

Abstract
The eukaryotic Hsp90 chaperone machinery comprises many co-chaperones and regulates the conformation of hundreds of cytosolic client proteins. Therefore, it is not surprising that the Hsp90 machinery has become an attractive therapeutic target for diseases such as cancer. The compounds used so far to target this machinery affect the entire Hsp90 system. However, it would be desirable to achieve a more selective targeting of Hsp90–co-chaperone complexes. To test this concept, in this-proof-of-principle study, we screened for modulators of the interaction between Hsp90 and its co-chaperone Aha1, which accelerates the ATPase activity of Hsp90. A FRET-based assay that monitored Aha1 binding to Hsp90 enabled identification of several chemical compounds modulating the effect of Aha1 on Hsp90 activity. We found that one of these inhibitors can abrogate the Aha1-induced ATPase stimulation of Hsp90 without significantly affecting Hsp90 ATPase activity in the absence of Aha1. NMR spectroscopy revealed that this inhibitory compound binds the N-terminal domain of Hsp90 close to its ATP-binding site and overlapping with a transient Aha1-interaction site. We also noted that this inhibitor does not dissociate the Aha1–Hsp90 complex but prevents the specific interaction with the N-terminal domain of Hsp90 required for catalysis. In consequence, the inhibitor affected the activation and processing of Hsp90–Aha1-dependent client proteins in vivo. We conclude that it is possible to abrogate a specific co-chaperone function of Hsp90 without inhibiting the entire Hsp90 machinery. This concept may also hold true for other co-chaperones of Hsp90.
Keywords: ATPase, biophysics, heat shock protein 90 (Hsp90), molecular chaperone, protein folding, co-chaperones
Introduction
Heat shock protein 90 (Hsp90)4 is one of the most abundant and evolutionary conserved molecular chaperones. Together with its co-chaperones, it plays a key role in stress and cell cycle control, as well as hormone signaling in the eukaryotic cytosol (1–3). Hsp90 is a flexible homodimer consisting of three domains per monomer. The N-terminal domain (NTD) contains the ATP-binding site, the middle domain (MD) has been implicated in client protein and co-chaperone binding, and the C-terminal domain is the site of dimerization (4–7) and binds several TPR-containing co-chaperones (8–10). Hsp90 undergoes large conformational changes from an open to an N-terminally closed state during its ATPase cycle (11–14). These rate-limiting steps are modulated by co-chaperones (1, 15, 16). Among them, Aha1 (activator of the Hsp90 ATPase) is a potent accelerator of the Hsp90 ATPase (17, 18).
Aha1 is a monomeric, two-domain protein that has two interaction sites with Hsp90: the Hsp90 MD binds to the Aha1 N-domain (19) and the Aha1 C-domain binds more dynamically to the N-domain of Hsp90 supporting the formation of the unfavorable and rate-limiting N-terminally closed state of Hsp90 (20, 21). In the chaperone cycle, Aha1 interaction precedes the formation of the fully closed state (22). Specifically, Aha1 accelerates the association of the two N-terminal domains (20). The activation mechanism is asymmetric because one Aha1 per Hsp90 dimer is sufficient to fully stimulate the Hsp90 ATPase (20). Aha1 is involved in the interaction of Hsp90 with specific client proteins like protein kinases and steroid hormone receptors (17, 23, 24). Furthermore, Aha1 together with Hsp90 has been shown to play an important role in the quality control of mutants of the cystic fibrosis transmembrane conductance regulator (CFTR) (21, 25, 26). Mutations in the CFTR represent the primary cause of cystic fibrosis. In particular, deletion of Phe508 in CFTR results in a loss of function and rapid degradation (25, 27–29). A knockdown of Aha1 led to increased levels of the CFTR ΔF508 mutant (26), suggesting that inhibition of the Hsp90–Aha1 interaction may be beneficial in this context.
Targeting the Hsp90–Aha1 interaction by chemical compounds is therefore attractive as a potential therapeutic approach, and at the same time it could provide insight into the possibility of targeting specific Hsp90–co-chaperone effects. Efforts to target the Hsp90 machinery by small molecules focused mainly on Hsp90. Promising Hsp90 inhibitors like the geldanamycin (GA) derivative 17-allylamino-17-demethoxygeldanamycin (17-AAG), IPI-504 (retaspimycin hydrochloride), and radicicol derivatives like NVP-AUY922 (luminespib) or STA-9090 (ganetespib) entered clinical trials (30–33). All these inhibitors bind to the ATP-binding site of Hsp90. Alternatively, other interaction sites in Hsp90, like the C-terminal domain, have been tried to target by inhibitors. In this context, the amino-coumarin antibiotic novobiocin binds to the C-terminal domain of Hsp90 and has shown promising results in prostate cancer cells (33, 34). In a previous proof of concept study, we showed that it is also possible to identify compounds that accelerate the ATPase cycle of Hsp90 (35), reminiscent of the function of the co-chaperone Aha1.
For the Aha1–Hsp90 interaction, it has been shown that it is in principle possible to interfere with complex formation by chemical compounds (36). Here we report in a proof-of-principle study identifying modulators that specifically interfere with the catalytic function of the co-chaperone Aha1 but do not affect complex formation with Hsp90 in general. The characterization of the mode of action of the key compound reveals that by binding to the Hsp90 N-domain, the interaction important for accelerating the ATPase is blocked.
Results
Identification of Hsp90–Aha1 modulators
We monitored the interaction between Hsp90 and Aha1 by FRET (22) to screen a compound library of ∼15,000 chemical compounds. Our FRET system consists of Hsp90 carrying a fluorescent acceptor dye (Atto 550) in the M-domain and of Aha1, which is randomly labeled with a donor dye (Alexa 488). Binding of Aha1 to Hsp90 results in an increase of the FRET efficiency (Fig. 1, A and B). Because Aha1 binds preferably to a closed conformation of Hsp90 that can be induced by the nonhydrolyzable ATP analog AMP-PNP (8, 11, 12), FRET kinetics were recorded after the addition of AMP-PNP to labeled Hsp90 and Aha1 (Fig. 1B). DMSO, added as a negative control, had no influence on the FRET kinetics. Because there are no specific Hsp90–Aha1 modulators known so far, the Hsp90 inhibitor GA served as a positive control. GA binds to the nucleotide-binding pocket of Hsp90 and keeps Hp90 in an open conformation (37). Accordingly, the FRET kinetics were significantly influenced. Having established that the Hsp90–Aha1 FRET system responds to perturbations of the interaction, we analyzed the effects of the library compounds. Representative kinetic traces for the screened compounds are depicted in Fig. 1B. Some compounds exhibited strong inhibitory effects on the Hsp90–Aha1 interaction and significantly slowed down the FRET kinetics. We also found activators of the Hsp90–Aha1 interplay as manifested by faster FRET kinetics. Compounds altering the FRET kinetics of the Hsp90–Aha1 complex formation were rescreened twice to exclude false positive hits. We identified ∼40 compounds as possible modulators of the Hsp90–Aha1 interaction. Fig. 1C depicts the structures of the six most effective Hsp90–Aha1 modulators (HAMs). The same library had been recently used to identify Hsp90 ATPase accelerators (35). Interestingly, there is no overlap of compounds identified in this study and the molecules uncovered by the Hsp90–Aha1 FRET system. This further implies that the compounds identified are specific for modulating the interaction of Hsp90 with Aha1.
