend shows negative correlation with that of autophagosomes (Fig. 6A to H; Fig. 7A to D). We further confirmed the TEM observations by examination of ATG12, SQSTM1/p62, LC3B and MFN2 (mitofusin 2) proteins in MEFs with western blot (Fig. 8A to G). Previously, it has been reported that STK11/LKB1 (serine/threonine kinase 11)-NUAK (NUAK family, SNF1-like kinase, 1) immobilize mitochondria for axonal branching. The axonal development is a high-energy-demanding process, which requires that mitochondria are available throughout the neuron, especially the axon, to provide a large amount of ATP.31 Thus, we hypothesized that upregulation of local Mir505–3p in axons turns down the ATG12 autophagy signal that digests substrate proteins and mitochondria, and then maintains mitochondria to promote axon specification, extension and branching (Fig. 8H).
Discussion
Neurons are binary cells with structural and functional differences between the somato-dendritic compartment and the axon. Under in vitro conditions, dissociated cortical neurons extend several neurites which are still indiscriminate before growing asymmetrically, then a single nascent neurite experiences rapid outgrowth and becomes the axon.42 During axon specification and concomitant elongation, axonal branching occurs. Disturbance of these processes is proven to cause neurodevelopmental disorders such as autism spectrum disorders.2 The developing axon undergoes organelle localization, including drastic changes in cytoskeletal structure,43,44 and membrane composition.45,46 These processes result from a balance between protein synthesis,47,48 and protein degradation.49,50
It is well known that axons and nerve terminals contain numerous microRNAs (miRNAs).51 miRNAs are short, noncoding RNAs that act as translational regulators, affecting both mRNA degradation and translational repression. Recent studies have shown miRNAs to be involved in C. elegans axon specification, X. laevis retinal axon pathfinding, and mouse axon extension and branching during development.52,53 However, the detailed mechanism of miRNAs in regulating axon development remains to be determined.
Previously, Mir505–3p has been identified as an indicator of tumor genesis and development. It is supposed to be one among potential early diagnostic markers for cervical carcinogenesis,54 a biomarker for local recurrence or distant metastasis of synovial sarcoma,55 and a prognostic biomarker for breast cancer.56 Besides, Mir505/Mir505–3p is proven to be a tumor repressor. It inhibits cell proliferation by inducing apoptosis in a drug-resistant breast cancer cell line MCF7-ADR.57 It regulates the oncogene PSMD10 (proteasome [prosome, macropain] 26S subunit, non-ATPase, 10) functional variant to reduce gastric cancer risk58 and it partially affects senescence and apoptosis by targeting SRSF1 under control of ZBTB7A/LRF (zinc finger and BTB domain containing 7a) in a MEF cell line.36 In addition, Mir505/Mir505–3p modulates angiogenic processes by targeting FGF18 (fibroblast growth factor 18), indicating that Mir505/Mir505–3p is a potential target for intervention of hypertension.59 However, most studies about Mir505/Mir505–3p focus on proliferation and apoptosis related pathways and tumor related physiological events, whether Mir505/Mir505–3p regulates potential target genes to mediate other physiological effects is still elusive.
The first reported functional target protein of Mir505/Mir505–3p in primary cultured MEF cells is SRSF1. SRSF1 is a prototypical serine/arginine-rich protein that modulates both constitutive and alternative splicing of pre-mRNA.60,61 SRSF1 is also known as an oncoprotein, as it is frequently overexpressed in tumors.62,63 It is reported that SRSF1 activates the MTOR pathway, phosphorylates RPS6KB1/S6K1 (ribosomal protein S6 kinase, polypeptide 1) and EIF4EBP1 (eukaryotic translation initiation factor 4E binding protein 1).64 As the MTOR pathway is responsible for axon outgrowth and regeneration, it is unavoidable to find out whether SRSF1 is involved in promotion of axon growth caused by Mir505–3p upregulation. Thus, we first tested mRNA level of Srsf1 with qPCR and examined the protein level of SRSF1 with immunoblotting in cortical neurons overexpressing Mir505–3p. However, no difference was observed in mRNA (Fig. 4C) and protein level (data not shown). Then, we used RPMC, an inhibitor of MTOR, to block the MTOR pathway to determine whether promotion of axonal development caused by Mir505–3p was dependent on this pathway. Surprisingly, compromising MTOR signaling by RPMC did not suppress promotion of axonal development caused by Mir505–3p, indicating that MTOR was not directly involved in the signaling pathway (data not shown).
In other previous studies, interfering autophagy by inhibiting ATG7 with siRNAs leads to total axonal length increase but has no effect on axon branching.65 In contrast, our observations show that downregulating autophagy via targeting Atg12 by a natural inhibitory molecule, Mir505–3p, promotes axon specification, extension and branching. To illustrate this distinction, there are 2 potential possibilities. First, the autophagy signaling as well as essential constitutive components, including ATG12 and ATG7, are responsible for axon extension while perhaps other Mir505–3p target genes still to be discovered, might induce branching. Otherwise, ATG12 contributes the majority of aspects of axonal development, exhibiting a total different regulatory mechanism compared with ATG7.
The establishment of polarity and regulation of axonal branching are critical for neuron to form neural circuitry in the brain. Our finding of the novel action of Mir505–3p and ATG12 provides a new direction for understanding the roles of microRNAs and autophagy signal in neural development.
