

Abstract
Tau is a microtubule-associated protein that functions in regulating cytoskeleton dynamics, especially in neurons. Misfolded and aggregated forms of tau produce pathological structures in a number of neurodegenerative diseases, including Alzheimer’s disease (AD) and tauopathy dementias. These disorders can present with a sporadic, such as AD, or familial etiology, such as in some cases of frontotemporal dementia with parkinsonism. Notably, the pathological features of tau pathology in these diseases can be very distinct. For example, the tau pathology in corticobasal degeneration is quite distinct from that of an AD patient. A wealth of evidence has emerged within the last decade to suggest that the misfolded tau in tauopathies possesses prion-like features, and that such features may explain the diverse features of tauopathies. The prion-like concept for tauopathies arose initially from the observation that the progressive accumulation of tau pathology as the symptoms of AD progress seemed to follow anatomically linked pathways. Subsequent studies in cell and animal models revealed that misfolded tau can propagate from cell-to-cell, and region-to-region in the brain through direct neuroanatomical connections. Studies in these cell and mouse models have demonstrated that experimentally propagated forms of misfolded tau can exist as conformationally distinct “strains” with unique biochemical, morphological and neuropathological characteristics. This review discusses the clinical, pathological, and genetic diversity of tauopathies; and the discoveries underlying the emerging view that the unique features of clinically distinct tauopathies may be a reflection of the “strain” of misfolded tau that propagates in each disease.
Keywords: Alzheimer’s disease, prion, strains, tau, transmission, animal models
Tauopathies are a spectrum of neurodegenerative diseases characterized by the brain accumulation of cellular proteinaceous inclusions predominantly comprised of tau protein (1–3). The most common of these disorders is Alzheimer’s disease (AD) where tau predominantly aggregates and accumulates in neurons as somatodendritic neurofibrillary tangles (NFTs) and neuropil threads, as well as in dystrophic neurites associated with extracellular deposits of amyloid-β peptides (Figure 1) (1–3). These tau aggregates (4–7) are comprised of structurally variable 8–20 nm twisted double-helical ribbons, referred to as paired helical filaments (8, 9) and less abundant 15 nm wide straight filaments (10, 11). Population based autopsy studies have suggested that tau pathological inclusions appear to spread in a predictable pattern that has been characterized by six Braak stages (I–VI) (12, 13). More recent data indicates that aberrant changes in tau (i.e. phosphorylation at specific epitopes) can occur in the locus ceruleus in a significant percent of young adults in their 20’s, and much earlier than the appearance of brain amyloid-β deposition (14). This early presentation of abnormal tau is reminiscent of the recently recognized occurrence of tau inclusions in the brains of cognitively normal individuals known as primary-aged tauopathy (15); however, it remains to be established if these accumulations of tau represent an early stage of AD or unrelated biological occurrences (16–18).
Figure 1.
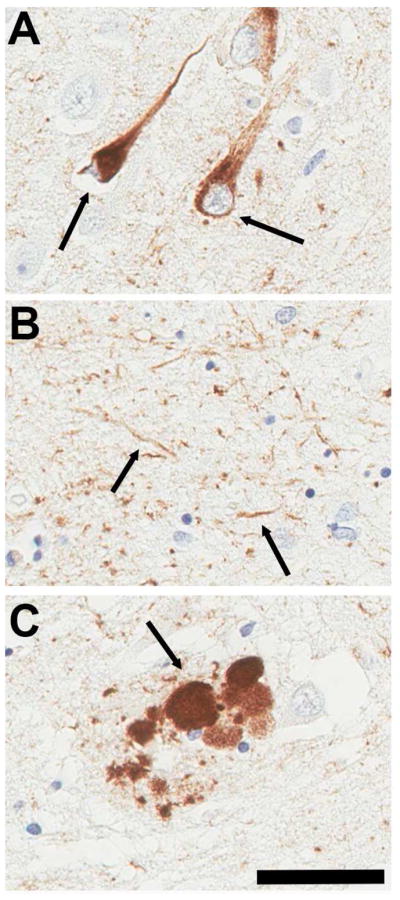
Representative images of (A) neurofibrillary tangles, (B) neuropil threads and (C) dystrophic neurites within a senile plaque in the hippocampus of patients with Alzheimer’s disease stained with anti-phospho tau antibody AT8. Bar = 50 μM.
Tauopathies also include many other types of dementias, such as corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), tangle-only dementia and Pick’s disease that occurs without a necessity for amyloid-β deposition (3). In some of these diseases, the type and distribution of tau pathological inclusion can be a defining feature. In CBD, tau inclusions are observed in the form of neuronal cytoplasmic inclusions and neuropil threads, as well as astrocytic plaques and oligodendrocytic coiled bodies (19–21). Tau pathology in PSP includes neuropil threads and classical flame-shaped NFTs or globose NFTs, but these structures are most prevalent in the basal ganglia, subthalamus and brainstem whereas in AD they are most abundant in the hippocampus and neocortex (19, 20). Furthermore, tau inclusions in PSP are prominent in glial cells as tufted astrocytes, thorn-shaped astrocytes, and oligodendroglial coiled bodies (19–21). Collectively, these observations illustrate the diversity in tau pathology that occurs in human diseases.
Tau gene, protein and function
Tau refers to microtubule (MT) associated proteins (22, 23) expressed from the MAPT gene located on chromosome 17q21-22 (24, 25). In human adult brain, 6 major tau isoforms ranging between 352 –441 amino acids in length are produced as a result of alternative RNA splicing of exons 2, 3 and 10 (Figure 2) (26, 27). The incorporation or exclusion of exon 2, or exons 2 and 3 (exon 3 is only ever included in tandem with exon 2), yields protein variants with 0 (0N), 29 (1N) or 58 (2N) amino acid inserts in the amino-terminal region. Similarly, exon 10 can be alternatively spliced to yield products containing either three (3R) or four (4R) tandem MT binding repeats of 31 or 32 amino acids. In normal adult human brain, 3R and 4R tau isoforms are present at approximately equal amounts while 2N tau-isoforms are significantly less abundant relative to the 0N or 1N isoforms (28, 29). In the CNS, tau is preferentially found in neurons (30, 31), but it can also be detected at lower levels in oligodendrocytes and astrocytes (31–33). Precisely how the diversity in tau isoforms and patterns of expression combine to produce the many clinical and pathological manifestations of tauopathy is an area of intense interest.
Figure 2.

Representative diagram of the 6 tau isoforms expressed in human adult brain due to alternative RNA splicing generated from the MAPT gene. Above is an alignment (not drawn to scale) corresponding to the MAPT exons that yield these various tau isoforms. Only the exons that be retained for expression in human brain are depicted and these are colored matched the protein regions. The amino acids are numbered according to the longest of these isoforms (441 amino acid in length). The amino-terminal inserts are identified as N1 and N2. The microtubule-binding repeats are depicted as R1–R4.
