

Abstract
The genomic sequences of 20 Leishmania infantum isolates collected in northeastern Brazil were compared with each other and with the available genomic sequences of 29 L. infantum/donovani isolates from Nepal and Turkey. The Brazilian isolates were obtained in the early 1990s or since 2009 from patients with visceral or non-ulcerating cutaneous leishmaniasis, asymptomatic humans, or dogs with visceral leishmaniasis. Two isolates were from the blood and bone marrow of the same visceral leishmaniasis patient. All 20 genomic sequences display 99.95% identity with each other and slightly less identity with a reference L. infantum genome from a Spanish isolate. Despite the high identity, analysis of individual differences among the 32 million base pair genomes showed sufficient variation to allow the isolates to be clustered based on the primary sequence. A major source of variation detected was in chromosome somy, with only four of the 36 chromosomes being predominantly disomic in all 49 isolates examined. In contrast, chromosome 31 was predominantly tetrasomic/pentasomic, consistent with its regions of synteny on two different disomic chromosomes of Trypanosoma brucei. In the Brazilian isolates, evidence for recombination was detected in 27 of the 36 chromosomes. Clustering analyses suggested two populations, in which two of the five older isolates from the 1990s clustered with a majority of recent isolates. Overall the analyses do not suggest individual sequence variants account for differences in clinical outcome or adaptation to different hosts. For the first known time, DNA of isolates from asymptomatic subjects were sequenced. Of interest, these displayed lower diversity than isolates from symptomatic subjects, an observation that deserves further investigation with additional isolates from asymptomatic subjects.
Keywords: Leishmania infantum, Intracellular parasite, Evolution, Parasite population structure, Copy number variation, Brazil
1. Introduction
Leishmania infantum is the etiological agent of most cases of visceral leishmaniasis (VL) in the American continents, Europe, and northern Africa (Deane and Deane, 1962; Gradoni et al., 1995). This pathogen was likely introduced into the Americas during European settlement (Maurício et al., 2000; Zemanová et al., 2007; Leblois et al., 2011). Prior to the early 1990s, VL was mostly endemic in rural regions of northeastern Brazil (Deane and Deane, 1962; Badaro et al., 1986a, b; Evans et al., 1992). However, due to increased migratory movements from rural to urban areas, outbreaks of VL began to appear in major metropolitan cities in Brazil (Costa et al., 1990; Jeronimo et al., 1994; Marzochi and Marzochi, 1994; Silva et al., 2001; Albuquerque et al., 2009). The disease is now endemic in all regions of Brazil and in northern Argentina (Salomón, 2009). Despite the expanding geographic endemic areas, the overall incidence in Brazil has not increased (Almeida and Werneck, 2014).
VL in Latin America and Europe is considered a zoonosis, with domestic dogs presumed to be the most important reservoir (for review see Roque and Jansen (2014)). This differs from the situation in the Indian subcontinent, where VL caused by the closely related species, Leishmania donovani, is an anthroponosis and humans are thought to be the most important reservoir (Bern et al., 2005). Interestingly, human VL epidemics appear to occur in a cyclical pattern (Franke et al., 2002; Bhunia et al., 2013). However, a possible role of humans as a reservoir for L. infantum infection in population-dense urban areas has not been studied, and would be difficult to detect in the presence of an animal reservoir (Costa et al., 2000; Maia-Elkhoury et al., 2008).
In recent years, genome-wide studies of different Leishmania spp. and/or isolates from Europe, Turkey, India, and Nepal have been used to assess genomic differences associated with geographically disperse populations, sodium stibogluconate resistance (Downing et al., 2011; Rogers et al., 2011), sexual reproduction and recombination patterns (Rogers et al., 2014). It has been postulated that the ability of L. donovani isolates to cause VL or cutaneous leishmaniasis (CL) may be determined by sequence polymorphisms and/or amplification of specific genes (Zhang et al., 2014). Furthermore, genetic diversity within a single gene locus from Indian L. donovani isolates was recently attributed to population expansion after a recent bottleneck, possibly the ‘Roll- Back Malaria’ campaign, which may account for drug resistance amongst isolates from Indian subjects with VL (Imamura et al., 2016). The results from this study were used as a basis for the development of a L. donovani genotyping methodology in Nepal and permitted an association to be made between the Leishmania genotype and the treatment applied to the patients (Rai et al., 2017). It might also be useful in identifying new drugs against leishmaniasis (Hefnawy et al., 2016). Although these studies have successfully examined intra-species genomic diversity and genes associated with visceral versus non-visceral forms of disease, an analysis of Leishmania genomes associated with different host phenotypes within a species is required to reveal possible sequence signatures or genomic structural markers associated with diverse clinical manifestations or potential host specificity.
Based on the hypothesis that distinct clinical outcomes caused by the same Leishmania sp. would be associated with genetically distinguishable isolates, we determined whole genome sequences of 20 different L. infantum isolates collected from infected symptomatic or asymptomatic humans, or from dogs, in the state of Rio Grande do Norte, northeastern Brazil. We analyzed the genetic variation among these isolates, taking into consideration the years when samples were isolated and their geographic locations. These analyses were compared with features of publicly available L. infantum/donovani genomes of isolates from countries on four different continents or sub-continents (Brazil, Nepal, Turkey, Spain) (Peacock et al., 2008; Downing et al., 2011; Rogers et al., 2014).
2. Materials and methods
2.1. Ethical considerations
The protocol used for work with human samples in this study was reviewed and approved by the Universidade Federal do Rio Grande do Norte Ethical Committee on Human Research (CAAE 12584513.1.0000.5537), Brazil. Each participant or their legal guardian provided written informed consent. The consent form was approved by the Ethics Committee. The protocol used for dogs with VL was reviewed and approved by the ethical committee on animal research at Federal University of Rio Grande do Norte (CEUA Protocol number 062/2014). CEUA-UFRN is registered with the Conselho Nacional de Controle de Experimentação Animal (CONCEA) in Brazil and follows international guidelines on the use of animals in research. Leishmania isolates obtained either from blood or bone marrow were kept frozen in the Immunogenetics Laboratory, Bioscience Center, Universidade Federal do Rio Grande do Norte.
2.1.1. Leishmania sample collection
Leishmania infantum strains were isolated from bone marrow or blood of individuals with symptomatic VL (n = 10), from blood of individuals with asymptomatic Leishmania infection (n = 4), from a skin lesion of a human subject (n = 1) or from spleens of dogs with VL (n = 5). All samples came from the state of Rio Grande do Norte, Brazil (Supplementary Fig. S1). This region has been endemic for VL since the early 1990s (Jeronimo et al., 1994). Five of the human isolates were obtained between 1991 and 1993; the remaining 15 were obtained between 2009 and 2013. Parasites from asymptomatic Leishmania subjects were isolated in culture from blood donated to the blood bank in Natal, Rio Grande do Norte. Subjects were healthy, with normal red blood cell counts. The asymptomatic cases were followed at the Giselda Trigueiro Hospital, Rio Grande do Norte outpatient clinic and they became negative for the presence of anti-Leishmania antibodies by PCR within 3–6 months after they were first recruited for study, but with a positive Montenegro Test positive, indicating a positive cellular response Miles et al. (2005). None of them developed VL. All isolates were cryopreserved until used for this study. All specimens were typed by isoenzymes in a reference World Health Organization laboratory (Fiocruz, Rio de Janeiro, RJ, Brazil) and by sequencing PCR amplification products derived from the ribosomal internal transcribed spacer and from heat shock protein (HSP) 70 (Kuhls et al., 2011; Schönian et al., 2011).
2.2. Parasite cloning, DNA extraction and sequencing
Cryopreserved isolates were thawed and expanded in HO-MEM media (Pearson and Steigbigel, 1980), diluted to 0.1 parasites/ml and seeded on semisolid agar plates. Single colonies were expanded in HO-MEM media supplemented with 10% FCS. Genomic DNA was extracted from 2 × 106 cloned parasites/ml in logarithmic growth phase. Briefly, parasites were washed in PBS, digested with proteinase K and incubated in RNAse A (65 °C for 10 min), and proteins were precipitated in ammonium acetate (7.5 M) and removed by centrifugation (15,700g) at 4°C. DNA was column-purified from the supernatant following the manufacturer’s protocol (Qiagen DNeasy blood and tissue kit, Qiagen, USA). DNA quality was checked by agarose gel electrophoresis. Genomic DNA from the 20 isolates was sequenced on an Illumina HiSEQ 2000 platform at the Iowa Institute of Human Genetics, Genomics Division, at the University of Iowa, USA. Approximately 60 million paired-end reads per genomic DNA isolate were generated in two separate runs for each sample, with an average read length of 101 bp.
2.3. Read mapping to reference genome
Reads were aligned to the reference genome L. infantum JPCM5 (Peacock et al., 2008; available at ftp://ftp.sanger.ac.uk/pub/project/pathogens/gff3/CURRENT) using BWA v. 0.7.5a-r405, with default parameters. Picard tools (version 1.109; http://picard.sourceforge.net) were used to convert files, sort and mark duplicates. The marking of PCR duplicates is an important step during the genome analysis toolkit (GATK) pipeline for single nucleotide polymorphism (SNP) discovery from whole genome sequencing (WGS) data, readily allowing the identification of duplicates, decreasing the risk of over-representation of areas amplified during PCRs, which would lead to introduction of bias during the variant calling.
