

Abstract
Neuronal inclusions of aggregated RNA‐binding protein fused in sarcoma (FUS) are hallmarks of ALS and frontotemporal dementia subtypes. Intriguingly, FUS's nearly uncharged, aggregation‐prone, yeast prion‐like, low sequence‐complexity domain (LC) is known to be targeted for phosphorylation. Here we map in vitro and in‐cell phosphorylation sites across FUS LC. We show that both phosphorylation and phosphomimetic variants reduce its aggregation‐prone/prion‐like character, disrupting FUS phase separation in the presence of RNA or salt and reducing FUS propensity to aggregate. Nuclear magnetic resonance spectroscopy demonstrates the intrinsically disordered structure of FUS LC is preserved after phosphorylation; however, transient domain collapse and self‐interaction are reduced by phosphomimetics. Moreover, we show that phosphomimetic FUS reduces aggregation in human and yeast cell models, and can ameliorate FUS‐associated cytotoxicity. Hence, post‐translational modification may be a mechanism by which cells control physiological assembly and prevent pathological protein aggregation, suggesting a potential treatment pathway amenable to pharmacologic modulation.
Keywords: amyotrophic lateral sclerosis, frontotemporal dementia, intrinsically disordered protein, prion, ribonucleoprotein granule
Subject Categories: Neuroscience, Protein Biosynthesis & Quality Control
Introduction
Protein aggregation is a common hallmark of neurodegenerative disease (Harper & Lansbury, 1997; Trojanowski et al, 1998; Koo et al, 1999; Aguzzi & O'Connor, 2010) although the mechanisms of toxicity remain poorly understood. Different neurodegenerative diseases, or their subtypes, are often distinguished by the specific patterns of protein aggregation. Subtypes of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), two related neurodegenerative diseases, are characterized by neuronal inclusions of the RNA‐binding protein fused in sarcoma (FUS; Kwiatkowski et al, 2009; Neumann et al, 2009; Vance et al, 2009; Mitchell et al, 2013). FUS is highly abundant (Beck et al, 2011; Uhlen et al, 2015) and involved in RNA processing, splicing, and transport (Sama et al, 2014). FUS consists of a N‐terminal serine/tyrosine/glycine/glutamine (SYGQ)‐rich low‐complexity domain (LC), several RGG‐rich domains, an RNA recognition motif (RRM), a zinc finger domain, and a PY nuclear localization signal (NLS). FUS is primarily localized in the nucleus, but can shuttle between the nucleus and the cytoplasm. Familial ALS mutations in FUS most often disrupt the nuclear localization signal, resulting in cytoplasmic accumulation and apparent gain‐of‐function toxicity (Scekic‐Zahirovic et al, 2016; Sharma et al, 2016).
An emerging hypothesis is that RNA‐binding protein aggregation in neurodegenerative diseases is seeded from cytoplasmic ribonucleoprotein (RNP) granules (Li et al, 2013), including stress granules (SGs), processing bodies (P‐bodies), and transport granules (Kiebler & Bassell, 2006; Thomas et al, 2011). Recent reports suggest these granules are membraneless organelles, partially or wholly stabilized by liquid–liquid phase separation (LLPS) where molecules self segregate (demix) to form two co‐existing liquids as observed for oil droplets in water (Courchaine et al, 2016). However, unlike oil–water emulsions, RNP granules formed by LLPS are not devoid of water and are thought to be stabilized by multivalent, dynamic protein:protein and protein:mRNA interactions, concentrating granule components into a dense and viscous compartment into which other molecules can partition and localizing their cellular function (Ryu et al, 2014; Burke et al, 2015; Di Salvio et al, 2015; Lenzi et al, 2015; Bowden & Dormann, 2016; Daigle et al, 2016; Nott et al, 2016). Indeed, FUS‐containing granules and FUS at sites of DNA damage have liquid‐like properties in cells (Patel et al, 2015). We recently showed that FUS LC maintains its primarily disordered structure within in vitro models of RNP granules (Burke et al, 2015). However, the high local concentration of proteins in RNP granules formed by LLPS may potentiate the conversion of liquid compartments into solid cytoplasmic aggregates and inclusions observed in disease (Murakami et al, 2015; Patel et al, 2015). This phenomenon may be enhanced by mutations in disease‐associated ribonuclear proteins that increase their cytoplasmic concentration (Vance et al, 2013) or aggregation propensity (Kim et al, 2013).
LLPS, FUS granule assembly, and FUS inclusion formation both in vitro and in cells require the aggregation‐prone FUS LC (Shelkovnikova et al, 2014; Patel et al, 2015). The FUS LC has garnered particular attention because its polar‐rich sequence resembles so‐called prion domains that form self‐propagating amyloid aggregates in yeast cells (Gitler & Shorter, 2011; Ju et al, 2011; Kryndushkin et al, 2011; Sun et al, 2011). Yeast prion domains contain a paucity of hydrophobic and charged residues, but are rich in polar residues, especially glutamine and asparagine (Ross & Toombs, 2010). Several human proteins including FUS, TDP‐43, hnRNPA1, and hnRNPA2 contain low‐complexity sequences that are compositionally similar to yeast prion domains. Note that yeast prion and mammalian prion‐like domains have no sequence resemblance nor homology to the mammalian PrP prion associated with “mad cow disease”. Human prion‐like domains have emerged as primary drivers of cytoplasmic protein aggregation in neurodegenerative disease and are also sites of mutations in genetic forms of these conditions (King et al, 2012). Importantly, mammalian domains categorized as prion‐like are not homologous to yeast prions, but do contain amino acids such as serine, threonine, and tyrosine, which are potential targets of cellular phosphorylation.
Low‐complexity domains associated with LLPS and granule formation are targets of phosphorylation, possibly to regulate assembly (Aguzzi & Altmeyer, 2016). Previous studies have found that FUS is phosphorylated by phosphoinositide 3‐kinase‐like kinases (PIKKs), especially DNA‐dependent protein kinase (DNA‐PK; Gardiner et al, 2008; Deng et al, 2014). PIKKs phosphorylate their protein targets at serine or threonine residues followed by glutamine (S/TQ; Kim et al, 1999; O'Neill et al, 2000). Indeed, the FUS LC domain has twelve S/TQ motifs (8 SQ, 4 TQ), but previous in vitro DNA‐PK phosphorylation of FUS identified only pS26, pS42, pS61, pS84, and pS131 (Gardiner et al, 2008; Han et al, 2012).
Although FUS phosphorylation is thought to increase cytoplasmic FUS partitioning (Deng et al, 2014), little is known about the effect of post‐translational modifications on FUS self‐assembly. Interestingly, in vitro phosphorylation of recombinant, isolated FUS LC reduces its ability to bind hydrogels formed from amyloid‐like fibrils of purified recombinant, unphosphorylated FUS LC (Han et al, 2012). However, the effect of FUS phosphorylation on LLPS has not yet been determined. Modification of FUS with poly(ADP‐ribose; PAR) at sites of DNA damage induces phase separation of FUS (Altmeyer et al, 2015), while arginine methylation of the nuage protein DDX4 reduces LLPS (Nott et al, 2015). More generally, many of the low‐complexity prion‐like domains associated with aggregation in disease have been shown by proteomic screens to harbor many sites of post‐translational modification. However, the significance of these sites remains largely unknown. Here, we combine nuclear magnetic resonance spectroscopy, in vitro phase separation and aggregation assays, in‐cell phosphorylation site identification by proteomic mass spectrometry, and cellular aggregation and toxicity assays to characterize the location and effect of LC phosphorylation on FUS structure, interactions, aggregation, and toxicity. Because FUS aggregation in the cytoplasm of motor neurons is linked to gain‐of‐function cytotoxicity, altering FUS phosphorylation could serve as a therapeutic strategy for diseases that currently have no effective therapeutics.
Results
FUS LC is multiply phosphorylated by DNA‐PK
The N‐terminal, 163‐amino acid, low‐complexity domain (LC) of FUS contains 12 conserved S/TQ sequence motifs that make up the primary recognition site of DNA‐dependent protein kinase (DNA‐PK; Fig 1A, gray lines, Appendix Fig S1A; Kim et al, 1999). However, DNA‐PK treatment in vitro followed by either tandem mass spectroscopy or Edman sequencing resulted in identification of only five phosphorylation sites in FUS: S26, S42, S61, S84, and S131 (Gardiner et al, 2008; Han et al, 2012). None of the four conserved TQ sites were detected. We and others have suspected that the complete lack of positively charged residues in FUS LC obscures phosphorylation detection by standard proteomic workflows due to lack of positive residues both for efficient positive ion formation for mass spectrometry and for trypsin cleavage sites (Knight et al, 2003). Therefore, to determine the full possible extent of the FUS LC phosphorylation by DNA‐PK, we used in vitro DNA‐PK phosphorylation as described previously (Gardiner et al, 2008; Han et al, 2012; Deng et al, 2014) and detected phosphosites by two‐dimensional solution NMR, which offers direct residue resolution. The appearance of many robust peaks in the phosphoserine and importantly in the phosphothreonine regions of the 1H 15N heteronuclear single quantum coherence (HSQC) spectrum immediately suggests FUS can be phosphorylated at more positions than the five previously reported serines (Fig 1B). By phosphorylating a series of single serine/threonine‐to‐alanine FUS LC variants and single alanine‐to‐serine/threonine variants in a 12 alanine FUS LC background, we assigned the phosphorylated resonances. We find that FUS LC is phosphorylated at all 12 distinct S/TQ sites. Observing the extent of phosphorylation over time in vitro by NMR shows that some residues (T7, T11, T19, S26, S42, S61, S84) are effectively fully phosphorylated as measured by a complete loss of intensity of the unphosphorylated peak (Fig 1C, Appendix Fig S1B). Other positions (S30, T68, S87, S117) appear to be incompletely phosphorylated even after extended incubation. However, the apparent halting of the reaction can be attributed to deactivation of DNA‐PK by auto‐phosphorylation that is almost complete by 30 min (Carter et al, 1990). The fact that the sum of the intensity of phosphorylated and unphosphorylated peaks does not add to 1 may be due to heterogeneity in the phosphorylation pattern increasing peak width and to alteration in spin relaxation properties. Based on the extent of phosphorylation observed, each FUS polypeptide must be multiply phosphorylated. Taken together, these data establish that DNA‐PK is able to phosphorylate all S/TQ positions in the FUS LC, including all 4 TQ positions, none of which had been identified in previous in vitro experiments.
Figure 1. FUS LC is multiply phosphorylated.
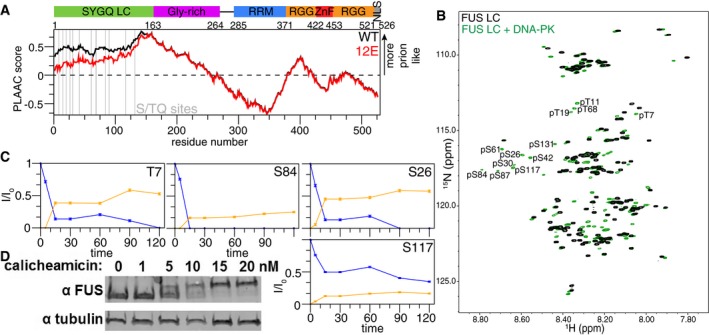
- FUS contains an SYQG‐rich low‐complexity (LC) domain, a glycine‐rich domain containing RGG motifs, an RNA recognition motif (RRM), two RGG‐rich domains separated by a zinc finger (ZnF), and a non‐canonical PY nuclear localization signal (NLS). PLAAC scores (Lancaster et al, 2014) indicating the degree of prion‐like character in WT (black) and phosphomimetic (12E, red) full‐length FUS show that phosphomimetic mutations reduce the prion‐like character of the low‐complexity domain. 12E refers to FUS containing 12 phosphomimetic (E, glutamate) substitutions at the S/TQ DNA‐PK consensus phosphorylation sites in the LC domain, indicated by a gray line.
