

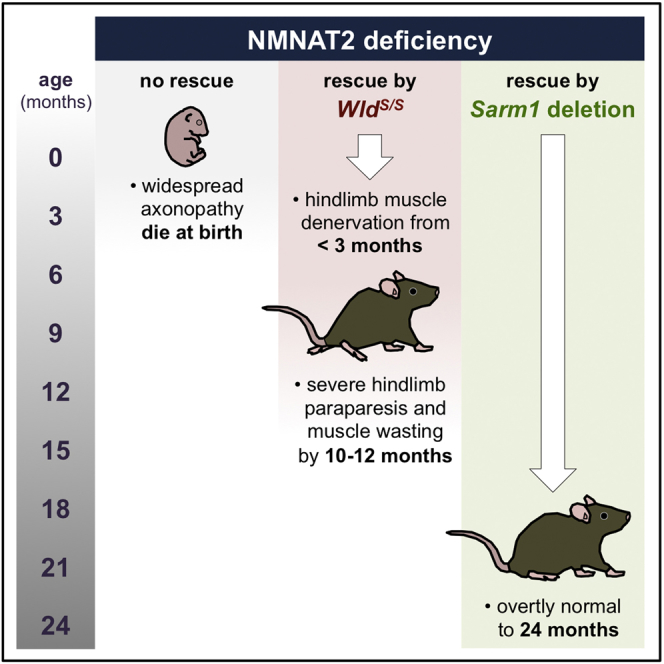
Summary
Studies with the WldS mutant mouse have shown that axon and synapse pathology in several models of neurodegenerative diseases are mechanistically related to injury-induced axon degeneration (Wallerian degeneration). Crucially, an absence of SARM1 delays Wallerian degeneration as robustly as WldS, but their relative capacities to confer long-term protection against related, non-injury axonopathy and/or synaptopathy have not been directly compared. While Sarm1 deletion or WldS can rescue perinatal lethality and widespread Wallerian-like axonopathy in young NMNAT2-deficient mice, we report that an absence of SARM1 enables these mice to survive into old age with no overt phenotype, whereas those rescued by WldS invariantly develop a progressive neuromuscular defect in their hindlimbs from around 3 months of age. We therefore propose Sarm1 deletion as a more reliable tool than WldS for investigating Wallerian-like mechanisms in disease models and suggest that SARM1 blockade may have greater therapeutic potential than WLDS-related strategies.
Keywords: Sarm1, WldS, axonopathy, synaptopathy, NMNAT2-deficient mice, aging, motor function, neuromuscular junction, disease model, neurodegeneration
Graphical Abstract
Highlights
-
•
Rescue of an axonopathy model by Sarm1 deletion or WldS compared in an aging study
-
•
Young adult NMNAT2-deficient mice rescued by WldS develop a hindlimb motor defect
-
•
NMNAT2-deficient mice rescued by Sarm1 deletion are overtly normal up to 24 months
-
•
SARM1 depletion/inhibition may have analytical and therapeutic advantages over WLDS
Both Sarm1 deletion and WldS prevent axonopathy and perinatal lethality in NMNAT2-deficient mice. Gilley et al. report that those rescued by WldS develop hindlimb motor problems as young adults, whereas Sarm1 deletion allows survival to 24 months with no overt defect. These findings have important analytical and therapeutic implications.
Introduction
WldS, a spontaneous mutant mouse allele encoding a fusion protein (WLDS) with nicotinamide mononucleotide adenylyltransferase (NMNAT) activity, robustly delays injury-induced axon and synapse degeneration (Wallerian degeneration) by locally substituting for loss of the endogenous NMNAT2 isoform (Mack et al., 2001, Gilley and Coleman, 2010, Cohen et al., 2012, Conforti et al., 2014). WldS has been the tool of choice for investigating the molecular basis of axon pathology in animal models of neurodegenerative diseases and has revealed an involvement of Wallerian-like mechanisms in several cases (Conforti et al., 2014). Key steps in this process are thus potential targets for intervention in patients.
Sterile alpha and TIR motif-containing protein 1 (SARM1) acts downstream of NMNAT2 loss to promote axon degeneration (Osterloh et al., 2012, Gerdts et al., 2013, Gilley et al., 2015, Loreto et al., 2015, Walker et al., 2017). Depletion of SARM1 is, to date, the only other manipulation that can delay Wallerian degeneration and related axon degeneration in mice as robustly as exogenous expression of WLDS or other NMNAT variants (Conforti et al., 2014), but its effectiveness in maintaining the long-term health of axons and synapses in mouse models of axonopathy and/or synaptopathy has not yet been directly compared to WldS. Such a comparison is needed to ascertain the relative usefulness of Sarm1 deletion in determining whether Wallerian-like mechanisms are involved in models of neurodegeneration and should be informative in terms of therapeutic strategies for those disorders.