Although multiple functions have been attributed to tau (1), its function in binding and promoting MT assembly, nucleation and bundling has been the most extensively studied (22, 34–39). Consistent with its interaction with MTs, it can influence the function of other MT-interacting proteins such as dynein and kinesin to regulate trafficking of organelles and axonal cargo transport (40–42). Surprisingly, at least in mice, tau is not essential for MT function as genetically engineered null mice are viable and do not present with an overt phenotype (43). The loss of tau in this model may be compensated or shadowed by the increased expression of other MT-binding proteins such as MAP1A (43, 44).
Tau interacts with MT via the carboxyl-terminal region containing the three or four MT-binding repeats (3R, 4R) (45–47). Each individual repeat can bind to MTs, although with lower affinities than when combined in the full-length protein, as each repeat contributes to the overall MT affinity (48, 49). Furthermore, MT binding is more complex than a simple linear array of binding sites, (35, 38, 49) as tau also has a proline-rich region upstream of the repeat region that strongly influences MT binding and assembly (35, 50). Nevertheless, 4R tau has a greater MT polymerization and binding capacity than 3R-tau (28, 46). While the amino-terminal inserts do not significantly contribute to the MT binding affinity of tau, they can influence bundling (46). It is also well established that tau phosphorylation can reduce its ability to bind and modulate MT assembly (34, 37, 51–53). The prevailing view in the field is that the loss of MT binding by tau may contribute to the formation of pathologic features in tauopathies.
Tau amyloid aggregation
Recent experimental data (discussed in detailed below) suggest that tau aggregation could propagate throughout the nervous system by a prion-like transmission mechanism (54, 55), but the biological changes involved in the initial aggregation events (i.e. seed formation), elongation and regulation of tau aggregation remain highly debated. Native tau is highly soluble and “natively unfolded” (56, 57), but it has a tendency to form a global hairpin fold (58) that is not permissive for aggregation. Purified human tau is largely refractory to aggregation, although under some conditions it can self-aggregate into amyloid fibrils (59). In vitro tau filament assembly can be greatly facilitated by the presence of long polyanionic molecules such as sulfated glycosaminoglycans, poly-glutamate and nucleic acids (60–62) that likely suppress local intra- or inter- molecular positive charge repulsion. Tau fibril polymerization also can be facilitated in vitro by fatty acids, such as arachidonic acid (63, 64). The aggregation of tau is thought to require conformational changes in tau monomers as well as the formation of stable oligomeric complexes that can act as nucleation units. As such, in vitro, the addition of a small amount of preformed amyloid tau assemblies accelerates the formation of tau amyloid fibrils through the recruitment of permissive tau monomers that polymerize onto preformed seeds (65). Similarly, under conditions that promote the cellular entry of preformed tau amyloid seeds, endogenous cellular tau in cultured HEK293 cells can be readily recruited into fibrillar aggregates with properties reminiscent of inclusions in human brain (66, 67). Collectively, these studies demonstrate that tau has the capacity for seeded aggregation that is clearly a property associated with prion-like proteins.
Aggregated tau isolated from AD brains consists of all six brain tau isoforms (68) that are heavily phosphorylated at more than 40 different Ser and Thr residues (69–71). Numerous kinases and phosphatases can modulate tau phosphorylation at these sites in vivo and/or in vitro but it is still unclear which enzymes are responsible for the elevated phosphorylation state of AD aggregated tau (70, 72). Since elevated tau phosphorylation is a prominent distinction between tau pathological inclusions and normal tau, it is reasonable to hypothesize that phosphorylation might be involved in the polymerization of tau to form amyloid fibrils; however, there is no conclusive evidence to support this model, and non-phosphorylated, recombinant tau has the capacity to assemble into filaments in vitro (59–62). In addition, fetal tau can be as highly phosphorylated as AD aggregated tau (73), although it has been suggested that some phosphorylation sites in AD tau may be unique (74). Soluble tau in normal tissue can be rapidly dephosphorylated following harvesting, which can lead to under-representation of many phosphorylation sites (75). Whether aggregated tau is less prone to dephosphorylation is unclear. Nevertheless, pathological findings indicate that hyperphosphorylation of tau, at least at some residues, occurs early in tau inclusion formation (76, 77). Even without directly affecting tau aggregation, elevated phosphorylated tau can increase the pool of MT-free tau (34, 37, 53, 78), which becomes available for aggregate formation. Indeed, the ability of aggregated tau isolated from human brains to promote MT polymerization is impaired and this loss of function can be at least partially recovered by dephosphorylation (79). It can also be argued that the increased phosphorylation of tau in pathological inclusions is simply due to an imbalance in the levels of phosphorylation/dephosphorylation compared to soluble tau, such that aggregated tau is protected from dephosphorylation or preferentially phosphorylated after aggregation (70). Additionally, the effect of tau phosphorylation on its ability to be seeded is still unknown (80, 81). Despite the unresolved issues for the role of tau phosphorylation in influencing its aggregation, the enormous number of permutations in the phosphorylated forms of tau forms can yield many variants that could contribute to the diversity of pathological and clinical features in tauopathies.
Aggregated tau in human pathological inclusions is also modified by many other post translational modifications, including tyrosine phosphorylation (71), ubiquitination (82), glycation (83, 84), glycosylation (85), nitration (86) and acetylation (87). Tau also can be proteolytically cleaved at several sites (eg. Glu391 and Asp421) associated with inclusion formation (88). The role of these modifications in aggregate formation is still largely debated. For example, ubiquitination likely occurs after aggregation, probably as an attempt by the cellular machinery to degrade the inclusion (77, 89). Glycation is a non-enzymatic addition of reduced carbohydrates, and the presence of this modification is likely to result from the slow turnover of aggregated tau; however, increasing evidence points to the importance of lysine acetylation in modulating tau aggregation (87). Prolyl-isomerization is also likely involved in tau aggregation and spread of pathology (90). Collectively, all these modifications can yield a large spectrum of tau structural variants that can recruit endogenous tau for elongation, thus yielding a diverse array of unique structural entities. Furthermore, differential tau isoforms resulting from altered splicing are likely to result in varied aggregate conformers as each isoform displays different aggregation propensities (91).