Materials and methods
Plasmids and constructs
The Mir505–3p (or Mir505–5p and scrambled) siRNA sequence was synthesized as oligonucleotide primers and manually annealed. Primers are the following:
Mir505–3p oligo forward:
5′-TGCTGCGTCAACACTTGCTGGTTTTCTGTTTTGGCCACTGACTGACAGAAAACCCAAGTGTTGACG-3′
Mir505–3p oligo reverse:
5′-CCTGCGTCAACACTTGGGTTTTCTGTCAGTCAGTGGCCAAAACAGAAAACCAGCAAGTGTTGACGC-3′
Mir505–5p oligo forward:
5′-TGCTGGGGAGCCAGGAAGTATTGATGTTGTTTTGGCCACTGACTGACAACATCAACTTCCTGGCTCCC-3′
Mir505–5p oligo reverse:
5′-CCTGGGGAGCCAGGAAGTTGATGTTGTCAGTCAGTGGCCAAAACAACATCAATACTTCCTGGCTCCCC-3′
Scrambled siRNA oligo forward:
5′-TGCTGAAATGTACTGCGCGTGGAGACGTTTTGGCCACTGACTGACGTCTCCACGCAGTACATTT-3′
Scrambled siRNA oligo reverse:
5′-CCTGAAATGTACTGCGTGGAGACGTCAGTCAGTGGCCAAAACGTCTCCACGCGCAGTACATTTC-3′
The Mir505–3p (or Mir505–5p and scrambled siRNA) sequence was inserted into FUGW (Addgene, 14883) to get Mir505–3p (or Mir505–5p and scrambled siRNA) plasmid. mmu-Mir505–3p mimics or inhibitors were purchased from GenePharma.com (customized designed). Mir505–3p inhibitors were 2-methylated-modified RNA oligos completely complementary to mmu-Mir505–3p and purified by high-performance liquid chromatography. The seed match region in 3′UTR of the candidate target gene was amplified with NEST PCR primers and then the NEST PCR product was diluted to 1:1000 to be used as template for 3′UTR region amplification. Primers are as the following:
Atg12 NEST forward: 5′-TTGCTTATTCAGGGGACCTC -3′
Atg12 NEST reverse: 5′- TGGTTCACTCTTCCTGCTCA-3′
Atg12 3′UTR forward: 5′-TAGGCGATCGCAGTTGGCAAAAAGGCTTGAC -3′
Atg12 3′UTR reverse: 5′- TTGCGGCCAGCGGCCCGCAAGAACCAGAAATG-
-ACA -3′
Map2k4 NEST forward: 5′- TGCTGGTCAGAGAGACCTCA-3′
Map2k4 NEST reverse: 5′- GTAGGGAATTTGGCTGAGCA-3′
Map2k4 3′UTR forward: 5′- TAGGCGATCGCACATATTCATGACGCGTGGA -3′
Map2k4 3′UTR reverse: 5′- TTGCGGCCAGCGGCCTGCAGAAGGCAATGTG-
-TCTC -3′
Srsf1 NEST forward: 5′- ATCTCGATCTCGAAGCCGTA-3′
Srsf1 NEST reverse: 5′- AAACATAAGAACTTCCCCCAAG-3′
Srsf1 3′UTR forward: 5′- TAGGCGATCGCTTACTCCCCAAGGAGAAGCA-3′
Srsf1 3′UTR reverse: 5′- TTGCGGCCAGCGGCCCCAAAGACATGAGGGGA
ATG -3′
Mecp2 NEST forward: 5′- AGCACAGTCAGGTTGAAGACC -3′
Mecp2 NEST reverse: 5′- TTTCTCCTGCATCACACCTC -3′
Mecp2 3′UTR forward: 5′- TAGGCGATCGCGGCCAGAAGTAGCTTTGCAC -3′
Mecp2 3′UTR reverse: 5′- GGCCGCTGGCCGCAATCCTTCAGATTGGGAGTT
GG -3′
Abl2 NEST forward: 5′- GTGCAATCCAGCAGTGAAGA -3′
Abl2 NEST reverse: 5′- TGAACAGGGCATCACTAGCA -3′
Abl2 3′UTR forward: 5′- TAGGCGATCGCCTACATGCTCGGCTTTTGGT -3′
Abl2 3′UTR reverse: 5′- GGCCGCTGGCCGCAAGCCACCTCCACATGATTT
TT -3′
Fmr1 NEST forward: 5′- AAATGGCAACAAACTGCACA -3′
Fmr1 NEST reverse: 5′-ACGGCTAACCTCCTTTGACA-3′
Fmr1 3′UTR forward: 5′- TAGGCGATCGCTTGGTACCTTGCACACATCA -3′
Fmr1 3′UTR reverse: 5′- GGCCGCTGGCCGCAATTGCATCAACATCAATTTA
GCA -3′
Rasa3 NEST forward: 5′- ACTACATCCGGCAGCAGTCT -3′
Rasa3 NEST reverse: 5′- CTGCGAGGGAACAGGTTTA -3′
Rasa3 3′UTR forward: 5′- TAGGCGATCGCACTACATCCGGCAGCAGTCT -3′
Rasa3 3′UTR reverse: 5′- GGCCGCTGGCCGCAATTGGTAGGACCCGTGTG
AAC -3′
Nck2 NEST forward: 5′- GGGAGATGGTGGTGAGAAAA -3′
Nck2 NEST reverse: 5′- GGAATGTGCCATCTCAAGACT -3′
Nck2 3′UTR forward: 5′- TAGGCGATCGCAAAAGAGGAAGGTGGCAACA -3′
Nck2 3′UTR reverse: 5′- GGCCGCTGGCCGCAAGTAGCTCCCCTTCCCAAG
AG -3′
Bcl11b NEST forward: 5′- TGGCAAGATGGCTTCTCTTT -3′
Bcl11b NEST reverse: 5′- GTTTTCAAGGCACGCACAT -3′
Bcl11b 3′UTR forward: 5′- TAGGCGATCGCCAAAGGAATCTCACCCGTTC -3′
Bcl11b 3′UTR reverse: 5′- TTGCGGCCAGCGGCCTCTGGGCCAAAGACAAGA-
-AT -3′
The Atg12 ORF sequence was inserted into FUGW to get the Atg12 ORF plasmid. The Atg12 ORF sequence was amplified from the Atg12 full-length cDNA plasmid which was purchased from OriGene.com (MG200886). Primers are as the following: Atg12 ORF forward, 5′-GGATCCATGTCGGAAGATTCAGAG-3′; and reverse, 5′- GAATTCTTCATT
TCTGGCTCATCC -3′.