Figure 1.

Identification of hits. A, Hsp90–Aha1 FRET system with donor-labeled yAha1 and acceptor-labeled yHsp90 was used to screen a 15,000-compound library for the identification of specific modulators of the Hsp90–Aha1 interaction. B, FRET kinetics of Hsp90–Aha1 with active compounds. Compounds (500 μg/ml; HAM-1 (green), HAM-2 (lilac), HAM-3 (dark blue), HAM-4 (pink), HAM-5 (blue), and HAM-6 (red)) were added to each 500 nm of Hsp90 and Aha1 and preincubated. The addition of 2 mm AMP-PNP allowed monitoring complex formation. DMSO (black) and GA (purple) served as negative and positive controls, respectively. C, structures of the HAMs.
The modulators alter the stimulatory effect of Aha1 on the Hsp90 ATPase
The identified modulators were further studied for their effects on the Hsp90 ATPase in the presence of Aha1. Aha1 is a strong accelerator of the Hsp90 ATPase cycle (17, 18, 20). In the presence of the identified compounds (Fig. 1C), the stimulatory effect of Aha1 on the Hsp90 ATPase was altered (Fig. 2A). Three compounds acted as inhibitors of the Aha1 stimulation on Hsp90, whereas three other compounds further stimulated catalysis. Compound HAM-1, the strongest inhibitor, is able to almost completely suppress the Aha1-mediated stimulation of Hsp90 (93 ± 1% inhibition). The modulators HAM-2 and HAM-3 inhibit the Aha1 stimulation by 27 ± 3 and 20 ± 1%, respectively. The activating compounds HAM-4 and HAM-5 increased the ATPase by 1.3- and 1.5-fold (130 ± 8 and 151 ± 8% of the Hsp90–Aha1 activity), respectively. HAM-6 did not have a significant effect on the ATPase.
Figure 2.

A, effect on the stimulatory potential of Aha1 on the Hsp90 ATPase. Normalized ATPase activities of Hsp90 with Aha1 at saturating concentrations. B, effects of the modulators on Hsp90 ATPase without Aha1. Compounds were added to the Hsp90–Aha1 complex in saturating conditions. C, complex formation of Hsp90 and Aha1 in the presence of HAM-1 and AMP-PNP. Black, Aha1; lilac, Hsp90–Aha1; red, Aha1 + 240 μm HAM-1; light blue, Hsp90–Aha1 + 240 μm HAM-1; pink, Hsp90–Aha1 + 120 μm HAM-1; green, Hsp90–Aha1 + 60 μm HAM-1; dark blue, Hsp90–Aha1/AMP-PNP + 500 μm HAM-1. D, SPR measurements to determine the Aha1 affinity to Hsp90 in the presence of HAM-1 and nucleotide (AMP-PNP). Black, Hsp90; red, Hsp90/AMP-PNP; blue, Hsp90/HAM-1; green, Hsp90/AMP-PNP/HAM-1.
The apparent binding constant (KD app) of each modulator to the Hsp90–Aha1 complex was determined by titration of the compounds in the ATPase assay. All compounds displayed affinities in the micromolar range (Table 1). HAM-1 has the highest affinity for Hsp90–Aha1 with a KD app of 24 ± 2 μm. For HAM-2, a KD app of 40 ± 5 μm was determined, whereas the affinity for HAM-3 was found to be higher than 100 μm (KD app > 100 μm). The activators, HAM-4 and HAM-5, have KD app values of 163 ± 42 and 145 ± 70 μm, respectively. It should be noted that because of the low affinity for HAM-4 and HAM-5, we could not reach saturation in the ATPase assays (supplemental Fig. S1).
Table 1.
Apparent affinity of the modulators for Hsp90/Aha1 and their effects on ATP turnover in the presence and absence of Aha1
ATPase measurements were performed as described under “Experimental procedures.” The KDapp for the modulators was determined by assaying ATP turnover at different modulator concentrations and nonlinear curve fitting. The relative Hsp90 ATPase activity was determined by measuring the ATPase activity in the presence of saturating concentrations of the modulator relative to the turnover in the absence of modulators in the presence of 1% DMSO.
| Compound | KDapp Hsp90–Aha1 | Relative Hsp90–Aha1 ATPase | Relative Hsp90 ATPase |
|---|---|---|---|
| μm | % | % | |
| 1% DMSO | – | 100 | 100 |
| HAM-1 | 24 ± 2 | 7 ± 1 | 78 ± 2 |
| HAM-2 | 40 ± 5 | 73 ± 3 | 206 ± 3 |
| HAM-3 | >100 | 81 ± 1 | 146 ± 12 |
| HAM-4 | 163 ± 42 | 130 ± 8 | 565 ± 8 |
| HAM-5 | 145 ± 70 | 151 ± 8 | 220 ± 5 |
| HAM-6 | >100 | 107 ± 9 | 126 ± 3 |
The assays described above do not tell us whether the modulators act on the Hsp90–Aha1 complex or on Hsp90 alone. To clarify this, we performed ATPase assays in the absence of Aha1 (Fig. 2B and Table 1). Here, the modulator HAM-1 did not have a significant influence on the Hsp90 ATPase activity without Aha1 (inhibition by HAM-1, 22 ± 2%; activation by HAM-6, 26 ± 3%). For HAM-2, surprisingly, we found that it activates the Hsp90 ATPase 2-fold when Aha1 is not present. Together with Aha1, HAM-2 acts as an inhibitor of the Hsp90–Aha1 chaperone system (inhibition by 27 ± 3%). This implies that HAM-2 is a unique type of modulator of the Hsp90 chaperone machinery. HAM-4 exhibited an almost 6-fold activation (565 ± 8%) of the Hsp90 ATPase activity. When Aha1 is present, HAM-4 has a much weaker stimulatory effect on the Hsp90 ATPase (30 ± 8% stimulation). HAM-5, screened as an activator of the Hsp90–Aha1 complex, also activated the Hsp90 ATPase 2-fold (220 ± 5%). These findings suggest that HAM-2, -4, and -5 might directly bind to Hsp90. Because HAM-1 and -6 do not significantly alter the Hsp90 ATPase activity, they may either affect Aha1 directly or the Aha1–Hsp90 interaction.
In the following, we focused on HAM-1, because this compound exhibited the most promising properties. Derivatives of HAM-1 (supplemental Fig. S2) were generated and characterized ( supplemental Figs. S3 and S6). However, none of these showed improved characteristics concerning the effects on Hsp90–Aha1.
Because HAM-1 had a profound effect on the stimulation of the Hsp90 ATPase by Aha1, it could interfere with the binding of Aha1 to Hsp90. Analytical ultracentrifugation was used to analyze complex formation between Hsp90 and Aha1 in the presence of HAM-1 (Fig. 2C). Under all conditions tested (+HAM-1, +AMP-PNP, or +HAM-1/AMP-PNP), the Hsp90–Aha1 complex was formed. Also, a titration of HAM-1 to Hsp90 and Aha1 did not reveal an influence on complex formation. Therefore, we conclude that HAM-1 does not abolish Aha1 association with Hsp90. To test binding of Aha1 to Hsp90 directly by an orthogonal method, SPR measurements were performed (Fig. 2D). Aha1 was titrated to Hsp90 in the presence of HAM-1. The affinity of Aha1 for Hsp90 is only marginally affected by the presence of HAM-1, both in the absence (from a KD of 3.12 ± 0.65 to 4.70 ± 0.74 μm) or presence of the non-hydrolyzable ATP analog AMP-PNP (from a KD of 2.55 ± 0.30 μm to a KD = 1.80 ± 0.30 μm) (Fig. 2D). These findings show that the inhibitor HAM-1 does not prevent binding of Aha1 to Hsp90, whereas at the same time, Aha1 is not able to stimulate Hsp90 efficiently anymore.