MAPT mutations and dementia
Frontotemporal dementias (FTDs) are a distinct class of neurodegenerative disorders characterized by the selective atrophy of the frontal and temporal lobes, and early changes in behavioral conduct, language difficulties, and cognitive disturbance (92). Several families were described with autosomal dominant hereditary neurodegenerative disorders characteristic of FTD behavioral changes and in some cases with parkinsonism that could be genetically linked to chromosome 17; these were termed FTD and parkinsonism linked to chromosome 17 (FTDP-17) (93, 94). For most of these initial kindreds, autosomal dominant mutations in the tau gene (MAPT) were shown to be causal of disease (95–97), which is now referred to as FTPD-17t. Some of these kindreds were later found to be unrelated to tau mutations and instead were found to be linked to mutations in the granulin (GRN) gene (FTDP-17 GRN), which is in close proximity to MAPT (98, 99). More than 50 different mutations in MAPT causal of FTDP-17t have been identified (95, 100) and this is irrefutable evidence for a pathogenic role of tau in neurodegeneration. Most, if not all, FTDP-17t families lack amyloid β-deposition and show tau deposits in neurons, with some also having glial pathology (97, 100–107).
Most of the mutations identified in the MAPT locus are within exon 9–13 or in the introns surrounding exon 10 (1–3, 95, 100) with the only exceptions being the R5H and R5L amino acid mutations (108, 109). Within the coding MAPT exons, missense, deletion and silent mutations are associated with FTDP-17t (1–3, 95, 100). In the majority of the intronic mutations, the exonic silent mutations, and even some of the missense exonic mutations, the primary defect caused by the mutation is to alter RNA processing to enhance the inclusion of exon 10 in the mature mRNA (1–3, 29, 95, 100). Alteration in the splicing of exon 10 results in an imbalance in the normal ratio of 3R/4R tau isoforms, which is normally ~1:1 in adult human brain (29). The mechanism by which changes in the 3R/4R-tau ratio lead to neuronal and, in some cases, glial dysfunction and death is still nebulous. Interestingly, the presence of exon 10 in tau mRNA is developmentally regulated with the expression of isoforms containing exon 10 being under-represented during early development (27). 4R-tau and 3R-tau may bind distinct sites on MTs (49) and the over-production of one group of tau isoforms may result in an increased pool of MT-unbound tau that may polymerize into filaments over time. It is also possible that a specific ratio of tau isoforms is required for the normal maintenance and function of MTs. Although speculative, the possibility that specific isoforms might have other, undetermined functions should not be overlooked.
The role for an imbalance in tau splicing isoforms in neurodegeneration is supported by the accumulation of predominant tau isoforms in specific neurodegenerative diseases. In both PSP and CBD, tau inclusions are predominantly comprised of 4R-tau isoforms (110). Conversely, in Pick’s disease, another FTD disease which is characterized by the presence of round-shaped neuronal inclusions composed of 10–20 nm diameter filamentous tau inclusions (Pick bodies), the aggregates are predominantly assembled from 3R-tau isoforms (111–113). The reason for the preferential aggregation of specific tau isoforms in these diseases is unknown, but a possible explanation is that cells expressing specific forms of tau are more vulnerable. For example, the expression of tau isoforms can be cell-type specific as shown for 3R-tau in the granule cell layer of the dentate gyrus (4). Alternatively, the splicing of tau could be subtly imbalanced in these diseases, or other factors could initiate an isoform specific induction and spread of tau pathology (see discussion below).
Many tau missense mutations causal of FTDP-17t have been shown to impair the ability of tau to bind to or promote MTs polymerization (1–3, 100, 114), perhaps providing a greater pool of MT-free tau that can be recruited to aggregate. In addition, reduced association with MT binding can reduce axonal transport, resulting in perikaryal accumulation. Some missense tau mutations such as P301L, V337M, and R406W can also potentiate tau aggregation (115, 116). Consistent with the location of these mutations within tau, aggregated tau from cases with mutations such as V337M or R406W that affect all isoforms are predominantly comprised of all six tau isoforms, while inclusions in patients with the P301L mutation, which is only present in 4R-tau isoforms, are predominantly comprised of 4R tau (29, 117). In addition, in vitro studies indicate that P301L tau preferentially aggregates with itself and not wild-type tau akin to the property of a misfolded conformer-specific templated misfolding (118, 119).
Propagation of tau pathology
As mentioned above, during AD progression, pathological inclusions of tau appear to spread in a predictable pattern throughout the brain along known neuroanatomical connections (12), similar to those observed for the infectious prion protein (120). To further study the spreading phenomenon of tau, many studies have undertaken both in vitro and in vivo approaches to identify which mechanisms are at play. Although tau is predominantly an intracellular protein, a small amount can be detected in the CNS interstitial fluid (121) and cerebrospinal fluid (122). Many non-mutually exclusive mechanisms including synaptic secretion, direct unconventional plasma membrane translocation and exosome release may be sources of extracellular tau (123, 124). In disease settings, cellular damage and demise could account for extracellular tau that could be taken up endocytosis (directly or via receptor). Tau transfer between cells cold also occur by exosome uptake or direct cellular transfer can occur via nanotubes (123, 124)(Figure 3). Interestingly, neuronal activity can promote tau cellular release (125) likely promoting the intraneuronal spread of tau (126).
Figure 3.

(A) Cartoon depicting the proposed mechanisms for the spread of tau inclusion including: tunneling nanotubes (1), endocytosis (2), plasma membrane translocation (3), receptor-mediated endocytosis (4), and exosome release and fusion (5). (B) Distinct conformations of tau strains seed aggregate formation of conformationally similar templates when exogenously administered to the CNS containing both 3R and 4R tau isoforms. All of the indicated strains could contain 0, 1, or 2 N-terminal repeats, as these regions have not been demonstrated to affect tau strain properties.
In experimental animal studies, it has been shown that misfolded tau has the capacity to propagate along neuroanatomical pathways. The model systems used to demonstrate propagation are copied from what has been used in the study of prion disease. Several studies have revealed that the direct brain injections of samples containing recombinant tau aggregated in vitro can accelerate the onset of tau pathology in transgenic mice that over-express human tau (127, 128). Similarly, aggregated tau derived from tissue preparations of transgenic mice or humans can seed tau pathology (54, 129–131). In both cases, the induced tau inclusions occur in anatomically connected brain regions, suggesting neuroanatomical transport or induction of tau pathology along these routes. Most of these studies have used mice that express human P301S tau; however, there are examples of “transmission” to mice expressing wild-type human tau (129). Some studies indicate that not all tau transgenic mice are highly permissive to prion-like induction of tau pathology (132). Others have reported that cerebral injection of human tauopathy brain lysates can induce local tau pathology, or pathology that spreads along connected brain regions, in wild-type nontransgenic mice (129, 133). The induction of endogenous mouse tau aggregation in these latter studies strongly supported the prion-like nature involved in tauopathies (129, 133).
In addition to these exogenously induced models of tau spread, transgenic mouse models were created to try to restrict the expression of the tau transgene to the entorhinal cortex and then follow brain propagation (134, 135). These studies supported the notion that tau pathology could initially develop in one brain region, and then spread to distant, neuroanatomically connected neuronal populations, where human tau was not expressed; however, these mouse models use complex vector systems and whether these systems are as tightly regulated as originally thought has been called into question (136).