Antibodies
Antibodies used in this study were as follows: goat anti-GFP (1:1000; Abcam, ab5450), rabbit anti-GFP (1:1000; Abcam, ab6556); pan-axonal neurofilament SMI312 (Abcam, ab24574; 1:1000); MAPT/Tau (Cell Signaling Technology, 4019; 1:1000 for immunofluorescence); ATG12 (Cell Signaling Technology, 2011; 1:500 for immunoblotting and 1:50 for immunofluorescence); LC3 (Novusbio, NB100–2220; 1:3000); SQSTM1/p62 (Abcam, ab56416; 1:1000); MFN2/Mitofusin-2 (Cell Signaling Technology, 11925; 1:1000); RBFOX3/NeuN (Abcam, ab104225; 1:400); ACTB/β-Actin (Cell Signaling Technology, 8457; 1:3000) and GAPDH (Abcam, ab8245; 1:10,000). Antibodies coupled to Alexa Fluor dyes (Thermo Fisher, goat anti-rabbit, Alexa Fluor 488, A-11034, 1:1000; rabbit anti-goat, Alexa Fluor 488, A-21222, 1:1000; goat anti-mouse Alexa Fluor 555, A32727, 1:1000; goat anti-rabbit Alexa Fluor 555, A-21430, 1:1000) were used as secondary antibodies.
Primary cell culture and transfection
Cortical tissues of E14.5 C57BL/6 mice were obtained under dissection. Tissue was minced and digested with 0.125% trypsin (Thermo Fisher, 15090046) to generate dissociated neurons.66 Dissociated neurons were electroporated in the suspension status with the AMAXA Nucleofector (AAD-1001S, Germany) in 6-well plates or lipofected with Lipofectamine 2000 in 12-well plates. For AMAXA Nucleofector electroporation, we used the AMAXA Mouse Neuron Nucleofector kit with Nucleofector Solution buffer to resuspend the neurons and with the “mouse, neuron, 0–005” program on the AMAXA nucleofector II device, in which the neuron viability was over 80%. Optimal condition was 3 × 106 cells with 3 μg of plasmid for each electroporation. For Lipofectamine 2000-mediated transfections, we used a considerably small quantity of both plasmid and Lipofectamine 2000 (0.1 μg plasmid with 0.1 μl Lipofectamine 2000 for 2 × 105 cells in one well of the 12-well-plate) to avoid toxic effects caused by Lipofectamine 2000. As a result, neuron viability was as high as untransfected neurons. Cells were fed with Neurobasal medium supplemented with 2% B27 (Life Technology, 17504044) and fixed at 3–5 DIV for immunofluorescence staining, or harvested at 3 DIV for immunoblotting, or harvested at 5 or 6 DIV for quantitative real-time PCR assays after Lentivirus infection. The lentivirus used in this study was made with 108 or 109 viral genome/ml by Obio Technology (Shanghai). Primary MEF cells were generated as previously reported.67 Dissociated MEF cells were cultured for 2 passages (one passage/day) in dishes to eliminate astrocytes and then seeded in 8-well chambers with low density (104 cells/well). MEF cells were fed with Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Life Technology, 15140122). MEF cells were transfected at 3 DIV, fed with the stimulation drugs and fixed at 5 DIV for immunofluorescence staining.
Quantitative real-time PCR
Total RNA was extracted from mouse cortical neurons using phenol-chloroform method. We used DNaseI (Sigma, AMPD1) to digest the remaining genomic DNA in the extracted total RNA to avoid genomic DNA interference with the cDNA amplification step. The cDNA was synthesized by poly-dT primers from 1 μg of purified RNA by iScript cDNA Synthesis Kit (Bio-Rad, 1708890EDU). Quantitative real-time PCR was performed with SYBR Premix ExTaq (Takara, RR001B) and the Rotor-Gene Q apparatus (QIAGEN, 9001685, Germany). Gapdh was used for normalization to provide an internal control. For mature miRNA testing, Mir16–1–5p was used for normalization. Before specific reverse transcription, primers for Mir16–1–5p were mixed with either Mir505–3p or Mir505–5p and experienced a gradient annealing to form a stable stem-loop structure. Data analysis was done by using the comparative CT method in software by QIAGEN. Measurement of precursor and mature miRNA levels were conducted as described previously.35
Primers used in quantitative real-time PCR assays were as follows:
Atg12 forward: 5′- AGTTGGCAAAAAGGCTTGAC-3′
Atg12 reverse: 5′- CGCAAGAACCAGAAATGACA-3′
Map2k4 forward: 5′- ACATATTCATGACGCGTGGA-3′
Map2k4 reverse: 5′- TGCAGAAGGCAATGTGTCTC-3′
Srsf1 forward: 5′- TTACTCCCCAAGGAGAAGCA-3′
Srsf1 reverse: 5′- CCAAAGACATGAGGGGAATG-3′
Bcl11b forward: 5′- CAAAGGAATCTCACCCGTTC-3′
Bcl11b reverse: 5′- TCTGGGCCAAAGACAAGAAT-3′
Rasa3 forward: 5′- ACTACATCCGGCAGCAGTCT-3′
Rasa3 reverse: 5′- TTGGTAGGACCCGTGTGAAC-3′
Nck2 forward: 5′- AAAAGAGGAAGGTGGCAACA-3′
Nck2 reverse: 5′- GTAGCTCCCCTTCCCAAGAG-3′
Abl2 forward: 5′- CTACATGCTCGGCTTTTGGT-3′
Abl2 reverse: 5′- GCCACCTCCACATGATTTTT-3′
Fmr1 forward: 5′- TTGGTACCTTGCACACATCA-3′
Fmr1 reverse: 5′- TTGCATCAACATCAATTTAGCA-3′
Mecp2 forward: 5′- GGCCAGAAGTAGCTTTGCAC-3′
Mecp2 reverse: 5′- TCCTTCAGATTGGGAGTTGG-3′
Pre-Mir505 forward: 5′-CGCGGGAGCCAGGAAGTAT-3′
Pre-Mir505 reverse: 5′- TGGCGGAGAAAACCAGCAAG-3′
Mir505–3p RT: 5′-GCGTCTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTG-
-AGACGCAGAAAACCAGC-3′
Mir505–3p forward: 5′- GCGAGCACCGTCAACACT -3′
Mir505–3p reverse: 5′- TGGTGTCGTGGAGTCGGC -3′
Mir505–5p RT: 5′- GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACT
GGATACGACAACATC -3′
Mir505–5p forward: 5′- GCCCGGGAGCCAGGAAGTAT -3′
Mir505–5p reverse: 5′- CAGGGTCCGAGGTATTCGCAC -3′
Mir16–1–5p RT:
Five′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATAC
GACCGCCAA -3′
Mir16–1–5p forward: 5′- CGCGCTAGCAGCACGTAAAT -3′
Mir16–1–5p reverse: 5′- GTGCAGGGTCCGAGGT -3′
Axon and minor neurite analysis