The mapping files were then indexed using SAMtools (version 1.109) (Li et al., 2009). Sequence reads of isolates from Turkey and Nepal were downloaded from the TriTRypDB (Aslett et al., 2009; Downing et al., 2011; Rogers et al., 2014), and applied to the same workflow as sequences from Brazilian isolates. Sequences generated by this study are available from National Center for Biotechnology Information, USA, sequence read archive (NCBI SRA) (accession numbers SRR5117893, SRR5117894, SRR5117895, SRR5117896, SRR5117897, SRR5117898, SRR5117899, SRR5117900, SRR5117901, SRR5117902, SRR5117903, SRR5117904, SRR5117905, SRR5117906, SRR5117907, SRR5117908, SRR5117909, SRR5117910, SRR5117911, SRR5117912).
2.4. Variant prediction calling, SNP filtering and principal component analysis (PCA)
The GATK v. 3.3 suite of tools (DePristo et al., 2011) was used to realign reads in regions with insertion/deletions (indels) and to perform the variant calling through HaplotypeCaller under diploid organism assumption. Since there is no training dataset to use as a parameter for SNP filtering, we used GATK hard filters to exclude false positives. For this purpose, the filters were applied as described by GATK Best Practices and the RMSMappingQuality option ≥30. After the SNP filtering step, the variant data were gathered in a single file using VCFtools package (Danecek et al., 2011). SNPRelate was used to remove SNPs in linkage disequilibrium, with a sliding window of 5000 nucleotides and a threshold of 2. This dataset was used for a PCA using the R package SNPRelate (Zheng et al., 2012). This dataset was also used to obtain supportive data for population structure using the program Admixture (Alexander et al., 2009). The snpEFF package (Cingolani et al., 2012) was used for SNP variant annotation, and genome annotation files were retrieved from GeneDB (Logan-Klumpler et al., 2012).
2.5. Genome alignment
After SNP filtering, consensus sequences from each variant file were extracted using house-made scripts written in Shell. Whole genomes were then aligned by the MAUVE algorithm (Darling et al., 2004) plugin on Geneious (v. 8.1.8) with default parameters. Genome alignments were manually edited to exclude low quality regions, many of which are N-rich regions in the reference genome. Manual editing of the alignment was required to ensure the proper alignment of genomic regions with repetitive sequences. Furthermore, the reference genome of L. infantum contains many positions at which the nucleotide is undetermined (i.e., “N”); this complicates the use of automated tools, necessitating manual curation of the alignment.
2.6. Nucleotide diversity and population analysis
To calculate nucleotide diversity, all samples were grouped according to clinical characteristics and date of isolation. The isolates from the 1990s were named “VLh90”, the symptomatic isolates from humans obtained in recent years were grouped as “VLh”, the cutaneous sample as “CLh”, the asymptomatic isolates as “Ah”, and the dog isolates as “VLd”. Population analysis was conducted as described by Mosca et al. (2012). Aligned sequences were analyzed using the DnaSAM program (Eckert et al., 2010). Sites that violated the infinite sites mutation model were excluded from subsequent calculations. For each group, genetic diversity was estimated with Θπ (Nei, 1987; Tajima, 1983) and neutrality was assessed using the Tajima’s D statistic (Tajima, 1989). The recombination analysis was performed by a PHI Test, which presents a very low false positive rate even in the presence of growth or constant size population (Bruen et al., 2006). The folded site frequency spectrum analysis was performed through the pegas package (Paradis, 2010) in R.
2.7. Chromosome number variation analysis
Read depths and chromosome somy values were estimated based on previously reported methodologies (Downing et al., 2011; Zhang et al., 2014). Briefly, the read depth per haploid genome in each chromosome was calculated according to the raw read depth divided by the median read depth in the entire chromosome. The read depth for each chromosome was in turn normalized to the median read depth of the genome. The median read depth values were calculated using VCFtools in the SAMtools suite (Li et al., 2009). Heat maps and isolate clustering based on chromosome somy were constructed using RColorBrewer and gplots R packages (R Core Team, 2015, A Language and Environment for Statistical Computing).
3. Results
3.1. Leishmania infantum isolation and initial characterization
Fifteen of the isolates were obtained from either symptomatic or asymptomatic humans and five from dogs. The 15 human isolates came from a geographic region with an approximate 180 km diameter in the state of Rio Grande do Norte, Brazil, whereas the dog isolates were from an area approximately 30 km wide in the metropolitan area of Natal (Supplementary Fig. S1). Of the human isolates, 10 were from people with symptomatic VL, one from a person with CL and four from people with asymptomatic Leishmania infection (Table 1). Nine of the human isolates were obtained from the bone marrow, five from blood, and one from skin, whereas the dog isolates were obtained from spleens of symptomatic animals. Five of the human isolates were obtained in the 1990s. Importantly, two isolates (19VLh and 20VLh) were obtained from specimens taken simultaneously from the blood and bone marrow, respectively of the same VL patient. All human isolates were typed as L. infantum and cloned prior to sequencing.
Table 1.
Information about the 20 Brazilian Leishmania isolates used in this study. Isolates are named 1–20 in the chronological order in which each isolate was collected during 1991– 2013.
| Isolate name | Isolation year | Status | Host | Organ/Tissue | City |
|---|---|---|---|---|---|
| 1VLh90 | 1991 | VL | Human | Bone Marrow | Natal |
| 2VLh90 | 1992 | VL | Human | Bone Marrow | Ielmo Marinho |
| 3VLh90 | 1992 | VL | Human | Bone Marrow | Macaiba |
| 4VLh90 | 1993 | VL | Human | Bone Marrow | Ceará Mirim |
| 5VLh90 | 1993 | VL | Human | Bone Marrow | São José Mipibú |
| 12VLh | 2012 | VL | Human | Bone Marrow | Extremoz |
| 13VLh | 2012 | VL | Human | Bone Marrow | Açu |
| 14VLh | 2012 | VL | Human | Bone Marrow | Macaiba |
| 19VLha | 2013 | VL | Human | Peripheral Blood | Sitio Novo |
| 20VLha | 2013 | VL | Human | Bone Marrow | Sítio Novo |
| 6CLh | 2009 | CL | Human | Skin | Macaiba |
| 8Ah | 2011 | Asymptomatic | Human | Peripheral Blood | Natal |
| 9Ah | 2011 | Asymptomatic | Human | Peripheral Blood | Touros |
| 10Ah | 2011 | Asymptomatic | Human | Peripheral Blood | Extremoz |
| 18Ah | 2012 | Asymptomatic | Human | Peripheral Blood | Natal |
| 7VLd | 2010 | VL | Dog | Spleen | Natal |
| 11VLd | 2011 | VL | Dog | Spleen | Natal |
| 15VLd | 2012 | VL | Dog | Spleen | Natal |
| 16VLd | 2012 | VL | Dog | Spleen | Natal |
| 17VLd | 2012 | VL | Dog | Spleen | Natal |
VL, visceral leishmaniasis; CL, cutaneous leishmaniasis; A, asymptomatic person; h, human; d, dog; 90, isolated in the 1990s.
19VLh and 20VLh are from the same person.
3.2. Chromosome copy number variation
Fig. 1A shows the estimated somy values for the 36 chromosomes of the 20 Brazilian isolates. Chromosome 31 is the only chromosome whose copy number is consistently higher than disomic in all isolates, similar to previous observations (Downing et al., 2011; Rogers et al., 2014). In isolate 7VLd, all chromosomes were disomic except chromosome 31, whereas all other isolates have additional chromosomes that display at least some degree of aneuploidy. For example, in Fig. 1A an estimated somy value between three and four is consistent with the possibility that half the cells are trisomic for that chromosome and half are tetrasomic. Thirteen of the 36 chromosomes are disomic or predominantly disomic in all 20 isolates, whereas all other chromosomes show variable somy patterns ranging from di- to pentasomic.
Fig. 1.

Aneuploidy in Leishmania Isolates. (A) Heatmap for aneuploidy status of Leishmania infantum chromosomes. Mosaic aneuploidy was found in the 20 Brazilian L. infantum isolates based on normalized chromosome read depth as shown in the key at the top left. The heatmap shows the estimated copy number of the 36 chromosomes (x-axis) in the isolates (y-axis) as disomic (value 2 in the key), pentasomic (value 5 in the key) or intermediate values (values between 2 and 5). The vertical bars on the left on the left of the heat map denote three groups of isolates formed by a hierarchical clustering analysis of the somy values. The 20 isolates indicated on the right are named as follows. Numbers 1–20 are the chronological order in which each isolate was collected during 1991–2013. “VL” isolates are from VL patients and from dogs; isolates with “A” in their name are from asymptomatic individuals; “CL” is an isolate from a CL patient; “h” isolates are from humans; d isolates are from dogs; “90” denotes isolates collected in the 1990s. Isolates 19VLh and 20VLh were collected from the blood and bone marrow, respectively, of the same individual. (B) Distributions of disomic chromosomes amongst three groups of L. infantum/Leishmania donovani. Chromosomes are shown that are predominately disomic in 20 L. infantum isolates from Brazil (this work), 17 L. donovani isolates from Nepal (Downing et al., 2011) and 12 L. infantum isolates from Turkey (Rogers et al., 2014). A total of 13 chromosomes in the Brazilian isolates, nine in the Nepalese isolates and six in the Turkish isolates were predominantly disomic. Four chromosomes (numbers 19, 28, 30 and 34) were predominantly disomic in all 49 isolates.