Some of the proposed mechanisms of tau cell-to-cell transmission involves secretion and uptake of the misfolded protein (Figure 3), thereby creating an extracellular pool of tau that could theoretically be targeted by immune therapies to prevent the spread of pathology. Indeed, active and passive immunization approaches have been carried out in tau transgenic mouse models with varying degrees of success (137–141). Together, these data support a prion-like spread of tau pathology by both a local cell-to-cell and distant axonal spread of inclusions. Whether such spread is a key feature of human disease is unknown and difficult to prove or disprove. The proof may emerge as immunotherapies developed in these model systems move into the clinic. Clearly, the timing of such therapies may be critical to success, and the bioavailability of antibodies to human CNS could limit efficacy, but if these model systems are replicating pathogenic events in humans then antibodies that are highly efficacious in these models should show significant efficacy in humans.
Existence of tau strains
A puzzling characteristics of prions was the observation that when passaging inocula containing the infectious prion protein, PrPSc, distinct and reproducible incubation periods and neuropathological features would appear (142, 143). Seeing as the PrPSc protein contained no nucleic acid, it was hypothesized and later revealed, that the tertiary structure of PrPSc could exist in multiple conformations and that these different conformations were capable of encoding strain-specific information (144–146). Among the human prion diseases there exists a wide variability in the clinical presentation of the disease, including progressive dementia, cerebellar ataxia, pyramidal signs, chorea, myoclonus, extrapyramidal features, pseudobulbar signs, seizures, and amyotrophic features (147). These differences are hypothesized to occur strictly based upon the conformation of the prion protein and although specific conformational changes have not been attributed to each distinct clinical feature, a number of studies have revealed biochemical differences in the infectious prion agents that give rise to certain forms of Creutzfeldt-Jakob Disease (CJD) (146, 148, 149). These findings set a precedent for the idea that distinct clinical features observed for a given neurodegenerative disease could be due to conformational variability, or strains, of a toxic protein.
As mentioned above, tauopathies comprise a group of neurodegenerative disorders that have diverse clinical features, tau deposition patterns, and cellular pathologies. There is now evidence to suggest that distinct conformations with strain-like properties govern these differences. Some of the first evidence came about by utilizing HEK293 cells stably expressing a tau reporter composed of a truncated form of 4R2N tau with both the P301L/V337M mutations and fused to a fluorescent reporter (150). Although unable to form inclusions on its own or following exposure to fibrils composed of proteins from other neurodegenerative proteins, upon exposure to tau fibrils, the truncated tau-YFP formed inclusions that revealed stable inheritance upon further passaging (150). Through successive passaging, the authors were able to establish 20 cell line clones that stably propagated morphologically distinct tau inclusions. These tau inclusion morphologies were characterized in a follow up study (151) in which the 20 clones were grouped into diffuse, large aggresome-like, nuclear, thread-like, and disordered inclusions, and those that changed from aggregate to diffuse over time, termed, mosaic. To determine whether tau inclusions were capable of templating their distinct pathology to naïve cells in a manner similar to prions, cells expressing the truncated tau-YFP were exposed to lysates from some of the clones. These studies revealed that the lysates were capable of inducing the same inclusion morphology of its associated progenitor, suggesting a process of templated conformational conversion for tau, strikingly similar to that characterized for prion strains. Moreover, these clones displayed distinct biochemical features, seeding activity, and toxicity (as measured by the growth clonal cell lines). Together, this data strongly supports the potential for tau to exist as distinct conformational strains.
The authors then went on to test the potential for these tau “strains” to induce distinct pathologies when injecting lysates from several of their tau cellular clones into the hippocampus of transgenic mice expressing the P301S human tau (150, 151). Although some lysates induced inclusions in vivo that were not morphologically similar to those that were observed in vitro, a subset of the lysates tested produced inclusions in mice that were morphologically and biochemically similar to what was produced in the cells, supporting the idea that distinct strains of misfolded tau could produce distinct pathologies. Central to the prion strain hypothesis is the stability of a given strain’s phenotypic and biochemical properties over multiple passages in mice. Not only did the authors demonstrate the in vivo stability of these tau strains, but they demonstrated that when brain lysates or immunopurified tau from these animals were added to tau-YFP expressing HEK293 cells, it induced an inclusion phenotype indistinguishable from the original strain injected into the mice. Taking their findings a step further, they sought to determine whether homogenates prepared from the brains of patients from a range of tauopathies, including AD, argyrophilic grain disease, CBD, Pick’s disease, and PSP would give rise to distinct pathologies upon exposure to their tau-YFP cell culture model (150). Indeed, each of these disorders induced a diverse pattern of tau phenotypes suggesting that different conformations of tau are responsible for the variable characteristics observed among tauopathies.
Further supporting the prion-like nature of tau and the existence of tau strains, there also appears to be a barrier preventing some strains of tau from seeding or inducing the aggregation of other tau variants. In transgenic mice that express 4R human tau, extracts containing aggregates of 4R tau induce pathology, whereas those containing 3R tau inclusions do not (54, 129, 152). Therefore, it is possible to envision a type of “species barrier” in which seeding only occurs when there is structural similarity between the seed and the template. To this point, other studies have indicated the effect of the seed and template on the degree and extent of seeding (67, 153). As discussed above, 3R and 4R tau isoforms are typically equally expressed in adult human brain, but in some tauopathies only one of these groups of isoforms preferentially forms inclusions (Figure 3). This specificity in isoform aggregation in an environment, in which all isoforms are present, further supports the notion that the molecular specificity of the seed and template can be critical determinants in the permissiveness of polymer elongation driving tau inclusion formation. It may be possible to block these template interactions with antibodies or small molecules, but the diversity in tau strains could be a harbinger of a difficult road to effective therapies.
Acknowledgments
This work was supported by a grant from the National Institute on Aging (AG047266), and by the SantaFe HealthCare Alzheimer’s Disease Research Center. We thank our colleagues in the Center for Translational Research in Neurodegenerative Disease for helpful discussions on the content of this review.