- FUS is phosphorylated at all 12 S/TQ sites in vitro by DNA‐PK, as identified by NMR.
- Change in intensity of phosphorylated (orange) and not phosphorylated (blue) peaks in NMR spectra quantified over time for select residues (time course of phosphorylation for the remaining residues can be found in Appendix Fig S1). Error bars indicate ± SD derived from uncertainty in NMR signal intensity in a single representative biochemical experiment.
- Treatment of HEK cells with calicheamicin, which causes double‐strand breaks, results in a change in the apparent mobility of FUS when analyzed by SDS–PAGE, consistent with previously published findings and reversible by subsequent phosphatase treatment (Deng et al, 2014).
Identification of FUS LC phosphorylation sites in cells
Although FUS LC is known to be a target of phosphorylation, site‐specific identification of in‐cell phosphorylation sites has not been reported using proteomic approaches due to the difficulties described above. Treatment of cells with calicheamicin causes double‐strand DNA breaks that activate DNA‐PK, which is proposed to phosphorylate FUS LC. We confirmed phosphorylation in cells induces a large SDS–PAGE mobility shift (Fig 1D) that collapses upon treatment with phosphatase (data not shown), as reported previously (Deng et al, 2014). A similar phosphorylation‐induced mobility shift was reported after treatment with calyculin A (Deng et al, 2014), a phosphatase inhibitor that stimulates DNA damage (Takeuchi et al, 1994). Using phosphopeptide enrichment approaches, we attempted to identify phosphorylation sites in the LC of endogenous FUS in human cells. Our initial efforts to identify the phosphosites by FUS immunoprecipitation and positive ion tandem mass spectroscopy of chymotrypsin digests were not successful. This was likely a result of the complete lack of positively charged residues in FUS LC, precluding trypsin digest and resulting in low ionization of highly negatively charged peptides (Schuller & Eick, 2016), placing FUS LC phosphopeptides in the noise. We therefore developed a strategy to remove easily ionizable peptides by an initial digest with trypsin, washing away tryptic fragments via centrifugal filtration leaving the undigested FUS LC intact, and then cleaving with chymotrypsin followed by TiO2 phosphopeptide enrichment. This procedure enabled identification of pS26 as well as three other non‐S/TQ sites pT71, pS77, and pS96 after calicheamicin treatment. Calyculin A treatment resulted in detection of phosphorylation at S/TQ sites pT7, pS26, pS30, pS42, and pS61, as well as the same non‐S/TQ sites (pT71, pS77, and pS96) among others (Appendix Fig S1C). We observed only mono‐phosphorylated chymotryptic peptide fragments. Multi‐phosphorylated peptides likely were present but were not detected due to low abundance or increasing net negative charge of the peptide. No FUS LC phosphopeptides were identified in control treatments with DMSO, which is consistent with none being previously reported under un‐stressed conditions (Gardiner et al, 2008; Deng et al, 2014). Combined with previous work showing that DNA‐PK activity is required for the mobility shift detected phosphorylation, these results demonstrate that FUS LC is cellularly phosphorylated both at S/TQ and other positions following DNA damage. Given the high specificity of DNA‐PK for S/TQ sites (Fig 1B), the phosphorylation we identified at non‐S/TQ positions likely arises from other kinases. In total, by mass spectrometry and NMR we have identified 17 putative phosphosites in FUS LC, suggesting phosphorylation could have a role in FUS function.
Phosphorylation and phosphomimetic variants reduce FUS LC phase separation and aggregation
FUS LC is rich in aromatic and glutamine residues, mediating self‐assembly into granules and inclusion formation in ALS and frontotemporal dementia. We next sought to test the hypothesis that phosphorylation could disrupt FUS self‐association and aggregation by altering the biophysical properties of the LC. One measure of the self‐assembly/aggregation propensity of disordered domains is a quantitative assessment of their “prion‐like” character, a name given to sequences enriched in polar residues and lacking charged and aliphatic residues as found in yeast prion proteins (Cascarina & Ross, 2014). Mimicking phosphorylation at S/TQ positions by substitution of serine or threonine with the negatively charged residue glutamic acid (glutamate) results in a marked decrease in the prion‐like propensity as measured by the PLAAC algorithm (Fig 1A; Lancaster et al, 2014), which identifies probable prion‐like protein segments. This result is consistent with changing the sequence composition character of the domain from nearly uncharged (only two negatively charged residues; no positively charged lysine, arginine, or histidine) to polyanionic and hence soluble and self‐repulsive by like‐charge interactions.
We tested the influence of charge on liquid–liquid phase separation (LLPS) via molecular simulations using a novel coarse‐grained model (one “bead” per residue) incorporating favorable interactions based on a common hydrophobicity scale and screened electrostatic potentials. We simulated FUS LC wild‐type and 12E (all 12 S/TQ sites substituted with glutamic acid, S/TQ→EQ) using a box of 22 chains and 150 mM NaCl. For wild‐type FUS LC, a marked peak in the heat capacity as a function of temperature (Fig 2B) is observed concomitant with phase separation into a single continuous phase (Fig 2B, upper left inset). At the same temperature and conditions, FUS LC 12E does not show a heat capacity peak or collapse (Fig 2B). Snapshots of simulated FUS LC polypeptides are shown within Fig 2B to illustrate the change from a single uniform phase to a phase‐separated condition; wild type is phase separated at temperatures below the phase transition (upper left), while 12E remains dispersed, not phase separated (bottom). To establish that the change in charge is sufficient to explain the difference between wild type and 12E, simulations isolating the contributions from electrostatic interactions were designed. Adding virtual negative charges to the 12 serine and threonine positions resulted in lack of phase separation mirroring 12E (Appendix Fig S2A), confirming that the disruption in phase separation observed in the model arises due to electrostatic repulsion.
Figure 2. FUS LC phosphorylation and phosphomimetic substitution reduce FUS phase separation and aggregation in vitro .
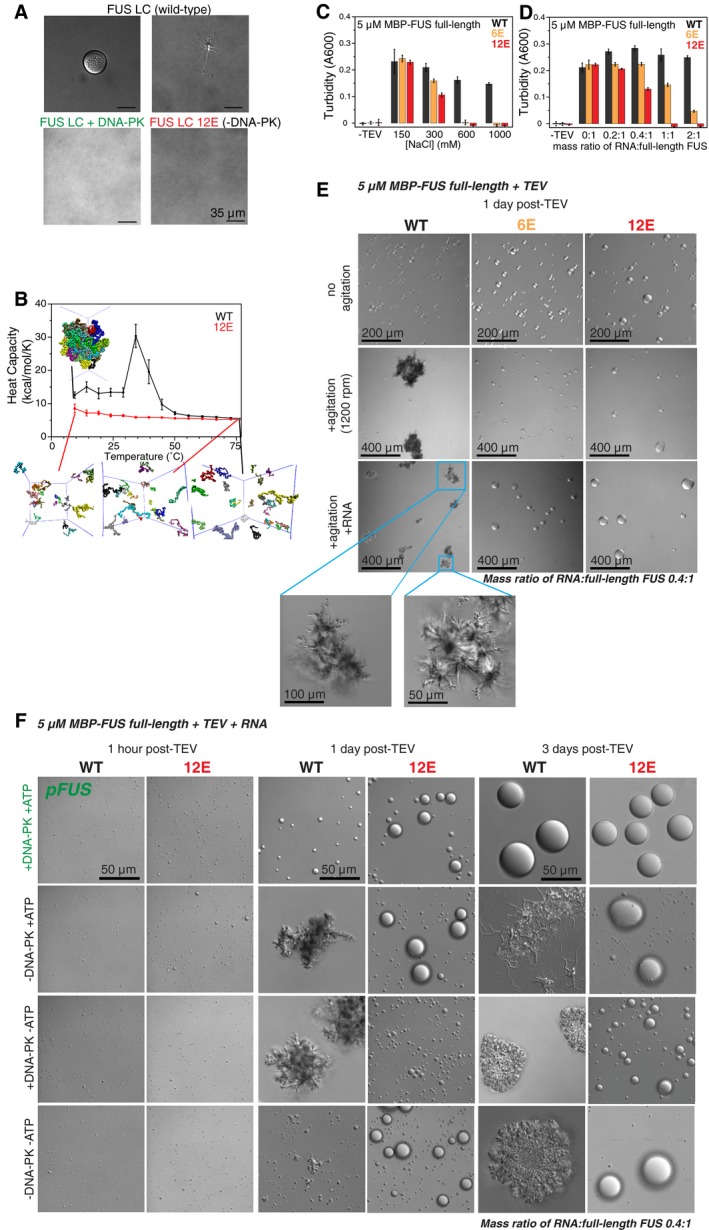
- WT FUS LC undergoes LLPS (top left) and aggregation (top right) under DNA‐PK reaction solution conditions (no DNA‐PK), while DNA‐PK‐treated WT FUS LC or phosphomimetic (12E) FUS LC does not (all samples incubated identically: 1 day at 25°C).
- Heat capacity curves for simulations of WT and 12E FUS LC show that WT undergoes a phase transition while, at the same conditions, 12E does not. At high temperatures, snapshots show both WT and 12E are disperse but at temperatures below the phase transition (7°C), WT is phase separated, while 12E remains disperse.
- Full‐length FUS 6E and 12E are much more sensitive to salt‐induced disruption of phase separation than FUS WT. FUS 6E and 12E exhibit decreased phase separation compared to WT at salt concentrations above 150 mM, and both show no phase separation in high salt conditions. Data shown is 45 min after the addition of TEV protease, which cleaves a maltose‐binding protein (MBP) solubility tag fused to the N‐terminus of FUS.
- Phase separation of full‐length FUS 6E and 12E is disrupted, not enhanced, by RNA. At mass ratios of RNA:full‐length FUS of 1:1 or higher, full‐length FUS WT phase separation persists whereas full‐length FUS 12E does not. Full‐length FUS 6E shows reduced phase separation compared to WT when RNA is added at mass ratios of RNA:full‐length FUS ranging from 0.2:1 to 2:1. Data shown are 45 min after addition of TEV protease.
- Full‐length FUS 6E and 12E do not form fibrous aggregates. Differential interference contrast microscopy shows that 5 μM full‐length FUS WT forms morphologically distinct assemblies as compared to 5 μM full‐length FUS 6E and 12E. After the addition of TEV protease to cleave the N‐terminal MBP tag, indicated samples were agitated (middle and bottom) for 1 day at 25°C. FUS WT forms fibrous aggregates in the presence and absence of RNA (0.4:1 full‐length FUS, by mass), whereas FUS 6E and FUS 12E assemblies remain spherical despite agitation. Larger images of fibrous WT aggregates are shown (bottom left, blue insets).
- Phosphorylation of full‐length FUS WT by DNA‐PK prevents formation of fibrous aggregates. DIC images taken 1 h after cleavage of the N‐terminal MBP tag show that full‐length FUS WT and 12E undergo LLPS following phosphorylation (row 1) and control treatments (rows 2–4). After agitation at 25°C for 1 day, phosphorylated full‐length FUS WT assemblies remain spherical, whereas unphosphorylated FUS WT begins to form fibrous aggregates. Full‐length FUS WT exhibits aggregation in all control treatments after 3 days of agitation at 25°C. See Appendix Fig S2 for additional images of samples 1 and 3 days post‐TEV.