An absence of NMNAT2 in mice causes widespread axon truncation during embryogenesis and perinatal lethality (Gilley et al., 2013). Early rescue by WldS (dose dependently) or by Sarm1 deletion has shown that outgrowth of NMNAT2-deficient axons stalls due to a Wallerian-like degenerative mechanism (Gilley et al., 2013, Gilley et al., 2015). Reduced NMNAT2 levels have already been linked to tauopathy in mice and to decreased cognitive function in humans (Ljungberg et al., 2012, Ali et al., 2016), but the severity of the phenotype in mice lacking NMNAT2 suggests a complete lack of the protein is unlikely to directly model any neurodegenerative conditions. Nevertheless, these mice represent a well-defined and robust system for comparing the longer-term protective effects of WldS and Sarm1 deletion against a severe Wallerian-like axonopathy. While the survival of NMNAT2-deficient mice homozygous for either WldS or a Sarm1 knockout allele up to 3 months of age with no overt problems initially suggested similarly robust rescue in each case (Gilley et al., 2013, Gilley et al., 2015), we now report striking age-dependent differences between the two lines, which are likely to have important experimental and therapeutic implications.
Results
Locomotor Defects and Muscle Atrophy in Nmnat2gtE/gtE;WldS/S Mice, but Not Nmnat2gtE/gtE;Sarm1−/− Mice
Mice homozygous for the Nmnat2gtE gene trap allele, lacking NMNAT2, that are additionally homozygous for WldS or a Sarm1 knockout allele (Nmnat2gtE/gtE;WldS/S or Nmnat2gtE/gtE;Sarm1−/− mice, respectively) are born at the expected frequencies and are outwardly indistinguishable from NMNAT2-expressing littermates up to 3 months of age (Table S1) (Gilley et al., 2013, Gilley et al., 2015). However, despite continued silencing of the trapped Nmnat2 alleles in each case (Figure S1A), further aging has revealed clear differences between the lines: Nmnat2gtE/gtE;Sarm1−/− mice remarkably survived for up to 2 years with no noticeable behavioral deficiency or phenotype, whereas Nmnat2gtE/gtE;WldS/S mice invariantly developed a conspicuous, progressive hindlimb defect from around 3–5 months of age.
The defect in Nmnat2gtE/gtE;WldS/S mice (male and female) first presented as a modest hindlimb gait abnormality during spontaneous locomotion, but this progressively deteriorated, resulting in mice invariantly dragging their hindlimbs regularly during locomotion from around 6 months onward as a result of worsening paraparesis (Movies S1, S2, S3, and S4). Consistent with this, locomotor ability of Nmnat2gtE/gtE;WldS/S mice in an accelerating Rotarod task deteriorated rapidly between 4 and 6 months (Figure 1A). Movement became so limited by 10–12 months that it impaired free access to food and water, so Nmnat2gtE/gtE;WldS/S mice were not aged further. In contrast, locomotor performance of Nmnat2gtE/gtE;Sarm1−/− mice did not decline during the same period (Figure 1A; Movies S5 and S6), and Nmnat2gtE/gtE;Sarm1−/− mice still performed as well as Sarm1−/− controls up to at least 15 months (Figure 1B).
Figure 1.

Progressive Locomotor Dysfunction and Hindlimb Muscle Atrophy in Nmnat2gtE/gtE;WldS/S Mice, but Not Nmnat2gtE/gtE;Sarm1−/− Mice
(A and B) Latency to fall in an accelerating Rotarod task for mice of the indicated genotypes and ages. Means ± SEM are plotted (maximum test duration, 300 s) for n = 7/8 male mice of each genotype (A) (∗∗∗p < 0.001 in two-way ANOVA with Dunnett’s multiple comparisons) and n = 7 female mice of each genotype (B) (NS, not significant [p = 0.29] in t test).
(C) Representative transverse sections of gastrocnemius muscle for mice of the selected genotypes and ages (as indicated) stained with H&E. Modest fiber atrophy is seen in Nmnat2gtE/gtE;WldS/S gastrocnemius at 10 weeks, and more severe atrophy, with centrally located nuclei, hypertrophic fibers (∗), and pyknotic nuclear clumps (arrow), is evident at 10 months. Images are representative of male and female mice at 10 weeks and 8–12 months but just female mice at 24 months.
(D and E) Gastrocnemius muscle weights for mice of the indicated genotypes and ages. Individual animal values with means ± SEM are plotted for n = 3–5 male mice per group (D) (∗p < 0.05 and ∗∗∗p < 0.001 in one-way ANOVA with Tukey’s multiple comparisons; NS, not significant) and n = 4–5 female mice per group (E) (∗p < 0.05 in t test).
See also Movies S1, S2, S3, S4, S5, and S6, Table S1, and Figure S1.