Footnotes
Financial Disclosures
The authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016;17:5–21. doi: 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- 2.Iqbal K, Liu F, Gong CX. Tau and neurodegenerative disease: the story so far. Nat Rev Neurol. 2016;12:15–27. doi: 10.1038/nrneurol.2015.225. [DOI] [PubMed] [Google Scholar]
- 3.Lee VM-Y, Goedert M, Trojanoswki JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 4.Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad of Sci U S A. 1988;85:4051–4055. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wischik CM, Novak M, Thogersen HC, Edwards PC, Runswick MJ, Jakes R, et al. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85:4506–4510. doi: 10.1073/pnas.85.12.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science. 1991;251:675–678. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 7.Kosik KS, Orecchio LD, Binder L, Trojanowski JQ, Lee VM, Lee G. Epitopes that span the tau molecule are shared with paired helical filaments. Neuron. 1988;1:817–825. doi: 10.1016/0896-6273(88)90129-8. [DOI] [PubMed] [Google Scholar]
- 8.Kidd M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature. 1963;197:192–193. doi: 10.1038/197192b0. [DOI] [PubMed] [Google Scholar]
- 9.Crowther RA, Wischik CM. Image reconstruction of the Alzheimer paired helical filament. EMBO J. 1985;4:3661–3665. doi: 10.1002/j.1460-2075.1985.tb04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crowther RA. Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc Natl Acad Sci U S A. 1991;88:2288–2292. doi: 10.1073/pnas.88.6.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yagishita S, Itoh Y, Nan W, Amano N. Reappraisal of the fine structure of Alzheimer’s neurofibrillary tangles. Acta Neuropath. 1981;54:239–246. doi: 10.1007/BF00687747. [DOI] [PubMed] [Google Scholar]
- 12.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 13.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del TK. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960–969. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- 15.Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128:755–766. doi: 10.1007/s00401-014-1349-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jellinger KA, Alafuzoff I, Attems J, Beach TG, Cairns NJ, Crary JF, et al. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol. 2015;129:757–762. doi: 10.1007/s00401-015-1407-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braak H, Del Tredici K. Are cases with tau pathology occurring in the absence of Abeta deposits part of the AD-related pathological process? Acta Neuropathol. 2014;128:767–772. doi: 10.1007/s00401-014-1356-1. [DOI] [PubMed] [Google Scholar]
- 18.Duyckaerts C, Braak H, Brion JP, Buee L, Del Tredici K, Goedert M, et al. PART is part of Alzheimer disease. Acta Neuropathol. 2015;129:749–756. doi: 10.1007/s00401-015-1390-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dickson DW. Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol. 1999;246(Suppl 2):II6–15. doi: 10.1007/BF03161076. [DOI] [PubMed] [Google Scholar]
- 20.Feany MB, Mattiace LA, Dickson DW. Neuropathologic overlap of progressive supranuclear palsy, Pick’s disease and corticobasal degeneration. J Neuropathol Exp Neurol. 1996;55:53–67. doi: 10.1097/00005072-199601000-00006. [DOI] [PubMed] [Google Scholar]
- 21.Chin SS, Goldman JE. Glial inclusions in CNS degenerative diseases. J Neuropathol Exp Neurol. 1996;55:499–508. doi: 10.1097/00005072-199605000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Cleveland DW, Hwo SY, Kirschner MW. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol. 1977;116:207–225. doi: 10.1016/0022-2836(77)90213-3. [DOI] [PubMed] [Google Scholar]
- 23.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neve RL, Harris P, Kosik KS, Kurnit DM, Donlon TA. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 1986;387:271–280. doi: 10.1016/0169-328x(86)90033-1. [DOI] [PubMed] [Google Scholar]
- 25.Andreadis A, Brown WM, Kosik KS. Structure and novel exons of the human tau gene. Biochemistry. 1992;31:10626–10633. doi: 10.1021/bi00158a027. [DOI] [PubMed] [Google Scholar]
- 26.Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 28.Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9:4225–4230. doi: 10.1002/j.1460-2075.1990.tb07870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 30.Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Migheli A, Butler M, Brown K, Shelanski ML. Light and electron microscope localization of the microtubule-associated tau protein in rat brain. JNeurosci. 1988;8:1846–1851. doi: 10.1523/JNEUROSCI.08-06-01846.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI. Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc Natl Acad Sci U S A. 1995;92:10369–10373. doi: 10.1073/pnas.92.22.10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tashiro K, Hasegawa M, Ihara Y, Iwatsubo T. Somatodendritic localization of phosphorylated tau in neonatal and adult rat cerebral cortex. Neuroreport. 1997;8:2797–2801. doi: 10.1097/00001756-199708180-00029. [DOI] [PubMed] [Google Scholar]
- 34.Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. 1992;3:1141–1154. doi: 10.1091/mbc.3.10.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanai Y, Chen J, Hirokawa N. Microtubule bundling by tau proteins in vivo: analysis of functional domains. EMBO J. 1992;11:3953–3961. doi: 10.1002/j.1460-2075.1992.tb05489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drubin DG, Kirschner MW. Tau protein function in living cells. J Cell Biol. 1986;103:2739–2746. doi: 10.1083/jcb.103.6.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993;10:1089–1099. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- 38.Preuss U, Biernat J, Mandelkow EM, Mandelkow E. The ‘jaws’ model of tau-microtubule interaction examined in CHO cells. J Cell Sci. 1997;110(Pt 6):789–800. doi: 10.1242/jcs.110.6.789. [DOI] [PubMed] [Google Scholar]
- 39.Brandt R, Lee G. Functional organization of microtubule-associated protein tau. Identification of regions which affect microtubule growth, nucleation, and bundle formation in vitro. J Biol Chem. 1993;268:3414–3419. [PubMed] [Google Scholar]
- 40.Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer’s disease. J Cell Biol. 1998;143:777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156:1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319:1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, et al. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–491. doi: 10.1038/369488a0. [DOI] [PubMed] [Google Scholar]
- 44.Takei Y, Teng J, Harada A, Hirokawa N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J Cell Biol. 2000;150:989–1000. doi: 10.1083/jcb.150.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Himmler A, Drechsel D, Kirschner MW, Martin DW., Jr Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol Cell Biol. 1989;9:1381–1388. doi: 10.1128/mcb.9.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Butner KA, Kirschner MW. Tau protein binds to microtubules through a flexible array of distributed weak sites. J Cell Biol. 1991;115:717–730. doi: 10.1083/jcb.115.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee G, Neve RL, Kosik KS. The microtubule binding domain of tau protein. Neuron. 1989;2:1615–1624. doi: 10.1016/0896-6273(89)90050-0. [DOI] [PubMed] [Google Scholar]