The modulator HAM-1 binds to the Hsp90 N-domain
To determine the binding site of the inhibitor HAM-1, we recorded 1H,15N NMR correlation spectra of 15N-labeled Hsp90 NTD or MD in the absence and presence of the inhibitor HAM-1. Although the MD shows minor differences in chemical shift upon addition of HAM-1 (supplemental Fig. S4A), the NTD exhibits pronounced chemical shift perturbations (Fig. 3A and supplemental Fig. S5). Residues with large chemical shift perturbations cluster around the nucleotide-binding site in the NTD of Hsp90 (Fig. 3B). Especially the NMR signals assigned to residues in helix α2 are strongly affected. Interestingly, this region overlaps partly with the previously determined second Aha1-binding site in the NTD (20) (Fig. 3B). To test whether HAM-1 affects ATP binding, we recorded 1H,15N correlation spectra of the Hsp90 NTD in the presence of ATP. Upon adding HAM-1 to the ATP-bound NTD, NMR chemical shifts move to the same position as observed for the NTD bound to HAM-1 in the absence of ATP. In addition, a number of signals with weaker intensity remains with chemical shifts of the ATP-bound NTD (Fig. 3A, blue spectrum). This indicates that binding of HAM-1 can release ATP from the nucleotide-binding pocket. Because NMR signals for both the HAM-1 and the ATP-bound state are visible, the exchange between these two states is slow on the NMR time scale. As control experiments, we tested binding of HAM-1 to the individual Aha1 domains. Here, the addition of HAM-1 to the Aha1 ND and CD domains showed no or very minor differences in chemical shifts, respectively, indicating no significant interaction (supplemental Fig. S4, B and C).
Figure 3.

Interaction of HAM-1 with Hsp90. A, zoomed views of a 1H,15N correlation spectrum of the Hsp90 NTD (black) superimposed with spectra of the complex with HAM-1 (red), the ATP-bound form (green), and the complex with HAM-1 in the presence of ATP (blue). B, chemical shift changes upon HAM-1 binding >0.08 ppm (red) and resonance frequency changes, which could not be traced (purple spheres) are highlighted on the structure of the full-length Hsp90 homodimer (Protein Data Bank code 2CG9). The bound nucleotide is colored in orange to mark the nucleotide-binding pocket.
HAM-1 effects Hsp90–Aha1 client processing in vivo
To test whether HAM-1 has an influence on Hsp90 client proteins in vivo, we used well established reporter assays in yeast (38–41). We first analyzed the modulator's effects on steroid hormone receptor (SHR) activity, specifically on the glucocorticoid receptor (GR) and the mineralocorticoid receptor (MR). Both are highly dependent on Hsp90 and its co-chaperones (42–45). Inhibition of Hsp90 with radicicol and GA showed reduced GR and MR activity in vivo (46, 47). In contrast to GR, MR seems to be also dependent on Aha1. When we added HAM-1 to the yeast cultures, we observed that HAM-1 exhibits a strong inhibitory effect on MR activity by decreasing its activity to 40 ± 6% in a concentration-dependent manner (Fig. 4A, black line), although the activity of GR was not affected (Fig. 4A, red line). Thus, we are able to specifically affect an Aha1-dependent client by HAM-1 in vivo. Determining the dose response of MR to HAM-1 resulted in an IC50 of 125.5 μm. To further investigate the effect of HAM-1 on MR activity, we tested its influence on MR in WT and ΔAha1 yeast strains (Fig. 4B). MR activity was not significantly altered in the ΔAha1 strain upon addition of HAM-1, whereas in the WT MR strain HAM-1 resulted in a pronounced inhibition. This finding underlines the selectivity of HAM-1 on the Hsp90–Aha1 interaction. Taken together, we could show that the identified compound is a potent modulator of Hsp90 function in vitro and in vivo. The selectivity of HAM-1 for MR, with a dependence on Aha1, demonstrates the Aha1 specificity of the effect.
Figure 4.

Effects of the modulator HAM-1 on SHR activity in Saccharomyces cerevisiae. SHR activity (mean ± S. D.) in yeast was monitored at different HAM-1 concentrations (0–1000 μm) by using the β-galactosidase reporter assay. A, effects of HAM-1 on MR (black) and GR (red) activity, respectively. B, effects of HAM-1 on MR activity on WT (black) compared with ΔAha1 (red). DMSO was included as negative control in ΔAha1 (blue).
HAM-1 alters CFTR degradation
Degradation of ΔF508 CFTR, which causes cystic fibrosis, was shown to be triggered by the Hsp90 machinery (21, 26). Because Aha1 has been described to be an important co-chaperone in this context (26), we tested whether the modulator identified in this study can also affect CFTR stability. To this end, we used a WT yeast strain (BY4741) expressing WT CFTR or ΔF508 CFTR, respectively (48). We followed CFTR degradation after addition of the translation inhibitor cycloheximide over time in the absence and presence of HAM-1 (Fig. 5). CFTR levels were corrected for variations of a loading control. After 5 min, a decrease of the overall CFTR levels can be detected. 10% of WT CFTR and 40% of the ΔF508 variant were degraded. The ΔF508 CFTR variant was degraded faster than WT CFTR, which is in agreement with the literature and manifests its instability (49, 50). In the presence of HAM-1, higher amounts of ΔF508 CFTR and thus a prolonged half-life could be observed (40% degradation compared with 70% without the modulator; Fig. 5B). This stabilization of the ΔF508 variant implies a specific effect of HAM-1 on the Hsp90–Aha1 interplay. Consistent with a weaker dependence of WT CFTR on Hsp90–Aha1 compared with the ΔF508 variant (26), HAM-1 did not have a strong effect on the stabilization on WT CFTR (Fig. 5A).
Figure 5.

Effect of HAM-1 on CFTR degradation. A and B, cycloheximide chase (0–40 min) of yeast cells expressing human HA-tagged CFTR WT (A) and ΔF508 (B) in the absence (black) and presence (red) of HAM-1 (200 μm), respectively. CFTR levels were detected by Western blot (α-HA) and levels corrected for PGK loading controls (representative Western blot). Five independent experiments for WT CFTR and seven independent experiments for ΔF508 CFTR were performed. The data represent means and standard error of the means.