A hierarchical clustering analysis grouped the samples according to their somy values. As observed in Fig. 1A, this analysis formed three groups of samples, indicated by the three vertical bars on the left of the heat map, two of which show little variation in somy amongst group isolates. No group showed a direct association with the isolates’ phenotype, except for the bottom of the three groups, which has three of the four asymptomatic samples (8Ah, 9Ah and 10Ah), as well as three others (6CLh, 12VLh and 15VLd). The two isolates from the same patient, 19VLh and 20VLh, display a somewhat different somy pattern.
The degree of chromosomal disomy varies amongst the 20 Brazilian isolates, 17 Nepalese isolates, and 12 Turkish isolates (49 isolates in total) of the L. infantum/L. donovani complex on which WGS has been conducted (Fig. 1B). Seven chromosomes of the Brazilian isolates share predominantly disomic status with the corresponding chromosomes of the Nepalese isolates (chromosomes 17, 19, 21, 28, 30, 34, and 36). The Nepalese and Turkish isolates do not share any predominantly disomic chromosomes that are not also disomic in the Brazilian isolates. However, four chromosomes are predominantly disomic in all 49 isolates from the three continents (chromosomes 19, 28, 30, and 34). The fact that the other 32 chromosomes display some degree of mosaic aneuploidy in at least one, and usually most, of the 49 isolates suggests an existing constraint on these four chromosomes that maintains their disomy.
3.3. Nucleotide diversity
The reads of the 20 L. infantum isolates were mapped to the reference genome L. infantum clone JPCM5, which was derived from the spleen of a naturally infected dog in the Madrid area of Spain in 1998 (Denise et al., 2006). After alignment and removal of duplicates, the genome coverage was more than 90-fold. The 20 Brazilian isolates have greater than 99.94% identity with the reference genome and are even more closely related to each other with 99.95% overall identity.
The total number of SNPs in the 20 isolates relative to the reference genome ranges from 1228 in isolate 5VLh90 to 1274 in isolate 6CLh (Table 2). A total of 2665 SNPs was identified. Isolate 13VLh has the highest number of SNPs within coding regions relative to the reference (99 total), whereas the lowest number was observed in 2VLh90 (83 total). Interestingly, only 7–8% of SNPs occur in coding regions, whereas 48% of the genome consists of coding regions (Ivens et al., 2005).
Table 2.
Isolate properties relative to JPCM5 reference sequence.
| Isolate Name | Indels | SNPs | Coding SNPs | Synonymous | Non-Synonymous | Fraction of CDS SNPs |
|---|---|---|---|---|---|---|
| 1VLh90 | 2473 | 1254 | 88 | 25 | 63 | 0.07 |
| 2VLh90 | 2487 | 1231 | 83 | 25 | 58 | 0.07 |
| 3VLh90 | 2395 | 1264 | 87 | 26 | 61 | 0.07 |
| 4VLh90 | 2512 | 1252 | 89 | 26 | 63 | 0.07 |
| 5VLh90 | 2422 | 1228 | 94 | 30 | 64 | 0.08 |
| 12VLh | 2407 | 1252 | 93 | 28 | 65 | 0.07 |
| 13VLh | 2399 | 1256 | 99 | 30 | 69 | 0.08 |
| 14VLh | 2416 | 1245 | 88 | 27 | 61 | 0.07 |
| 19VLh | 2453 | 1246 | 90 | 30 | 60 | 0.07 |
| 20VLh | 2435 | 1232 | 86 | 27 | 59 | 0.07 |
| 6CLh | 2430 | 1274 | 91 | 28 | 63 | 0.07 |
| 7VLd | 2432 | 1236 | 93 | 30 | 63 | 0.08 |
| 11VLd | 2420 | 1266 | 93 | 28 | 65 | 0.07 |
| 15VLd | 2452 | 1245 | 91 | 30 | 61 | 0.07 |
| 16VLd | 2435 | 1259 | 93 | 31 | 62 | 0.07 |
| 17VLd | 2455 | 1240 | 84 | 26 | 58 | 0.07 |
| 8Ah | 2462 | 1245 | 89 | 27 | 62 | 0.07 |
| 9Ah | 2440 | 1261 | 92 | 28 | 64 | 0.07 |
| 10Ah | 2444 | 1271 | 91 | 30 | 61 | 0.07 |
| 18Ah | 2416 | 1252 | 89 | 26 | 63 | 0.07 |
Numbers in isolate names indicate the chronological order in which the isolates were collected; VL, visceral leishmaniasis; CL, cutaneous leishmaniasis; A = asymptomatic person; h, human; d, dog; 90, isolated in the 1990s Isolates 19VLh and 20VLh are from the same person.
Indels, insertion/deletions; SNP, single nucleotide polymorphism; CDS, coding sequence.
Even though the 20 isolates have 99.95% identity with each other, a matrix analysis of individual differences among them shows considerable variation. For example, two asymptomatic isolates, 6CLh and 10Ah, have the fewest SNPs between them at 242, whereas isolates 4VLh90 and 11VLd have the most at 603 (Supplementary Table S1). The nucleotide differences were assigned to their respective chromosomes and the nucleotide diversity (Θπ) for each chromosome was calculated for each group of isolates. When considered as a group, the four asymptomatic isolates display lower nucleotide diversity among each other than any other groupings, including those isolated from dogs, humans during the 1990s, or the other VL patients. All 36 chromosomes in the asymptomatic and dog groups display less diversity than do those in the two VLh groups (P < 0.0001 comparing VLd with VLh or VLh90, Wilcoxon rank test, Fig. 2). Asymptomatic isolates also differ significantly from VLh and from VLh90 (P < 0.0001, Wilcoxon rank test). Interestingly, amongst the human VL isolates (the VLh90 and VLh + CLh groups) chromosomes 12, 15, 24 and 26, and the larger chromosomes 32–36, have increased diversity relative to the dog VL and asymptomatic groups of isolates. According to the folded Site Frequency Spectrum (SFS) analysis there is a predominance of single variants in the whole population (data not shown), which could be indicative of expansion from a recent bottleneck.
Fig. 2.

Nucleotide diversity among Leishmania Infantum chromosomes. Nucleotide diversity and Tajimas’s D test for each chromosome according to isolate grouping characteristics. Chromosomes highlighted in grey bars are those that have a P value ≤0.05 for recombination under the PHI Test (Φw); those without bars (chromosomes 5,6, 8, 9, 14,17, 25, 26 and 31) showed no sign of recombination.
We calculated Tajima’s D for all groups of isolates across all chromosomes to determine whether there was evidence of deviations from neutrality or a constant population size. Specifically, recent population contractions or balancing selection produce positive Tajima’s D values, whereas population expansion after a recent bottleneck or selective sweeps produce negative Tajima’s D values (Fig. 2). None of the Tajima’s D values were significantly different from zero, consistent with a neutrally evolving population of constant size.
The Φw was used to identify possible signatures of recombination in the dataset. Using this test, we identified 27 chromosomes with significant signatures of intrachromosomal recombination (P ≤ 0.05, Fig. 2).
3.4. Individual clustering
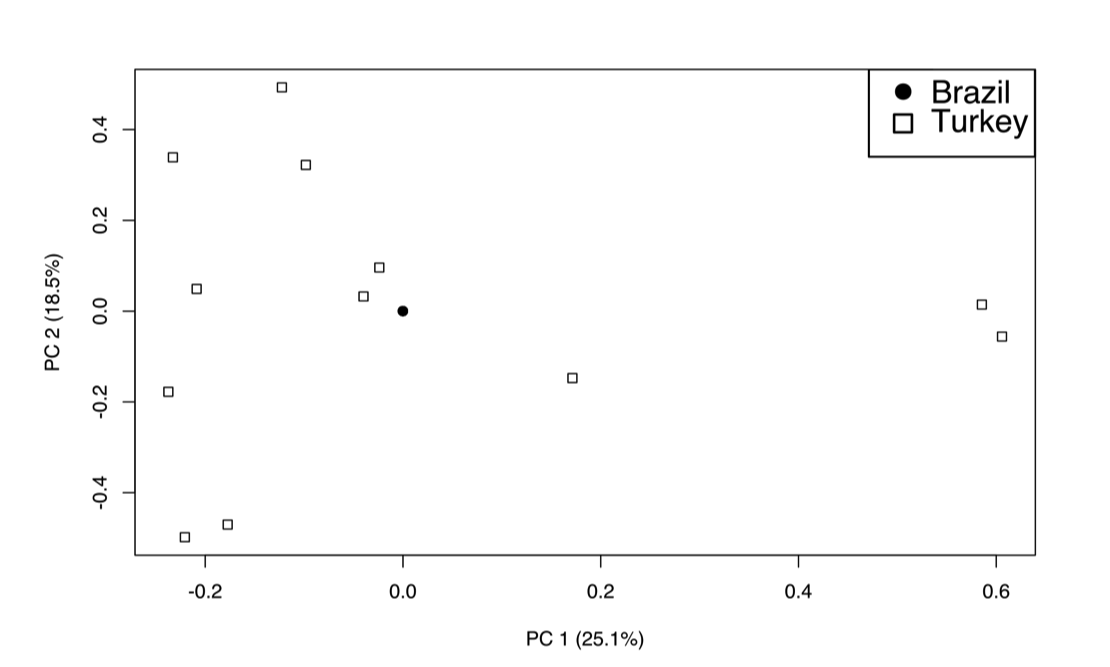
After removing SNPs in linkage disequilibrium, 653 SNPs remained in the data set and were used for clustering analysis. A principal component analysis of the SNPs in linkage equilibrium identified two main components that, combined, comprise 34.5% of the variation of the samples (21.0% and 13.5% for the first and second components, respectively). Interestingly, 11 of 20 samples cluster very closely in PC1 and PC2 (circled in Fig. 3). The nine remaining samples include three isolates from the VLh90 group, four VLh isolates and two VLd isolates. The two VLh isolates from the same patient (19 and 20VLh) are among these non-clustered samples. The two isolates from the same patient and 13VLh were collected from the two geographically most distant locations among the 20 isolates (Supplementary Fig. S1). In contrast, the three isolates outside of the main cluster from the VLh90 group were collected within a smaller area encompassing Natal and nearby areas. Plots of the first 10 principal components are shown in Supplementary Fig. S2. We tested the significance of each principal component using the Trace-Widom test, and only PC1 reached statistical significance (Supplementary Table S2). In contrast, combined PCA of the 12 Turkish and 20 Brazilian isolates showed much greater variation among the 12 Turkish isolates than the 20 Brazilian ones (Supplementary Fig. S3; see also Supplementary Table S3). Notably, all 20 Brazil isolates overlapped at single point, consistent with a recent common ancestry amongst Brazilian isolates (Maurício et al., 2000; Zemanová et al., 2007; Leblois et al., 2011). This is consistent with the Admixture analysis (Supplementary Fig. S4) where, at K = 2, all of the Brazilian isolates cluster together, with evidence of ancestry from within the Turkish isolates. Of interest, in this Admixture analysis, it is the Brazilian isolates that show substructure into two populations at K = 3, before some substructure is observed in the Turkish isolates at K = 4. The separation of Brazilian isolates into two subpopulations is also observed in Admixture analysis of the Brazilian isolates alone (Supplementary Fig. S4D), and involves the same subset of isolates (1VLh90, 2VLh90, 4VLh90, 13VLh, 19VLh, and 20VLh) that appear to be closer in origin to the Turkish isolates than the remaining Brazilian isolates.