- 48.Ennulat DJ, Liem RK, Hashim GA, Shelanski ML. Two separate 18-amino acid domains of tau promote the polymerization of tubulin. J Biol Chem. 1989;264:5327–5330. [PubMed] [Google Scholar]
- 49.Goode BL, Feinstein SC. Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J Cell Biol. 1994;124:769–782. doi: 10.1083/jcb.124.5.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goode BL, Denis PE, Panda D, Radeke MJ, Miller HP, Wilson L, et al. Functional interactions between the proline-rich and repeat regions of tau enhance microtubule binding and assembly. Mol Biol Cell. 1997;8:353–365. doi: 10.1091/mbc.8.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–163. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- 52.Yoshida H, Ihara Y. Tau in paired helical filaments is functionally distinct from fetal tau: assembly incompetence of paired helical filament-tau. J Neurochem. 1993;61:1183–1186. doi: 10.1111/j.1471-4159.1993.tb03642.x. [DOI] [PubMed] [Google Scholar]
- 53.Lindwall G, Cole RD. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem. 1984;259:5301–5305. [PubMed] [Google Scholar]
- 54.Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frost B, Ollesch J, Wille H, Diamond MI. Conformational diversity of wild-type Tau fibrils specified by templated conformation change. J Biol Chem. 2009;284:3546–3551. doi: 10.1074/jbc.M805627200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cleveland DW, Hwo SY, Kirschner MW. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol. 1977;116:227–247. doi: 10.1016/0022-2836(77)90214-5. [DOI] [PubMed] [Google Scholar]
- 57.Schweers O, Schonbrunn-Hanebeck E, Marx A, Mandelkow E. Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J Biol Chem. 1994;269:24290–24297. [PubMed] [Google Scholar]
- 58.Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006;45:2283–2293. doi: 10.1021/bi0521543. [DOI] [PubMed] [Google Scholar]
- 59.Crowther RA, Olesen OF, Smith MJ, Jakes R, Goedert M. Assembly of Alzheimer-like filaments from full-length tau protein. FEBS Lett. 1994;337:135–138. doi: 10.1016/0014-5793(94)80260-2. [DOI] [PubMed] [Google Scholar]
- 60.Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–553. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- 61.Kampers T, Friedhoff P, Biernat J, Mandelkow EM, Mandelkow E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996;399:344–349. doi: 10.1016/s0014-5793(96)01386-5. [DOI] [PubMed] [Google Scholar]
- 62.Perez M, Valpuesta JM, Medina M, Montejo dG, Avila J. Polymerization of tau into filaments in the presence of heparin: the minimal sequence required for tau-tau interaction. J Neurochem. 1996;67:1183–1190. doi: 10.1046/j.1471-4159.1996.67031183.x. [DOI] [PubMed] [Google Scholar]
- 63.King ME, Gamblin TC, Kuret J, Binder LI. Differential assembly of human tau isoforms in the presence of arachidonic acid. J Neurochem. 2000;74:1749–1757. doi: 10.1046/j.1471-4159.2000.0741749.x. [DOI] [PubMed] [Google Scholar]
- 64.Wilson DM, Binder LI. Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer’s disease. Am J Pathol. 1997;150:2181–2195. [PMC free article] [PubMed] [Google Scholar]
- 65.Friedhoff P, von Bergen M, Mandelkow EM, Davies P, Mandelkow E. A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc Natl Acad Sci U S A. 1998;95:15712–15717. doi: 10.1073/pnas.95.26.15712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286:15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M. Seeded aggregation and toxicity of {alpha}-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem. 2010;285:34885–34898. doi: 10.1074/jbc.M110.148460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159–168. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- 69.Hanger DP, Betts JC, Loviny TL, Blackstock WP, Anderton BH. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer’s disease brain using nanoelectrospray mass spectrometry. J Neurochem. 1998;71:2465–2476. doi: 10.1046/j.1471-4159.1998.71062465.x. [DOI] [PubMed] [Google Scholar]
- 70.Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15:112–119. doi: 10.1016/j.molmed.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 71.Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: implications for Alzheimer’s disease. Neurochem Int. 2011;58:458–471. doi: 10.1016/j.neuint.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 72.Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997;323(Pt 3):577–591. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kenessey A, Yen SH. The extent of phosphorylation of fetal tau is comparable to that of PHF-tau from Alzheimer paired helical filaments. Brain research. 1993;629:40–46. doi: 10.1016/0006-8993(93)90478-6. [DOI] [PubMed] [Google Scholar]
- 74.Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Watanabe A, et al. Hyperphosphorylation of tau in PHF. Neurobiol Aging. 1995;16:365–371. doi: 10.1016/0197-4580(95)00027-c. discussion 371–380. [DOI] [PubMed] [Google Scholar]
- 75.Matsuo ES, Shin RW, Billingsley ML, Van deVoorde A, O’Connor M, Trojanowski JQ, et al. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron. 1994;13:989–1002. doi: 10.1016/0896-6273(94)90264-x. [DOI] [PubMed] [Google Scholar]
- 76.Braak E, Braak H, Mandelkow EM. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol(Berl) 1994;87:554–567. doi: 10.1007/BF00293315. [DOI] [PubMed] [Google Scholar]
- 77.Bancher C, Grundke-Iqbal I, Iqbal K, Fried VA, Smith HT, Wisniewski HM. Abnormal phosphorylation of tau precedes ubiquitination in neurofibrillary pathology of Alzheimer disease. Brain Res. 1991;539:11–18. doi: 10.1016/0006-8993(91)90681-k. [DOI] [PubMed] [Google Scholar]
- 78.Gustke N, Steiner B, Mandelkow EM, Biernat J, Meyer HE, Goedert M, et al. The Alzheimer-like phosphorylation of tau protein reduces microtubule binding and involves Ser-Pro and Thr-Pro motifs. FEBS Lett. 1992;307:199–205. doi: 10.1016/0014-5793(92)80767-b. [DOI] [PubMed] [Google Scholar]
- 79.Iqbal K, Zaidi T, Bancher C, Grundke-Iqbal I. Alzheimer paired helical filaments. Restoration of the biological activity by dephosphorylation. FEBS letters. 1994;349:104–108. doi: 10.1016/0014-5793(94)00650-4. [DOI] [PubMed] [Google Scholar]
- 80.Guo JL, Buist A, Soares A, Callaerts K, Calafate S, Stevenaert F, et al. The Dynamics and Turnover of Tau Aggregates in Cultured Cells: Insights into Therapies for Tauopathies. J Biol Chem. 2016;291:13175–13193. doi: 10.1074/jbc.M115.712083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Falcon B, Cavallini A, Angers R, Glover S, Murray TK, Barnham L, et al. Conformation determines the seeding potencies of native and recombinant Tau aggregates. J Biol Chem. 2015;290:1049–1065. doi: 10.1074/jbc.M114.589309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Titani K, Ihara Y. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron. 1993;10:1151–1160. doi: 10.1016/0896-6273(93)90063-w. [DOI] [PubMed] [Google Scholar]