To test the hypothesis that phosphorylation alters FUS LC assembly in vitro, we evaluated the phase separation of wild‐type FUS LC (with and without DNA‐PK treatment) and FUS LC 12E, carrying the 12 phosphomimetic substitutions at the consensus DNA‐PK sites. We performed differential interference contrast (DIC) microscopy of FUS LC incubated for 1 day where wild‐type FUS LC undergoes LLPS to form round micron‐sized droplets (Fig 2A, top left). Some of these droplets convert to fibrous aggregates during this time (Fig 2A, top right). After the same incubation period, DNA‐PK‐phosphorylated FUS LC and FUS LC 12E (phosphomimetic) showed no LLPS or aggregation (Fig 2A, bottom).
Given that phosphorylation and phosphomimetics of FUS LC alter self‐assembly and aggregation of the isolated LC, we hypothesized that LLPS would be disrupted for full‐length FUS bearing phosphomimetic substitutions within the LC. We previously established that wild‐type, full‐length FUS undergoes LLPS after TEV cleavage of a solubilizing N‐terminal fusion (Burke et al, 2015). Contrary to our expectations, full‐length FUS 12E and a partial phosphomimetic FUS 6E (specific residues are described in Appendix Supplementary Methods) phase separated in vitro much like the wild‐type protein. The phase‐separated states appeared similarly when observed by DIC microscopy within 1 day (pH 7.5 Tris, 150 mM NaCl 25°C) following initial cleavage of the solubilizing tag (Fig 2E, top row). However, we noticed that increasing ionic strength dramatically reduced phase separation of full‐length FUS 6E and FUS 12E (Fig 2C, Appendix Fig S2C), while wild‐type FUS was significantly less affected. Unlike unmodified full‐length FUS, the strong salt dependence of FUS 6E and FUS 12E suggests that electrostatic interactions between the phosphomimetic negatively charged N‐terminus and the arginine‐rich domains could stabilize LLPS. In summary, phosphomimetic substitution in the LC suppresses full‐length FUS LLPS at high salt.
FUS is an avid and promiscuous RNA binder (Schwartz et al, 2013; Wang et al, 2015), so we tested how mimicking the cytoplasmic environment by adding RNA would affect FUS phase separation. We and others have previously shown that addition of substoichiometric amounts of RNA can enhance LLPS (Burke et al, 2015) or fibrillization (Schwartz et al, 2013) of wild‐type, full‐length FUS. Importantly, we found addition of RNA to full‐length FUS 6E and 12E disrupted LLPS as observed by turbidity and DIC microscopy, while LLPS appeared consistent for wild‐type FUS at all RNA conditions (Fig 2D, Appendix Fig S2B and C). LLPS was dramatically suppressed for FUS 6E with increasing RNA levels, and no LLPS was observed for FUS 12E at high RNA:FUS ratios (Fig 2D). These data suggest that for phase separation of phosphomimetic FUS, RNA might compete with the negatively charged LC for contacts with positively charged regions of FUS. The low dependence on both salt and RNA suggests that wild‐type FUS LLPS is not primarily mediated by these charged interactions, rather by intermolecular LC‐LC interactions.
It was previously shown that liquid droplets of full‐length FUS can convert into fibrillar aggregates over time (Patel et al, 2015). We thus examined phase separation of full‐length FUS, 6E, and 12E by monitoring changes in morphology of protein assemblies over longer time periods or with orbital agitation (Fig 2E), using a method adapted from a protocol previously shown to induce fibrillization of hnRNPA2B1 and hnRNPA1 (Kim et al, 2013). Non‐agitated FUS, 6E, and 12E formed similar liquid‐like droplets, as observed by DIC microscopy up to 1 day after cleavage from the solubility tags (Fig 2E). However, agitation (1,200 rpm, 25°C, 1 day) induces wild‐type to form irregular aggregates, while assemblies of 6E and 12E remained spherical, with or without RNA (0.4:1 RNA:FUS, by mass; Fig 2E).
We hypothesized phosphorylation of recombinant protein would have similar effects as phosphomimetic substitutions on full‐length FUS aggregation. Both wild‐type FUS and 12E were treated with DNA‐PK, and their phase separation and aggregation were monitored over 3 days by DIC microscopy. Liquid droplets of phosphorylated and unmodified full‐length wild‐type FUS were indistinguishable 1 h after TEV cleavage of the MBP solubility tag; however, dramatic morphological differences emerged within 1 day of incubation (1,200 rpm, 25°C; Fig 2F). In addition to monitoring samples incubated with neither DNA‐PK nor ATP, we tested two distinct phosphorylation‐negative controls: one without DNA‐PK but with ATP and one with DNA‐PK but without ATP to ensure ATP alone was not inducing fibrillization and that DNA‐PK was not acting as a chaperone in the absence of ATP, respectively. After 3 days of agitation, all three unphosphorylated wild‐type FUS control samples exhibited irregular aggregates, whereas phosphorylated wild‐type FUS and all phosphomimetic 12E assemblies remained spherical (Fig 2F, Appendix Fig S2D). Thus, only phosphorylation by addition of DNA‐PK and ATP, and not a chaperone effect of DNA‐PK or alteration of solution conditions by adding ATP, was sufficient to eliminate the transition of wild‐type FUS from liquid‐like droplets into solid aggregates. Furthermore, assemblies of phosphomimetic and phosphorylated wild‐type FUS grew larger over time, suggesting that droplets became larger by fusion and/or by monomers redistributing from smaller to larger assemblies via Ostwald ripening.
The continued presence of round droplets that grow in size and the lack of solid aggregates suggests LLPS persists in phosphorylated and phosphomimetic full‐length FUS samples up to 3 days post‐TEV cleavage (Fig 2F, Appendix Fig S2D). However, full‐length FUS droplets appear much more viscous and heterogeneous than previously characterized FUS LC droplets (Burke et al, 2015), as suggested by incomplete recovery after photobleaching at time points as early as 45 min after TEV cleavage. Full‐length wild‐type FUS droplets failed to completely recover within 6 min of a partial photobleach, though this observation may be due to static forms adsorbed to the glass slide (which is within the confocal volume) as nearby droplets readily fused as liquids (Movie EV1). While it is still unknown to what degree assemblies of phosphorylated or phosphomimetic FUS become more static and/or viscous over time, phase‐separated assemblies of unmodified full‐length FUS convert much more readily into fibrillar aggregates. Therefore, phosphorylation and phosphomimetic substitution in the LC domain suppress the aggregation or liquid‐to‐solid transition at conditions where LLPS occurs regardless of phosphorylation state. Taken together, these data on isolated FUS LC and full‐length FUS demonstrate that the addition of negatively charged phosphate groups or phosphomimetic substitutions impedes LC domain self‐association and subsequent aggregation.
Phosphorylation and phosphomimetics do not alter the disordered structure of FUS LC
To understand the molecular details of the differences in LLPS and aggregation, we chose to characterize FUS LC 12E due to the difficulty in obtaining complete uniformly DNA‐PK phosphorylated FUS LC. We applied the solution nuclear magnetic resonance (NMR) approach we previously used for FUS LC wild type (Burke et al, 2015) to determine whether the introduction of negatively charged phosphorylated or phosphomimetic residues changed the structure or local motions of FUS LC. The two‐dimensional “fingerprint” NMR spectra (HSQC) of both phosphorylated FUS LC (Fig 1B) and FUS LC 12E (Appendix Fig S3A) show narrow 1H chemical shift dispersion and are similar to that of unmodified/wild‐type FUS LC, consistent with structural disorder. Chemical shift deviations consistent with large‐scale conformational changes (e.g., folding) are not observed.
Secondary structure (e.g., α‐helix, β‐sheet) propensities as determined from full backbone/Cβ NMR resonance assignment are consistent with disorder and no major structural differences between wild‐type and 12E LC (Appendix Fig S3B). To confirm similar structural disorder in wild‐type and 12E, we used NMR spin relaxation experiments sensitive to local reorientational motions that show changes if structure is formed/unformed (Conicella et al, 2016; Fig 3A, Appendix Fig S4A). Longitudinal spin relaxation rate constants, R 1, and heteronuclear Overhauser effect values, hetNOE, for wild type and 12E are uniform and similar, indicating there are very few structural differences between the domains. However, transverse relaxation rate constants, R 2, for wild type are slightly higher than for 12E at both 850 and 500 MHz 1H Larmor frequency (Fig 3A, Appendix Fig S4A), suggesting that the wild‐type domain experiences either slower reorientational motions or greater contribution to R 2 from conformational exchange. Essentially, the same values of R 2 are observed at half the concentration (Fig 3A, gray), ruling out exchange contribution to R 2 from intermolecular contacts at these concentrations. Spectral density mapping—using the data from both fields (500 and 850 MHz) simultaneously (Farrow et al, 1995)—was performed to further characterize the molecular motions of FUS and 12E. The higher observed R 2 for wild‐type FUS relative to 12E can likely be accounted for by a combination of small contributions from both slower motions and greater intramolecular conformational exchange (Appendix Fig S4B). This suggests there is a higher propensity for wild‐type FUS to visit a transiently collapsed state.
Figure 3. Phosphomimetic mutations in FUS LC preserve disordered LC domain structure but disrupt transient intramolecular collapse and intermolecular interactions.
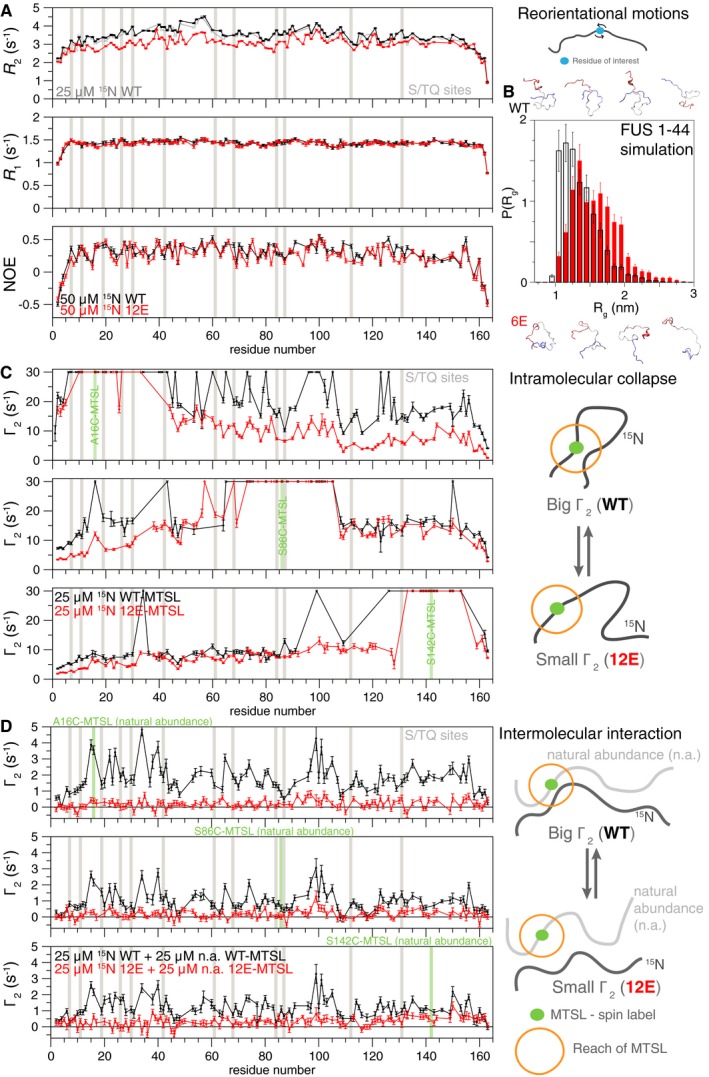
- Residue‐specific backbone 15N spin relaxation parameters (R 2, R 1, heteronuclear NOE) measured at 850 MHz 1H Larmor frequency are consistent with structural disorder for both FUS LC WT (black) and 12E (red). FUS LC WT (black) shows elevated transverse relaxation, R 2, even at half concentration (gray) consistent with slower motions or increased contribution from conformational exchange. See Appendix Fig S4. Errors bars represent ± SD calculated from NMR signal uncertainty in a single representative experiment.