Fig. 3.

Principal component analysis of the single nucleotide polymorphism dataset. Principal component analysis of genomic variation in the five Leishmania Infantum isolates from visceral leishmaniasis patients in the 1990s, five isolates from visceral leishmaniasis patients in 2012–13, four isolates from asymptomatic humans in 2011–12, five isolates from visceral leishmaniasis dogs in 2010–12 and one isolate from a cutaneous leishmaniasis patient in 2009. The two most significant PCs are related to 34.5% (21 + 13.5%) of the variation for all isolates. The 11 most closely related isolates, according to PCA, are indicated within the oval.
3.5. Leishmania infantum/L. donovani-specific genes
The L. infantum/L. donovani complex contains 19 to 25 genes that are either absent or present as pseudogenes in CL-causing Leishmania major and Leishmania braziliensis (Peacock et al., 2008; Zhang and Matlashewski, 2010; Rogers et al., 2011). The functions of some of these 25 genes can be predicted by sequence similarities, although the majority encode hypothetical proteins with no known function. At least some of these genes likely contribute to visceralization, including the well-characterized gene family encoding protein A2 that is primarily comprised of 10-aminoacid repeats (Zhang et al., 2003). The 12 L. infantum isolates from Turkey whose genomes have been sequenced cause CL, not VL (Rogers et al., 2014). These Turkish isolates were shown to be derived from a single cross of two diverse strains, one similar to the Spanish L. infantum JPCM5 reference strain and one not. One possibility is that differences in some of the 25 genes derived from the non-JPCM5-like parental strain are responsible for the fact that these Turkish isolates cause CL rather than VL. Thus, we compared the sequences of these 25 genes in the JPCM5 reference genome with those in our 20 Brazilian L. infantum isolates and those in the 12 Turkish L. infantum isolates that cause CL.
As expected, the 25 genes in the Brazilian isolates were much more similar to the reference strain than they are in the Turkish isolates. Indeed, only five of the 25 genes in all the Turkish isolates were identical to the JPCM5 reference genome. Another five genes were identical to the reference in some but not all Turkish isolates, whilst the other 15 genes in the Turkish isolates were different in at least one location from those of the reference (not shown).
In contrast, 21 of the 25 genes in all the Brazilian isolates were identical to the reference genome. Isolate 6CLh does not have any unique changes in any of these 25 genes that distinguish it from the other 19 Brazilian isolates. This isolate had only one variation inside a coding region (Linj_33_3230) encoding a conserved hypothetical protein of 2554 amino acid residues. This SNP changed histidine to glutamine at position 1145 (H1145Q). The four genes that did exhibit SNP difference(s) in all or some of the Brazilian isolates were LinJ28.0340 (hypothetical protein; R41L), LinJ36.2060 (hypothetical protein; 1–2 bp deletions destroying the reading frame), LinJ24.1510 (hypothetical protein; T266I) and LinJ22.067, the multi-copy gene family encoding the 10-amino-acid-repeat protein A2 (Zhang et al., 2014). In the A2 gene family the SNPs occurred in seven different locations and many were heterozygous, reflecting both the multi-copy nature of the genes and the presence of internal repeats whose sequences can vary (Supplementary Figs. S5, S6). Since this gene family is large, repetitive, and difficult to sequence reliably, it is possible that isolate 6CLh had multiple copy differences and/or nucleotide changes in this region that we could not detect and that correlate with the association of 6CLh with CL rather than VL. Aside from this possibility, however, other factors seem to be responsible for the involvement of 6CLh in a CL presentation.
3.6. RagC gene
Another gene of interest is RagC on chromosome 36, which encodes a ras-like GTPase protein that in higher eukaryotic cells participates in regulation of the mTOR pathway. This gene was originally identified by comparing two naturally occurring isolates in Sri Lanka of the closely related species, L. donovani, which typically causes VL. The study found that one L. donovani isolate caused CL and the other caused VL (Zhang et al., 2014). One of the SNP differences between these two isolates occurs in RagC and converts an arginine at position 231 of the GTPase in the Sri Lanka VL strain to a cysteine in the Sri Lanka CL strain (R231C). It was found that recombinant expression of the VL RagC gene in the CL strain increased the ability of the CL strain to survive in visceral organs of experimental animals by seven- to 40-fold, i.e., strong evidence for involvement of this GTPase in visceralization of CL-causing Leishmania spp. (Zhang et al., 2014).
We inspected RagC (LinJ_36_6140) in the 20 L. infantum isolates described here for the presence of this R231C mutation, but no isolates had it and no other mutations were found in this gene. Similarly, the 12 L. infantum CL isolates from Turkey (Rogers et al., 2014) and the Spanish reference L. infantum genome JPCM5 do not have this mutation. These findings suggest R231C may not be associated with CL disease caused by L. infantum, unlike the Sri Lankan L. donovani case.
3.7. Comparison of two isolates from the same patient
The two isolates cloned from samples taken at the same time from the peripheral blood (19VLh) and bone marrow (20VLh) of the same VL patient offered an opportunity to compare the genomic DNA sequences and ploidy of two cloned isolates simultaneously infecting a single individual. A total of 280 SNP differences were found between these two genomes (Supplementary Table S1). These 280 SNPs were then compared with each other, as well as SNPs in the other 18 genomes and the reference JPCM5 genome. These comparisons show that 19VLh has 23 unique SNPs (all in intergenic regions) not found in any of the other 19 isolates including 20VLh, or in the reference genome, whereas 20VLh had 24 unique SNPs (22 in intergenic regions; two in coding regions) not found in the other 19 isolates or the reference. These SNPs distinguish the two isolates from each other and the other 18 isolates. However, these two isolates share 84 SNPs (79 and five in intergenic and coding regions, respectively) not found in the other 18 isolates, which distinguish them as a pair from the other isolates. The exact positions of these SNPs in each of the affected chromosomes are listed in Supplementary Table S4.
As controls, this pair-wise analysis was also conducted on all possible combinations of the 20 isolates. For example, 6CLh and 10Ah are the two isolates that have the fewest overall SNP differences (242 differences; Supplementary Table S1). Isolate 6CLh has 13 (12 + 1) unique SNPs and 10Ah has 18 (17 + 1) unique SNPs. However, in contrast to the 19/20VLh pair, these two isolates do not share any SNPs that are absent in all other 18 isolates. Similarly, 4VLh90 and 11VLd have the most overall SNPs differences at 603 (Supplementary Table S1). Each of these two isolates had 82 and 45 unique SNPs, respectively, but as a pair, they do not share any SNPs not found in the other 18 isolates. Similar results were obtained with all the other pair-wise comparisons, i.e., each isolate of the pair had many more unique SNPs than it had SNPs shared with the other member of the pair, consistent with the lack of deviation of Tajima’s D from zero value (shown in Fig. 2). These results were in dramatic contrast to the 19/20VLh pair that share 84 SNPs not found in the other 18 isolates.
Two additional pairs of isolates deserve mention, 1VLh90 and 2VLh90, and 13VLh and 4VLh90, which showed greater numbers of shared SNPs than other pairs, both non-coding and coding (24 + 1 and 16 + 1, respectively). This suggests these two pairs share more recent common ancestors than other pairs, other than 19 and 20, even though members of one pair were isolated more than a decade apart. The above pairs of isolates sharing SNPs also appear outside the main cluster in the PCA shown in Fig. 3.