- 83.Ledesma MD, Bonay P, Avila J. Tau protein from Alzheimer’s disease patients is glycated at its tubulin-binding domain. J Neurochem. 1995;65:1658–1664. doi: 10.1046/j.1471-4159.1995.65041658.x. [DOI] [PubMed] [Google Scholar]
- 84.Ledesma MD, Bonay P, Colaco C, Avila J. Analysis of microtubule-associated protein tau glycation in paired helical filaments. J Biol Chem. 1994;269:21614–21619. [PubMed] [Google Scholar]
- 85.Wang JZ, Grundke-Iqbal I, Iqbal K. Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer’s disease. NatMed. 1996;2:871–875. doi: 10.1038/nm0896-871. [DOI] [PubMed] [Google Scholar]
- 86.Horiguchi T, Uryu K, Giasson BI, Ischiropoulos H, LightFoot R, Bellmann C, et al. Nitration of tau protein is linked to neurodegeneration in tauopathies. Am J Pathol. 2003;163:1021–1031. doi: 10.1016/S0002-9440(10)63462-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cook C, Stankowski JN, Carlomagno Y, Stetler C, Petrucelli L. Acetylation: a new key to unlock tau’s role in neurodegeneration. Alzheimers Res Ther. 2014;6:29. doi: 10.1186/alzrt259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Basurto-Islas G, Luna-Munoz J, Guillozet-Bongaarts AL, Binder LI, Mena R, Garcia-Sierra F. Accumulation of aspartic acid421- and glutamic acid391-cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol. 2008;67:470–483. doi: 10.1097/NEN.0b013e31817275c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Iwatsubo T, Hasegawa M, Esaki Y, Ihara Y. Lack of ubiquitin immunoreactivities at both ends of neuropil threads. Possible bidirectional growth of neuropil threads. Am J Pathol. 1992;140:277–282. [PMC free article] [PubMed] [Google Scholar]
- 90.Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. 2015;523:431–436. doi: 10.1038/nature14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhong Q, Congdon EE, Nagaraja HN, Kuret J. Tau isoform composition influences rate and extent of filament formation. J Biol Chem. 2012;287:20711–20719. doi: 10.1074/jbc.M112.364067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol. 2001;58:1803–1809. doi: 10.1001/archneur.58.11.1803. [DOI] [PubMed] [Google Scholar]
- 93.Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D’Amato CJ, Gilman S. Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Conference Participants. Ann Neurol. 1997;41:706–715. doi: 10.1002/ana.410410606. [DOI] [PubMed] [Google Scholar]
- 94.Reed LA, Wszolek ZK, Hutton M. Phenotypic correlations in FTDP-17. Neurobiol Aging. 2001;22:89–107. doi: 10.1016/s0197-4580(00)00202-5. [DOI] [PubMed] [Google Scholar]
- 95.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 96.Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 97.Spillantini MG, Crowther RA, Kamphorst W, Heutink P, van Swieten JC. Tau pathology in two Dutch families with mutations in the microtubule-binding region of tau. Am J Pathol. 1998;153:1359–1363. doi: 10.1016/S0002-9440(10)65721-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 99.Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 100.Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. 2015;41:24–46. doi: 10.1111/nan.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Iijima M, Tabira T, Poorkaj P, Schellenberg GD, Trojanowski JQ, Lee VM, et al. A distinct familial presenile dementia with a novel missense mutation in the tau gene. Neuroreport. 1999;10:497–501. doi: 10.1097/00001756-199902250-00010. [DOI] [PubMed] [Google Scholar]
- 102.Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MR, Ghetti B. Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci U S A. 1997;94:4113–4118. doi: 10.1073/pnas.94.8.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sima AA, Defendini R, Keohane C, D’Amato C, Foster NL, Parchi P, et al. The neuropathology of chromosome 17-linked dementia. Ann Neurol. 1996;39:734–743. doi: 10.1002/ana.410390609. [DOI] [PubMed] [Google Scholar]
- 104.Reed LA, Schmidt ML, Wszolek ZK, Balin BJ, Soontornniyomkij V, Lee VM, et al. The neuropathology of a chromosome 17-linked autosomal dominant parkinsonism and dementia (“pallido-ponto-nigral degeneration”) J Neuropathol Exp Neurol. 1998;57:588–601. doi: 10.1097/00005072-199806000-00006. [DOI] [PubMed] [Google Scholar]
- 105.Goedert M, Spillantini MG, Crowther RA, Chen SG, Parchi P, Tabaton M, et al. Tau gene mutation in familial progressive subcortical gliosis. Nat Med. 1999;5:454–457. doi: 10.1038/7454. [DOI] [PubMed] [Google Scholar]
- 106.D’Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, et al. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci U S A. 1999;96:5598–5603. doi: 10.1073/pnas.96.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sumi SM, Bird TD, Nochlin D, Raskind MA. Familial presenile dementia with psychosis associated with cortical neurofibrillary tangles and degeneration of the amygdala. Neurology. 1992;42:120–127. doi: 10.1212/wnl.42.1.120. [DOI] [PubMed] [Google Scholar]
- 108.Hayashi S, Toyoshima Y, Hasegawa M, Umeda Y, Wakabayashi K, Tokiguchi S, et al. Late-onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann Neurol. 2002;51:525–530. doi: 10.1002/ana.10163. [DOI] [PubMed] [Google Scholar]
- 109.Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H, et al. An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol. 2002;52:511–516. doi: 10.1002/ana.10340. [DOI] [PubMed] [Google Scholar]
- 110.Sergeant N, Wattez A, Delacourte A. Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusively “exon 10” isoforms. J Neurochem. 1999;72:1243–1249. doi: 10.1046/j.1471-4159.1999.0721243.x. [DOI] [PubMed] [Google Scholar]
- 111.Delacourte A, Sergeant N, Wattez A, Gauvreau D, Robitaille Y. Vulnerable neuronal subsets in Alzheimer’s and Pick’s disease are distinguished by their tau isoform distribution and phosphorylation. Ann Neurol. 1998;43:193–204. doi: 10.1002/ana.410430209. [DOI] [PubMed] [Google Scholar]
- 112.Sergeant N, David JP, Lefranc D, Vermersch P, Wattez A, Delacourte A. Different distribution of phosphorylated tau protein isoforms in Alzheimer’s and Pick’s diseases. FEBS letters. 1997;412:578–582. doi: 10.1016/s0014-5793(97)00859-4. [DOI] [PubMed] [Google Scholar]
- 113.Rewcastle NB, Ball MJ. Electron microscopic structure of the “inclusion bodies” in Pick’s disease. Neurology. 1968;18:1205–1213. doi: 10.1212/wnl.18.12.1205. [DOI] [PubMed] [Google Scholar]
- 114.Fontaine SN, Sabbagh JJ, Baker J, Martinez-Licha CR, Darling A, Dickey CA. Cellular factors modulating the mechanism of tau protein aggregation. Cell Mol life Sci. 2015;72:1863–1879. doi: 10.1007/s00018-015-1839-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Arrasate M, Perez M, Armas-Portela R, Avila J. Polymerization of tau peptides into fibrillar structures. The effect of FTDP-17 mutations. FEBS Lett. 1999;446:199–202. doi: 10.1016/s0014-5793(99)00210-0. [DOI] [PubMed] [Google Scholar]