- Simulations of FUS LC subpeptide spanning residues 1–44 of both WT (black) and the phosphomimetic variant (red) are consistent with disordered conformations. Slightly larger radius of gyration, R g, for the phosphomimetic variant is observed. Error bars indicate ± SEM derived from 10 equal, non‐overlapping divisions of the equilibrated ensemble.
- Intramolecular paramagnetic relaxation enhancement (PRE) NMR experiments for MTSL spin label placed at three single engineered cysteine sites (A16C, S86C, S142C—green). Higher PRE values, Γ2, consistent with a greater degree of transient intramolecular interactions are observed for WT than 12E, particularly when probing at the N‐terminus, where most of the S/T to E mutations are. Experiments were conducted with 25 μM NMR visible (15N isotopic labeled), spin‐labeled peptide FUS LC. Errors bars represent ± SD calculated from NMR signal uncertainty in a single representative experiment.
- Intermolecular PRE NMR experiments sensitive to inter‐protein contacts demonstrate WT stabilizes transient intermolecular interactions compared to 12E. Experiments were conducted with 25 μM NMR visible (15N isotopic labeled) FUS LC combined with 25 μM paramagnetic spin‐labeled NMR silent (natural isotopic abundance) FUS LC. Errors bars represent ± SD calculated from NMR signal uncertainty in a single representative experiment.
We next used the validated atomistic molecular simulation approaches (Zerze et al, 2015) we previously used for interrogating the primarily disordered C‐terminal domain of the related protein TDP‐43 (Conicella et al, 2016) to help interpret the structural differences between wild‐type FUS and 12E observed by experiment. FUS 1–44 contains half of the SQ/TQ sites (6 S/TQ sites) and was chosen for its tractable length for rigorous sampling of peptide structure and dynamics. As observed by experiment, the FUS 1–44 wild‐type and phosphomimetic are both primarily disordered with very little change in secondary structure (Appendix Fig S3C). However, phosphomimetic substitutions result in a slight increase in the distribution of the radius of gyration (Fig 3B) and slightly faster local motions as measured by calculation of spectral density values and NMR spin relaxation parameters (Appendix Fig S4C and D). Taken together, these data suggest that FUS LC remains globally dynamic after phosphorylation and phosphomimetic substitution. However, phosphomimetic substitutions result in small changes in chain collapse and motions. These changes in intramolecular behavior provide insight into how phosphorylation could suppress both LLPS and aggregation.
Phosphomimetic substitutions in FUS LC decrease intramolecular collapse and transient intermolecular interactions
To investigate why the disordered phosphorylated and phosphomimetic FUS LC undergo LLPS and aggregation less readily than the unmodified FUS LC, we compared the propensity of FUS LC wild type and 12E to form transient interactions that must precede LLPS. At conditions where both proteins are in a single, dispersed phase (25 μM FUS LC, 150 mM NaCl, 25°C), we performed both intra and intermolecular paramagnetic relaxation enhancement (PRE) experiments sensitive to intramolecular collapse and transient intermolecular interactions, respectively. Individual PRE experiments are analogous to a full series of ensemble Förster resonance energy transfer (FRET) experiments, where the donor fluorophore is replaced by a small (184 Da) stabilized nitroxide radical label and the acceptor fluorophore is replaced by all hydrogen atoms in the protein, providing measures of close approach of the spin‐labeled region to every residue in the protein simultaneously. PREs have a steep dependence on distance, making them a sensitive probe of transient structure (Clore, 2015). Higher intramolecular PREs, corresponding to closer approach of the labeled region, for wild type compared to 12E suggest that wild‐type LC stabilizes transient collapsed conformations more than 12E LC (Fig 3C). When the spin label is placed in the first 42 residues (where six of the S/TQ sites are found) or near the domain center, large PREs are observed up to 100 residues away for wild‐type FUS LC, but not 12E (Fig 3C, top and middle). When the spin label is placed near the C‐terminus of the domain, where there are few phosphomimetic substitutions, wild‐type intramolecular PREs, while still consistently higher, deviate less sharply from 12E intramolecular PREs (Fig 3C, bottom). Higher intramolecular PREs for wild type compared to 12E suggest that wild‐type LC populates transient collapsed conformations more than 12E LC. The higher PREs for wild‐type FUS LC compared to 12E are consistent with the slightly smaller radius of gyration, R g, observed by simulation (Fig 3B).
Furthermore, intermolecular PRE experiments performed by mixing unmodified, NMR‐active FUS LC with spin‐labeled FUS LC show that residues throughout wild‐type FUS make frequent transient intermolecular contacts. Conversely, residues within 12E FUS LC make few detectable intermolecular contacts at these conditions (Fig 3D), comparable with a control with MTSL in free solution (Appendix Fig S5A). These observed intermolecular interactions for wild‐type FUS LC vary in magnitude but are similar regardless of the label position, consistent with the repetitive low‐complexity sequence. Importantly, full suppression of the interactions requires phosphomimetic substitutions on both the NMR visible and the spin‐labeled peptide (Appendix Fig S5B), further suggesting that the phosphomimetic substitutions decrease these intermolecular interactions by mutual repulsion. In other words, these contacts lack bias to form LC subregion‐specific contacts; however, they are significantly disrupted by addition of negatively charged residues. Together, the PRE data suggest that amino acid substitutions mimicking phosphorylation suppress both transient intra‐ and intermolecular LC contacts.
Phosphomimetic substitutions in FUS LC reduce aggregation propensity in cells
We next sought to test the ability of phosphomimetic substitutions to alter the aggregation of full‐length FUS in a cell model of FUS pathology. The yeast Saccharomyces cerevisiae has been used for evaluating the misfolding and aggregation of many human disease‐linked proteins (Khurana & Lindquist, 2010; Kryndushkin et al, 2011). In yeast, ectopically expressed human FUS forms cytoplasmic aggregates that are toxic and mildly detergent‐resistant, features reminiscent of FUS inclusions from FUS‐associated ALS motor neurons (Fushimi et al, 2011; Kryndushkin et al, 2011; Sun et al, 2011). Significantly, the yeast model provides a crowded, eukaryotic cytoplasm to observe how specific modifications to a given protein alter aggregation potential. To test the effect of phosphomimetic substitution on intracellular cytoplasmic aggregation, a series of variants of full‐length FUS incorporating increasing numbers of S/TQ to EQ substitutions were constructed and expressed in the wild‐type yeast strain W303 (the substitutions are defined in the Appendix Supplementary Methods section). FUS subtypes of ALS are most often caused by mutations in the FUS nuclear localization signal; yeast models naturally lack the importin recognizing the FUS PY‐NLS and collect ectopic FUS in the cytoplasm, allowing direct testing of the effect of sequence variants on cytoplasmic aggregation. Immunofluorescence microscopy of fixed cells showed three distinct localization patterns for wild‐type FUS, 6E, and 12E (Fig 4A). Quantification of punctate, intermediate, and diffuse localization patterns demonstrated that FUS 12E consistently had a more diffuse pattern of fluorescence than wild‐type FUS (Fig 4B, Appendix Fig S6A). 6E had an aggregation pattern intermediate between wild type and 12E (Fig 4B). Further, both 6E and 12E were less likely to accumulate in the insoluble fraction of yeast lysate than wild‐type FUS (Fig 4C and Appendix Fig S6B). This decrease in aggregation propensity was consistent for both low‐copy and high‐copy plasmids (i.e., intermediate and high expression levels; Fig 4C). Phosphomimetic variants formed less SDS‐resistant species than wild‐type FUS (Fig 4D).
Figure 4. Phosphomimetic substitutions in the FUS LC domain reduce cytoplasmic aggregation.
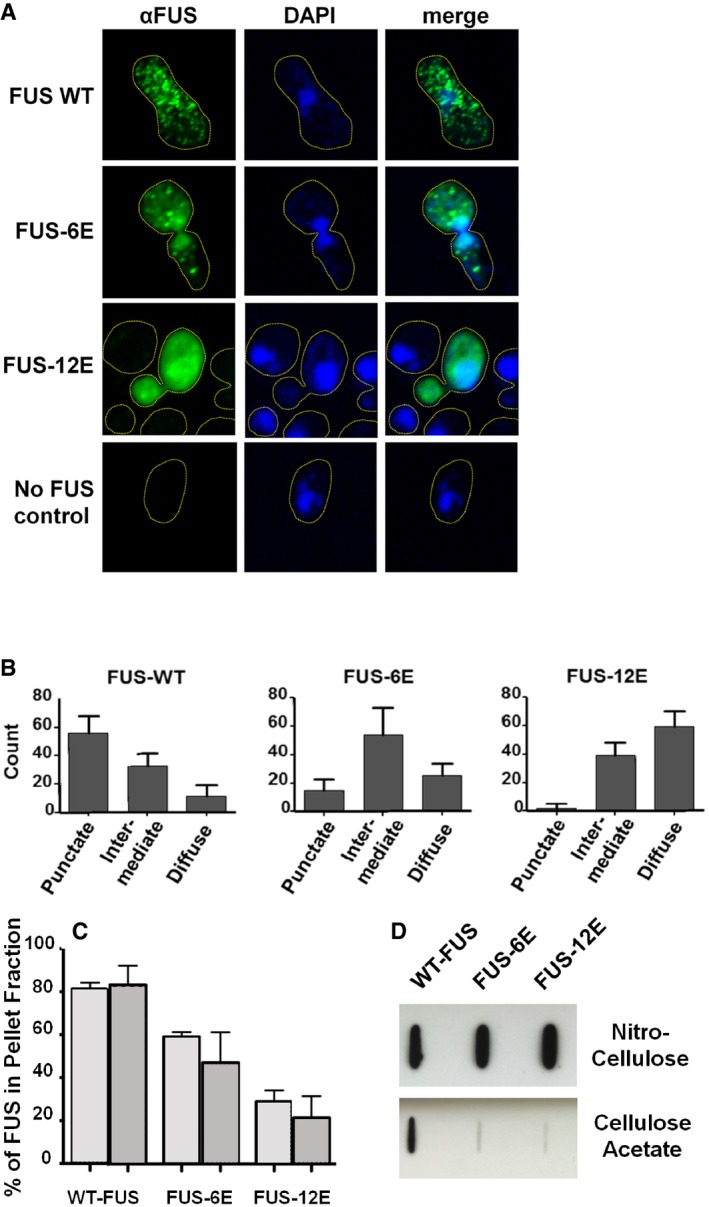
- Immunofluorescence microscopy of fixed yeast cells shows typical localization patterns of FUS, FUS‐6E, and FUS‐12E, where FUS WT appears punctate, consistent with aggregation, while FUS 12E is diffuse throughout cytoplasm, consistent with a soluble protein. FUS 6E is consistent with an intermediate pattern.