Discussion
Our findings demonstrate that it is in principle possible to selectively target the influence of Aha1 on Hsp90 by chemical compounds. Screening a 15,000-compound-spanning library resulted in the identification of several modulators for the Hsp90–Aha1 interaction. Six chemical compounds showed promising effects and characteristic profiles in vitro. HAM-1 was the most effective inhibitor in terms of binding and the potential to alter the Aha1-mediated stimulation of Hsp90. The presence of HAM-1 resulted in a roughly 90% inhibition of the stimulatory effect of Aha1 on Hsp90, whereas the effect on the Hsp90 ATPase in the absence of Aha1 was moderate. We would have expected that this inhibitor abolishes complex formation of Hsp90 with Aha1, which was surprisingly not the case. Analytical ultracentrifugation and SPR measurements showed that the Hsp90–Aha1 complex is formed in the presence of HAM-1. However, this complex seems to be inefficient in progressing through the Hsp90 ATPase cycle because Aha1 cannot potently stimulate the N-terminal association of Hsp90 anymore. Modulation of the Hsp90 chaperone machinery by HAM-1 therefore seems to be specific for Aha1. These results may be counterintuitive at first glance because only the function of Aha1 is affected but not its interaction with Hsp90. However, one has to bear in mind that the interaction of Aha1 with Hsp90 is complex (Fig. 6), with the two domains of Aha1 interacting with two different domains of Hsp90: the Aha1 N-domain stably interacts with the Hsp90 M-domain (6, 19, 51). The interaction of this domain alone does not strongly affect the Hsp90 ATPase cycle. Only together with the Aha1 C-domain, which interacts at a specific point of the conformational transitions with the Hsp90 N-terminal domain, has an acceleration been observed (17, 20, 21). This transient interaction occurs only for a short period of time in the reaction cycle of Hsp90 to stabilize a specific closed state the formation of which is rate-limiting (12, 20). Thus, we assumed that HAM-1 may interfere with this interaction. Our NMR results are consistent with this notion: HAM-1 interacts with the Hsp90 N-domain. This binding site has a significant overlap with the Aha1 binding site in the Hsp90 N-domain. Because Aha1 can still bind to Hsp90, our results suggest that the Aha1 N-domain is not affected in its interaction with the Hsp90 M-domain. In contrast, the Aha1 C-domain exhibits an impaired interaction with the Hsp90 N-domain in the presence of HAM-1 and is no longer able to stimulate the Hsp90 ATPase to its full potential. Thus, HAM-1 seems to exert its function by sterically blocking the interaction of Aha1 with the Hsp90 N-domains required to accelerate the formation of an N-terminally closed state, which is rate-limiting for ATP hydrolysis (Fig. 6). Furthermore, HAM-1 does not change Aha1 affinity for Hsp90 significantly as determined by SPR measurements (Fig. 4D). This further supports a mechanism where HAM-1 affects more the transient interaction between Hsp90 and Aha1, which is crucial for the accelerating effect of Aha1 on the Hsp90 cycle. In other words, in the presence of HAM1, Aha1 is still bound to Hsp90, but it is no longer functional as an accelerator of the ATPase, because the second, “catalytic” interaction can no longer occur.
Figure 6.
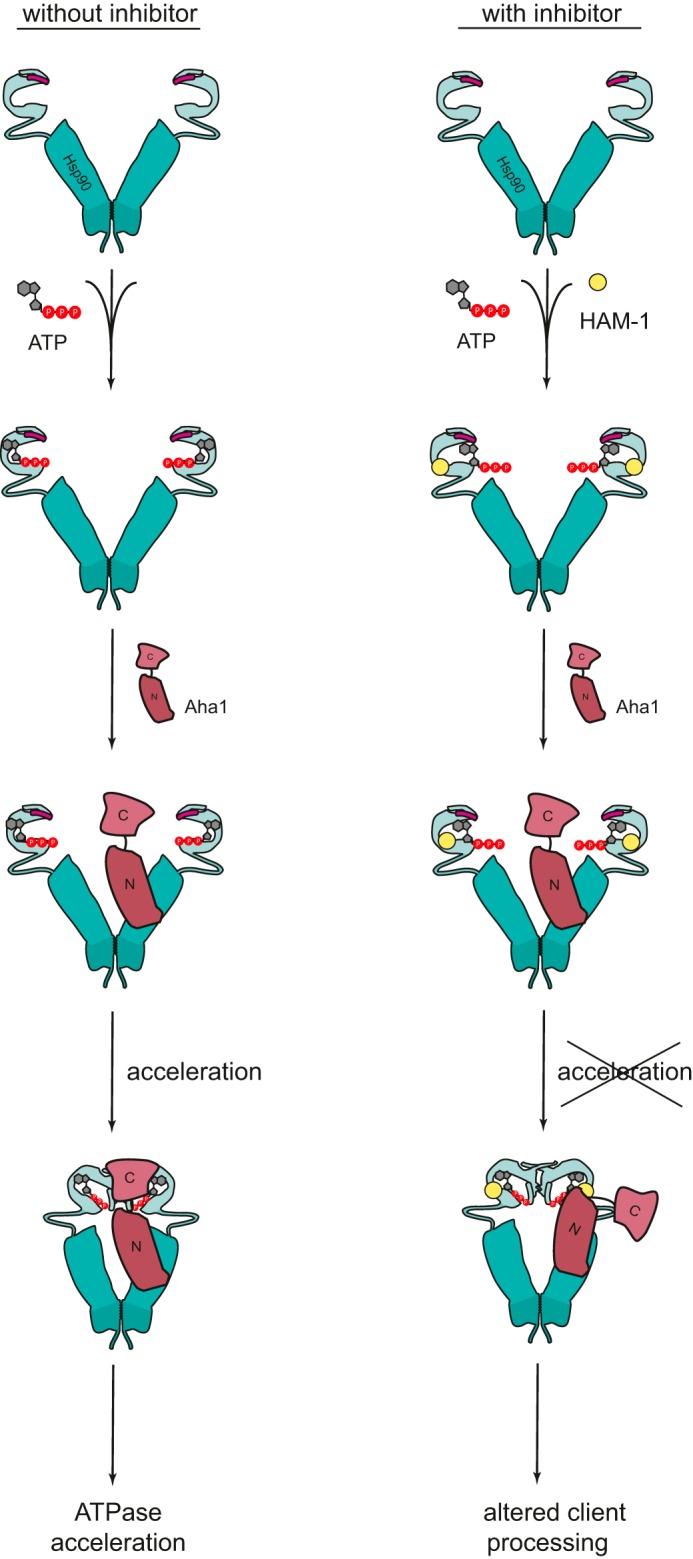
Model for the mode of interaction of HAM-1 with Hsp90–Aha1.
This impeded interaction also influences the processing of different Hsp90 clients in vivo. SHR such as GR are highly dependent on Hsp90 (52), and the inhibition of Hsp90 by RA or GA results in reduced GR activity in vivo (46, 47). In all cases investigated so far, the activity of GR is not completely abolished, which may be due to spontaneous folding or assistance by the Hsp70 chaperone system. Interestingly, HAM-1 did not inhibit the processing and activation of GR in vivo. Because Aha1 is not required for GR processing and because HAM-1 only slightly affects the Hsp90 ATPase, these results point toward a selectivity of HAM-1 on the Hsp90–Aha1 interplay. In contrast, the activation of MR, a client that requires Aha1, was impaired by 40%. The fact that the MR activity was not altered by HAM-1 in the ΔAha1 strain further supports a mechanism where HAM-1 requires the presence of Aha1 to unfold its inhibitory potential. Additionally, MR processing in vivo was inhibited by HAM-1 in a dose-dependent manner, resulting in an IC50 of 125.5 μm, which is lower than the affinity of HAM-1 for Hsp90–Aha1 measured in vitro (23.5 ± 1.7 μm). In this context, it should be noted that we do not know the effective inhibitor concentration in the yeast cell and have to assume that the uptake of the compound is counteracted by efflux pumps (53). Similarly, when a well established general inhibitor of the Hsp90 ATPase (RA) was used to inhibit Hsp90 in yeast, much higher concentrations were required to abrogate the effect of Hsp90 on MR and GR than for the inhibition of Hsp90 in vitro (54). In this case, the Hsp90-dependent activation of MR and GR was affected by ∼80%, which may reflect the higher affinity of RA for Hsp90, differences in uptake/efflux or differences in mechanism because RA is inhibiting the entire Hsp90 system, and HAM-1 affects a specific aspect.
