

Abstract
Palladium-catalyzed methylene β-C(sp3)–H arylation of aliphatic ketones using a transient directing group is developed. The use of α-benzyl β-alanine directing group that forms a six-membered chelation with palladium is crucial for promoting the methylene C(sp3)–H bond activation.
Keywords: C–H activation, arylation, transient directing group, β-amino acid, ketone, palladium
Graphical Abstract
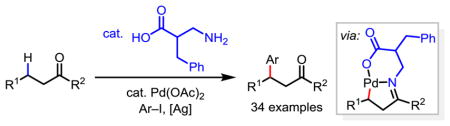
Ketones are abundant in natural products and drug molecules. In addition, carbonyls serve as extremely versatile functional group handles for a wide range of transformations. Reactions of aliphatic ketones typically rely on the reactivity of the carbonyl group or the acidic α-carbon centers.1 Therefore, β-C–H functionalization via directed C–H activation2 would extend the versatility of ketones by rendering the inert β-carbon center reactive.3 Directed β-C–H activation of ketones via metal insertion has been investigated using pre-installed oxime or imine directing groups.4–6 To omit the installation and removal steps, we have extensively exploited the transient directing group (TDG) strategy7 to achieve β-C(sp3)–H activation of ketones and aldehydes. Our investigations regarding the utility of bidentate monoprotected amino acid (MPAA) ligands in transition metal catalyzed C–H functionalizations8,9 has led us to develop the Pd-catalyzed C(sp3)–H functionalization of ketones using a catalytic amino acid as a transient directing group (Scheme 1A).10,11 Subsequently, the Ge group reported that β-amino acids could serve as transient directing groups that promote palladium-catalyzed β-C–H arylation of aliphatic aldehydes .12 Despite these advances, the activation of β-methylene C–H bonds of acyclic ketones or aldehydes using either pre-installed or transient directing groups has remained an unsolved challenge. Herein we report the development of a modified β-amino acid directing group that enables palladium catalyzed methylene β-C(sp3)–H arylation of linear aliphatic ketones (Scheme 1B). Extensive modification of the β-amino acids revealed that the efficiency of the transient directing group in this transformation is critically dependent on the structure of the amino acid. The 6-membered chelation of the β-amino acid directing group with the Pd(II) catalyst is crucial for this reaction.
Scheme 1.

Pd(II)-Catalyzed C(sp3)–H Arylation Using a Transient Directing Group Strategy
β-C(sp3)–H activation of ketones and aldehydes using either pre-installed or transient directing groups is largely limited to primary C–H bonds (Scheme 1A).10–12 For example, palladium catalyzed C–H arylation of the β-methylene C–H bonds on an acyclic aldehyde has been reported to provide only 25% yield when employing a transient directing group strategy.12 This is unsurprising as methylene C–H bonds are significantly more resistant to palladium insertion than primary C–H bonds due to the steric hindrance. Indeed, while glycine was demonstrated to be an optimal transient directing group in our prior work, β-arylation of acyclic ketone (1) using this transient directing group provided the desired product in only 24% NMR yield with severe decomposition of the substrate (Table 1, TDG-1 with AgTFA as the additive). In our extensive search for ligands that can accelerate methylene C–H insertion, we have recently shown both experimentally and computationally that six-membered chelation with Pd(II) is significantly more effective than the five-membered chelation.13 We therefore envisioned that TDG based on β-amino acid could form the 6-membered chelation in the precursor and promote the formation of the desired 5,6-fused palladacycle intermediate (Scheme 1B).
Table 1.

|
Standard conditions: 0.2 mmol of 1, 2.0 equiv. of methyl 4-iodobenzoate, 10 mol% of Pd(OAc)2, 30 mol% of TDG, 1.0 equiv. of AgTFA, 2.0 equiv. of AgOAc, 1.5 mL of HFIP, 0.5 mL of acetic acid, 120 °C, under air, 48 h.
The yield was determined by 1H NMR analysis of the crude product using CH2Br2 as the internal standard.
3.0 equiv. of AgTFA was employed as the additive.
ArF = 4-(CF3)C6F4.
Reaction was conducted for 72 h.