4. Discussion
Leishmania infantum can infect many mammalian species, some of which serve as asymptomatic reservoirs maintaining the infection in endemic regions, and others (e.g. dogs, humans) who additionally develop symptomatic disease (Jeronimo et al., 2000; Wilson et al., 2005; Lima et al., 2012). Manifestations of human infection are heterogeneous, resulting most commonly in asymptomatic infection which is detected by a positive skin test or by serology (Lima et al., 2012). Symptomatic L. infantum infection most commonly manifests as VL, a disease that is usually fatal if untreated. Recently, non-ulcerating cutaneous lesions due to L. infantum have been reported, which are different from CL due to other species of Leishmania, which ulcerate before spontaneous healing (Alger et al., 1996). Canine leishmaniasis is often slowly progressive with heavy skin involvement, making this an ideal reservoir for human infection (Ready, 2014; Kaszak et al., 2015). Here we examined the genomic sequences of (i) four isolates from different asymptomatic individuals, (ii) 15 isolates collected over 21 years (1991–2012) from subjects with symptomatic VL, of which two were from the same individual and five were from dogs, and (iii) one isolate obtained from a subject with a non-ulcerating cutaneous lesion.
We identified substantial chromosome copy-number variation in the 20 Brazilian L. infantum isolates. This was not unexpected since several Leishmania spp., including the L. donovani/infantum complex, exhibit similar ‘mosaic aneuploidy’, a phenomenon in which the number of chromosome copies per cell varies in a given population (reviewed by Sterkers et al. (2012)). WGS of 17 L. donovani isolates from Nepal identified nine of the 36 chromosomes as predominantly disomic, whereas the other 27 chromosomes displayed some degree of higher order somy in one or more isolates (Downing et al., 2011). Similarly, WGS of 12 L. infantum isolates from Turkey showed that six of the 36 chromosomes were predominantly disomic, and the others had a higher copy number in at least one isolate (Rogers et al., 2014). In most cases the copy number was not a whole integer above diploid, suggesting the cultured Leishmania population was mixed with respect to how many copies of a given chromosome were present in each cell (i.e., mosaic aneuploidy).
We used read alignments against the reference JPCM5 genome to assess chromosome copy number variation in the 20 Brazilian isolates. Each of the original wild isolates was cloned and grown for only a limited number of cell divisions before DNA isolation, since somy changes have been observed in Leishmania spp. cultures (Sterkers et al., 2011). Therefore, we anticipated any possible monosomic chromosomes would display few or no heterozygous SNPs. Despite the 99.95% sequence identity among the 20 isolates, at least several di-heterozygous SNPs were detected in each of the chromosomes with the lowest somy, therefore indicating that the baseline chromosome somy was disomic.
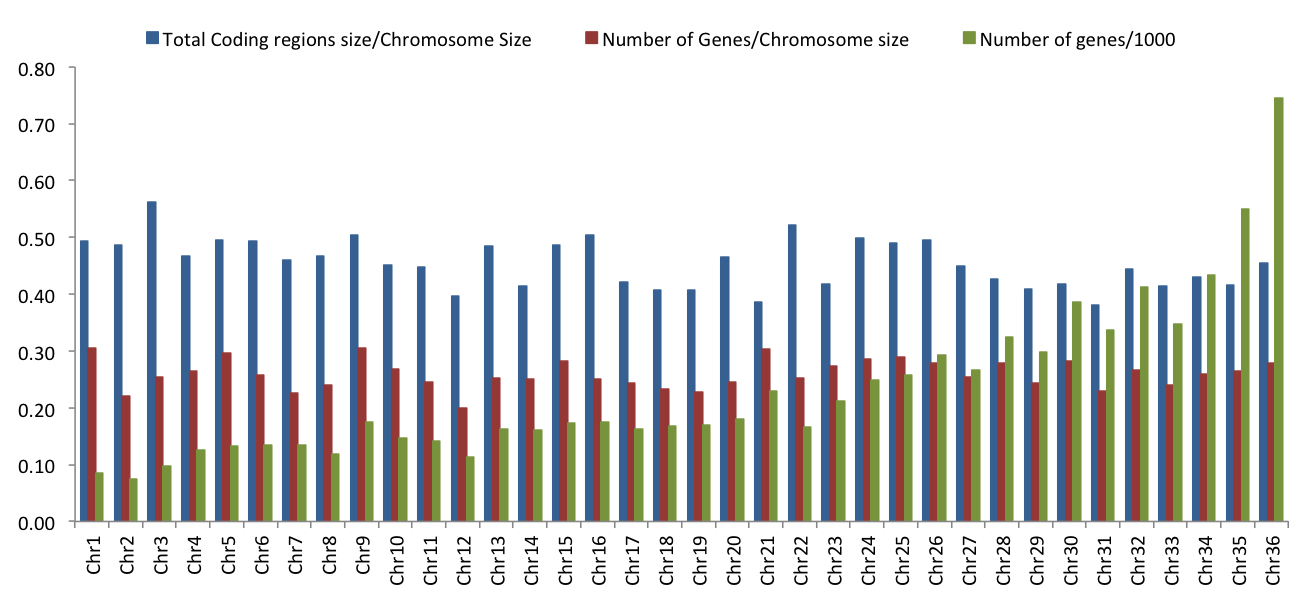
Our comparison of the Brazilian isolates with those from Turkey and Nepal revealed that chromosome 31 is aneuploid in all 49 isolates. We also observed that the other larger chromosomes appeared more likely to be predominantly disomic than were smaller chromosomes. For example, among the 15 smallest chromosomes, only 20% were predominantly disomic in one of the three isolate groups (3/15; of chromosomes 1–15, only 3, 7 and 10 were disomic) (Fig. 1B). In contrast, among the 21 largest chromosomes, 67% were predominantly disomic in one or more of the three isolate groups (14/21 within chromosomes 16–36). Four of these large chromosomes – chromosomes 19, 28, 30 and 34 – were disomic in all 49 isolates from Brazil, Nepal and Turkey. It is currently unclear why some chromosomes appear to be constrained to two copies whereas others are not. One of several possibilities is that these chromosomes contain one or more genes or regions for which increased expression is deleterious to the cell. This possibility is increased for the larger chromosomes since they harbour more genes than the smaller chromosomes. Furthermore, if more genes are on a chromosome, these genes may also interact in more gene networks; thus, changes in somy might be expected to affect more cellular pathways. Chromosome 31 would be the exception to this hypothesis. Perhaps its tetra- to pentasomic status across all isolates is an adaptive increase in expression levels above those produced by disomic chromosomes. Determination of the ratio of the coding versus non-coding regions of each of the 36 chromosomes in the reference JPCM genome (Supplementary Fig. S7) revealed that chromosome 31 has the lowest percentage of coding region at 38% (chromosome 3 has the highest at 56%). This may be related to its aneuploid tendency. Other possibilities for the disomic exclusivity of specific chromosomes not based on gene expression levels are chromosomal differences in the mechanisms of sister chromatid segregation during cell division or differences in replication from the chromosomes’ replication origins (Walton, 2014).
In addition to rampant copy number variation amongst L. infantum chromosomes, we identified the presence of intrachromosomal recombination on 27 of the 36 chromosomes using the PHI test statistic (Φw) (Bruen et al., 2006). These results are consistent with the amount of linkage encountered in preparation of the Brazilian isolate sequence data for PCA (out of 2665 total SNPs, 653 were in linkage equilibrium and retained for PCA). This is not surprising since recombination has been found in other Leishmania studies. For example, Rogers et al. (2014) reported that Leishmania genomes from Turkey were the product of a single cross between L. donovani and L. infantum, and that the distribution of introgressed haplotypes in the isolate genomes reflects subsequent back-crossing and sexual recombination. Our findings support the developing viewpoint that recombination in Leishmania occurs, albeit likely infrequently, and, as hypothesised by others, possibly only during infection of the insect vector (Rougeron et al., 2015).
The nucleotide diversity displayed by both groups of isolates from symptomatic humans (VLh and VLh90) is greater than diversity of most chromosomes of the other groups, i.e., asymptomatic humans and symptomatic dogs (Fig. 2). This is even more evident in an analysis performed at 1 kb intervals across the 36 chromosomes (Supplementary Fig. S8), which shows that the VLh90 isolates have regions of greater diversity more often than the others. A positive Tajima’s D value is associated with a recent population contraction or balancing selection, whilst a negative value reflects population expansion after a recent bottleneck or a selective sweep. Tajima’s D values did not differ significantly from zero for all groups of isolates in all chromosomes, an unexpected result given the dynamic nature of VL in Brazil, changing in distribution between rural and urban regions over the most recent decades (Jeronimo et al., 2000, 1994). Although a Tajima’s D of zero is consistent with a neutrally evolving population of constant size, we hypothesise that the lack of significant deviation of Tajima’s D from zero in our study may reflect the limited sample size.
Taking into consideration the SNP differences among the isolates, we can hypothesise that we are dealing with at least two different populations. In the analyses that consider genetic sequences (nucleotide diversity, PCA and phylogenetics; Figs. 2 and 3 and Supplementary Fig. S9, respectively), three samples isolated in the 1990s (1VLh90, 2VLh90 and 4VLh90) always appear collectively more divergent than the others in all analyses, followed by the two samples from the same individual (19 and 20 VLh). Notably, the other two VLh90 isolates (3VLh90 and 5VLh90) were always associated, in the same analyses, with the most recent asymptomatic human, visceral human and dog isolates. The close relationship between L. infantum strains obtained from dogs and humans is well documented in Brazilian isolates (Segatto et al., 2012) as well as in other countries where leishmaniasis outbreaks occur frequently (Kuhls et al., 2011). Thus, when the nucleotide diversity, principal components and phylogenetic analysis are considered collectively, it is possible to suggest that during the 1990s there were at least two different lineages of L. infantum isolates giving rise to most of the isolates in the current study. This could explain why only two of the VLh90 isolates are clustered with most of the recent VLh, Ah and VLd isolates. This hypothesis is supported by a population structure analysis in which the Brazilian isolates are compared with the Turkish L. infantum isolates (Supplementary Fig. S4). This analysis suggests a hierarchical structuring of the isolates into Brazilian and Turkish isolates first (at K = 2), and then a subdivision of Brazilian isolates second (at K = 3).