Given its specificity for the effect of Aha1, HAM-1 could have a beneficial effect in the context of cystic fibrosis where the misfolded ΔF508 CFTR protein, which most commonly causes the childhood disease cystic fibrosis, is an Hsp90 client (25, 26). Together with the quality control system of the endoplasmic reticulum, the misfolded ΔF508 CFTR is targeted by the Hsp90 machinery for degradation (55–57). Interestingly, the interplay between Hsp90 and Aha1 seems to be crucial for the ERAD-associated degradation of the ΔF508 CFTR because the down-regulation of Aha1 was shown to promote the stability of the ΔF508 channel activity (26–28, 59). Our results further support this scenario because in the presence of HAM-1, a prolonged life time of ΔF508 CFTR was observed. The inhibition of the Hsp90–Aha1 interplay in this context might thus provide a novel approach to target the folding problem of the CFTR in cystic fibrosis. In a previous study by the Obermann lab (36), compounds were identified that, in combination with the VX-809 corrector, could support the activity of ΔF508 CFTR. In this case, the mechanism seems to be different here because the dissociation of the Aha1–Hsp90 complex may be the underlying principle (36). Because Hsp90 and Aha1 are involved in the processing of several other disease-related clients like the kinases Raf and c-Abl (23, 60), modulating the Hsp90–Aha1 interaction might also have beneficial effects in the context of cancer and other diseases. This concept may also be applicable to other Hsp90 co-chaperone interactions.
Experimental procedures
Protein purification and labeling
Yeast Hsp90 and Aha1 were expressed and purified as described previously (61). For NMR experiments, the Hsp90 NTD and MD, as well as the Aha1 ND and CD, were 15N-labeled. Fluorescent labeling of the yeast Hsp90 cysteine variant Q385C with Atto 550 (Atto-Tec, Siegen, Germany) was performed as described earlier (12). Yeast Aha1 was labeled randomly via cysteine residues with Alexa Fluor 488 C5 maleimide dye (Life Technologies) according to the manufacturer's protocol. Free label was separated as previously described (22).
FRET-based screening
For the identification of specific modulators of the Hsp90–Aha1 interaction, a compound library composed of ∼15,000 small molecules (New Chemistry and Discovery Chemistry Collection, ChemDiv, San Diego, CA) was screened. We made use of an Hsp90–Aha1 FRET system (22). To this end, 500 nm of labeled Hsp90 (yHsp90 Q385C* Atto 550) and 500 nm of labeled Aha1 (yAha1*Alexa 488) were mixed in low salt buffer (40 mm HEPES, 20 mm KCl, 5 mm MgCl2, pH 7.5) to allow formation of the FRET complex. This complex was incubated in a 96-well plate with the compounds of the library (dissolved in DMSO; final concentration, 50 μg/ml; 1% DMSO). The conformational changes leading to FRET were induced by the addition of 2 mm AMP-PNP (Roche). DMSO and 100 μm of the Hsp90 inhibitor GA (Sigma–Aldrich) were used as negative and positive controls, respectively. Changes in acceptor fluorescence were recorded for 20 min at 25 °C in a Wallac EnVision Xcite multilabel plate reader (Perkin Elmer). A photometric 570/8 emission filter (center wavelength, 570 nm; bandwidth, 8 nm) and excitation at 490 nm (FITC, 485/14; center wavelength, 485 nm; bandwidth, 14 nm) were used. The Wallac Envision Manager Software 1.12 was used to overlay and evaluate the recorded kinetic traces. The Z-factor of the assay was calculated based on the following equation.
| (Eq. 1) |
The Z-factor is at least 0.4 for the selection procedure to determine hits based on the kinetics of the closing reaction, which was used as primary screen and 0.7 for the ATPase reaction, which was used as secondary screen.
ATPase activity and affinity measurements
ATPase assays, using an ATP-regenerating enzyme system, were performed as previously described (62). For testing the effects of the modulators on the Hsp90–Aha1 complex, an Hsp90 concentration of 1 μm and an Aha1 concentration of 20 μm was used. Compounds were titrated to the Hsp90–Aha1 complex in varying concentrations from 0 to 0.1 mg/ml (1% DMSO, final concentration). To determine the ATPase activity of Hsp90 in the absence of Aha1, assays were performed with 3 μm Hsp90. Measurements were carried out in low salt buffer (40 mm HEPES, 20 mm KCl, 5 mm MgCl2, pH 7.5) at 30 °C in a Cary 50 UV-visible spectrophotometer (Varian Inc.; Agilent Technologies, Santa Clara, CA). Assays were performed with 2 mm ATP (Roche). The recorded kinetics were analyzed using the OriginPro software version 8.6 (OriginLab Corporation, Northhampton, MA) to determine the catalytic activity of Hsp90. Apparent binding affinities (KD app) were determined by applying a Michaelis–Menten fit.
The apparent binding affinities of Aha1 to Hsp90 in the presence of the modulators (final concentration, 0.01 mg/ml) were measured under varying concentrations of Aha1 (0–30 μm). To determine the KD app of ATP to Hsp90 and Hsp90–Aha1, 0–3 mm ATP was titrated to 3 μm Hsp90 and 1 μm Hsp90/20 μm Aha1, respectively. All ATPase assays were measured at least three times. The results are expressed as the mean catalytical activity and KD app, respectively, ± standard deviation.
Surface plasmon resonance
SPR analysis was carried out in a Biacore X100 system (GE Healthcare Life Sciences). Hsp90 was coupled to a CM5 sensor chip using amine coupling (500 RU) following the manufacturer's instructions. Direct binding of Aha1 (0–5 μm) was measured in low salt buffer. Binding of Aha1 to Hsp90 was addressed under different conditions, adding nucleotide (2 mm AMP-PNP) and/or the inhibitor HAM-1 (60 μm) to the measurement buffer. The recorded sensorgrams were analyzed with the Biacore X100 software (GE Healthcare Life Sciences). For the calculation of binding affinities, steady-state affinity was determined. The steady-state response against concentration was derived from the recorded sensorgrams and then fitted with the Biacore software to obtain the KD.
Analytical ultracentrifugation sedimentation velocity experiments
Analytical ultracentrifugation was carried out with a ProteomLab XL-I (Beckman, Krefeld, Germany) supplied with absorbance optics. 450 μl of the samples were loaded into assembled cells with sapphire windows and 12-mm-path length charcoal-filled Epon double-sector centerpieces and centrifuged at 42,000 rpm in an eight-hole Beckman-Coulter AN50-Ti rotor. Sedimentation was monitored with an UV-visible spectrophotometer, equipped with a monochromator, at 280 nm. Data analysis was carried out with the program Sedfit (63), using a non–model-based continuous Svedberg distribution method (c(S)), with time and radial invariant noise on.