We defined the SMI312- and MAPT-positive process with length above 5 times that of the average diameter of the soma of a neuron as an axon. The remaining processes were defined as minor neurites (Fig. S8F). The experiments for measuring morphology of axons or minor neurites were measured, with specimen identification blinded to the reader, as the following: Briefly, 3 persons participated in this analysis. After transfection, samples were randomly labeled and recorded by person A (Fig. S8A). Then, target neurons that were randomly selected (Fig. S8B) were captured using a microscope (Nikon 80i, MBA75020, Japan) by person B (Fig. S8C). For image capturing, a 20 × or 40 × objective lens was used to image a whole neuron with proper exposure time. The axon or minor neurite morphology analysis was conducted by person C (Fig. S8D). Last, data from each procedure matched to each other to generate the final data (Fig. S8E). The total length or branch number of all processes was analyzed using Fiji software. All quantifications were tested with a Student t test and expressed as SEM. Results were considered significant, if P < 0.05. At least 3 independent experiments were performed.
In utero electroporation
E14.5 C57BL/6 mice were used for IUE according to previously reported methods.68 Pregnant mice were anesthetized with 0.7% sodium pentobarbital (10 ml/kg). Uteruses were exposed using an aseptic technique. FUGW empty plasmid, Mir505–3p-FUGW plasmid and a mixture of Atg12 ORF plasmid with Mir505–3p-FUGW plasmid (2 μg/μl) were injected by microinjection into the lateral ventricle of embryos, indicated by Fast Green (2 mg/ml; Sigma, F7252). Electric pulses were produced by a T830 electroporator (BTX Molecular Delivery Systems, Holliston, MA, USA) and applied 5 repeats of 30 V for 50 ms with an interval of 1 sec for each embryo. After perfusion,69 brains were selected under a fluorescent dissecting microscope (Nikon 80i, MBA75020, Japan) and only those with EGFP expression were maintained for further immunofluorescence staining.
Luciferase assay
A ∼200-bp sequence including seed match region of candidate target genes was inserted into the 3′UTR of the Renilla luciferase reporter gene of the dual-luciferase vector psi-CHECK-2, with a recombination reaction kit (Novoprotein.com, NR001). The psi-CHECK-2 dual-luciferase kit was purchased from Promega.com (E1910). This construct (0.3 μg) and Mir505–3p-containing vector (0.7 μg) were cotransfected into human embryonic kidney 293 cell in 12-well plate. Twenty-four h after transfection, lysis buffer was used to harvest cells under cold conditions. Luciferase activity was detected using the Berthold Detection Systems Sirius (Germany).
Transmission electron microscopy assay
MEF cells were scratched and collected after fixing by 2.5% glutaraldehyde in phosphate-buffered saline (PBS; Corning, R21–040-CV). Cells were centrifuged to the bottom of the tube and then, the cells were embedded with agar. The agar was cut into 1-mm3 size samples for further treatment with 1% osmic acid in PBS and gradient dehydration with alcohol. The sample was treated with a mixture of epoxypropane (Sinopharm Chemical Reagent, 80059118) and resin (Electron Microscopy Sciences, 14900), followed by embedding with pure resin. Then the sample was sliced with a diamond tool bit. For the cortex tissue sample, the P0 mouse was perfused with 1% glutaraldehyde and 4% paraformaldehyde in PBS. Then the cortex was dissected and the sample was treated with the same strategy as MEF cells. For cultured neurons, the neuron was seeded on glass precoated with poly-D-lysine. Care was taken to ensure that neurons were not scratched, to maintain natural morphology of individual neurons. The glass where neurons were grown was picked up with sharp ophthalmic forceps and put into 12-well plates for further treatment. Target cells that were randomly selected were captured with a transmission electron microscope (Nippon Tekno, JEOL-1230, Japan).
CRISPR/Cas9-based mir505 knockout mouse
Two sgRNAs (sgRNA1: ccaggttagcgtcaacacttgct, sgRNA2: acacttgctggttttctctctgg) were designed against the coding sequence of the mouse Mir505 gene (Fig. 3A). The regions including the sgRNAs directing sites were amplified by primers (Genotyping-F: ACAAACAGAATCCACGAACTTG, Genotyping-R: CGGTAGTAAATTGATGCAC CCA), to distinguish Mir505–3p-deletion mice from hetero-deletion and wild-type littermates, and were amplified by primers (Seq-F: TAGGAGCA TGGGAGAGAGGAGATGC, Seq-R: GGCTTCTACTCCCTGGTGATCTACAGG) for sequencing. Both 2 sgRNAs were injected into mouse zygotes. We excluded off-target effect of the mir505 KO mice from CRISPR/Cas9 system. First, we performed bioinformatic analysis with Off-Spotter system70 and classified the potential off-target genes into 3 categories (see Table S2), including the potential off-target site in the intron, in the UTR of the exon and in the coding region of the exon. Then, we focused on the genes containing off-target sites in their coding region. We designed primers to amplify the proximal region of off-target sites followed by sequencing (see Table S2 and Fig. S5A and B). We examined these sites in all the 3 founder mice (numbered as #12, #17 and #19), using WT mice as control. Indeed, no sequence alterations of these sites were observed in all founders. Thus, no off-target effects were detected in the mir505 KO mice. The predicted off-targets genes and specific amplification primers are described in Table S2.