Deteriorating locomotor function in Nmnat2gtE/gtE;WldS/S mice coincided with progressive and widespread wasting of hindlimb muscles (Figure S1B). A specific analysis of gastrocnemius muscle revealed evidence of some muscle fiber atrophy and slightly reduced muscle weight even at 10 weeks in Nmnat2gtE/gtE;WldS/S mice, before the onset of overt locomotor dysfunction, but this had progressed to severe muscle fiber atrophy and loss of mass by 10 months (Figures 1C and 1D). In contrast, no muscle fiber atrophy or loss of mass was seen in Nmnat2gtE/gtE;Sarm1−/− gastrocnemius up to 24 months (Figures 1C–1E).
Although Nmnat2gtE/gtE;WldS/S mice did not lose body weight between 10 weeks and 10–12 months, they failed to gain weight as expected (Figure S1C). This presumably reflected loss of muscle mass in the hindlimb being broadly matched by normal weight gain in the upper torso and forelimbs, which appeared largely unaffected. In contrast, developmental weight gain in Nmnat2gtE/gtE;Sarm1−/− mice was comparable to Sarm1−/− controls up to 24 months (Figure S1D).
Neuromuscular Denervation in Nmnat2gtE/gtE;WldS/S Mice, but Not Nmnat2gtE/gtE;Sarm1−/− Mice
Changes in neuromuscular junction (NMJ) innervation indicated that the muscle defect in Nmnat2gtE/gtE;WldS/S mice is neurogenic. Motor endplate occupancy in Nmnat2gtE/gtE;WldS/S gastrocnemius was found to be moderately reduced at 10 weeks, but consistent with the timing of gastrocnemius muscle fiber atrophy and weight loss in these mice, by 10 months, denervation was extensive, with only around 10% of endplates showing normal innervation (Figures 2A and 2B).
Figure 2.

Progressive Loss of NMJ Innervation in Gastrocnemius Muscles of Nmnat2gtE/gtE;WldS/S Mice, but Not Nmnat2gtE/gtE;Sarm1−/− Mice
(A) Percentage of fully occupied (full), partially occupied (partial), and denervated endplates in gastrocnemius muscles from mice of the indicated genotypes and ages. Endplate occupancy was determined by assessing signal overlap between α-bungarotoxin labeling of acetylcholine receptors in the motor endplate and βIII-tubulin immunostaining of axon terminals (see Experimental Procedures). Total numbers of NMJs analyzed in muscles from 3–5 mice per genotype (male and female) are listed at the base of each column. Means ± SEM of occupancy per animal are plotted (NS, not significant [p > 0.05] or ∗∗∗p < 0.001 in one-way ANOVA with Tukey’s multiple comparisons of full innervation, selected comparisons).
(B) Representative confocal z series projections of fixed gastrocnemius (Gn) muscle preparations showing merged α-bungarotoxin (red) and βIII-tubulin (green) signals. Endplates in the images are marked as fully occupied (asterisks), partially occupied (arrows), or denervated (arrowheads).
We also investigated motor endplate occupancy in a more distal hindlimb muscle, flexor digitorum brevis (FDB), at 10 months. Although denervation was also evident in this muscle (Figure 3A), it was less severe than in gastrocnemius at the same age. Clear regional variation in the pattern of endplate occupancy was seen in FDB, with discrete zones of normal innervation being found adjacent to zones of complete denervation (Figures 3B and 3C). Isometric tension recordings indicated that this distinctive pattern of NMJ innervation reflects discrete loss of entire motor units (Figures 3D and 3E), with apparently normal function of remaining motor units (Figure S2).
Figure 3.

Localized Loss of NMJ Innervation from Distinct Motor Unit Groups in FDB Muscle of Nmnat2gtE/gtE;WldS/S Mice
(A) Percentage of fully occupied (full), partially occupied (partial), and denervated endplates in FDB muscles of 10-month-old mice of the indicated genotypes (method as in Figure 2A). Total numbers of NMJs analyzed in muscles from 5–9 mice per genotype (male and female) are listed at the base of each column. Means ± SEM of occupancy per animal are plotted (NS, not significant [p > 0.05] or ∗∗p < 0.01 in one-way ANOVA with Tukey’s multiple comparisons of full innervation, selected comparisons).
(B) Percentage of full occupancy of endplates per imaged field revealed a zonal pattern of denervation in Nmnat2gtE/gtE;WldS/S FDB at 10 months, with more widespread denervation in gastrocnemius (Gn). Fields with normal levels of full occupancy (80%–100%) were relatively common in FDB (7/18) but absent in gastrocnemius (average of 16.9 and 11.6 endplates per field, respectively).
(C) Representative confocal z series projections of fixed FDB muscle preparations showing merged α-bungarotoxin (red) and βIII-tubulin (green) signals, highlighting regional variability in Nmnat2gtE/gtE;WldS/S endplate occupancy at 10 months. In addition to regions with widespread full occupancy (top panel) or denervation (middle panel), there were areas containing degenerated endplates (bottom panel, not included in quantifications).