To test this hypothesis, β-alanine (TDG-2) was first examined under the same conditions (AgTFA as the additive). The yield was increased to 41% with improved mass balance. To rule out the enone as a possible intermediate, we have also treated the corresponding enone with the standard arylation conditions and no product was obtained. An extensive survey of palladium sources, silver additives, and solvents was then performed (see supporting information). The use of a 3:1 mixture of HFIP and acetic acid as the solvent is the optimal media for this reaction. Palladium precursors other than Pd(OAc)2 gave similar results. When only AgTFA was used as the additive, a large amount of homocoupling product (Ar–Ar) was generated along with the rapid deactivation of the catalyst. Notably, the use of binary silver salts (a 1:2 mixture of AgTFA and AgOAc) as the additives suppressed the formation of the homocoupling product, allowing formation of the desired product in 57% yield. Significantly lower conversion and yields were observed when AgOAc or another silver salt was used alone. Next, derivatives of β-alanine, such as ester and amides, were synthesized and evaluated (TDG-3 to 5). Loss of reactivity with these TDGs suggests the 6-membered imino-carboxyl chelation is crucial for the methylene C–H insertion. To further improve the reactivity, we investigated the influence of the substituents on β-alanine. We found that substituents at the β-position significantly reduce the reactivity (TDG-6 and 7), presumably due to slowing the imine formation. While the presence of an α-methyl group has negligible effect, moderate-sized α-substitution generally enhanced the yield (TDG-9 to 12), and TDG-11 with a benzyl substituent was optimal. As a comparison, the analogous L-phenylalanine gave only 8% yield under the same conditions. The bulky tert-butyl and 1-adamantyl group, as well as aryl groups, affect the reaction adversely (TDG-13 to 16). Further increasing temperature lead to poor material balance. Finally, 84% NMR yield was obtained when the reaction was extended to 72 hours with 10 mol% of Pd(OAc)2, 30 mol% of TDG-11, 2.0 equiv. of methyl 4-iodobenzoate, 1.0 equiv. of AgTFA and 2.0 equiv. of AgOAc in HFIP/AcOH at 120 °C.
With the optimal TDG and reaction conditions in hand, we then surveyed the scope of aryl iodides for the methylene β-C(sp3)–H arylation using 2-decanone (1) as the model substrate. As shown in Table 2, a wide range of aryl iodides with substituents were employed as the coupling partners. Compared with our previous study,10a the scope of aryl iodide for C(sp3)–H arylation of ketones was significantly expanded. Electron-deficient aryl iodides bearing ester, trifluoromethyl and nitro groups at the meta- or para-positions were well tolerated under the standard conditions, providing consistently good yields (2a–2d, 2f), although the yield slightly dropped to 66% with para-nitro substitution (2e). Similar results were also observed with fluoro-substituted aryl iodides (2g and 2h). Notably, chloro- and bromo-substituted aryl iodides afforded the corresponding products in high yield, preserving the halogen groups for further synthetic elaborations via cross-coupling reactions (2i and 2j). Electron-neutral iodides, such as iodobenzene and iodotoluenes, were equally effective coupling reagents (2k–2m). However, the reaction with 4-iodobiphenyl gave lower yield of the desired product (48%). Electron-rich aryl iodides are weaker oxidants to convert the alkyl Pd(II) species into the corresponding Pd(IV) complex. As a consequence, a reduced yield was obtained with 3-iodoanisole (2o). While both meta- and para-substituted aryl iodides were suitable coupling partners, ortho-substituted aryl iodides are less effective due to the steric hindrance (2p and 2q). Lastly, a heteroaryl iodide was examined under the standard conditions. The reaction efficiency was considerably influenced by the coordination of the pyridine nitrogen with the palladium catalyst. Arylation with 2-trifluoromethyl-2-iodopyridine gave the desired product in 35% yield (2r).
Table 2.

|
Conditions: 0.2 mmol of 1, 2.0 equiv. of aryl iodide, 10 mol% of Pd(OAc)2, 30 mol% of TDG-11, 1.0 equiv. of AgTFA, 2.0 equiv. of AgOAc, 1.5 mL of HFIP, 0.5 mL of acetic acid, 120 °C, under air, 72 h.
Isolated yields.