The SNP data from isolates 19VLh and 20VLh from the same patient demonstrate they are much more closely related to each other than are any other pair of isolates, consistent with phylogenetic analyses based on all nucleotide differences described here (Supplementary Fig. S9). The data do not, however, completely resolve the question of whether (i) this patient was co-infected with at least two closely related strains or (ii) the SNPs that distinguish between 19VLh and 20VLh arose in the patient after the initial infection or during the cloning/culturing of the isolates. One consideration is that a human infection can last for as long as 4– 14 months before VL symptoms appear (Chagas, 1936; Jeronimo et al., 2000), which might be enough time for point changes in the parasites’ DNA to occur. This possibility is balanced by an observation of this work that shows the approximate number of nucleotide differences of these 20 isolates compared with the reference JPCM genome has not changed in 20 years, i.e., the number of differences in VLh90 isolates is about the same as in the isolates from 2012 to 2013 (Table 2, Supplementary Table S1). Still another consideration is that there are somy differences of some chromosomes in these two isolates, as shown in Fig. 1A. It has been shown, however, that somy differences can occur during in vitro culturing (Sterkers et al., 2012), and probably are not associated with the SNP differences seen here. Thus, on balance it seems most likely that the patient was co-infected with two highly related strains, although this conclusion cannot be definitively stated. WGS of more cloned isolates from individual patients will be necessary to address the issue.
Variations in somy appear to be the major source of the genomic diversity in the L. infantum isolates sequenced herein, as well as the combined analysis of Brazilian isolates and L. donovani/L. infantum isolates from Turkey and Nepal. Increased somy is, in general, inversely related to chromosome size but is not related to disease status, time of isolation, or the host. The exception is chromosome 31, which has the highest somy but lowest proportion of coding regions to chromosome size. Remarkably, portions of two chromosomes of disomic Trypanosoma brucei (chromosomes 4 and 8) are syntenic with the entirety of L. major chromosome 31 (El-Sayed et al., 2005), effectively rendering this sequence also tetrasomic in T. brucei. Thus, a tetrasomic L. major chromosome 31 is equivalent in terms of gene content to two disomic chromosomal regions of T. brucei. This, in turn, suggests that there is a functional reason for chromosome 31 aneuploidy, possibly explaining the stability of chromosome 31 somy across isolates. It may also be worth noting that the entire chromosome 31 sequence in Leishmania and its two equivalents in T. brucei are transcribed as a single very large polycistronic transcript (El-Sayed et al., 2005), although the significance of this feature is unclear. It is also unclear how the stable aneuploidy of chromosome 31 relates to the mosaic aneuploidy seen in the other chromosomes. It remains to be determined if aneuploidy across the chromosomes is a pleiotropic effect of the necessity for aneuploidy at chromosome 31.
These analyses revealed that the genomes of L. infantum in northeastern Brazil are remarkably similar at a sequence level, perhaps reflecting that they are derived from a limited number of strains introduced into this region of South America in the past several hundred years. There is, nonetheless, increased diversity in isolates from subjects with symptomatic VL compared with isolates from asymptomatically infected subjects, a subgroup that has not previously been available for sequence comparisons. There was similarly lower diversity in the isolates sequenced from dogs. One explanatory hypothesis for this observation is that some parasite strains are more likely than others to result in asymptomatic infection. However, all conclusions need to be validated with additional genomic sequences from isolates in each of the groups examined in this study.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This work was supported in part by grants P50 AI-30639 (SMBJ, MEW, RDP) and R01 AI076233 (MEW, JMB) from the US National Institutes of Health. The study was performed while JED (CNPQ 401546/2013-6) and DGT were supported by Science Without Borders from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brazil. This research was supported in part through computational resources provided by the University of Iowa, USA, and Universidade Federal do Rio Grande do Norte, Natal, RN, Brazil.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.ijpara.2017.04.004.
Footnotes
Note: New sequences reported here have been submitted to the National Center for Biotechnology Information, USA, sequence read archive (NCBI SRA) under accession numbers accession numbers SRR5117893, SRR5117894, SRR5117895, SRR5117896, SRR5117897, SRR5117898, SRR5117899, SRR5117900, SRR5117901, SRR5117902, SRR5117903, SRR5117904, SRR5117905, SRR5117906, SRR5117907, SRR5117908, SRR5117909, SRR5117910, SRR5117911, SRR5117912.
References
- Albuquerque PLMM, Da Silva GB, Júnior, Freire CCF, Oliveira SBDC, Almeida DM, Da Silva HF, Cavalcante MDS, Sousa ADQ. Urbanization of visceral leishmaniasis (kala-azar) in Fortaleza, Ceará, Brazil. Rev Panam Salud Publ. 2009;26:330–333. doi: 10.1590/s1020-49892009001000007. http://dx.doi.org/10.1590/S1020-49892009001000007. [DOI] [PubMed] [Google Scholar]
- Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. http://dx.doi.org/10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alger J, Acosta MC, Lozano C, Velasquez C, Labrada LA. Stained smears as a source of DNA. Mem. Inst Oswaldo Cruz. 1996;91:589–591. doi: 10.1590/s0074-02761996000500009. http://dx.doi.org/10.1590/S0074-02761996000500009. [DOI] [PubMed] [Google Scholar]
- Almeida AS, Werneck GL. Prediction of high-risk areas for visceral leishmaniasis using socioeconomic indicators and remote sensing data. Int J Health Geogr. 2014;13:13. doi: 10.1186/1476-072X-13-13. http://dx.doi.org/10.1186/1476-072X-13-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, Depledge DP, Fischer S, Gajria B, Gao X, Gardner MJ, Gingle A, Grant G, Harb OS, Heiges M, Hertz-Fowler C, Houston R, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Logan FJ, Miller JA, Mitra S, Myler PJ, Nayak V, Pennington C, Phan I, Pinney DF, Ramasamy G, Rogers MB, Roos DS, Ross C, Sivam D, Smith DF, Srinivasamoorthy G, Stoeckert CJ, Subramanian S, Thibodeau R, Tivey A, Treatman C, Velarde G, Wang H. TriTrypDB: a functional genomic resource for the trypanosomatidae. Nucleic Acids Res. 2009;38:457–462. doi: 10.1093/nar/gkp851. http://dx.doi.org/10.1093/nar/gkp851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badaro R, Jones TC, Carvalho EM, Sampaio D, Reed SG, Barral A, Teixeira R, Johnson WD. New perspectives on a subclinical form of visceral leishmaniasis. J Infect Dis. 1986a;154:1003–1011. doi: 10.1093/infdis/154.6.1003. http://dx.doi.org/10.1093/infdis/154.6.1003. [DOI] [PubMed] [Google Scholar]
- Badaro R, Jones TC, Lorenco R, Cerf BJ, Sampaio D, Carvalho EM, Rocha H, Teixeira R, Johnson WD. A prospective-study of visceral leishmaniasis in an endemic area of Brazil. J Infect Dis. 1986b;154:639–649. doi: 10.1093/infdis/154.4.639. [DOI] [PubMed] [Google Scholar]
- Bern C, Hightower AW, Chowdhury R, Ali M, Amann J, Wagatsuma Y, Haque R, Kurkjian K, Vaz LE, Begum M, Akter T, Cetre-Sossah CB, Ahluwalia IB, Dotson E, Secor WE, Breiman RF, Maguire JH. Risk factors for kalaazar in Bangladesh. Emerg Infect Dis. 2005;11:655–662. doi: 10.3201/eid1105.040718. http://dx.doi.org/10.3201/eid1105.040718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhunia GS, Kesari S, Chatterjee N, Kumar V, Das P. Spatial and temporal variation and hotspot detection of kala-azar disease in Vaishali district (Bihar) India BMC Infect Dis. 2013;13:64. doi: 10.1186/1471-2334-13-64. http://dx.doi.org/10.1186/1471-2334-13-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruen TC, Philippe H, Bryant D. A simple and robust statistical test for detecting the presence of recombination. Genetics. 2006;172:2665–2681. doi: 10.1534/genetics.105.048975. http://dx.doi.org/10.1534/genetics.105.048975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chagas E. Visceral leishmaniasis in Brazil. Science (80-) 1936;84:397–398. doi: 10.1126/science.84.2183.397-a. http://dx.doi.org/10.1126/science.84.2183.397-a. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Ruden DM, Lu X. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:1–13. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa CH, Gomes RB, Silva MR, Garcez LM, Ramos PK, Santos RS, Shaw JJ, David JR, Maguire JH. Competence of the human host as a reservoir for Leishmania chagasi. J Infect Dis. 2000;182:997–1000. doi: 10.1086/315795. http://dx.doi.org/10.1086/315795. [DOI] [PubMed] [Google Scholar]