(D) Isometric twitch tension recordings for representative Nmnat2gtE/gtE;WldS/S and Nmnat2gtE/gtE;Sarm1−/− FDB muscle-tibial nerve preparations showing progressive, discrete recruitment of motor units (incremental steps in twitch tension) during continuously graded stimulation of the nerve.
(E) Motor unit numbers in FDB muscles from 10-month-old male mice of the indicated genotypes (n = 4 or 6 muscles from 2 or 3 mice as shown). Individual values and means ± SEM are plotted (NS, not significant [p > 0.99] or ∗∗∗p < 0.001 in one-way ANOVA with Tukey’s multiple comparisons, selected comparisons).
See also Figure S2.
In contrast, and consistent with the lack of locomotor problems, no significant endplate denervation and/or motor unit loss was evident in either gastrocnemius or FDB muscles from 10-month-old Nmnat2gtE/gtE;Sarm1−/− mice (Figures 2, 3A, 3D, 3E, and S2) and innervation remained comparable to Sarm1−/− controls at 24 months, despite modest age-dependent denervation in both (Figure 2A).
WldS/S and Sarm1−/− mice both performed maximally in locomotor tests, and neither showed signs of neuromuscular denervation at the ages studied. Although a direct comparison with wild-type mice will be needed to establish whether more subtle differences in motor function exist in either line, our data suggest that both have broadly normal neuromuscular function. The defect in Nmnat2gtE/gtE;WldS/S mice thus appears to be specific to a declining inability of WLDS to counter the lack of NMNAT2 in older mice, rather than other intrinsic differences.
No Loss of Myelinated Tibial Nerve Axons in Either Nmnat2gtE/gtE;WldS/S or Nmnat2gtE/gtE;Sarm1−/− Mice
Despite progressive denervation of motor endplates in hindlimb muscles from Nmnat2gtE/gtE;WldS/S mice, no concurrent loss of myelinated axons was seen in the tibial nerve (Figure 4A) and axons remained morphologically normal (Figure 4B). A gross assessment revealed that most hindlimb muscles became extensively atrophied in these mice (Figure S1B), so significantly reduced numbers of axons would have been expected, even in a mixed nerve such as this, if motor axon loss was the underlying cause. The age-dependent neuromuscular denervation in Nmnat2gtE/gtE;WldS/S muscles thus appears to result from selective loss of the distal ends of motor axons and/or their terminals. This mirrors the age-dependent loss of protection of synapses at NMJs after axotomy in homozygous WldS mice, despite continued protection of the main body of the transected axon (Gillingwater et al., 2002).
Figure 4.

No Loss of Myelinated Tibial Nerve Axons in Either Nmnat2gtE/gtE;WldS/S or Nmnat2gtE/gtE;Sarm1−/− Mice
(A) Numbers of myelinated axons in tibial nerve (mid-calf level) from mice of the indicated genotypes and ages (WldS/S control groups include some Nmnat2+/gtE;WldS/S mice that were indistinguishable from WldS/S mice). Individual values (n = 3–8, as shown, male and female) and means ± SEM are plotted. No statistically significant differences were identified between groups (one-way ANOVA with Tukey’s multiple comparisons).
(B) FluoroMyelin red-stained tibial nerve cross-sections from 10-month-old WldS/S and Nmnat2gtE/gtE;WldS/S mice (representative of n = 5 each genotype). No structural differences are evident, even though Nmnat2gtE/gtE;WldS/S mice have an advanced neuromuscular defect at this age.
Counts of myelinated axons in Nmnat2gtE/gtE;Sarm1−/− tibial nerves remained comparable to those of Sarm1−/− controls, even up to 24 months (Figure 4A), with no age-related axon loss in either group up to this age. We also found no significant axon loss in a separate cohort of wild-type mice (on a related background) up to 24 months (1,514 ± 22 myelinated axons at 1.5 months, compared to 1,473 ± 40 at 24 months). This contrasts a previous study that reported significant loss of myelinated tibial nerve axons by 24 months in wild-type mice (Valdez et al., 2010), although this could simply reflect strain differences.
Discussion
To date, WldS has been the preferred tool for assessing the involvement of Wallerian-like axon and synapse degeneration in rodent models of neurodegeneration (Conforti et al., 2014). However, the relatively short-term preservation of neuromuscular innervation by WldS in hindlimb muscles of Nmnat2gtE/gtE mice raises the possibility that this strategy might have greatly underestimated the involvement of Wallerian-like mechanisms in some models. Likely candidates are wabbler-lethal (Atp8a2wl/wl) and gracile axonal dystrophy (Uchl1gad/gad) mice, in which WldS robustly protects (proximal) axons but does not rescue neuromuscular dysfunction (Mi et al., 2005, Zhu et al., 2012). Human SOD1G37R, SOD1G85R, and SOD1G93A transgenic mouse models of amyotrophic lateral sclerosis (ALS) are also candidates, although WldS largely fails to protect axons in these models, suggesting that unrelated degenerative mechanisms contribute substantially to disease signs (Vande Velde et al., 2004, Fischer et al., 2005). Because SARM1 deficiency confers longer-lasting preservation of NMJ innervation in NMNAT2-deficient mice than WldS, it could confer a better outcome in these or related models.