1.0 equiv. 2-iodo-6-(trifluoromethyl)pyridine was used.
The optimal C–H arylation conditions were then applied to a variety of aliphatic ketones with methyl 4-iodobenzoate as the coupling partner (Table 3), demonstrating the feasibility of this protocol as a solution for β-C(sp3)–H arylation of ketones. In the presence of a primary γ-C–H bond, 2-pentanone 3a underwent methylene C–H arylation predominantly at the β-position, furnishing the desired product 4a in 74% isolated yield. Substrates containing bulky groups at the β-position, such as cyclohexyl and aryl groups, provided 4b and 4c in 39% and 41% yield, respectively.14 The steric hindrance of a cyclohexyl group at the γ-position has little impact on the reaction efficiency (72%, 4d). C–H arylation of the ketones with γ-aryl substitution proceeded smoothly to afford useful 1,2-diarylated compounds15 in moderate yields (4e–4h), and considerable amounts of the starting materials (25–37%) were retrieved in these cases (see supporting information). Functional groups such as ether, acetate and amide were well tolerated under the reaction conditions and the desired products were obtained in moderate to good yields (4j–4l). Sterically encumbered ketones other than methyl ketones are less effective presumably due to the sluggish formation of the ketimines. The use of 60 mol% transient directing group restored the reactivity of these ketones and gave synthetically useful yields (4m and 4n). The use of symmetrical 4-heptanone bearing two reactive C–H sites led to a mixture of mono- and di-arylated products in 54% combined yield (4o). In the presence of two methylene carbon centers, the less hindered methylene C–H bond was arylated preferentially accompanied by a small amount of di-arylation (4p). In all cases, no diastereoselectivity was observed (4n, 4odi and 4pdi ). Lastly, α-branched ketones, especially those bearing primary β-C–H bonds, led to mixtures of multi-arylated products with low yields.
Table 3.

|
Conditions: 0.2 mmol of 3, 2.0 equiv. of methyl 4-iodobenzoate, 10 mol% of Pd(OAc)2, 30 mol% of TDG-11, 1.0 equiv. of AgTFA, 2.0 equiv. of AgOAc, 1.5 mL of HFIP, 0.5 mL of acetic acid, 120 °C, under air, 72 h.
Isolated yields.
60 mol% of TDG-11 was employed.
In summary, the methylene β-C(sp3)–H arylation of aliphatic ketones was developed using α-benzyl β-alanine as a transient directing group. β-Amino acid directing groups adopting six-membered chelation with the palladium catalyst are found to be advantageous in promoting the methylene C(sp3)–H bond insertion. Further efforts to render this reaction enantioselective using chiral β-amino acids and their derivatives are currently ongoing.
Supplementary Material
Acknowledgments
We gratefully acknowledge The Scripps Research Institute and the NIH (NIGMS, 2R01GM084019) for financial support. H.P. thanks the Korea Foundation for Advanced Studies for a predoctoral fellowship.
Footnotes
ASSOCIATED CONTENT
Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.(a) Mukaiyama T. Angew Chem Int Ed. 2004;43:5590–5614. doi: 10.1002/anie.200300641. [DOI] [PubMed] [Google Scholar]; (b) Evans DA, Vogel E, Nelson JV. J Am Chem Soc. 1979;101:6120–6123. [Google Scholar]; (c) Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J Am Chem Soc. 1997;119:6496–6511. [Google Scholar]; (d) Cowden CJ, Paterson I. Org React. 1997;51:1–200. [Google Scholar]; (e) Cano R, Zakarian A, McGlacken GP. Angew Chem Int Ed. 2017;56:2–15. doi: 10.1002/anie.201703079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For selected reviews, see: Kakiuchi F, Murai S. Acc Chem Res. 2002;35:826–834. doi: 10.1021/ar960318p.Chen X, Engle KM, Wang DH, Yu JQ. Angew Chem Int Ed. 2009;48:5094–5115. doi: 10.1002/anie.200806273.Daugulis O, Do HQ, Shabashov D. Acc Chem Res. 2009;42:1074–1086. doi: 10.1021/ar9000058.Ackermann L, Vicente R, Kapdi AR. Angew Chem Int Ed. 2009;48:9792–9826. doi: 10.1002/anie.200902996.Lyons TW, Sanford MS. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e.Colby DA, Bergman RG, Ellman JA. Chem Rev. 2010;110:624–655. doi: 10.1021/cr900005n.Wencel-Delord J, Dröge T, Liu F, Glorius F. Chem Soc Rev. 2011;40:4740–4761. doi: 10.1039/c1cs15083a.