- Quantification of different cytoplasmic localization patterns of FUS, FUS‐6E, and FUS‐12E indicates that phosphomimetic substitutions in the FUS LC domain decrease puncta formation; 67 cells expressing FUS, FUS‐6E, or FUS‐12E were randomly selected and scored from field images.
- Densitometry analysis of Western blot repeated in triplicate indicates the relative amounts of protein in the pellet fractions (light gray bars = single‐copy plasmid expression; dark gray bars = high‐copy plasmid expression). Error bars represent standard deviation.
- A filter retardation slot blot of crude yeast lysates in 1% SDS passed through a cellulose acetate membrane (binds only large aggregates) indicates partial SDS resistance of aggregates composed of wild‐type FUS, but not FUS‐6E or FUS‐12E. Total FUS is comparable in all samples as shown by slot blots on nitrocellulose (binds all protein forms) of the same lysates in 0% SDS.
We next evaluated how phosphomimetic substitutions in LC would affect cytoplasmic aggregation of FUS in mammalian cells. We constructed GFP‐FUS transfection plasmids with the R495X ALS‐linked nonsense mutation. The premature stop codon eliminates FUS's carboxy‐terminal nuclear localization signal, confining GFP‐FUS to the cytoplasm of transfected cells. Phosphomimetic substitutions (6E and 12E) were introduced into GFP‐FUS(R495X). In HEK293T cells imaged with identical exposure and processing, GFP‐FUS(R495X) formed large cytoplasmic inclusions, while the 12E variant showed predominately diffusely localized protein and the 6E variant had an intermediate appearance (Fig 5A, Appendix Fig S7). Western blotting indicated the proteins are expressed at similar levels (Fig 5B). We also performed a biochemical fractionation assay on endogenously phosphorylated FUS following calicheamicin treatment of H4 neuroglioma cells. Relative to endogenous unphosphorylated FUS, endogenous phosphorylated FUS had decreased propensity to accumulate in the pellet fraction following incubation and centrifugation of a cell lysate at 4°C (Fig 5C). This was consistent with observations of phosphomimetic FUS expressed in yeast, suggesting phosphorylation of LC lowers FUS's potential for forming aggregates in a cellular environment.
Figure 5. Low‐complexity domain phosphomimetic substitutions and phosphorylation reduce FUS propensity to form insoluble species in human cell lines.
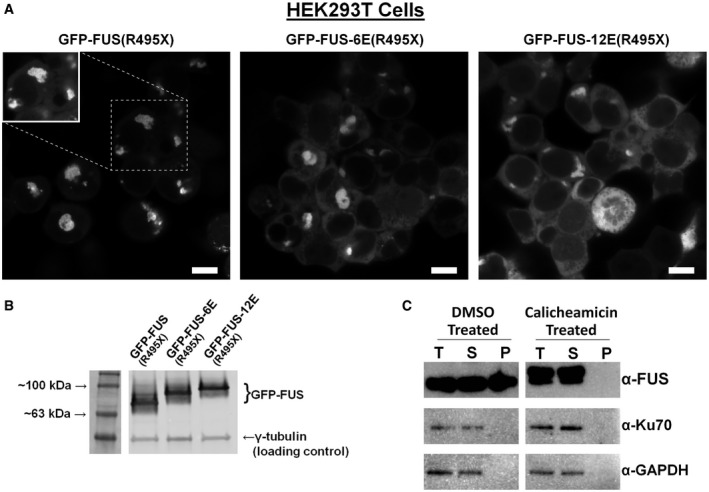
- Phosphomimetic substitutions in the LC domain reduce GFP‐FUS propensity to accumulate in large cytoplasmic aggregates in HEK293T cells. Cells were transfected with plasmids encoding GFP‐FUS(R495X), with 0, 6, or 12 glutamate substitutions at serines/threonines within the LC domain. Images were collected 16 h after transfection (scale bar = 10 μm) with identical exposure and processing parameters to visualize the differences in cytoplasmic distribution of GFP‐FUS(R495X) variants. Enhanced brightness (inset) shows that GFP‐FUS(R495X) without phosphomimetic glutamate substitutions is excluded from the nucleus (dark regions in cell center); see Appendix Fig S7.
- Relative expression levels of GFP‐FUS(R495X) were confirmed by Western blotting.
- Lysate fractionation of H4 neuroglioma cells shows that FUS from cells treated with calicheamicin does not accumulate in the insoluble fraction, while FUS from untreated cells does (T = total; S = supernatant; P = pellet). Cytoplasmic (GAPDH) and nuclear (Ku70) controls are shown to demonstrate complete lysis of the cell and nuclear membranes.
Phosphomimetic substitutions in FUS LC reduce FUS‐linked toxicity
Previous work in the yeast model supports a view that aggregation is integral to FUS toxicity (Kryndushkin et al, 2011). To test whether phosphomimetic substitution can alter FUS toxicity and aggregation, several variants of full‐length FUS with phosphomimetic substitutions in the LC were expressed from high‐copy plasmids in yeast cells spotted in serial dilutions on solid growth medium. Relative to wild‐type FUS, increasing the number of phosphomimetic substitutions strongly correlated with reduced growth inhibition (Fig 6A). The phosphomimetic substitutions were also evaluated under lower expression conditions that result in milder toxicity (Kryndushkin et al, 2011). The modified FUS proteins were expressed from single‐copy plasmids in yeast in liquid culture, and growth (≤ 8 cell divisions) was quantified by absorbance (Fig 6C). As the number of phosphomimetic substitutions increased, the growth inhibition caused by FUS markedly decreased. Western blotting revealed that all FUS variants were expressed to similar levels (Fig 6B and D), and thus, the reduced toxicity associated with phosphomimetic substitutions was not due to differences in protein expression. Together, these results suggest that phosphomimetic substitutions in the FUS LC reduce not only its cytoplasmic aggregation, but also disrupt FUS toxicity.
Figure 6. FUS‐linked toxicity in yeast is reduced by phosphomimetic substitutions in the LC domain.
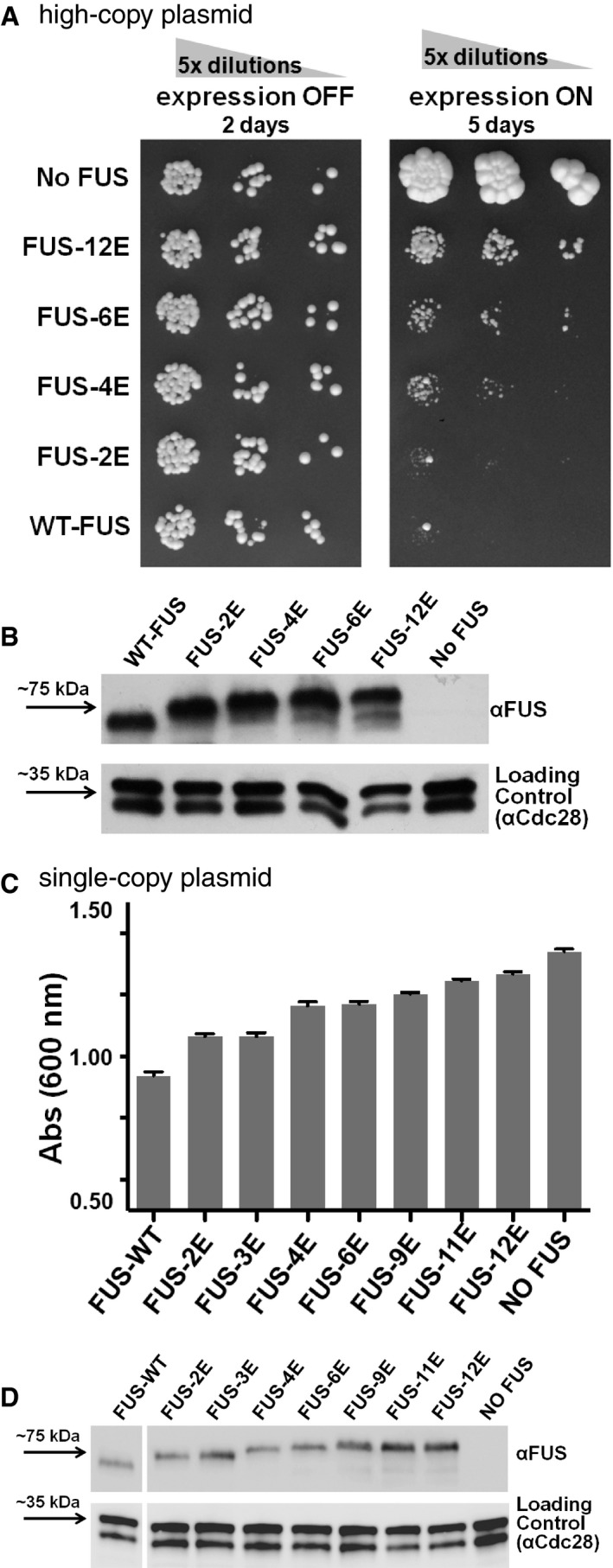
- Serial dilutions of yeast strain W303 expressing wild‐type or variant forms of human FUS from high‐copy plasmids show diminished toxicity as measured by reduced growth inhibition with greater number of phosphomimetic substitutions in the LC domain (FUS expression off, control = glucose medium, left; FUS expression on = galactose medium, right).
- Western blotting confirms comparable levels of FUS expression from high‐copy plasmids following overnight expression in yeast grown in galactose medium regardless of the number of phosphomimetic substitutions. Following immunoblotting, the membrane was stripped and re‐probed with anti‐Cdc28p, which serves as a loading control.
- Increasing the number of phosphomimetic substitutions in FUS expressed from single‐copy plasmids in yeast strain W303 in liquid culture (96‐well plates; 200 μl/well) is associated with reduced toxicity as measured by less growth inhibition relative to wild‐type FUS. Experiments were performed on three biological replicates. Errors bars represent ± SD.
- Western blotting confirms comparable relative levels of FUS expressed from single‐copy plasmids following overnight expression in yeast grown in galactose medium regardless of the number of phosphomimetic substitutions.
Discussion
Low‐complexity domains of RNA‐binding proteins have garnered much recent attention due to their role in RNA‐binding protein self‐assembly and disease‐associated aggregation. Recent work strongly points to the ability of these domains to form functional interactions with specific sets of proteins bearing similar low‐complexity (or prion‐like) domains (Kato et al, 2012; Molliex et al, 2015). Currently, many questions remain about the molecular nature of these interactions inside RNP granules—that is, where each specific membraneless organelle falls on a continuum of molecular architectures (Aguzzi & Altmeyer, 2016) ranging from structured cross‐β amyloids to disordered polymer chains (Patel et al, 2015; Xiang et al, 2015; Boke et al, 2016). However, it is clear that low‐complexity domains specify and mediate functional protein–protein interactions and pathological self‐aggregation. Low‐complexity domains are well‐established targets of post‐translational modifications (Martin et al, 2016), and their sequences specify homotypic and heterotypic interactions mediating liquid–liquid phase separation (LLPS; Lin et al, 2015; Pak et al, 2016). Hence, cells might employ post‐translational modification to “change” the amino acid sequence of prion‐like domains and thus disrupt self‐interaction. Here we suggest that phosphorylation of the aggregation‐prone FUS LC can dynamically control protein interactions, assembly, and in‐cell aggregation.