We consider that prolonged preservation of Nmnat2gtE/gtE;Sarm1−/− motor axon terminals, compared to those in Nmnat2gtE/gtE;WldS/S mice, might reflect that local availability of WLDS, which is required for protection (Beirowski et al., 2009, Cohen et al., 2012), is likely to be subject to a variety of influences, whereas protection conferred by an absence of SARM1 will be invariant. Global expression of WLDS in homozygous WldS mice does not diminish significantly with age up to 12 months (Gillingwater et al., 2002), but we propose that normal changes in physiology, from as young as 2 months of age, could alter the stability, delivery, or activity of WLDS in Nmnat2gtE/gtE;WldS/S motor axon terminals, or otherwise alter the local environment, such that it can no longer effectively substitute for the lack of NMNAT2 to promote survival. More widespread NMJ denervation in gastrocnemius compared to FDB in Nmnat2gtE/gtE;WldS/S mice suggests that changes specific to different muscle or motor unit types are more critical to the loss of WLDS-mediated protection than those relating to axon length (FDB being more distal). These considerations will similarly apply to the age-dependent loss of protection of motor axon terminals after axotomy (and the resulting NMNAT2 loss) in homozygous WldS mice (Gillingwater et al., 2002). Annulospiral (sensory) nerve endings in the muscle remained protected in older mice in that context, suggesting a motor-specific defect (Oyebode et al., 2012). Although we have seen qualitative preservation of annulospiral endings in Nmnat2gtE/gtE;WldS/S FDB at 10 months (not shown), a comprehensive analysis of sensory innervation will be required to determine whether sensory endings in general are better preserved than motor axon terminals.
A model in which sustaining the effective potency of WLDS locally is required for its protective effects leaves open the possibility that Sarm1 deletion may be more effective than WldS at rescuing symptoms in models of other types of neurodegenerative disease, not just those with early neuromuscular symptoms. If the disease-causing defect in a given model additionally reduces the activity or concentration of WLDS within axons or synapses in some way, then its protective capacity might be diminished. This could apply to disorders of axonal transport, protein synthesis, or protein turnover, among others.
Our findings have therapeutic implications for human disorders. Specifically, they suggest that strategies directed at blockade of SARM1 function have the potential to be more effective than WLDS-related therapies in neuromuscular synaptopathies and potentially in a broader group of neurodegenerative disorders. In addition, the remarkable survival and health of Nmnat2gtE/gtE;Sarm1−/− mice into old age suggests that even long-term therapeutic interventions based on blocking SARM1 function might be both effective and well tolerated by patients.
This study confirms SARM1 as a key regulator of degeneration caused by a NMNAT2 deficiency. Sarm1 deletion appears to block this process indefinitely without affecting long-term survival, despite the predicted substantial reduction in nicotinamide adenine dinucleotide (NAD)-synthesizing capacity (Gilley et al., 2015). SARM1 has been shown to possess NADase activity that promotes injury-induced axon or synapse degeneration and can be inhibited by NMNAT activity (Gerdts et al., 2015, Sasaki et al., 2016, Essuman et al., 2017). Therefore, a model in which survival depends on NMNAT-dependent NAD production balancing NAD consumption, including any resulting from constitutive SARM1 NADase activity, is attractively simple. However, NAD consumption in uninjured Sarm1−/− axons has been shown to be comparable to wild-type consumption, suggesting that SARM1 NADase activity under normal conditions is minimal (Sasaki et al., 2016). Instead, a situation in which a loss of NMNAT activity results in the upregulation of SARM1 NADase activity via intermediate signals and/or interactions to trigger degeneration is more consistent with current findings (Sasaki et al., 2016, Di Stefano et al., 2017).
Finally, the similarity between the progressive phenotype in Nmnat2gtE/gtE;WldS/S mice and some mouse models of ALS is intriguing. While there is, as yet, no established link between NMNAT2 and ALS, the SARM1 locus has been associated with sporadic ALS (Fogh et al., 2014), hinting at involvement of Wallerian-like mechanisms. The neuromuscular defect in Nmnat2gtE/gtE;WldS/S mice could thus model some aspects of ALS disease pathogenesis. Even if the underlying mechanisms are unrelated, Nmnat2gtE/gtE;WldS/S mice would be a useful tool for assessing reversibility of ALS-like symptoms, because silencing of the Nmnat2gtE gene trap allele is reversible (Gilley et al., 2013).