- 3.For β-C(sp3)–H functionalizations of ketones via either enamine or enone intermediates, see: Pirnot MT, Rankic DA, Martin DBC, MacMillan DWC. Science. 2013;339:1593–1596. doi: 10.1126/science.1232993.Huang Z, Dong G. J Am Chem Soc. 2013;135:17747–17750. doi: 10.1021/ja410389a.Jie X, Shang Y, Zhang X, Su W. J Am Chem Soc. 2016;138:5623–5633. doi: 10.1021/jacs.6b01337.Hu X, Yang X, Dai XJ, Li CJ. Adv Synth Catal. 2017;359:2402–2406.Chen Y, Huang D, Zhao Y, Newhouse TR. Angew Chem Int Ed. 2017;56:8258–8262. doi: 10.1002/anie.201704874.
- 4.For selected examples of stoichiometric reactions: Constable AG, McDonald WS, Sawkins LC, Shaw BL. J Chem Soc Chem, Commun. 1978:1061–1062.Carr K, Sutherland JK. J Chem Soc Chem, Commun. 1984:1227–1228.Baldwin JE, Nájera C, Yus M. J Chem Soc Chem, Commun. 1985:126–127.Baldwin JE, Jones RH, Nájera C, Yus M. Tetrahedron. 1985;41:699–711.
- 5.(a) Desai LV, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]; (b) Thu HY, Yu WY, Che CM. J Am Chem Soc. 2006;128:9048–9049. doi: 10.1021/ja062856v. [DOI] [PubMed] [Google Scholar]; (c) Kang T, Kim Y, Lee D, Wang Z, Chang S. J Am Chem Soc. 2014;136:4141–4144. doi: 10.1021/ja501014b. [DOI] [PubMed] [Google Scholar]; (d) Gao P, Guo W, Xue J, Zhao Y, Yuan Y, Xia Y, Shi Z. J Am Chem Soc. 2015;137:12231–12240. doi: 10.1021/jacs.5b06758. [DOI] [PubMed] [Google Scholar]; (e) Peng J, Chen C, Xi C. Chem Sci. 2016;7:1383–1387. doi: 10.1039/c5sc03903g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For Cu-mediated hydroxylation using imino-pyridine directing group, see: Schönecker B, Zheldakova T, Liu Y, Kötteritzsch M, Gunther W, Görls H. Angew Chem Int Ed. 2003;42:3240–3244. doi: 10.1002/anie.200250815.See YY, Herrmann AT, Aihara Y, Baran PS. J Am Chem Soc. 2015;137:13776–13779. doi: 10.1021/jacs.5b09463.Trammell R, See Y, Herrmann AT, Xie N, Díaz DE, Siegler MA, Baran PS, Garcia-Bosch Isaac. J Org Chem. 2017;82:7887–7904. doi: 10.1021/acs.joc.7b01069.