- 116.Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, Hutton M, et al. Accelerated filament formation from tau protein with specific FTDP-17 missence. FEBS Lett. 1999;447:195–199. doi: 10.1016/s0014-5793(99)00294-x. [DOI] [PubMed] [Google Scholar]
- 117.Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, et al. Pathogenic implications of mutations in the tau gene in pallido-ponto- nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci U S A. 1998;95:13103–13107. doi: 10.1073/pnas.95.22.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Aoyagi H, Hasegawa M, Tamaoka A. Fibrillogenic nuclei composed of P301L mutant tau induce elongation of P301L tau but not wild-type tau. J Biol Chem. 2007;282:20309–20318. doi: 10.1074/jbc.M611876200. [DOI] [PubMed] [Google Scholar]
- 119.Rizzu P, Joosse M, Ravid R, Hoogeveen A, Kamphorst W, van Swieten JC, et al. Mutation-dependent aggregation of tau protein and its selective depletion from the soluble fraction in brain of P301L FTDP-17 patients. Hum Mol Geneti. 2000;9:3075–3082. doi: 10.1093/hmg/9.20.3075. [DOI] [PubMed] [Google Scholar]
- 120.Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yamada K, Cirrito JR, Stewart FR, Jiang H, Finn MB, Holmes BB, et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 2011;31:13110–13117. doi: 10.1523/JNEUROSCI.2569-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Johnson GV, Seubert P, Cox TM, Motter R, Brown JP, Galasko D. The tau protein in human cerebrospinal fluid in Alzheimer’s disease consists of proteolytically derived fragments. J Neurochem. 1997;68:430–433. doi: 10.1046/j.1471-4159.1997.68010430.x. [DOI] [PubMed] [Google Scholar]
- 123.Mohamed NV, Herrou T, Plouffe V, Piperno N, Leclerc N. Spreading of tau pathology in Alzheimer’s disease by cell-to-cell transmission. Eur J Neurosci. 2013;37:1939–1948. doi: 10.1111/ejn.12229. [DOI] [PubMed] [Google Scholar]
- 124.Guo JL, Lee VM. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med. 2014;20:130–138. doi: 10.1038/nm.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013;14:389–394. doi: 10.1038/embor.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. 2016;19:1085–1092. doi: 10.1038/nn.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci. 2013;33:1024–1037. doi: 10.1523/JNEUROSCI.2642-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Peeraer E, Bottelbergs A, Van Kolen K, Stancu IC, Vasconcelos B, Mahieu M, et al. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis. 2015;73:83–95. doi: 10.1016/j.nbd.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110:9535–9540. doi: 10.1073/pnas.1301175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jackson SJ, Kerridge C, Cooper J, Cavallini A, Falcon B, Cella CV, et al. Short Fibrils Constitute the Major Species of Seed-Competent Tau in the Brains of Mice Transgenic for Human P301S Tau. J Neurosci. 2016;36:762–772. doi: 10.1523/JNEUROSCI.3542-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H, et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 2014;127:667–683. doi: 10.1007/s00401-014-1254-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chakrabarty P, Hudson VJ, III, Sacino AN, Brooks MM, D’Alton S, Lewis J, et al. Inefficient induction and spread of seeded tau pathology in P301L mouse model of tauopathy suggests inherent physiological barriers to transmission. Acta Neuropathol. 2015;130:303–305. doi: 10.1007/s00401-015-1444-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Guo JL, Narasimhan S, Changolkar L, He Z, Stieber A, Zhang B, et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med. 2016;213:2635–2654. doi: 10.1084/jem.20160833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, et al. Trans-synaptic spread of tau pathology in vivo. PloS one. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yetman MJ, Lillehaug S, Bjaalie JG, Leergaard TB, Jankowsky JL. Transgene expression in the Nop-tTA driver line is not inherently restricted to the entorhinal cortex. Brain Struct Funct. 2016;221:2231–2249. doi: 10.1007/s00429-015-1040-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci. 2010;30:16559–16566. doi: 10.1523/JNEUROSCI.4363-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H, et al. Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem. 2011;286:34457–34467. doi: 10.1074/jbc.M111.229633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bi M, Ittner A, Ke YD, Gotz J, Ittner LM. Tau-targeted immunization impedes progression of neurofibrillary histopathology in aged P301L tau transgenic mice. PloS one. 2011;6:e26860. doi: 10.1371/journal.pone.0026860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, et al. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013;80:402–414. doi: 10.1016/j.neuron.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.McEwan WA, Falcon B, Vaysburd M, Clift D, Oblak AL, Ghetti B, et al. Cytosolic Fc receptor TRIM21 inhibits seeded tau aggregation. Proc Nat l Acad Sci U S A. 2017;114:574–579. doi: 10.1073/pnas.1607215114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fraser H, Dickinson AG. The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol. 1968;78:301–311. doi: 10.1016/0021-9975(68)90006-6. [DOI] [PubMed] [Google Scholar]
- 143.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 144.Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. Journal of virology. 1994;68:7859–7868. doi: 10.1128/jvi.68.12.7859-7868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Caughey B, Raymond GJ, Bessen RA. Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J Biol Chem. 1998;273:32230–32235. doi: 10.1074/jbc.273.48.32230. [DOI] [PubMed] [Google Scholar]
- 146.Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 147.Kovacs GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RS, Budka H. Mutations of the prion protein gene phenotypic spectrum. J Neurol. 2002;249:1567–1582. doi: 10.1007/s00415-002-0896-9. [DOI] [PubMed] [Google Scholar]
- 148.Parchi P, Giese A, Capellari S, Brown P, Schulz-Schaeffer W, Windl O, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46:224–233. [PubMed] [Google Scholar]
- 149.Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 150.Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271–1288. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kaufman SK, Sanders DW, Thomas TL, Ruchinskas AJ, Vaquer-Alicea J, Sharma AM, et al. Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron. 2016;92:796–812. doi: 10.1016/j.neuron.2016.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Woerman AL, Aoyagi A, Patel S, Kazmi SA, Lobach I, Grinberg LT, et al. Tau prions from Alzheimer’s disease and chronic traumatic encephalopathy patients propagate in cultured cells. Proc Natl Acad Sci U S A. 2016;113:E8187–E8196. doi: 10.1073/pnas.1616344113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Dinkel PD, Siddiqua A, Huynh H, Shah M, Margittai M. Variations in filament conformation dictate seeding barrier between three- and four-repeat tau. Biochemistry. 2011;50:4330–4336. doi: 10.1021/bi2004685. [DOI] [PubMed] [Google Scholar]