Experimental Procedures
Mouse Breeding and Maintenance
Animal work was performed in accordance with the 1986 Animals (Scientific Procedures) Act under Project License PPL 70/7620 following an appropriate ethical review process at the Babraham Institute. Genotyping for the Nmnat2gtE, WldS, and Sarm1 knockout alleles was performed as described previously (Gilley et al., 2013, Gilley et al., 2015). Littermates were used where possible. The ages and genders of mice used in individual experiments are described in the figure legends.
RT-PCR
Semiquantitative endpoint RT-PCR was used to confirm Nmnat2 gene silencing in the brains of Nmnat2gtE/gtE;WldS/S and Nmnat2gtE/gtE;Sarm1−/− mice aged 10–12 months essentially as described previously (Gilley et al., 2013).
Accelerating Rotarod Task
Locomotor performance was tested on an accelerating Rotarod (Ugo Basile, Model 7650, Varese, Italy). Mice were familiarized with the apparatus (two 5 min runs at 10 rpm) one day before testing. At each test age, mice performed three 5 min trials (3 to 30 rpm) separated by 30 min rests. Latency to fall (max 300 s) was recorded. Only involuntary falls were scored. Mice dismounting voluntarily were placed back onto the apparatus once, but the run was excluded from the analysis if repeated. Best trial performance was used for statistical analyses.
H&E Staining
Transverse cryosections of gastrocnemius muscles snap frozen in liquid nitrogen-chilled isopentane (8 μm thickness) or fixed in 4% paraformaldehyde (20 μm thickness) were stained with H&E as previously described (Gilley et al., 2013). Images were captured using a MicroPublisher camera (QImaging) on an Olympus BX50 microscope (20× objective). Staining of snap-frozen muscle sections was optimal for visualization of muscle structure without the artifactual muscle fiber separation seen on fixed sections.
NMJ Innervation
Innervation of NMJs in gastrocnemius and FDB muscles was assessed by immunofluorescent staining. Staining was performed essentially as described previously (Krieger et al., 2013) on whole-mount muscles or longitudinal cryosections (60 μm thickness). Confocal z stack series were acquired using Olympus FV1000 or Leica SPE scanning laser confocal microscopes (20× or 40× objectives). Multiple z stack series were acquired for each muscle, and z projections were generated for analysis. Endplate occupancy was determined by assessing the extent of overlap or direct abuttal of βIII-tubulin staining (axon terminal) with α-bungarotoxin staining (endplate). Endplates were scored as denervated when essentially none of the endplate (less than ∼5%) was deemed to be occupied by the axon terminal, fully innervated with complete (greater than ∼95%) occupancy, and partially innervated with intermediate occupancy (observer determined, scored blind). Original z stack series were examined to exclude chance overlay of proximal axon segments and endplates in non-adjacent focal planes.
Isometric Muscle Tension Recordings
Force measurements for FDB muscle were made from FDB muscle-tibial nerve preparations as described previously (Beirowski et al., 2009), except that the proximal tendon was connected to a MLT0202 (0–25 g) isometric force transducer (AD Instruments, Oxford, UK) and the tibial nerve was stimulated using 0.1–0.2 ms pulses of up to 10 V using a Digitimer DS2 isolated stimulator (Digitimer, Welwyn Garden City, UK) triggered via a Powerlab 26T interface. Tension responses were digitized at 1 kHz using Chart 7 or Scope 4 software (all ADInstruments).
Counts of Myelinated Tibial Nerve Axons
Transverse sections (20 μm) of fixed calf (from mid-way between knee and ankle) were stained with FluoroMyelin red according to the manufacturer’s instructions (Life Technologies). Images of tibial nerves were captured on an Olympus FV1000 point scanning confocal microscope imaging system (40× objective). Axon counts (inferred from numbers of myelin sheaths) were performed blind using the multi-point selection tool in ImageJ.
Statistical Analysis
Appropriate statistical testing of data was performed using Prism (GraphPad Software, La Jolla, USA). Tests are described in the figure legends. A p value < 0.05 was considered significant.
Author Contributions
Conceptualization, J.G. and M.P.C.; Methodology, J.G. and R.R.R.; Investigation, J.G. and R.R.R.; Writing – Original Draft, J.G.; Writing – Review & Editing, J.G., R.R.R., and M.P.C.; Visualization, J.G.; Funding Acquisition, M.P.C. and R.R.R.; Supervision, M.P.C.
Acknowledgments
We thank Robert Chou for technical help optimizing NMJ staining, Andrea Loreto for comments on the manuscript, Babraham Institute animal facility staff for providing movies of mice, and Dr. Anne Segonds-Pichon for help with statistical analyses. This work was funded by an Institute Strategic Programme grant from the Biotechnology and Biological Sciences Research Council, Medical Research Council grants MR/N004582/1 and MR/M024075/1, and Motor Neurone Disease Association (MNDA) grant 838-791.
Published: October 3, 2017
Footnotes
Supplemental Information includes two figures, one table, and six movies and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.09.027.