- Costa CHN, Pereira HF, Araújo MV. Epidemia de Leishmaniose visceral no Piauí, Brasil, 1980–1986*. Rev Saude Publica 1980–1986. 1990 doi: 10.1590/s0034-89101990000500003. [DOI] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. http://dx.doi.org/10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling ACE, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394– 1403. doi: 10.1101/gr.2289704. http://dx.doi.org/10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane LM, Deane MP. Visceral leishmaniasis in Brazil: geographical distribution and trnsmission. Rev Inst Med Trop Sao Paulo. 1962;4:198–212. [PubMed] [Google Scholar]
- Denise H, Poot J, Jiménez M, Ambit A, Herrmann DC, Vermeulen AN, Coombs GH, Mottram JC. Studies on the CPA cysteine peptidase in the Leishmania infantum genome strain JPCM5. BMC Mol Biol. 2006;7:42. doi: 10.1186/1471-2199-7-42. http://dx.doi.org/10.1186/1471-2199-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin RE, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. http://dx.doi.org/10.1038/ng.806.A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing T, Imamura H, Decuypere S, Clark TG, Coombs GH, Cotton JA, Hilley JD, de Doncker S, Maes I, Mottram JC, Quail MA, Rijal S, Sanders M, Schönian G, Stark O, Sundar S, Vanaerschot M, Hertz-Fowler C, Dujardin J-C, Berriman M. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 2011;21:2143–2156. doi: 10.1101/gr.123430.111. http://dx.doi.org/10.1101/gr.123430.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert AJ, Liechty JD, Tearse BR, Pande B, Neale DB. DnaSAM: Software to perform neutrality testing for large datasets with complex null models. Mol Ecol Resour. 2010;10:542–545. doi: 10.1111/j.1755-0998.2009.02768.x. http://dx.doi.org/10.1111/j.1755-0998.2009.02768.x. [DOI] [PubMed] [Google Scholar]
- El-Sayed NM, Myler PJ, Blandin G, Berriman M, Crabtree J, Aggarwal G, Caler E, Renauld H, Worthey EA, Hertz-Fowler C, Ghedin E, Peacock C, Bartholomeu DC, Haas BJ, Tran AN, Wortman JR, Alsmark UCM, Angiuoli S, Anupama A, Badger J, Bringaud F, Cadag E, Carlton JM, Cerqueira GC, Creasy T, Delcher AL, Djikeng A, Embley TM, Hauser C, Ivens AC, Kummerfeld SK, Pereira-Leal JB, Nilsson D, Peterson J, Salzberg SL, Shallom J, Silva JC, Sundaram J, Westenberger S, White O, Melville SE, Donelson JE, Andersson B, Stuart KD, Hall N. Comparative genomics of trypanosomatid parasitic protozoa. Science. 2005;309:404–409. doi: 10.1126/science.1112181. http://dx.doi.org/10.1126/science.1112181. [DOI] [PubMed] [Google Scholar]
- Evans TG, Teixeira MJ, McAuliffe IT, Vasconcelos IDB, Vasconcelos AW, Sousa AD, Lima JWD, Pearson RD. Epidemiology of visceral leishmaniasis in northeast Brazil. J Infect Dis. 1992;166:1124–1132. doi: 10.1093/infdis/166.5.1124. [DOI] [PubMed] [Google Scholar]
- Franke CR, Staubach C, Ziller M, Schluter H. Trends in the temporal and spatial distribution of visceral and cutaneous leishmaniasis in the state of Bahia, Brazil, from 1985 to 1999. Trans R Soc Trop Med Hyg. 2002;96:236–241. doi: 10.1016/s0035-9203(02)90087-8. [DOI] [PubMed] [Google Scholar]
- Gradoni L, Bryceson A, Desjeux P. Treatment of mediterranean visceral leishmaniasis. Bull World Health Organ. 1995;73:191–197. [PMC free article] [PubMed] [Google Scholar]
- Hefnawy A, Berg M, Dujardin J-C, De Muylder G. Exploiting Knowledge on Leishmania Drug Resistance to Support the Quest for New Drugs. Trends Parasitol. 2016:1–13. doi: 10.1016/j.pt.2016.11.003. http://dx.doi.org/10.1016/j.pt.2016.11.003. [DOI] [PubMed]
- Imamura H, Downing T, van den Broeck F, Sanders MJ, Rijal S, Sundar S, Mannaert A, Vanaerschot M, Berg M, de Muylder G, Dumetz F, Cuypers B, Maes I, Domagalska M, Decuypere S, Rai K, Uranw S, Bhattarai NR, Khanal B, Prajapati VK, Sharma S, Stark O, Schönian G, de Koning HP, Settimo L, Vanhollebeke B, Roy S, Ostyn B, Boelaert M, Maes L, Berriman M, Dujardin JC, Cotton JA. Evolutionary genomics of epidemic visceral leishmaniasis in the Indian subcontinent. Elife. 2016;5:1–39. doi: 10.7554/eLife.12613. http://dx.doi.org/10.7554/eLife.12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivens AC, Peacock CS, Worthey EA, Murphy L, Aggarwal G, Berriman M, Sisk E, Rajandream M, Adlem E, Aert R, Anupama A, Apostolou Z, Attipoe P, Bason N, Bauser C, Beck A, Beverley SM, Bianchettin G, Borzym K, Bothe G, Bruschi CV, Collins M, Cadag E, Ciarloni L, Clayton C, Coulson RMR, Cronin A, Cruz AK, Davies RM, De Gaudenzi J, Dobson DE, Duesterhoeft A, Fazelina G, Fosker N, Frasch AC, Fraser A, Fuchs M, Gabel C, Goble A, Goffeau A, Harris D, Hertz-Fowler C, Hilbert H, Horn D, Huang Y, Klages S, Knights A, Kube M, Larke N, Litvin L, Lord A, Louie T, Marra M, Masuy D, Matthews K, Michaeli S, Mottram JC, Müller-Auer S, Munden H, Nelson S, Norbertczak H, Oliver K, O’neil S, Pentony M, Pohl TM, Price C, Purnelle B, Quail MA, Rabbinowitsch E, Reinhardt R, Rieger M, Rinta J, Robben J, Robertson L, Ruiz JC, Rutter S, Saunders D, Schäfer M, Schein J, Schwartz DC, Seeger K, Seyler A, Sharp S, Shin H, Sivam D, Squares R, Squares S, Tosato V, Vogt C, Volckaert G, Wambutt R, Warren T, Wedler H, Woodward J, Zhou S, Zimmermann W, Smith DF, Blackwell JM, Stuart KD, Barrell B, Myler PJ. The genome of the kinetoplastid parasite, Leishmania major. Science. 2005;309:436–442. doi: 10.1126/science.1112680. http://dx.doi.org/10.1126/science.1112680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo SM, Oliveira RM, Mackay S, Costa RM, Sweet J, Nascimento ET, Luz KG, Fernandes MZ, Jernigan J, Pearson RD. An urban outbreak of visceral leishmaniasis in Natal, Brazil. Trans R Soc Trop Med Hyg. 1994;88:386–388. doi: 10.1016/0035-9203(94)90393-x. http://dx.doi.org/10.1016/0035-9203(94)90393-X. [DOI] [PubMed] [Google Scholar]
- Jeronimo SM, Teixeira MJ, Sousa AD, Thielking P, Pearson RD, Evans TG. Natural history of Leishmania (Leishmania) chagasi infection in Northeastern Brazil: long-term follow-up. Clin Infect Dis. 2000;30:608–609. doi: 10.1086/313697. http://dx.doi.org/10.1086/313697. [DOI] [PubMed] [Google Scholar]
- Kaszak I, Planellas M, Dworecka-Kaszak B. Canine leishmaniosis – an emerging disease. Ann Parasitol. 2015;61:69–76. [PubMed] [Google Scholar]
- Kuhls K, Alam MZ, Cupolillo E, Ferreira GEM, Mauricio IL, Oddone R, Feliciangeli MD, Wirth T, Miles Ma, Schönian G. Comparative microsatellite typing of new world Leishmania infantum reveals low heterogeneity among populations and its recent old world origin. PLoS Negl Trop Dis. 2011;5:1–16. doi: 10.1371/journal.pntd.0001155. http://dx.doi.org/10.1371/journal.pntd.0001155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblois R, Kuhls K, François O, Schonian G, Wirth T. Guns, germs and dogs: On the origin of Leishmania chagasi. Infect Genet Evol. 2011;11:1091–1095. doi: 10.1016/j.meegid.2011.04.004. http://dx.doi.org/10.1016/j.meegid.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. http://dx.doi.org/10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima ID, Queiroz JW, Lacerda HG, Queiroz PVS, Pontes NN, Barbosa JDA, Martins DR, Weirather JL, Pearson RD, Wilson ME, Jeronimo SMB. Leishmania infantum chagasi in Northeastern Brazil: asymptomatic infection at the urban perimeter. Am J Trop Med Hyg. 2012;86:99–107. doi: 10.4269/ajtmh.2012.10-0492. http://dx.doi.org/10.4269/ajtmh.2012.10-0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan-Klumpler FJ, De Silva N, Boehme U, Rogers MB, Velarde G, McQuillan JA, Carver T, Aslett M, Olsen C, Subramanian S, Phan I, Farris C, Mitra S, Ramasamy G, Wang H, Tivey A, Jackson A, Houston R, Parkhill J, Holden M, Harb OS, Brunk BP, Myler PJ, Roos D, Carrington M, Smith DF, Hertz-Fowler C, Berriman M. GeneDB–an annotation database for pathogens. Nucleic Acids Res. 2012;40:D98–D108. doi: 10.1093/nar/gkr1032. http://dx.doi.org/10.1093/nar/gkr1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia-Elkhoury ANS, Alves WA, Sousa-Gomes ML, De Sena JM, De Luna EA. Visceral leishmaniasis in Brazil: trends and challenges. Cad saude publica/Minist da Saude, Fund Oswaldo Cruz, Esc Nac Saude Publ. 2008;24:2941– 2947. doi: 10.1590/S0102-311X2008001200024. [DOI] [PubMed] [Google Scholar]