Low‐complexity domain phosphorylation—an “off switch” for prion‐like domain phase separation
Post‐translational modification of low‐complexity domains has been proposed as a means cells use to regulate the formation of membraneless organelles. For example, methylation of RG/RGG repeats suppresses phase separation of the highly charged domains of nuage protein DDX4 (Nott et al, 2015). Phosphorylation of the low‐complexity domains of maternal‐effect germline defective (MEG) proteins 1 and 3 promotes processing granule disassembly in Caenorhabditis elegans embryos (Wang et al, 2014). Therefore, some low‐complexity modifications may alter LLPS by disrupting the interactions of negative/positive charged residue patterns (Nott et al, 2015; Lee et al, 2016). However, unlike some low‐complexity domains, FUS LC is not patterned with charged residues—there are only two charged amino acids (D5 and D46) across the 163 residue SYGQ‐rich LC domain. Unlike methylation that preserves the net positive charge of arginine, serine/threonine phosphorylation imparts a net negative charge to an uncharged amino acid. In the case of FUS, multisite phosphorylation of the LC dramatically increases the effective number of negatively charged residues in the domain. Complementing previous findings that DNA‐PK treatment disrupts FUS LC recruitment to hydrogel models of granules (Han et al, 2012), here we demonstrate that phosphorylation and phosphomimetic substitution of FUS LC disrupts domain self‐interaction and LLPS. The LC is required for nuclear self‐association of FUS into chromatin‐binding dynamic puncta (Yang et al, 2014) and cytoplasmic granules (Shelkovnikova et al, 2014), as well as robust recruitment at sites of DNA damage (Mastrocola et al, 2013; Altmeyer et al, 2015). Therefore, cells might flip a chemical “off switch” by using phosphorylation to regulate FUS function via disrupting FUS self‐assembly.
How does phosphorylation disrupt phase separation of FUS? We observe no significant changes in LC secondary structure (i.e., folding) upon multiple phosphorylation, as shown for other heavily phosphorylated disordered domains (Martin et al, 2016). However, increasing negative charge at S/T sites across FUS LC decreases the propensity of those regions to form transient collapsed states and intermolecular contacts (Fig 3). Previously, phosphorylation has been shown to alter the complex coacervation (e.g., phase separation of a mixture of two oppositely charged species) of negatively charged RNA and short positively charged peptides (Aumiller & Keating, 2016). However, in the case of FUS, the phosphorylated LC contains no positively charged residues and phase separation is not based on its direct interaction with RNA (Burke et al, 2015). Importantly, we find that inclusion of either RNA to replicate the in vivo RNA‐bound state of FUS (Wang et al, 2015) or increased salt dramatically decreases the phase separation of phosphomimetic FUS, while wild‐type FUS is qualitatively unchanged. These data suggest that the interactions leading to the observed LLPS of phosphomimetic FUS are electrostatic, between the negatively charged phosphomimetic residues and the positively charged RNA‐binding regions, and that they are different from those stabilizing wild‐type FUS LLPS. Therefore, care must be taken in designing and interpreting in vitro LLPS assays as the results may depend strongly on the solution conditions and addition of binding partners like RNA. Further efforts are needed to determine how phase separation is governed in cells where a multitude of components make up physiological granules. FUS‐containing membraneless organelles are not homogeneous structures composed of one protein and one RNA (Kato et al, 2012; Jain et al, 2016). The addition of other granule components and molecular chaperones in vivo may decrease the dominant role of charge–charge repulsion in the effect on phase separation observed in vitro. Other post‐translational modifications may also play a role in regulating FUS phase separation. Fifteen additional FUS phosphorylation sites outside of LC have been identified by proteomic methods (see PhosphoSitePlus database record for FUS) that may function in regulation of FUS assembly, interaction, and function. FUS is also known to be heavily methylated in its RGG domains (Sama et al, 2014), which may further reduce FUS phase separation as was observed for DDX4 (Nott et al, 2015).
Low‐complexity domain phosphorylation—turning off prion‐like domain aggregation and toxicity
Low‐complexity domains with yeast prion‐like sequence composition are emerging as primary drivers of protein inclusion formation and are frequently the sites of disease‐causing missense mutations (King et al, 2012). However, it is unclear how the cell limits the aggregation propensity of these domains and if therapies targeting these domains could be developed. Efforts to enhance the ability of cells to disassemble inclusions using disaggregase chaperones show promise (Jackrel & Shorter, 2015; March et al, 2016). We thought post‐translational modifications would be an alternative and potentially more pharmacologically tractable strategy to disrupt assembly directly at its source. Therefore, we tested if post‐translational modifications could disrupt the assembly of prion‐like domains. Indeed, phosphorylation and phosphomimetic substitutions of FUS LC decrease the aggregation‐prone, prion‐like sequence character (Fig 1A). Thus, phosphorylation of LC prevents inter‐protein interactions that facilitate irreversible interactions and pathological aggregation. Phosphorylation may mechanistically alter disease aggregation in two ways. First, phosphorylation can increase solubility and the net charge of the domain. Second, if disease inclusions are in fact formed of amyloid fibrils, even incomplete phosphorylation may introduce sequence heterogeneity and disrupt the homotypic interactions required for taking on a repeating amyloid fibril conformation (Nomura et al, 2014; Shelkovnikova et al, 2014), including the in‐register parallel cross‐β structures commonly expected for these types of sequences (Shewmaker et al, 2006, 2009). A strategy for disrupting aggregation by altering post‐translational modifications is attractive as FUS has numerous phosphorylation sites, and such an intervention would not require stoichiometric delivery of an agent to bind each molecule of FUS and could be pharmacologically targeted via endogenous cellular kinases and signaling pathways. Therefore, it will be of importance to determine the detailed FUS phosphorylation state in ALS‐FUS cell models and FUS‐associated ALS patient tissue as well as healthy controls.
Here we show that phosphomimetic mutations of FUS reduce cytotoxicity and cytoplasmic aggregation. The effect of the increased charge repulsion in FUS LC is dose dependent: Increasing the number of phosphomimetic mutations proportionally decreases the amount of cytotoxicity and frequency of the punctate aggregates we and others have previously characterized (Fushimi et al, 2011; Kryndushkin et al, 2011). Aggregation and toxicity are reduced, even though the modifications are only in the LC of FUS, which comprises less than a third of the whole protein. Truncations excluding the RNA‐binding domains and modifications disrupting RRM RNA‐binding have been shown to suppress FUS toxicity in this model and similar models (Kryndushkin et al, 2011; Sun et al, 2011; Daigle et al, 2013), demonstrating that FUS toxicity involves RNA‐binding. The phosphomimetic substitutions explored here do not perturb RNA‐binding, only FUS aggregation. Therefore, we find that the aggregation‐prone, prion‐like nature of the unmodified LC is also required for toxicity. Our results are consistent with a model where cytoplasmic FUS gain‐of‐function toxicity arises from sequestration of RNA into inclusions, not simply from aberrant binding of cytoplasmic RNA, which would occur even if the protein remained soluble. Therefore, our results directly implicate FUS LC aggregation in FUS toxicity and demonstrate that post‐translational modifications can alter FUS's propensity to aggregate (Fig 7).
Figure 7. Model for FUS phosphorylation leading to reduced phase separation and aggregation.
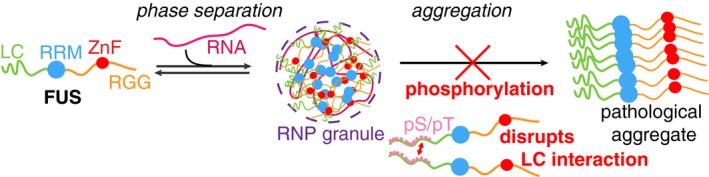
- In the cytoplasm, FUS and other prion‐like domain containing proteins and mRNA can partition into RNP granules. These granules concentrate FUS and hence may seed FUS inclusion formation in ALS and frontotemporal dementia. By discouraging LC‐LC intermolecular interactions of FUS, phosphorylation of FUS LC may regulate FUS/RNA phase separation into granules and disrupt FUS self‐aggregation into disease inclusions.
Phosphorylation may also have clinical implications by providing a mechanism for attenuating protein aggregation. Deletions of serines (S57 and S96) in FUS LC are associated with subtypes of ALS (Belzil et al, 2009; Yan et al, 2010). We identified S96 as a site phosphorylated in FUS. Therefore, diminished capacity for phosphorylation could enhance FUS aggregation during the decades‐long period in which disease develops. Of note, this is opposite of the current paradigm for tau aggregation, which is believed to be potentiated by hyper‐phosphorylation. In contrast, previous studies on other prion‐like, aggregation‐prone proteins suggest that phosphorylation may be associated with a reduction in self‐association (Li et al, 2011; Tenreiro et al, 2014).
Materials and Methods
Expression and purification of FUS
Protein expression of FUS wild type (WT) and phosphomimetic (6E, 12E) was performed in Escherichia coli as described previously (Burke et al, 2015). Details are provided in Appendix Supplementary Methods. MBP‐tagged (Peti & Page, 2007) FUS LC and FUS full‐length constructs were purified by immobilized metal ion affinity chromatography followed by gel filtration. MBP‐LCs were cleaved by TEV protease and further purified to leave FUS LC with no remaining tags. Samples for NMR spectroscopy were produced in M9 minimal media with 15N and 13C precursors as appropriate for the experiment. PRE values were measured by conjugating the paramagnetic compound MTSL to a series of FUS LC variants incorporating a single engineered cysteine position.
Turbidity and microscopy
WT and 12E MBP‐FUS full‐length protein was diluted to 5 μM into 20 mM Tris pH 7.4 with the NaCl concentration described for each experiment. Torula yeast RNA extract (Sigma, R6625) was desalted into 20 mM Tris 150 mM NaCl pH 7.4. Turbidity was assessed by measuring the optical density at 600 nm after the addition of TEV protease to induce phase separation by liberation of MBP solubility tag. Differential interference contrast (DIC) microscopy was conducted in the same conditions. 20 μl of samples was spotted onto a glass coverslip for imaging.
NMR spectroscopy
NMR experiments were recorded on Bruker Avance 850 or 500 MHz 1H Larmor frequency spectrometers with HCN TCI z‐gradient cryoprobes and sample temperature of 25°C. Experimental sweep widths, acquisition times, and the number of transients were optimized for the necessary resolution, experiment time, and signal‐to‐noise ratio as detailed in the Appendix Supplementary Methods. Backbone amide resonances assignments for 12E as well as spin relaxation experiments sensitive to backbone motions for WT and 12E were performed using standard experiments. Inter‐ and intramolecular PREs were quantified by explicit measurement of backbone amide 1HN R 2 values using a multiple time point interleaved experiment for paramagnetic and diamagnetic samples (Fawzi et al, 2010). All LC NMR experiments were performed in 50 mM MES, 150 mM NaCl at pH 5.5. See Appendix Supplementary Methods for full details. As we showed previously (Burke et al, 2015), this pH is chosen only to facilitate quantitative relaxation experiments (especially PRE experiments which rely upon slow amide hydrogen exchange with water) and does not affect the 1H 15N NMR resonance positions (spectra at pH 5.5 and pH 7.0 have the same chemical shifts), and hence, the structural ensemble of FUS LC is essentially unchanged.
Simulations
Parallel tempering metadynamics were performed for FUS 1–44 WT and phosphomimetic variant in well‐tempered ensemble simulations with explicit TIP4P/2005 water and the Amber ff03ws protein force field (Best et al, 2014). Coarse grain simulations were performed in LAMMPS (Plimpton et al, 2007) using an in‐house developed coarse grain model.