NMR titrations
1H,15N-HSQC NMR spectra were acquired on a Bruker AVIII 600 MHz spectrometer with a cryogenic triple resonance gradient probe. NMR samples of the yeast Hsp90 NTD (residues 1–210) or MD (residues 217–529) were dissolved in 20 mm sodium phosphate buffer (pH 6.5), 100 mm sodium chloride, 2 mm EDTA, 1 mm DTT, and 5% D2O. NMR samples of the yeast Aha1 ND (residues 1–156) and CD (residues 156–356) were prepared accordingly. For the measurements with the Aha1 ND and CD and the Hsp90 MD, protein concentrations of 200, 167, and 200 μm, respectively, and a HAM-1 concentration of 200 μm were used. For the NTD of Hsp90 95 μm protein and 285 μm of HAM-1 were used. Binding was also observed at lower concentrations of HAM-1 (data not shown) but not to saturation. For the measurements of the NTD in complex with ATP, 6 mm magnesium sulfate, and 4 mm of ATP were added to a 100 μm protein sample. Later on, 600 μm of HAM-1 were added. All spectra were processed with NMRPipe/Draw (64) and analyzed with CcpNMR (65).
β-Galactosidase assay
The influence of the modulators on client processing in vivo was tested with a β-galactosidase reporter assay system (39–41). The wild-type yeast strain BY4741 was used to test for SHR activity (MR, GR) in the presence of the modulators. The BY4741 ΔAha1 strain was also included to test the effects of the modulators in the absence of Aha1. Compounds were titrated with concentrations ranging from 0 to 1000 μm to a 10-μl overnight culture of yeast cells and supplemented with 190 μl of selection medium (Ura-His). The GR- and MR-dependent transcription was induced with 10 mm 11-deoxycorticosterone (Sigma–Aldrich) and aldosterone (Sigma–Aldrich), respectively. Cultures were incubated overnight at 30 °C. 50 μl of the yeast culture was centrifuged, and the pellet was lysed with 150 μl of SDS buffer (82 mm Na2HPO4, 12 mm NaH2PO4, 0.1% SDS (w/v), pH 7.5) for 15 min on a shaker at room temperature. The β-galactosidase activity was determined in triplicates after the addition of 50 μl of o-nitrophenyl-β-d-galactopyranosid (ONPG) (Sigma–Aldrich) (4 mg/ml stock in SDS buffer) by measuring the absorption in a Tecan Sunrise Absorbance Reader (Tecan, Mainz, Germany) at 405 nm. Differences in cell amounts were corrected by the respective A600. At least three independent experiments were performed in triplicates, and their mean ± S.D. was determined.
CFTR degradation assay
To assess the effect of the identified modulators on CFTR stability in vivo, we used a yeast strain (BY4741) expressing HA-tagged CFTR WT and ΔF508, respectively (the plasmids were a kind gift from Jeff Brodsky). CFTR degradation assays (48) were performed with yeast cells grown to log phase in the presence and absence of the modulator HAM-1 (200 μm). The assays were performed at least five times in triplicate. Protein synthesis was arrested by the addition of cycloheximide (50 μg/ml). For the analysis of the CFTR stability over time, samples were taken after 0, 5, 15, 25, and 40 min and prepared for Western blot with the TCA extraction protocol (48, 58). Samples were run on a gradient gel and transferred to a PVDF membrane. For the detection of CFTR, an anti-HA antibody (monoclonal anti-HA antibody, clone HA-7 (mouse); Sigma–Aldrich) was used. As controls, antibodies against PGK, yHsp90, yAha1, and yHsp70 (Dr. Pineda, Berlin, Germany) were employed. Primary antibodies were detected with sheep anti-mouse IgG horseradish peroxidase-conjugated or goat anti-rabbit horseradish peroxidase-conjugated antibody (Sigma–Aldrich) and an ECL Western blotting system (Western-Bright ECL Spray, Advansta, Menlo Park, CA) according to the manufacturer's specifications. Quantification of the Western blots was performed with the ImageJ software. Means of at least five experiments and the standard error of the means were calculated.
Author contributions
S. C. S. and J. B. designed the study; S. C. S., M. R., and V. S. K. planned and performed the experiments; and S. C. S., M. R., C. J., V. S. K., M. S., and J. B. analyzed data and contributed to writing the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Jeffrey L. Brodsky and Jennifer Lynn Goeckler-Fried for CFTR plasmids and protocols, Priyanka Sahasrabudhe for helpful advice and protocols for SHR experiments, and Klaus Richter for experimental advice and discussions.
This work was supported by a grant from the BMBF Project ProNet T3 (to J. B.), Deutsche Forschungsgemeinschaft Grant SFB 1035 (to J. B., M. S., and S. A. S.), and a doctoral scholarship from the Studienstiftung des deutschen Volkes (to S. C. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental text and Figs. S1–S6.
- Hsp90
- heat shock protein 90
- HAM
- Hsp90–Aha1 modulator
- CFTR
- cystic fibrosis transmembrane conductance regulator
- GA
- geldanamycin
- RA
- radicicol
- SPR
- surface plasmon resonance
- NTD
- N-terminal domain
- MD
- middle domain
- AMP-PNP
- adenosine 5′-(β,γ-imido)triphosphate
- SHR
- steroid hormone receptor
- GR
- glucocorticoid receptor
- MR
- mineralocorticoid receptor.