- Marzochi MC, Marzochi KB. Tegumentary and visceral leishmaniases in Brazil: emerging anthropozoonosis and possibilities for their control. Cad saude publica/Minist da Saude, Fund Oswaldo Cruz, Esc Nac Saude Publ. 1994;10(Suppl 2):359–375. doi: 10.1590/S0102-311X1994000800014. [DOI] [PubMed] [Google Scholar]
- Maurício IL, Stothard JR, Miles MA. The strange case of Leishmania chagasi. Parasitol. Today. 2000;16:188–189. doi: 10.1016/s0169-4758(00)01637-9. http://dx.doi.org/10.1016/S0169-4758(00)01637-9. [DOI] [PubMed] [Google Scholar]
- Miles SA, Conrad SM, Alves RG, Jeronimo SM, Mosser DM. A role for IgG immune complexes during infection with the intracellular pathogen Leishmania. J Exp Med. 2005;201:747–754. doi: 10.1084/jem.20041470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca E, Eckert AJ, Liechty JD, Wegrzyn JL, La Porta N, Vendramin GG, Neale DB. Contrasting patterns of nucleotide diversity for four conifers of Alpine European forests. Evol Appl. 2012;5:762–775. doi: 10.1111/j.1752-4571.2012.00256.x. http://dx.doi.org/10.1111/j.1752-4571.2012.00256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. Molecular Evolutionary Genetics. Columbia University Press; New York, USA: 1987. [Google Scholar]
- Paradis E. Pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics. 2010;26:419–420. doi: 10.1093/bioinformatics/btp696. http://dx.doi.org/10.1093/bioinformatics/btp696. [DOI] [PubMed] [Google Scholar]
- Peacock CS, Seeger K, Harris D, Murphy L, Ruiz JC, Quail MA, Peters N, Adlem E, Tivey A, Aslett M, Ivens A, Fraser A, Rajandream M, Carver T, Norbertczak H, Chillingworth T, Hance Z, Jagels K, Moule S, Ormond D, Rutter S, Squares R, Whitehead S, Rabbinowitsch E, Arrowsmith C, White B, Thurston S, Bringaud F, Baldauf SL, Faulconbridge A, Jeffares D, Depledge DP, Oyola SO, James D. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet. 2008;39:839–847. doi: 10.1038/ng2053. http://dx.doi.org/10.1038/ng2053.Comparative. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson RD, Steigbigel RT. Mechanism of lethal effect of human serum upon Leishmania donovani. J Immunol. 1980;125:2195–2201. [PubMed] [Google Scholar]
- Rai K, Bhattarai NR, Vanaerschot M, Imamura H, Gebru G, Khanal B, Rijal S, Boelaert M, Pal C, Karki P, Dujardin J-C, Van der Auwera G. Single locus genotyping to track Leishmania donovani in the Indian subcontinent: Application in Nepal. PLoS Negl Trop Dis. 2017;11:e0005420. doi: 10.1371/journal.pntd.0005420. http://dx.doi.org/10.1371/journal.pntd.0005420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ready PD. Epidemiology of visceral leishmaniasis. Clin Epidemiol. 2014;6:147– 154. doi: 10.2147/CLEP.S44267. http://dx.doi.org/10.2147/CLEP.S44267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers MB, Downing T, Smith BA, Imamura H, Sanders M, Svobodova M, Volf P, Berriman M, Cotton JA, Smith DF. Genomic Confirmation of Hybridisation and Recent Inbreeding in a Vector-Isolated Leishmania Population. PLoS Genet. 2014:10. doi: 10.1371/journal.pgen.1004092. http://dx.doi.org/10.1371/journal.pgen.1004092. [DOI] [PMC free article] [PubMed]
- Rogers MB, Hilley JD, Dickens NJ, Wilkes J, Bates PA, Depledge DP, Harris D, Her Y, Herzyk P, Imamura H, Otto TD, Sanders M, Seeger K, Dujardin J-C, Berriman M, Smith DF, Hertz-Fowler C, Mottram JC. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011;21:2129–2142. doi: 10.1101/gr.122945.111. http://dx.doi.org/10.1101/gr.122945.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roque ALR, Jansen AM. Wild and synanthropic reservoirs of Leishmania species in the Americas. Int J Parasitol Parasites Wildl. 2014;3:251–262. doi: 10.1016/j.ijppaw.2014.08.004. http://dx.doi.org/10.1016/j.ijppaw.2014.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougeron V, De Meeûs T, Bañuls A-L. A primer for Leishmania population genetic studies. Trends Parasitol. 2015;31:52–59. doi: 10.1016/j.pt.2014.12.001. http://dx.doi.org/10.1016/j.pt.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Salomón OD. Vectores De Leishmaniasis En Las Américas. Gaz Médica da Bahia. 2009;79:3–15. [Google Scholar]
- Schönian G, Kuhls K, Mauricio IL. Molecular approaches for a better understanding of the epidemiology and population genetics of Leishmania. Parasitology. 2011;138:405–425. doi: 10.1017/S0031182010001538. http://dx.doi.org/10.1017/S0031182010001538. [DOI] [PubMed] [Google Scholar]
- Segatto M, Ribeiro LS, Costa DL, Costa CHN, de Oliveira MR, Carvalho SFG, Macedo AM, Valadares HMS, Dietze R, de Brito CFA, Lemos EM. Genetic diversity of Leishmania infantum field populations from Brazil. Mem. Inst. Oswaldo Cruz. 2012;107:39–47. doi: 10.1590/s0074-02762012000100006. [DOI] [PubMed] [Google Scholar]
- Silva ES, Gontijo CM, Pacheco RS, Fiuza VO, Brazil RP. Visceral leishmaniasis in the Metropolitan Region of Belo Horizonte, State of Minas Gerais, Brazil. Mem Inst Oswaldo Cruz. 2001;96:285–291. doi: 10.1590/s0074-02762001000300002. http://dx.doi.org/10.1590/S0074-02762001000300002. [DOI] [PubMed] [Google Scholar]
- Sterkers Y, Lachaud L, Bourgeois N, Crobu L, Bastien P, Pagès M. Novel insights into genome plasticity in Eukaryotes: mosaic aneuploidy in Leishmania. Mol Microbiol. 2012;86:15–23. doi: 10.1111/j.1365-2958.2012.08185.x. http://dx.doi.org/10.1111/j.1365-2958.2012.08185.x. [DOI] [PubMed] [Google Scholar]
- Sterkers Y, Lachaud L, Crobu L, Bastien P, Pagès M. FISH analysis reveals aneuploidy and continual generation of chromosomal mosaicism in Leishmania major. Cell Microbiol. 2011;13:274–283. doi: 10.1111/j.1462-5822.2010.01534.x. http://dx.doi.org/10.1111/j.1462-5822.2010.01534.x. [DOI] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. 6628982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton EL. The Leishmania chromosome lottery. Microbes Infect. 2014;16:2–5. doi: 10.1016/j.micinf.2013.11.008. http://dx.doi.org/10.1016/j.micinf.2013.11.008. [DOI] [PubMed] [Google Scholar]
- Wilson ME, Jeronimo SMB, Pearson RD. Immunopathogenesis of infection with the visceralizing Leishmania species. Microb Pathog. 2005;38:147–160. doi: 10.1016/j.micpath.2004.11.002. http://dx.doi.org/10.1016/j.micpath.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Zemanová E, Jirků M, Mauricio IL, Horák A, Miles MA, Lukeš J. The Leishmania donovani complex: Genotypes of five metabolic enzymes (ICD, ME, MPI, G6PDH, and FH), new targets for multilocus sequence typing. Int J Parasitol. 2007;37:149–160. doi: 10.1016/j.ijpara.2006.08.008. http://dx.doi.org/10.1016/j.ijpara.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Zhang W-W, Matlashewski G. Screening Leishmania donovani-specific genes required for visceral infection. Mol Microbiol. 2010;77:505–517. doi: 10.1111/j.1365-2958.2010.07230.x. http://dx.doi.org/10.1111/j.1365-2958.2010.07230.x. [DOI] [PubMed] [Google Scholar]
- Zhang WW, Mendez S, Ghosh A, Myler P, Ivens A, Clos J, Sacks DL, Matlashewski G. Comparison of the A2 gene locus in Leishmania donovani and Leishmania major and its control over cutaneous infection. J Biol Chem. 2003;278:35508–35515. doi: 10.1074/jbc.M305030200. http://dx.doi.org/10.1074/jbc.M305030200. [DOI] [PubMed] [Google Scholar]
- Zhang WW, Ramasamy G, McCall L-I, Haydock A, Ranasinghe S, Abeygunasekara P, Sirimanna G, Wickremasinghe R, Myler P, Matlashewski G. Genetic Analysis of Leishmania donovani Tropism Using a Naturally Attenuated Cutaneous Strain. PLoS Pathog. 2014;10:e1004244. doi: 10.1371/journal.ppat.1004244. http://dx.doi.org/10.1371/journal.ppat.1004244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28:3326–3328. doi: 10.1093/bioinformatics/bts606. http://dx.doi.org/10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.