Statistical methods
Student t test was used in this study, as indicated in the figure legends. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Supplementary Material
Abbreviations
- 3′UTR
3 prime untranslated region
- Abl2
v-abl Abelson murine leukemia viral oncogene 2 (arg, Abelson-related gene)
- Atg
autophagy related
- Bcl11b
B cell leukemia/lymphoma 11B
- Cas9
CRISPR-associated system
- CQ
chloroquine
- CRISPR
clustered regularly interspaced short palindromic repeats
- DIV
day in vitro
- E14.5
embryonic d 14.5
- Fmr1
fragile X mental retardation syndrome 1
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GFP
green fluorescent protein
- IUE
in utero electroporation
- KO
knockout
- MAP1lC3B/LC3B
microtubule-associated protein 1 light chain 3 β
- Map2k4
mitogen-activated protein kinase kinase 4
- Mecp2
methyl CpG binding protein 2
- MEF
mouse embryonic fibroblast
- miRNA
microRNA
- MTOR
mechanistic target of rapamycin (serine/threonine kinase)
- Nck2
noncatalytic region of tyrosine kinase adaptor protein 2
- ORF
open reading frame
- P0
postnatal d 0
- PBS
phosphate-buffered saline
- q-PCR
quantitative polymerase chain reaction
- Rasa3
RAS p21 protein activator 3
- RPMC
rapamycin
- sgRNA
small guide RNA
- siRNA
small interfering RNA
- SQSTM1/p62
sequestosome1
- Srsf1
serine/arginine-rich splicing factor 1
- TAL
total axonal length
- TEM
transmission electron microscopy
- TMNL
total minor neurite length
- WT
wild type
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Yuefang Zhang for her management of experimental animals. We thank Dr. Shifang Shan for her technical assistance. We thank Drs. Jin Xu, Rong-Gui Hu and Zhi-xue Liu for sharing materials and valuable discussion. We thank Drs. Yu Kong, Bin Zhang and Lijun Pan for their technical assistance in TEM assays. We thank Drs. Qian Hu and Dan Xiang for their technical assistance in IF assays. We thank Drs. Song-lin Qian and Wei-fang Jiang for their assistance in molecular biologic assays.
Funding
This work was supported by CAS Strategic Priority Research Program (XDB02050400), NSFC Grants (#91432111, #31625013), National Key Scientific Instrument and Equipment Development Program of China (2012YQ03026008) to Z.Q, and NSFC Grants (#31371257), the Key Project of Science and Technology Commission of Shanghai Municipality (#14140900502) to Y.Z, and CUSF (DH-D-2014049) to K.Y.
References
- [1].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27-42. https://doi.org/ 10.1016/j.cell.2007.12.018. PMID:18191218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zoghbi HY, Bear MF. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb Perspect Biol. 2012;4, pii: a009886. https://doi.org/ 10.1101/cshperspect.a009886. PMID:22258914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al.. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880-4. https://doi.org/ 10.1038/nature04723. PMID:16625205 [DOI] [PubMed] [Google Scholar]
- [4].Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al.. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885-9. https://doi.org/ 10.1038/nature04724. PMID:16625204 [DOI] [PubMed] [Google Scholar]
- [5].Komatsu M, Waguri S, Koike M, Sou Y, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al.. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149-63. https://doi.org/ 10.1016/j.cell.2007.10.035. PMID:18083104 [DOI] [PubMed] [Google Scholar]
- [6].Ban BK, Jun MH, Ryu HH, Jang DJ, Ahmad ST, Lee JA. Autophagy negatively regulates early axon growth in cortical neurons. Mol Cell Biol. 2013;33:3907-19. https://doi.org/ 10.1128/MCB.00627-13. PMID:23918799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Walczak M, Martens S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy. 2013;9:424-5. https://doi.org/ 10.4161/auto.22931. PMID:23321721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tekirdag KA, Korkmaz G, Ozturk DG, Agami R, Gozuacik D. MIR181A regulates starvation- and rapamycin-induced autophagy through targeting of ATG5. Autophagy. 2013;9:374-85. https://doi.org/ 10.4161/auto.23117. PMID:23322078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. A protein conjugation system essential for autophagy. Nature. 1998;395:395-8. https://doi.org/ 10.1038/26506. PMID:9759731 [DOI] [PubMed] [Google Scholar]
- [10].Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T. Mouse Apg16L, a novel WD-repeat protein, targets to the autdphagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116:1679-88. https://doi.org/ 10.1242/jcs.00381. PMID:12665549 [DOI] [PubMed] [Google Scholar]
- [11].Murrow L, Malhotra R, Debnath J. ATG12-ATG3 interacts with Alix to promote basal autophagic flux and late endosome function. Nat Cell Biol. 2015;17:300-+. https://doi.org/ 10.1038/ncb3112. PMID:25686249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C, Debnath J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell. 2010;142:590-600. https://doi.org/ 10.1016/j.cell.2010.07.018. PMID:20723759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell. 2011;44:698-709. https://doi.org/ 10.1016/j.molcel.2011.10.014. PMID:22152474 [DOI] [PubMed] [Google Scholar]