Mammalian cell culture
HEK293T cells were grown at 37°C and 5% CO2 in DMEM with 10% fetal bovine serum and 1× penicillin–streptomycin–l‐glutamine. To induce phosphorylation of FUS, calicheamicin (Pfizer) and calyculin A (Sigma) were dissolved in DMSO and added to cultures at indicated concentration. Cell lysis and Western blots were performed by conventional methods. See Appendix Supplementary Methods for full details. For transfection and live‐imaging experiments, cells were grown to ~60–80% confluency and transfected with 1 μg GFP‐FUS(R495X) plasmids (derivatives of mEGFP‐C1 plasmid—kindly provided by Michael Davidson, Addgene #54759—with wild‐type, 6E, or 12E LC domains) using Lipofectamine 2000 and OptiMEM medium (Invitrogen) following the manufacturer's protocol. After 8 h of incubation at 37°C, the transfection medium was removed and fresh medium was added. Cells were exchanged into serum‐free medium (DMEM, Gibco) 16 h after transfection and viewed using a Nikon A1R+ Resonant Scanning Confocal Microscope. The cells were maintained at 37°C during imaging.
Yeast
Wild‐type FUS and its phosphomimetic variants were subcloned from the described pET vectors into the multiple‐cloning sites (MCS) of yeast expression plasmids pFPS425 (CEN LEU2 P GAL1) and pFPS426 (2μ LEU2 P GAL1) using NdeI/XhoI. Yeast expression plasmids were transformed into strain W303 (MATa leu2 ade2‐1 ura3 can1 trp1 his3 gal+) and grown in synthetic‐complete glucose medium lacking leucine (SC‐leu). To induce FUS expression, galactose was replaced as the carbon source (SCgal‐leu). Growth assays were performed by spotting dilution series on solid, or growth in a 96‐well plate containing liquid, SCgal‐leu medium. Growth at 30°C was measured at 24 and 48 h. For fluorescence microscopy, approximately 2 × 108 cells were fixed and then centrifuged. Spheroplasts were generated, mounted on slides, and probed with anti‐FUS antibodies. Cells were scored for diffuse, intermediate, and punctate FUS localization patterns. See Appendix Supplementary Methods for full details.
Data availability
The NMR chemical shift assignments for FUS LC 12E from this publication have been deposited to the BMRB database (http://www.bmrb.wisc.edu/) and assigned the accession number BMRB: 27125. The chemical shift assignments for FUS LC wild type were previously deposited and available, BMRB 26672.
Author contributions
ZM, SNR, and FS designed, performed, and analyzed in cell experiments. VHR, AMJ, KAB, AEC, FS, and NLF designed, performed, and analyzed in vitro experiments. VHR, ZM, AMJ, FS, and NLF wrote manuscript. RO'M and RNC designed and performed mass spectrometry experiments on samples provided by ZM, and analyzed the related data. GHZ, GLD, and JM designed and performed computational/simulation experiments and analyzed the related data. WZ and RBB participated in the development of the coarse‐grained model.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Movie EV1
Review Process File
Acknowledgements
We thank Michael Clarkson for helpful discussions. We thank Geoff Williams at the Leduc Bioimaging Facility at Brown University for microscopy assistance. We would like to thank Drs. Rachel Cox, USU Department of Biochemistry, and Dennis McDaniel, USU Biomedical Instrumentation Center, for help with fluorescence microscopy, and Mr. Michael Panagos, USU Department of Pharmacology, for help with project organization. Research reported in this publication was supported in part by the National Institute Of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under Award Numbers R01GM118530 (to N.L.F) and R35GM119790 (to F.S.), a subproject as part of an Institutional Development Award (IDeA) from NIGMS (P20GM104937), a grant from the DEARS Foundation (to N.L.F.), and Medical Research Grant no. 20133966 from the Rhode Island Foundation (to N.L.F). V.H.R. was supported in part by an NIMH training grant to the Neuroscience Graduate Program at Brown University (T32 MH020068). A.E.C. was supported in part by an NIGMS training grant to the graduate program in Molecular Biology, Cell Biology and Biochemistry (MCB) at Brown University (T32 GM07601) and a BIBS Graduate Award in Brain Science from the Brown Institute for Brain Science Reisman Fund. Work at Lehigh University was supported by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), Division of Material Sciences and Engineering, under Award DE‐SC0013979 (to J.M.). Use of the high‐performance computing capabilities of the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (NSF) Grant TG‐MCB‐120014, is gratefully acknowledged. This research used resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE‐AC02‐05CH11231. This research is based in part on data obtained at the Brown University Structural Biology Core Facility supported by the Division of Biology and Medicine, Brown University. We thank Christoph Schorl and the Brown Genomics Core Facility supported by NIGMS P30GM103410, NCRR P30RR031153, P20RR018728, and S10RR02763, National Science Foundation EPSCoR 0554548. This research is based in part upon work conducted using the Rhode Island NSF/EPSCoR Proteomics Share Resource Facility, which is supported in part by the National Science Foundation EPSCoR Grant No. 1004057, National Institutes of Health Grant No. 1S10RR020923, S10RR027027, a Rhode Island Science and Technology Advisory Council grant, and the Division of Biology and Medicine, Brown University. We are grateful to Pfizer for generously providing calicheamicin. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
See also: J Shorter (October 2017)
Contributor Information
Frank Shewmaker, Email: fshewmaker@usuhs.edu.
Nicolas L Fawzi, Email: nicolas_fawzi@brown.edu.
References
- Aguzzi A, O'Connor T (2010) Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov 9: 237–248 [DOI] [PubMed] [Google Scholar]
- Aguzzi A, Altmeyer M (2016) Phase separation: linking cellular compartmentalization to disease. Trends Cell Biol 26: 547–558 [DOI] [PubMed] [Google Scholar]
- Altmeyer M, Neelsen KJ, Teloni F, Pozdnyakova I, Pellegrino S, Grofte M, Rask MB, Streicher W, Jungmichel S, Nielsen ML, Lukas J (2015) Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP‐ribose). Nat Commun 6: 8088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aumiller WM Jr, Keating CD (2016) Phosphorylation‐mediated RNA/peptide complex coacervation as a model for intracellular liquid organelles. Nat Chem 8: 129–137 [DOI] [PubMed] [Google Scholar]
- Beck M, Schmidt A, Malmstroem J, Claassen M, Ori A, Szymborska A, Herzog F, Rinner O, Ellenberg J, Aebersold R (2011) The quantitative proteome of a human cell line. Mol Syst Biol 7: 549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belzil VV, Valdmanis PN, Dion PA, Daoud H, Kabashi E, Noreau A, Gauthier J, SD Team , Hince P, Desjarlais A, Bouchard JP, Lacomblez L, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA (2009) Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology 73: 1176–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best RB, Zheng W, Mittal J (2014) Balanced protein‐water interactions improve properties of disordered proteins and non‐specific protein association. J Chem Theory Comput 10: 5113–5124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boke E, Ruer M, Wuhr M, Coughlin M, Lemaitre R, Gygi SP, Alberti S, Drechsel D, Hyman AA, Mitchison TJ (2016) Amyloid‐like self‐assembly of a cellular compartment. Cell 166: 637–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden HA, Dormann D (2016) Altered mRNP granule dynamics in FTLD pathogenesis. J Neurochem 138(Suppl 1): 112–133 [DOI] [PubMed] [Google Scholar]
- Burke KA, Janke AM, Rhine CL, Fawzi NL (2015) Residue‐by‐residue view of in vitro FUS granules that bind the C‐terminal domain of RNA polymerase II. Mol Cell 60: 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter T, Vancurova I, Sun I, Lou W, DeLeon S (1990) A DNA‐activated protein kinase from HeLa cell nuclei. Mol Cell Biol 10: 6460–6471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascarina SM, Ross ED (2014) Yeast prions and human prion‐like proteins: sequence features and prediction methods. Cell Mol Life Sci 71: 2047–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clore GM (2015) Practical aspects of paramagnetic relaxation enhancement in biological macromolecules. Methods Enzymol 564: 485–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conicella AE, Zerze GH, Mittal J, Fawzi NL (2016) ALS mutations disrupt phase separation mediated by alpha‐helical structure in the TDP‐43 low‐complexity C‐terminal domain. Structure 24: 1537–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchaine EM, Lu A, Neugebauer KM (2016) Droplet organelles? EMBO J 35: 1603–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle JG, Lanson NA Jr, Smith RB, Casci I, Maltare A, Monaghan J, Nichols CD, Kryndushkin D, Shewmaker F, Pandey UB (2013) RNA‐binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS‐linked mutations. Hum Mol Genet 22: 1193–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle JG, Krishnamurthy K, Ramesh N, Casci I, Monaghan J, McAvoy K, Godfrey EW, Daniel DC, Johnson EM, Monahan Z, Shewmaker F, Pasinelli P, Pandey UB (2016) Pur‐alpha regulates cytoplasmic stress granule dynamics and ameliorates FUS toxicity. Acta Neuropathol 131: 605–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Holler CJ, Taylor G, Hudson KF, Watkins W, Gearing M, Ito D, Murray ME, Dickson DW, Seyfried NT, Kukar T (2014) FUS is phosphorylated by DNA‐PK and accumulates in the cytoplasm after DNA damage. J Neurosci 34: 7802–7813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Salvio M, Piccinni V, Gerbino V, Mantoni F, Camerini S, Lenzi J, Rosa A, Chellini L, Loreni F, Carri MT, Bozzoni I, Cozzolino M, Cestra G (2015) Pur‐alpha functionally interacts with FUS carrying ALS‐associated mutations. Cell Death Dis 6: e1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrow NA, Zhang O, Szabo A, Torchia DA, Kay LE (1995) Spectral density function mapping using 15N relaxation data exclusively. J Biomol NMR 6: 153–162 [DOI] [PubMed] [Google Scholar]
- Fawzi NL, Ying J, Torchia DA, Clore GM (2010) Kinetics of amyloid beta monomer‐to‐oligomer exchange by NMR relaxation. J Am Chem Soc 132: 9948–9951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushimi K, Long C, Jayaram N, Chen X, Li L, Wu JY (2011) Expression of human FUS/TLS in yeast leads to protein aggregation and cytotoxicity, recapitulating key features of FUS proteinopathy. Protein Cell 2: 141–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner M, Toth R, Vandermoere F, Morrice NA, Rouse J (2008) Identification and characterization of FUS/TLS as a new target of ATM. Biochem J 415: 297–307 [DOI] [PubMed] [Google Scholar]
- Gitler AD, Shorter J (2011) RNA‐binding proteins with prion‐like domains in ALS and FTLD‐U. Prion 5: 179–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, Chen M, Xie Y, Allen J, Xiao G, McKnight SL (2012) Cell‐free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149: 768–779 [DOI] [PubMed] [Google Scholar]
- Harper JD, Lansbury PT Jr (1997) Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time‐dependent solubility of amyloid proteins. Annu Rev Biochem 66: 385–407 [DOI] [PubMed] [Google Scholar]
- Jackrel ME, Shorter J (2015) Engineering enhanced protein disaggregases for neurodegenerative disease. Prion 9: 90–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, Parker R (2016) ATPase‐modulated stress granules contain a diverse proteome and substructure. Cell 164: 487–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju S, Tardiff DF, Han H, Divya K, Zhong Q, Maquat LE, Bosco DA, Hayward LJ, Brown RH Jr, Lindquist S, Ringe D, Petsko GA (2011) A yeast model of FUS/TLS‐dependent cytotoxicity. PLoS Biol 9: e1001052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, Grishin NV, Frantz DE, Schneider JW, Chen S, Li L, Sawaya MR, Eisenberg D, Tycko R, McKnight SL (2012) Cell‐free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149: 753–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana V, Lindquist S (2010) Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker's yeast? Nat Rev Neurosci 11: 436–449 [DOI] [PubMed] [Google Scholar]