References
- 1. Röhl A., Rohrberg J., and Buchner J. (2013) The chaperone Hsp90: changing partners for demanding clients. Trends Biochem. Sci. 38, 253–262 [DOI] [PubMed] [Google Scholar]
- 2. Taipale M., Jarosz D. F., and Lindquist S. (2010) HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 11, 515–528 [DOI] [PubMed] [Google Scholar]
- 3. Richter K., Haslbeck M., and Buchner J. (2010) The heat shock response: life on the verge of death. Mol. Cell 40, 253–266 [DOI] [PubMed] [Google Scholar]
- 4. Ratzke C., Nguyen M. N., Mayer M. P., and Hugel T. (2012) From a ratchet mechanism to random fluctuations evolution of Hsp90's mechanochemical cycle. J. Mol. Biol. 423, 462–471 [DOI] [PubMed] [Google Scholar]
- 5. Pearl L. H., and Prodromou C. (2006) Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem. 75, 271–294 [DOI] [PubMed] [Google Scholar]
- 6. Meyer P., Prodromou C., Hu B., Vaughan C., Roe S. M., Panaretou B., Piper P. W., and Pearl L. H. (2003) Structural and functional analysis of the middle segment of Hsp90: implications for ATP hydrolysis and client protein and cochaperone interactions. Mol. Cell 11, 647–658 [DOI] [PubMed] [Google Scholar]
- 7. Nemoto T., Ohara-Nemoto Y., Ota M., Takagi T., and Yokoyama K. (1995) Mechanism of dimer formation of the 90-kDa heat-shock protein. Eur. J. Biochem. 233, 1–8 [DOI] [PubMed] [Google Scholar]
- 8. Li J., and Buchner J. (2013) Structure, function and regulation of the hsp90 machinery. Biomed. J. 36, 106–117 [DOI] [PubMed] [Google Scholar]
- 9. Scheufler C., Brinker A., Bourenkov G., Pegoraro S., Moroder L., Bartunik H., Hartl F. U., and Moarefi I. (2000) Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70–Hsp90 multichaperone machine. Cell 101, 199–210 [DOI] [PubMed] [Google Scholar]
- 10. Prodromou C., Siligardi G., O'Brien R., Woolfson D. N., Regan L., Panaretou B., Ladbury J. E., Piper P. W., and Pearl L. H. (1999) Regulation of Hsp90 ATPase activity by tetratricopeptide repeat (TPR)-domain co-chaperones. EMBO J. 18, 754–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ali M. M., Roe S. M., Vaughan C. K., Meyer P., Panaretou B., Piper P. W., Prodromou C., and Pearl L. H. (2006) Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 440, 1013–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hessling M., Richter K., and Buchner J. (2009) Dissection of the ATP-induced conformational cycle of the molecular chaperone Hsp90. Nat. Struct. Mol. Biol. 16, 287–293 [DOI] [PubMed] [Google Scholar]
- 13. Mickler M., Hessling M., Ratzke C., Buchner J., and Hugel T. (2009) The large conformational changes of Hsp90 are only weakly coupled to ATP hydrolysis. Nat. Struct. Mol. Biol. 16, 281–286 [DOI] [PubMed] [Google Scholar]
- 14. Weikl T., Muschler P., Richter K., Veit T., Reinstein J., and Buchner J. (2000) C-terminal regions of Hsp90 are important for trapping the nucleotide during the ATPase cycle. J. Mol. Biol. 303, 583–592 [DOI] [PubMed] [Google Scholar]
- 15. Prodromou C. (2012) The “active life” of Hsp90 complexes. Biochim. Biophys. Acta 1823, 614–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li J., Soroka J., and Buchner J. (2012) The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta 1823, 624–635 [DOI] [PubMed] [Google Scholar]
- 17. Panaretou B., Siligardi G., Meyer P., Maloney A., Sullivan J. K., Singh S., Millson S. H., Clarke P. A., Naaby-Hansen S., Stein R., Cramer R., Mollapour M., Workman P., Piper P. W., Pearl L. H., et al. (2002) Activation of the ATPase activity of Hsp90 by the stress-regulated cochaperone Aha1. Mol. Cell 10, 1307–1318 [DOI] [PubMed] [Google Scholar]
- 18. Richter K., Soroka J., Skalniak L., Leskovar A., Hessling M., Reinstein J., and Buchner J. (2008) Conserved conformational changes in the ATPase cycle of human Hsp90. J. Biol. Chem. 283, 17757–17765 [DOI] [PubMed] [Google Scholar]
- 19. Meyer P., Prodromou C., Liao C., Hu B., Roe S. M., Vaughan C. K., Vlasic I., Panaretou B., Piper P. W., and Pearl L. H. (2004) Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. EMBO J. 23, 1402–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Retzlaff M., Hagn F., Mitschke L., Hessling M., Gugel F., Kessler H., Richter K., and Buchner J. (2010) Asymmetric activation of the Hsp90 dimer by its cochaperone Aha1. Mol. Cell 37, 344–354 [DOI] [PubMed] [Google Scholar]
- 21. Koulov A. V., LaPointe P., Lu B., Razvi A., Coppinger J., Dong M.-Q., Matteson J., Laister R., Arrowsmith C., Yates J. R., and Balch W. E. (2010) Biological and structural basis of Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol. Biol. Cell 21, 871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li J., Richter K., Reinstein J., and Buchner J. (2013) Integration of the accelerator Aha1 in the Hsp90 co-chaperone cycle. Nat. Struct. Mol. Biol. 20, 326–331 [DOI] [PubMed] [Google Scholar]
- 23. Holmes J. L., Sharp S. Y., Hobbs S., and Workman P. (2008) Silencing of HSP90 cochaperone AHA1 expression decreases client protein activation and increases cellular sensitivity to the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res. 68, 1188–1197 [DOI] [PubMed] [Google Scholar]
- 24. Harst A., Lin H., and Obermann W. M. (2005) Aha1 competes with Hop, p50 and p23 for binding to the molecular chaperone Hsp90 and contributes to kinase and hormone receptor activation. Biochem. J. 387, 789–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Loo M. A., Jensen T. J., Cui L., Hou Y., Chang X. B., and Riordan J. R. (1998) Perturbation of Hsp90 interaction with nascent CFTR prevents its maturation and accelerates its degradation by the proteasome. EMBO J. 17, 6879–6887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang X., Venable J., LaPointe P., Hutt D. M., Koulov A. V., Coppinger J., Gurkan C., Kellner W., Matteson J., Plutner H., Riordan J. R., Kelly J. W., Yates J. R. 3rd, and Balch W. E. (2006) Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 127, 803–815 [DOI] [PubMed] [Google Scholar]
- 27. Qu B. H., Strickland E. H., and Thomas P. J. (1997) Localization and suppression of a kinetic defect in cystic fibrosis transmembrane conductance regulator folding. J. Biol. Chem. 272, 15739–15744 [DOI] [PubMed] [Google Scholar]
- 28. Riordan J. R. (2005) Assembly of functional CFTR chloride channels. Annu. Rev. Physiol. 67, 701–718 [DOI] [PubMed] [Google Scholar]
- 29. Turnbull E. L., Rosser M. F., and Cyr D. M. (2007) The role of the UPS in cystic fibrosis. BMC Biochem. 8, S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Neckers L., and Workman P. (2012) Hsp90 molecular chaperone inhibitors: are we there yet? Clin. Cancer Res. 18, 64–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin T. Y., Bear M., Du Z., Foley K. P., Ying W., Barsoum J., and London C. (2008) The novel HSP90 inhibitor STA-9090 exhibits activity against Kit-dependent and -independent malignant mast cell tumors. Exp. Hematol. 36, 1266–127718657349 [Google Scholar]
- 32. Eccles S. A., Massey A., Raynaud F. I., Sharp S. Y., Box G., Valenti M., Patterson L., de Haven Brandon A., Gowan S., Boxall F., Aherne W., Rowlands M., Hayes A., Martins V., Urban F., et al. (2008) NVP-AUY922: a novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 68, 2850–2860 [DOI] [PubMed] [Google Scholar]