- [14].Haller M, Hock AK, Giampazolias E, Oberst A, Green DR, Debnath J, Ryan KM, Vousden KH, Tait SW. Ubiquitination and proteasomal degradation of ATG12 regulates its proapoptotic activity. Autophagy. 2014;10:2269-78. https://doi.org/ 10.4161/15548627.2014.981914. PMID:25629932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhu WS, Fan ZP, Zhang C, Guo ZX, Zhao Y, Zhou YX, Li K, Xing Z, Chen G, Liang Y, et al.. A dominant X-linked QTL regulating pubertal timing in mice found by whole genome scanning and modified interval-specific congenic strain analysis. Plos One. 2008;3:e3021. https://doi.org/ 10.1371/journal.pone.0003021. PMID:18725948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bauters M, Frints SG, Van Esch H, Spruijt L, Baldewijns MM, de Die-Smulders CE, Fryns JP, Marynen P, Froyen G. Evidence for increased SOX3 dosage as a risk factor for X-linked hypopituitarism and neural tube defects. Am J Med Genet A. 2014;164A:1947-52. https://doi.org/ 10.1002/ajmg.a.36580. PMID:24737742 [DOI] [PubMed] [Google Scholar]
- [17].Hol FA, Schepens MT, van Beersum SE, Redolfi E, Affer M, Vezzoni P, Hamel BC, Karnes PS, Mariman EC, Zucchi I. Identification and characterization of an Xq26-q27 duplication in a family with spina bifida and panhypopituitarism suggests the involvement of two distinct genes. Genomics. 2000;69:174-81. https://doi.org/ 10.1006/geno.2000.6327. PMID:11031100 [DOI] [PubMed] [Google Scholar]
- [18].Solomon NM, Ross SA, Morgan T, Belsky JL, Hol FA, Karnes PS, Hopwood NJ, Myers SE, Tan AS, Warne GL, et al.. Array comparative genomic hybridisation analysis of boys with X linked hypopituitarism identifies a 3.9 Mb duplicated critical region at Xq27 containing SOX3. J Med Genet. 2004;41:669-78. https://doi.org/ 10.1136/jmg.2003.016949. PMID:15342697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lagerstrom-Fermer M, Sundvall M, Johnsen E, Warne GL, Forrest SM, Zajac JD, Rickards A, Ravine D, Landegren U, Pettersson U. X-linked recessive panhypopituitarism associated with a regional duplication in Xq25-q26. Am J Hum Genet. 1997;60:910-6. PMID:9106538 [PMC free article] [PubMed] [Google Scholar]
- [20].Woods KS, Cundall M, Turton J, Rizotti K, Mehta A, Palmer R, Wong J, Chong WK, Al-Zyoud M, El-Ali M, et al.. Over- and underdosage of SOX3 is associated with infundibular hypoplasia and hypopituitarism. Am J Hum Genet. 2005;76:833-49. https://doi.org/ 10.1086/430134. PMID:15800844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ojeda SR, Lomniczi A, Loche A, Matagne V, Kaidar G, Sandau US, Dissen GA. The transcriptional control of female puberty. Brain Res. 2010;1364:164-74. https://doi.org/ 10.1016/j.brainres.2010.09.039. PMID:20851111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE, et al.. Mammalian microRNAs: Experimental evaluation of novel and previously annotated genes. Genes Dev. 2010;24:992-1009. https://doi.org/ 10.1101/gad.1884710. PMID:20413612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lewis TL Jr, Courchet J, Polleux F. Cell biology in neuroscience: Cellular and molecular mechanisms underlying axon formation, growth, and branching. J Cell Biol. 2013;202:837-48. https://doi.org/ 10.1083/jcb.201305098. PMID:24043699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang T, Liu Y, Xu XH, Deng CY, Wu KY, Zhu J, Fu XQ, He M, Luo ZG. Lgl1 activation of rab10 promotes axonal membrane trafficking underlying neuronal polarization. Dev Cell. 2011;21:431-44. https://doi.org/ 10.1016/j.devcel.2011.07.007. PMID:21856246 [DOI] [PubMed] [Google Scholar]
- [25].Zhang Y, Ueno Y, Liu XS, Buller B, Wang X, Chopp M, Zhang ZG. The MicroRNA-17-92 cluster enhances axonal outgrowth in embryonic cortical neurons. J Neurosci. 2013;33:6885-94. https://doi.org/ 10.1523/JNEUROSCI.5180-12.2013. PMID:23595747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: Critical roles of GSK-3beta and its upstream regulators. Cell. 2005;120:123-35. https://doi.org/ 10.1016/j.cell.2004.12.033. PMID:15652487 [DOI] [PubMed] [Google Scholar]
- [27].Yi JJ, Barnes AP, Hand R, Polleux F, Ehlers MD. TGF-beta signaling specifies axons during brain development. Cell. 2010;142:144-57. https://doi.org/ 10.1016/j.cell.2010.06.010. PMID:20603020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Barnes AP, Polleux F. Establishment of axon-dendrite polarity in developing neurons. Annu Rev Neurosci. 2009;32:347-81. https://doi.org/ 10.1146/annurev.neuro.31.060407.125536. PMID:19400726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang CL, Zhang L, Zhou Y, Zhou J, Yang XJ, Duan SM, Xiong ZQ, Ding YQ. Activity-dependent development of callosal projections in the somatosensory cortex. J Neurosci. 2007;27:11334-42. https://doi.org/ 10.1523/JNEUROSCI.3380-07.2007. PMID:17942728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zuccaro E, Bergami M, Vignoli B, Bony G, Pierchala BA, Santi S, Cancedda L, Canossa M. Polarized expression of p75(NTR) specifies axons during development and adult neurogenesis. Cell Rep. 2014;7:138-52. https://doi.org/ 10.1016/j.celrep.2014.02.039. PMID:24685135 [DOI] [PubMed] [Google Scholar]