- 7.(a) Jun CH, Lee H, Hong JB. J Org Chem. 1997;62:1200–1201. [Google Scholar]; (b) Bedford RB, Coles SJ, Hursthouse MB, Limmert ME. Angew Chem Int Ed. 2003;42:112–114. doi: 10.1002/anie.200390037. [DOI] [PubMed] [Google Scholar]; (c) Tan PW, Juwaini NAB, Seayad J. Org Lett. 2013;15:5166–5169. doi: 10.1021/ol402145m. [DOI] [PubMed] [Google Scholar]; (d) Mo F, Dong G. Science. 2014;345:68–72. doi: 10.1126/science.1254465. [DOI] [PubMed] [Google Scholar]
- 8.Shi BF, Maugel N, Zhang YH, Yu JQ. Angew Chem Int Ed. 2008;47:4882–4886. doi: 10.1002/anie.200801030. [DOI] [PubMed] [Google Scholar]
- 9.Gong W, Zhang G, Liu T, Giri R, Yu JQ. J Am Chem Soc. 2014;136:16940–16946. doi: 10.1021/ja510233h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Zhang FL, Hong K, Li TJ, Park H, Yu JQ. Science. 2016;351:252–256. doi: 10.1126/science.aad7893. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu Y, Chen YQ, Liu T, Eastgate MD, Yu JQ. J Am Chem Soc. 2016;138:14554–14557. doi: 10.1021/jacs.6b09653. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu XH, Park H, Hu JH, Hu Y, Zhang QL, Wang BL, Sun B, Yeung KS, Zhang FL, Yu JQ. J Am Chem Soc. 2017;139:888–896. doi: 10.1021/jacs.6b11188. [DOI] [PubMed] [Google Scholar]
- 11.For recent examples of C–H functionalization using a transient directing group or an in situ-formed directing group, see: Ma F, Lei M, Hu L. Org Lett. 2016;18:2708–2711. doi: 10.1021/acs.orglett.6b01170.Liu Y, Ge H. Nat Chem. 2016;9:26–32.Xu Y, Young MC, Wang C, Magness DM, Dong G. Angew Chem Int Ed. 2016;55:9084–9087. doi: 10.1002/anie.201604268.Xu Y, Young MC, Dong G. J Am Chem Soc. 2017;139:5716–5719. doi: 10.1021/jacs.7b02020.Yada A, Liao W, Sato Y, Murakami M. Angew Chem Int Ed. 2017;56:1073–1076. doi: 10.1002/anie.201610666.St John-Campbell S, White AJP, Bull JA. Chem Sci. 2017;8:4840–4847. doi: 10.1039/c7sc01218g.Chen XY, Ozturk S, Sorensen EJ. Org Lett. 2017;19:1140–1143. doi: 10.1021/acs.orglett.7b00161.Xu J, Liu Y, Wang Y, Li Y, Xu X, Jin Z. Org Lett. 2017;19:1562–1565. doi: 10.1021/acs.orglett.7b00363.Yao QJ, Zhang S, Zhan BB, Shi BF. Angew Chem Int Ed. 2017;56:6617–6621. doi: 10.1002/anie.201701849.Mu D, Wang X, Chen G, He G. J Org Chem. 2017;82:4497–4503. doi: 10.1021/acs.joc.7b00531.
- 12.Yang K, Li Q, Liu Y, Li G, Ge H. J Am Chem Soc. 2016;138:12775–12778. doi: 10.1021/jacs.6b08478. [DOI] [PubMed] [Google Scholar]
- 13.(a) Chen G, Gong W, Zhuang Z, Andra MS, Chen YQ, Hong X, Yang YF, Liu T, Houk KN, Yu JQ. Science. 2016;353:1023–1027. doi: 10.1126/science.aaf4434. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang YF, Chen G, Hong X, Yu JQ, Houk KN. J Am Chem Soc. 2017;139:8514–8521. doi: 10.1021/jacs.7b01801. [DOI] [PubMed] [Google Scholar]
- 14.Substrate 3c was recovered in 47% yield. Substrate 3b was not recovered since this ketone is difficult to visualize on TLC. In the crude 1H NMR, 40% of 3b was observed using CH2Br2 as the internal standard.
- 15.(a) Miller WH, Manley PJ, Cousins RD, Erhard KF, Heerding DA, Kwon C, Ross ST, Samanen JM, Takata DT, Uzinskas IN, Yuan CC, Haltiwanger RC, Gress CJ, Lark MW, Hwang SM, James IE, Rieman DJ, Willette RN, Yue TL, Azzarano LM, Salyers KL, Smith BR, Ward KW, Johanson KO, Huffman WF. Bioorg Med Chem Lett. 2003;13:1483–1486. doi: 10.1016/s0960-894x(03)00102-1. [DOI] [PubMed] [Google Scholar]; (b) Wang F, Li J, Sinn AL, Knabe WE, Khanna M, Jo I, Silver JM, Oh K, Li L, Sandusky GE, Sledge GW, Nakshatri H, Jones DR, Pollok KE, Meroueh SO. J Med Chem. 2011;54:7193–7205. doi: 10.1021/jm200782y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.