- 33. Burlison J. A., and Blagg B. S. (2006) Synthesis and evaluation of coumermycin A1 analogues that inhibit the Hsp90 protein folding machinery. Org. Lett. 8, 4855–4858 [DOI] [PubMed] [Google Scholar]
- 34. Eskew J. D., Sadikot T., Morales P., Duren A., Dunwiddie I., Swink M., Zhang X., Hembruff S., Donnelly A., Rajewski R. A., Blagg B. S., Manjarrez J. R., Matts R. L., Holzbeierlein J. M., and Vielhauer G. A. (2011) Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. BMC Cancer 11, 468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zierer B. K., Weiwad M., Rübbelke M., Freiburger L., Fischer G., Lorenz O. R., Sattler M., Richter K., and Buchner J. (2014) Artificial accelerators of the molecular chaperone Hsp90 facilitate rate-limiting conformational transitions. Angew. Chem. Int. Ed. Engl. 53, 12257–12262 [DOI] [PubMed] [Google Scholar]
- 36. Ihrig V., and Obermann W. M. (2017) Identifying inhibitors of the Hsp90–Aha1 protein complex, a potential target to drug cystic fibrosis, by Alpha Technology. SLAS Discov. 22, 923–928 [DOI] [PubMed] [Google Scholar]
- 37. Stebbins C. E., Russo A. A., Schneider C., Rosen N., Hartl F. U., and Pavletich N. P. (1997) Crystal structure of an Hsp90–geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell 89, 239–250 [DOI] [PubMed] [Google Scholar]
- 38. Susek R. E., and Lindquist S. (1990) Transcriptional derepression of the Saccharomyces cerevisiae HSP26 gene during heat shock. Mol. Cell Biol. 10, 6362–6373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smale S. T. (2010) β-Galactosidase assay. Cold Spring Harb. Protoc. 2010, pdb.prot5423 [DOI] [PubMed] [Google Scholar]
- 40. Louvion J. F., Warth R., and Picard D. (1996) Two eukaryote-specific regions of Hsp82 are dispensable for its viability and signal transduction functions in yeast. Proc. Natl. Acad. Sci. U.S.A. 93, 13937–13942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Johnson J. L., and Craig E. A. (2000) A role for the Hsp40 Ydj1 in repression of basal steroid receptor activity in yeast. Mol. Cell Biol. 20, 3027–3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lorenz O. R., Freiburger L., Rutz D. A., Krause M., Zierer B. K., Alvira S., Cuéllar J., Valpuesta J. M., Madl T., Sattler M., and Buchner J. (2014) Modulation of the Hsp90 chaperone cycle by a stringent client protein. Mol. Cell 53, 941–953 [DOI] [PubMed] [Google Scholar]
- 43. Kirschke E., Goswami D., Southworth D., Griffin P. R., and Agard D. A. (2014) Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 157, 1685–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bruner K. L., Derfoul A., Robertson N. M., Guerriero G., Fernandes-Alnemri T., Alnemri E. S., and Litwack G. (1997) The unliganded mineralocorticoid receptor is associated with heat shock proteins 70 and 90 and the immunophilin FKBP-52. Recept. Signal Transduct. 7, 85–98 [PubMed] [Google Scholar]
- 45. Faresse N., Ruffieux-Daidie D., Salamin M., Gomez-Sanchez C. E., and Staub O. (2010) Mineralocorticoid receptor degradation is promoted by Hsp90 inhibition and the ubiquitin-protein ligase CHIP. Am. J. Physiol. Renal Physiol. 299, F1462–F1472 [DOI] [PubMed] [Google Scholar]
- 46. Bamberger C. M., Wald M., Bamberger A. M., and Schulte H. M. (1997) Inhibition of mineralocorticoid and glucocorticoid receptor function by the heat shock protein 90-binding agent geldanamycin. Mol. Cell Endocrinol. 131, 233–240 [DOI] [PubMed] [Google Scholar]
- 47. Rosenhagen M. C., Young J. C., Wochnik G. M., Herr A. S., Schmidt U., Hartl F. U., Holsboer F., and Rein T. (2001) Synergistic inhibition of the glucocorticoid receptor by radicicol and benzoquinone ansamycins. Biol. Chem. 382, 499–504 [DOI] [PubMed] [Google Scholar]
- 48. Zhang Y., Michaelis S., and Brodsky J. L. (2002) CFTR Expression and ER-associated degradation in yeast. In Cystic Fibrosis Methods and Protocols, pp. 257–266, Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 49. Riordan J. R., Rommens J. M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., and Chou J. L. (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245, 1066–1073 [DOI] [PubMed] [Google Scholar]
- 50. Chong P. A., Kota P., Dokholyan N. V., and Forman-Kay J. D. (2013) Dynamics intrinsic to cystic fibrosis transmembrane conductance regulator function and stability. Cold Spring Harb. Perspect. Med. 3, a009522–a009522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lotz G. P., Lin H., Harst A., and Obermann W. M. (2003) Aha1 binds to the middle domain of Hsp90, contributes to client protein activation, and stimulates the ATPase activity of the molecular chaperone. J. Biol. Chem. 278, 17228–17235 [DOI] [PubMed] [Google Scholar]
- 52. Echeverria P. C., and Picard D. (2010) Molecular chaperones, essential partners of steroid hormone receptors for activity and mobility. Biochim. Biophys. Acta. 1803, 641–649 [DOI] [PubMed] [Google Scholar]
- 53. Jungwirth H., and Kuchler K. (2006) Yeast ABC transporters: a tale of sex, stress, drugs and aging. FEBS Lett. 580, 1131–1138 [DOI] [PubMed] [Google Scholar]
- 54. Sahasrabudhe P., Rohrberg J., Biebl M., Rutz D., and Buchner J. (2017) The plasticity of the Hsp90 co-chaperone system. Mol. Cell 10.1016/j.molcel.2017.08.004 [DOI] [PubMed] [Google Scholar]
- 55. Pind S., Riordan J. R., and Williams D. B. (1994) Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 269, 12784–12788 [PubMed] [Google Scholar]
- 56. Farinha C. M., and Amaral M. D. (2005) Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol. Cell Biol. 25, 5242–5252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Amaral M. D. (2006) Therapy through chaperones: sense or antisense? Cystic fibrosis as a model disease. J. Inherit. Metab. Dis. 29, 477–487 [DOI] [PubMed] [Google Scholar]
- 58. Brodsky J. L., Lawrence J. G., and Caplan A. J. (1998) Mutations in the cytosolic DnaJ homologue, YDJ1, delay and compromise the efficient translation of heterologous proteins in yeast. Biochemistry 37, 18045–18055 [DOI] [PubMed] [Google Scholar]
- 59. Nishikawa S., Brodsky J. L., and Nakatsukasa K. (2005) Roles of molecular chaperones in endoplasmic reticulum (ER) quality control and ER-associated degradation (ERAD). J. Biochem. 137, 551–555 [DOI] [PubMed] [Google Scholar]
- 60. Dunn D. M., Woodford M. R., Truman A. W., Jensen S. M., Schulman J., Caza T., Remillard T. C., Loiselle D., Wolfgeher D., Blagg B. S., Franco L., Haystead T. A., Daturpalli S., Mayer M. P., Trepel J. B., et al. (2015) c-Abl mediated tyrosine phosphorylation of Aha1 activates its co-chaperone function in cancer cells. Cell Rep. 12, 1006–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Buchner J., Weikl T., Bügl H., Pirkl F., and Bose S. (1998) Purification of Hsp90 partner proteins Hop/p60, p23, and FKBP52. Methods Enzymol. 290, 418–429 [DOI] [PubMed] [Google Scholar]
- 62. Richter K., Muschler P., Hainzl O., and Buchner J. (2001) Coordinated ATP hydrolysis by the Hsp90 dimer. J. Biol. Chem. 276, 33689–33696 [DOI] [PubMed] [Google Scholar]
- 63. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 65. Skinner S. P., Fogh R. H., Boucher W., Ragan T. J., Mureddu L. G., and Vuister G. W. (2016) CcpNmr AnalysisAssign: a flexible platform for integrated NMR analysis. J. Biomol. NMR. 66, 111–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.