- [31].Courchet J, Lewis TL Jr, Lee S, Courchet V, Liou DY, Aizawa S, Polleux F. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell. 2013;153:1510-25. https://doi.org/ 10.1016/j.cell.2013.05.021. PMID:23791179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Huang TN, Chuang HC, Chou WH, Chen CY, Wang HF, Chou SJ, Hsueh YP. Tbr1 haploinsufficiency impairs amygdalar axonal projections and results in cognitive abnormality. Nat Neurosci. 2014;17:240-7. https://doi.org/ 10.1038/nn.3626. PMID:24441682 [DOI] [PubMed] [Google Scholar]
- [33].Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15-20. https://doi.org/ 10.1016/j.cell.2004.12.035. PMID:15652477 [DOI] [PubMed] [Google Scholar]
- [34].Hancock ML, Preitner N, Quan J, Flanagan JG. MicroRNA-132 is enriched in developing axons, locally regulates Rasa1 mRNA, and promotes axon extension. J Neurosci. 2014;34:66-78. https://doi.org/ 10.1523/JNEUROSCI.3371-13.2014. PMID:24381269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cheng TL, Wang Z, Liao Q, Zhu Y, Zhou WH, Xu W, Qiu Z. MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev Cell. 2014;28:547-60. https://doi.org/ 10.1016/j.devcel.2014.01.032. PMID:24636259 [DOI] [PubMed] [Google Scholar]
- [36].Verduci L, Simili M, Rizzo M, Mercatanti A, Evangelista M, Mariani L, Rainaldi G, Pitto L. MicroRNA (miRNA)-mediated Interaction between leukemia/lymphoma-related factor (LRF) and alternative splicing factor/splicing factor 2 (ASF/SF2) affects mouse embryonic fibroblast senescence and apoptosis. J Biol Chem. 2010;285:39551-63. https://doi.org/ 10.1074/jbc.M110.114736. PMID:20923760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491-502. https://doi.org/ 10.1016/j.biocel.2004.02.005. PMID:15325587 [DOI] [PubMed] [Google Scholar]
- [38].Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3:181-206. https://doi.org/ 10.4161/auto.3678. PMID:17224625 [DOI] [PubMed] [Google Scholar]
- [39].Mizushima N. Methods for monitoring autophagy using GFP-LC3 transgenic mice. Methods Enzymol. 2009;452:13-23. https://doi.org/ 10.1016/S0076-6879(08)03602-1. PMID:19200873 [DOI] [PubMed] [Google Scholar]
- [40].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313-26. https://doi.org/ 10.1016/j.cell.2010.01.028. PMID:20144757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kovacs AL, Eldib A, Telbisz A. Autophagy in hepatocytes and erythropoietic cells isolated from the twenty-one day old rat embryo. Acta Biol Hung. 2001;52:417-33. https://doi.org/ 10.1556/ABiol.52.2001.4.7. PMID:11693992 [DOI] [PubMed] [Google Scholar]
- [42].Higgins D, Burack M, Lein P, Banker G. Mechanisms of neuronal polarity. Curr Opin Neurobiol. 1997;7:599-604. https://doi.org/ 10.1016/S0959-4388(97)80078-5. PMID:9384542 [DOI] [PubMed] [Google Scholar]
- [43].Shelly M, Cancedda L, Heilshorn S, Sumbre G, Poo MM. LKB1/STRAD promotes axon initiation during neuronal polarization. Cell. 2007;129:565-77. https://doi.org/ 10.1016/j.cell.2007.04.012. PMID:17482549 [DOI] [PubMed] [Google Scholar]
- [44].Barnes AP, Lilley BN, Pan YA, Plummer LJ, Powell AW, Raines AN, Sanes JR, Polleux F. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell. 2007;129:549-63. https://doi.org/ 10.1016/j.cell.2007.03.025. PMID:17482548. [DOI] [PubMed] [Google Scholar]
- [45].Wu KY, He M, Hou QQ, Sheng AL, Yuan L, Liu F, Liu WW, Li G, Jiang XY, Luo ZG. Semaphorin 3A activates the guanosine triphosphatase Rab5 to promote growth cone collapse and organize callosal axon projections. Sci Signal. 2014;7:ra81. https://doi.org/ 10.1126/scisignal.2005334. PMID:25161316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Xu XH, Deng CY, Liu Y, He M, Peng J, Wang T, Yuan L, Zheng ZS, Blackshear PJ, Luo ZG. MARCKS regulates membrane targeting of Rab10 vesicles to promote axon development. Cell Res. 2014;24:576-94. https://doi.org/ 10.1038/cr.2014.33. PMID:24662485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Li YH, Werner H, Puschel AW. Rheb and mTOR regulate neuronal polarity through Rap1B. J Biol Chem. 2008;283:33784-92. https://doi.org/ 10.1074/jbc.M802431200. PMID:18842593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Abe N, Borson SH, Gambello MJ, Wang F, Cavalli V. Mammalian target of rapamycin (mTOR) activation increases axonal growth capacity of injured peripheral nerves. J Biol Chem. 2010;285:28034-43. https://doi.org/ 10.1074/jbc.M110.125336. PMID:20615870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang QJ, Ding Y, Kohtz DS, Mizushima N, Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N, Yue Z. Induction of autophagy in axonal dystrophy and degeneration. J Neurosci. 2006;26:8057-68. https://doi.org/ 10.1523/JNEUROSCI.2261-06.2006. PMID:16885219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Komatsu M, Wang QJ, Holstein GR, Friedrich VL Jr., Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104:14489-94. https://doi.org/ 10.1073/pnas.0701311104. PMID:17726112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Aschrafi A, Kar AN, Natera-Naranjo O, MacGibeny MA, Gioio AE, Kaplan BB. MicroRNA-338 regulates the axonal expression of multiple nuclear-encoded mitochondrial mRNAs encoding subunits of the oxidative phosphorylation machinery. Cell Mol Life Sci. 2012;69:4017-27. https://doi.org/ 10.1007/s00018-012-1064-8. PMID:22773120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Olsson-Carter K, Slack FJ. A developmental timing switch promotes axon outgrowth independent of known guidance receptors. PLoS Genet. 2010;6(8), pii: e1001054. https://doi.org/ 10.1371/journal.pgen.1001054. PMID:20700435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Dajas-Bailador F, Bonev B, Garcez P, Stanley P, Guillemot F, Papalopulu N. microRNA-9 regulates axon extension and branching by targeting Map1b in mouse cortical neurons. Nat Neurosci. 2012; 15:697-99. https://doi.org/ 10.1038/nn.3082. PMID:22484572. [DOI] [PubMed] [Google Scholar]