Data and Software Availability
Source data for graphs can be found in the the University of Cambridge Repository (Apollo) at https://doi.org/10.17863/CAM.13389.
Supplemental Information
Movie highlighting the impaired hindlimb function in Nmnat2gtE/gtE;WldS/S mice but not Nmnat2gtE/gtE;Sarm1−/− mice or controls.
{kind=link}
Movie highlighting the impaired hindlimb function in Nmnat2gtE/gtE;WldS/S mice but not Nmnat2gtE/gtE;Sarm1−/− mice or controls.
{kind=link}
Movie highlighting the impaired hindlimb function in Nmnat2gtE/gtE;WldS/S mice but not Nmnat2gtE/gtE;Sarm1−/− mice or controls.
{kind=link}
Movie highlighting the impaired hindlimb function in Nmnat2gtE/gtE;WldS/S mice but not Nmnat2gtE/gtE;Sarm1−/− mice or controls.
{kind=link}
Movie highlighting the impaired hindlimb function in Nmnat2gtE/gtE;WldS/S mice but not Nmnat2gtE/gtE;Sarm1−/− mice or controls.
{kind=link}
Movie highlighting the impaired hindlimb function in Nmnat2gtE/gtE;WldS/S mice but not Nmnat2gtE/gtE;Sarm1−/− mice or controls.
{kind=link}
References
- Ali Y.O., Allen H.M., Yu L., Li-Kroeger D., Bakhshizadehmahmoudi D., Hatcher A., McCabe C., Xu J., Bjorklund N., Taglialatela G. NMNAT2:HSP90 complex mediates proteostasis in proteinopathies. PLoS Biol. 2016;14:e1002472. doi: 10.1371/journal.pbio.1002472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B., Babetto E., Gilley J., Mazzola F., Conforti L., Janeckova L., Magni G., Ribchester R.R., Coleman M.P. Non-nuclear Wld(S) determines its neuroprotective efficacy for axons and synapses in vivo. J. Neurosci. 2009;29:653–668. doi: 10.1523/JNEUROSCI.3814-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M.S., Ghosh A.K., Kim H.J., Jeon N.L., Jaffrey S.R. Chemical genetic-mediated spatial regulation of protein expression in neurons reveals an axonal function for wld(s) Chem. Biol. 2012;19:179–187. doi: 10.1016/j.chembiol.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L., Gilley J., Coleman M.P. Wallerian degeneration: an emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 2014;15:394–409. doi: 10.1038/nrn3680. [DOI] [PubMed] [Google Scholar]
- Di Stefano M., Loreto A., Orsomando G., Mori V., Zamporlini F., Hulse R.P., Webster J., Donaldson L.F., Gering M., Raffaelli N. NMN deamidase delays Wallerian degeneration and rescues axonal defects caused by NMNAT2 deficiency in vivo. Curr. Biol. 2017;27:784–794. doi: 10.1016/j.cub.2017.01.070. [DOI] [PubMed] [Google Scholar]
- Essuman K., Summers D.W., Sasaki Y., Mao X., DiAntonio A., Milbrandt J. The SARM1 Toll/interleukin-1 receptor domain possesses intrinsic NAD+ cleavage activity that promotes pathological axonal degeneration. Neuron. 2017;93:1334–1343. doi: 10.1016/j.neuron.2017.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer L.R., Culver D.G., Davis A.A., Tennant P., Wang M., Coleman M., Asress S., Adalbert R., Alexander G.M., Glass J.D. The WldS gene modestly prolongs survival in the SOD1G93A fALS mouse. Neurobiol. Dis. 2005;19:293–300. doi: 10.1016/j.nbd.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Fogh I., Ratti A., Gellera C., Lin K., Tiloca C., Moskvina V., Corrado L., Sorarù G., Cereda C., Corti S., SLAGEN Consortium and Collaborators A genome-wide association meta-analysis identifies a novel locus at 17q11.2 associated with sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014;23:2220–2231. doi: 10.1093/hmg/ddt587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J., Summers D.W., Sasaki Y., DiAntonio A., Milbrandt J. Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J. Neurosci. 2013;33:13569–13580. doi: 10.1523/JNEUROSCI.1197-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J., Brace E.J., Sasaki Y., DiAntonio A., Milbrandt J. SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science. 2015;348:453–457. doi: 10.1126/science.1258366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J., Coleman M.P. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J., Adalbert R., Yu G., Coleman M.P. Rescue of peripheral and CNS axon defects in mice lacking NMNAT2. J. Neurosci. 2013;33:13410–13424. doi: 10.1523/JNEUROSCI.1534-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J., Orsomando G., Nascimento-Ferreira I., Coleman M.P. Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep. 2015;10:1974–1981. doi: 10.1016/j.celrep.2015.