- Kiebler MA, Bassell GJ (2006) Neuronal RNA granules: movers and makers. Neuron 51: 685–690 [DOI] [PubMed] [Google Scholar]
- Kim ST, Lim DS, Canman CE, Kastan MB (1999) Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem 274: 37538–37543 [DOI] [PubMed] [Google Scholar]
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN et al (2013) Mutations in prion‐like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495: 467–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King OD, Gitler AD, Shorter J (2012) The tip of the iceberg: RNA‐binding proteins with prion‐like domains in neurodegenerative disease. Brain Res 1462: 61–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, Schilling B, Row RH, Kenski DM, Gibson BW, Shokat KM (2003) Phosphospecific proteolysis for mapping sites of protein phosphorylation. Nat Biotechnol 21: 1047–1054 [DOI] [PubMed] [Google Scholar]
- Koo EH, Lansbury PT Jr, Kelly JW (1999) Amyloid diseases: abnormal protein aggregation in neurodegeneration. Proc Natl Acad Sci USA 96: 9989–9990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryndushkin D, Wickner RB, Shewmaker F (2011) FUS/TLS forms cytoplasmic aggregates, inhibits cell growth and interacts with TDP‐43 in a yeast model of amyotrophic lateral sclerosis. Protein Cell 2: 223–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak‐Vance MA, Yan J, Ticozzi N et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323: 1205–1208 [DOI] [PubMed] [Google Scholar]
- Lancaster AK, Nutter‐Upham A, Lindquist S, King OD (2014) PLAAC: a web and command‐line application to identify proteins with prion‐like amino acid composition. Bioinformatics 30: 2501–2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A, Maxwell BA, Kim NC, Temirov J, Moore J, Kolaitis RM, Shaw TI, Bai B, Peng J, Kriwacki RW, Taylor JP (2016) C9orf72 dipeptide repeats impair the assembly, dynamics, and function of membrane‐less organelles. Cell 167: 774–788.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenzi J, De Santis R, de Turris V, Morlando M, Laneve P, Calvo A, Caliendo V, Chio A, Rosa A, Bozzoni I (2015) ALS mutant FUS proteins are recruited into stress granules in induced pluripotent stem cell‐derived motoneurons. Dis Model Mech 8: 755–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HY, Yeh PA, Chiu HC, Tang CY, Tu BP (2011) Hyperphosphorylation as a defense mechanism to reduce TDP‐43 aggregation. PLoS One 6: e23075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YR, King OD, Shorter J, Gitler AD (2013) Stress granules as crucibles of ALS pathogenesis. J Cell Biol 201: 361–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Protter DS, Rosen MK, Parker R (2015) Formation and maturation of phase‐separated liquid droplets by RNA‐binding proteins. Mol Cell 60: 208–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- March ZM, King OD, Shorter J (2016) Prion‐like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res 1647: 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin EW, Holehouse AS, Grace CR, Hughes A, Pappu RV, Mittag T (2016) Sequence determinants of the conformational properties of an intrinsically disordered protein prior to and upon multisite phosphorylation. J Am Chem Soc 138: 15323–15335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastrocola AS, Kim SH, Trinh AT, Rodenkirch LA, Tibbetts RS (2013) The RNA‐binding protein fused in sarcoma (FUS) functions downstream of poly(ADP‐ribose) polymerase (PARP) in response to DNA damage. J Biol Chem 288: 24731–24741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JC, McGoldrick P, Vance C, Hortobagyi T, Sreedharan J, Rogelj B, Tudor EL, Smith BN, Klasen C, Miller CC, Cooper JD, Greensmith L, Shaw CE (2013) Overexpression of human wild‐type FUS causes progressive motor neuron degeneration in an age‐ and dose‐dependent fashion. Acta Neuropathol 125: 273–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163: 123–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Qamar S, Lin JQ, Schierle GS, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FT, Michel CH, Kronenberg‐Versteeg D, Li Y, Yang SP, Wakutani Y, Meadows W, Ferry RR, Dong L, Tartaglia GG, Favrin G, Lin WL et al (2015) ALS/FTD mutation‐induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88: 678–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR (2009) A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132: 2922–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Watanabe S, Kaneko K, Yamanaka K, Nukina N, Furukawa Y (2014) Intranuclear aggregation of mutant FUS/TLS as a molecular pathomechanism of amyotrophic lateral sclerosis. J Biol Chem 289: 1192–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett‐Jones DP, Pawson T, Forman‐Kay JD, Baldwin AJ (2015) Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell 57: 936–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott TJ, Craggs TD, Baldwin AJ (2016) Membraneless organelles can melt nucleic acid duplexes and act as biomolecular filters. Nat Chem 8: 569–575 [DOI] [PubMed] [Google Scholar]
- O'Neill T, Dwyer AJ, Ziv Y, Chan DW, Lees‐Miller SP, Abraham RH, Lai JH, Hill D, Shiloh Y, Cantley LC, Rathbun GA (2000) Utilization of oriented peptide libraries to identify substrate motifs selected by ATM. J Biol Chem 275: 22719–22727 [DOI] [PubMed] [Google Scholar]
- Pak CW, Kosno M, Holehouse AS, Padrick SB, Mittal A, Ali R, Yunus AA, Liu DR, Pappu RV, Rosen MK (2016) Sequence determinants of intracellular phase separation by complex coacervation of a disordered protein. Mol Cell 63: 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, Pozniakovski A, Poser I, Maghelli N, Royer LA, Weigert M, Myers EW, Grill S, Drechsel D, Hyman AA, Alberti S (2015) A liquid‐to‐solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162: 1066–1077 [DOI] [PubMed] [Google Scholar]
- Peti W, Page R (2007) Strategies to maximize heterologous protein expression in Escherichia coli with minimal cost. Protein Expr Purif 51: 1–10 [DOI] [PubMed] [Google Scholar]
- Plimpton S, Crozier P, Thompson A (2007) LAMMPS‐large‐scale atomic/molecular massively parallel simulator. Sandia National Laboratories http://lammps.sandia.gov
- Ross ED, Toombs JA (2010) The effects of amino acid composition on yeast prion formation and prion domain interactions. Prion 4: 60–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu HH, Jun MH, Min KJ, Jang DJ, Lee YS, Kim HK, Lee JA (2014) Autophagy regulates amyotrophic lateral sclerosis‐linked fused in sarcoma‐positive stress granules in neurons. Neurobiol Aging 35: 2822–2831 [DOI] [PubMed] [Google Scholar]
- Sama RR, Ward CL, Bosco DA (2014) Functions of FUS/TLS from DNA repair to stress response: implications for ALS. ASN Neuro 6: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scekic‐Zahirovic J, Sendscheid O, El Oussini H, Jambeau M, Sun Y, Mersmann S, Wagner M, Dieterle S, Sinniger J, Dirrig‐Grosch S, Drenner K, Birling MC, Qiu J, Zhou Y, Li H, Fu XD, Rouaux C, Shelkovnikova T, Witting A, Ludolph AC et al (2016) Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J 35: 1077–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller R, Eick D (2016) Getting access to low‐complexity domain modifications. Trends Biochem Sci 41: 894–897 [DOI] [PubMed] [Google Scholar]
- Schwartz JC, Wang X, Podell ER, Cech TR (2013) RNA seeds higher‐order assembly of FUS protein. Cell Rep 5: 918–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC, Mentis GZ, Shneider NA (2016) ALS‐associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun 7: 10465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelkovnikova TA, Robinson HK, Southcombe JA, Ninkina N, Buchman VL (2014) Multistep process of FUS aggregation in the cell cytoplasm involves RNA‐dependent and RNA‐independent mechanisms. Hum Mol Genet 23: 5211–5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewmaker F, Wickner RB, Tycko R (2006) Amyloid of the prion domain of Sup35p has an in‐register parallel beta‐sheet structure. Proc Natl Acad Sci USA 103: 19754–19759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shewmaker F, Kryndushkin D, Chen B, Tycko R, Wickner RB (2009) Two prion variants of Sup35p have in‐register parallel beta‐sheet structures, independent of hydration. Biochemistry 48: 5074–5082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Diaz Z, Fang X, Hart MP, Chesi A, Shorter J, Gitler AD (2011) Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol 9: e1000614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Nakajima M, Morimoto K (1994) Calyculin A, a non‐phorbol ester type tumor promotor, induced oxidative DNA damage in stimulated human neutrophil‐like cells. Biochem Biophys Res Commun 205: 1803–1807 [DOI] [PubMed] [Google Scholar]
- Tenreiro S, Eckermann K, Outeiro TF (2014) Protein phosphorylation in neurodegeneration: friend or foe? Front Mol Neurosci 7: 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MG, Loschi M, Desbats MA, Boccaccio GL (2011) RNA granules: the good, the bad and the ugly. Cell Signal 23: 324–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski JQ, Goedert M, Iwatsubo T, Lee VM (1998) Fatal attractions: abnormal protein aggregation and neuron death in Parkinson's disease and Lewy body dementia. Cell Death Differ 5: 832–837 [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T et al (2015) Proteomics. Tissue‐based map of the human proteome. Science 347: 1260419 [DOI] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al‐Saraj S, Al‐Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323: 1208–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Scotter EL, Nishimura AL, Troakes C, Mitchell JC, Kathe C, Urwin H, Manser C, Miller CC, Hortobagyi T, Dragunow M, Rogelj B, Shaw CE (2013) ALS mutant FUS disrupts nuclear localization and sequesters wild‐type FUS within cytoplasmic stress granules. Hum Mol Genet 22: 2676–2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Smith J, Chen BC, Schmidt H, Rasoloson D, Paix A, Lambrus BG, Calidas D, Betzig E, Seydoux G (2014) Regulation of RNA granule dynamics by phosphorylation of serine‐rich, intrinsically disordered proteins in C. elegans . Elife 3: e04591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Schwartz JC, Cech TR (2015) Nucleic acid‐binding specificity of human FUS protein. Nucleic Acids Res 43: 7535–7543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang S, Kato M, Wu LC, Lin Y, Ding M, Zhang Y, Yu Y, McKnight SL (2015) The LC domain of hnRNPA2 adopts similar conformations in hydrogel polymers, liquid‐like droplets, and nuclei. Cell 163: 829–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Deng HX, Siddique N, Fecto F, Chen W, Yang Y, Liu E, Donkervoort S, Zheng JG, Shi Y, Ahmeti KB, Brooks B, Engel WK, Siddique T (2010) Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 75: 807–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Gal J, Chen J, Zhu H (2014) Self‐assembled FUS binds active chromatin and regulates gene transcription. Proc Natl Acad Sci USA 111: 17809–17814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerze GH, Best RB, Mittal J (2015) Sequence‐ and temperature‐dependent properties of unfolded and disordered proteins from atomistic simulations. J Phys Chem B 119: 14622–14630 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Movie EV1
Review Process File
Data Availability Statement
The NMR chemical shift assignments for FUS LC 12E from this publication have been deposited to the BMRB database (http://www.bmrb.wisc.edu/) and assigned the accession number BMRB: 27125. The chemical shift assignments for FUS LC wild type were previously deposited and available, BMRB 26672.