- [54].Sharma S, Hussain S, Soni K, Singhal P, Tripathi R, Ramachandran VG, Sharma S, Das S, Pillai B, Bharadwaj M.. Novel MicroRNA signatures in HPV-mediated cervical carcinogenesis in Indian women. Tumour Biol. 2016;37(4):4585-95. [DOI] [PubMed] [Google Scholar]
- [55].Fricke A, Ullrich PV, Heinz J, Pfeifer D, Scholber J, Herget GW, Hauschild O, Bronsert P, Stark GB, Bannasch H, et al.. Identification of a blood-borne miRNA signature of synovial sarcoma. Mol Cancer. 2015;14:151. https://doi.org/ 10.1186/s12943-015-0424-z. PMID:26250552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jonsdottir K, Janssen SR, Da Rosa FC, Gudlaugsson E, Skaland I, Baak JP, Janssen EA. Validation of expression patterns for nine miRNAs in 204 lymph-node negative breast cancers. Plos One. 2012;7:e48692. https://doi.org/ 10.1371/journal.pone.0048692. PMID:23144930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yamamoto Y, Yoshioka Y, Minoura K, Takahashi RU, Takeshita F, Taya T, Horii R, Fukuoka Y, Kato T, Kosaka N, et al.. An integrative genomic analysis revealed the relevance of microRNA and gene expression for drug-resistance in human breast cancer cells. Mol Cancer. 2011;10:135. https://doi.org/ 10.1186/1476-4598-10-135. PMID:22051041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Liu Y, Xu JZ, Jiang M, Ni LN, Chen Y, Ling Y. Association between functional PSMD10 Rs111638916 variant regulated by Mir505 and gastric cancer risk in a Chinese population. Cell Physiol Biochem. 2015;37:1010-7. https://doi.org/ 10.1159/000430227. PMID:26394032 [DOI] [PubMed] [Google Scholar]
- [59].Yang QB, Jia CL, Wang PW, Xiong MQ, Cui JG, Li L, Wang W, Wu Q, Chen Y, Zhang T. MicroRNA-505 identified from patients with essential hypertension impairs endothelial cell migration and tube formation. Int J Cardiol. 2014;177:925-34. https://doi.org/ 10.1016/j.ijcard.2014.09.204. PMID:25449503 [DOI] [PubMed] [Google Scholar]
- [60].Twyffels L, Gueydan C, Kruys V. Shuttling SR proteins: More than splicing factors. FEBS J. 2011;278:3246-55. https://doi.org/ 10.1111/j.1742-4658.2011.08274.x. PMID:21794093 [DOI] [PubMed] [Google Scholar]
- [61].Mount SM. Genetic depletion reveals an essential role for an SR protein splicing factor in vertebrate cells. Bioessays. 1997;19:189-92. https://doi.org/ 10.1002/bies.950190302. PMID:9080768 [DOI] [PubMed] [Google Scholar]
- [62].Anczukow O, Akerman M, Clery A, Wu J, Shen C, Shirole NH, Raimer A, Sun S, Jensen MA, Hua Y, et al.. SRSF1-regulated alternative splicing in breast cancer. Mol Cell. 2015;60:105-17. https://doi.org/ 10.1016/j.molcel.2015.09.005. PMID:26431027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Oltean S, Bates DO. Hallmarks of alternative splicing in cancer. Oncogene. 2014;33:5311-8. https://doi.org/ 10.1038/onc.2013.533. PMID:24336324 [DOI] [PubMed] [Google Scholar]
- [64].Karni R, Hippo Y, Lowe SW, Krainer AR. The splicing-factor oncoprotein SF2/ASF activates mTORC1. Proc Natl Acad Sci U S A. 2008;105:15323-7. https://doi.org/ 10.1073/pnas.0801376105. PMID:18832178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ban BK, Jun MH, Ryu HH, Jang DJ, Ahmad ST, Lee JA. Autophagy negatively regulates early axon growth in cortical neurons. Mol Cell Biol. 2013;33:3907-19. https://doi.org/ 10.1128/MCB.00627-13. PMID:23918799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Beaudoin GMJ, Lee SH, Singh D, Yuan Y, Ng YG, Reichardt LF, Arikkath J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc. 2012;7:1741-54. https://doi.org/ 10.1038/nprot.2012.099. PMID:22936216 [DOI] [PubMed] [Google Scholar]
- [67].Jain K, Verma PJ, Liu J. Isolation and handling of mouse embryonic fibroblasts. Methods Mol Biol. 2014;1194:247-52. https://doi.org/ 10.1007/978-1-4939-1215-5_13. PMID:25064107 [DOI] [PubMed] [Google Scholar]
- [68].Saito T. In vivo electroporation in the embryonic mouse central nervous system. Nat Protoc. 2006;1:1552-8. https://doi.org/ 10.1038/nprot.2006.276. PMID:17406448 [DOI] [PubMed] [Google Scholar]
- [69].Guyette JP, Gilpin SE, Charest JM, Tapias LF, Ren X, Ott HC. Perfusion decellularization of whole organs. Nat Protoc. 2014;9:1451-68. https://doi.org/ 10.1038/nprot.2014.097. PMID:24874812 [DOI] [PubMed] [Google Scholar]
- [70].Pliatsika V, Rigoutsos I. “Off-Spotter:” very fast and exhaustive enumeration of genomic lookalikes for designing CRISPR/Cas guide RNAs. Biol Direct. 2015;10:4. https://doi.org/ 10.1186/s13062-015-0035-z. PMID:25630343. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.