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingwater T.H., Thomson D., Mack T.G., Soffin E.M., Mattison R.J., Coleman M.P., Ribchester R.R. Age-dependent synapse withdrawal at axotomised neuromuscular junctions in Wld(s) mutant and Ube4b/Nmnat transgenic mice. J. Physiol. 2002;543:739–755. doi: 10.1113/jphysiol.2002.022343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger F., Elflein N., Ruiz R., Guerra J., Serrano A.L., Asan E., Tabares L., Jablonka S. Fast motor axon loss in SMARD1 does not correspond to morphological and functional alterations of the NMJ. Neurobiol. Dis. 2013;54:169–182. doi: 10.1016/j.nbd.2012.12.010. [DOI] [PubMed] [Google Scholar]
- Ljungberg M.C., Ali Y.O., Zhu J., Wu C.S., Oka K., Zhai R.G., Lu H.C. CREB-activity and nmnat2 transcription are down-regulated prior to neurodegeneration, while NMNAT2 over-expression is neuroprotective, in a mouse model of human tauopathy. Hum. Mol. Genet. 2012;21:251–267. doi: 10.1093/hmg/ddr492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loreto A., Di Stefano M., Gering M., Conforti L. Wallerian degeneration is executed by an NMN-SARM1-dependent late Ca(2+) influx but only modestly influenced by mitochondria. Cell Rep. 2015;13:2539–2552. doi: 10.1016/j.celrep.2015.11.032. [DOI] [PubMed] [Google Scholar]
- Mack T.G., Reiner M., Beirowski B., Mi W., Emanuelli M., Wagner D., Thomson D., Gillingwater T., Court F., Conforti L. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- Mi W., Beirowski B., Gillingwater T.H., Adalbert R., Wagner D., Grumme D., Osaka H., Conforti L., Arnhold S., Addicks K. The slow Wallerian degeneration gene, WldS, inhibits axonal spheroid pathology in gracile axonal dystrophy mice. Brain. 2005;128:405–416. doi: 10.1093/brain/awh368. [DOI] [PubMed] [Google Scholar]
- Osterloh J.M., Yang J., Rooney T.M., Fox A.N., Adalbert R., Powell E.H., Sheehan A.E., Avery M.A., Hackett R., Logan M.A. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyebode O.R., Hartley R., Singhota J., Thomson D., Ribchester R.R. Differential protection of neuromuscular sensory and motor axons and their endings in Wld(S) mutant mice. Neuroscience. 2012;200:142–158. doi: 10.1016/j.neuroscience.2011.10.020. [DOI] [PubMed] [Google Scholar]
- Sasaki Y., Nakagawa T., Mao X., DiAntonio A., Milbrandt J. NMNAT1 inhibits axon degeneration via blockade of SARM1-mediated NAD(+) depletion. eLife. 2016;5:e19749. doi: 10.7554/eLife.19749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez G., Tapia J.C., Kang H., Clemenson G.D., Jr., Gage F.H., Lichtman J.W., Sanes J.R. Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc. Natl. Acad. Sci. USA. 2010;107:14863–14868. doi: 10.1073/pnas.1002220107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Velde C., Garcia M.L., Yin X., Trapp B.D., Cleveland D.W. The neuroprotective factor Wlds does not attenuate mutant SOD1-mediated motor neuron disease. Neuromolecular Med. 2004;5:193–203. doi: 10.1385/NMM:5:3:193. [DOI] [PubMed] [Google Scholar]
- Walker L.J., Summers D.W., Sasaki Y., Brace E.J., Milbrandt J., DiAntonio A. MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2. eLife. 2017;6:e22540. doi: 10.7554/eLife.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X., Libby R.T., de Vries W.N., Smith R.S., Wright D.L., Bronson R.T., Seburn K.L., John S.W. Mutations in a P-type ATPase gene cause axonal degeneration. PLoS Genet. 2012;8:e1002853. doi: 10.1371/journal.pgen.1002853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie highlighting the impaired hindlimb function in Nmnat2gtE/gtE;WldS/S mice but not Nmnat2gtE/gtE;Sarm1−/− mice or controls.
Movie highlighting the impaired hindlimb function in Nmnat2gtE/gtE;WldS/S mice but not Nmnat2gtE/gtE;Sarm1−/− mice or controls.
Movie highlighting the impaired hindlimb function in Nmnat2gtE/gtE;WldS/S mice but not Nmnat2gtE/gtE;Sarm1−/− mice or controls.
Movie highlighting the impaired hindlimb function in Nmnat2gtE/gtE;WldS/S mice but not Nmnat2gtE/gtE;Sarm1−/− mice or controls.
Movie highlighting the impaired hindlimb function in Nmnat2gtE/gtE;WldS/S mice but not Nmnat2gtE/gtE;Sarm1−/− mice or controls.
Movie highlighting the impaired hindlimb function in Nmnat2gtE/gtE;WldS/S mice but not Nmnat2gtE/gtE;Sarm1−/− mice or controls.