

Abstract
Nonsense‐mediated mRNA decay (NMD) is a cellular surveillance pathway that recognizes and degrades mRNAs with premature termination codons (PTCs). The mechanisms underlying translation termination are key to the understanding of RNA surveillance mechanisms such as NMD and crucial for the development of therapeutic strategies for NMD‐related diseases. Here, we have used a fully reconstituted in vitro translation system to probe the NMD proteins for interaction with the termination apparatus. We discovered that UPF3B (i) interacts with the release factors, (ii) delays translation termination and (iii) dissociates post‐termination ribosomal complexes that are devoid of the nascent peptide. Furthermore, we identified UPF1 and ribosomes as new interaction partners of UPF3B. These previously unknown functions of UPF3B during the early and late phases of translation termination suggest that UPF3B is involved in the crosstalk between the NMD machinery and the PTC‐bound ribosome, a central mechanistic step of RNA surveillance.
Keywords: nonsense‐mediated mRNA decay, translation termination, UPF3B
Subject Categories: Protein Biosynthesis & Quality Control, RNA Biology
Introduction
Nonsense‐mediated mRNA decay (NMD) is a eukaryotic surveillance mechanism that controls the expression of aberrant mRNAs, degrading transcripts with premature termination codons (PTCs). PTCs can be introduced into mRNAs by mutations, transcriptional errors and aberrant splicing, but are also contained in 5–15% of normal transcripts (Mendell et al, 2004; Nguyen et al, 2014; Karousis et al, 2016). By modulating the expression of physiological target mRNAs, NMD serves as a post‐transcriptional regulator of gene expression and thus controls important cellular and organismal processes in development, cellular stress responses, immunity and neuronal differentiation (Linder et al, 2015; Lykke‐Andersen & Jensen, 2015; Kurosaki & Maquat, 2016; Ottens & Gehring, 2016). NMD is also of medical importance as it limits the production of truncated proteins that may otherwise exert dominant negative functions but can also result in loss of function when mRNAs encoding (partially) functional truncated proteins are degraded (Bhuvanagiri et al, 2010; Nguyen et al, 2014). Mutations or copy number variations in NMD factors are linked to genetic diseases, specifically to neurodevelopmental disorders and intellectual disabilities (Nguyen et al, 2014; Linder et al, 2015).
Conceptually, NMD can be divided into a translation termination phase and an mRNA degradation phase. During the past two decades, a wealth of information has accumulated documenting the interplay between the core NMD factors and decay enzymes that enable the recognition and degradation of NMD substrates (Schweingruber et al, 2013; Fatscher et al, 2015). However, the mechanism by which translation termination at a PTC is distinguished from termination at a normal termination codon (NTC) is still poorly understood. Two prevailing models, the “downstream marker model” and the “faux 3′UTR model”, have been proposed to explain the difference between normal and aberrant termination (reviewed in Bhuvanagiri et al, 2010; He & Jacobson, 2015). The “downstream marker model” posits the formation of an aberrant termination complex at a PTC consisting of the terminating ribosome, the central NMD effector UPF1, the SMG1‐8‐9 kinase complex and the release factors eRF1 and eRF3. This so‐called SURF complex (Kashima et al, 2006) is thought to delay translation termination and to sense the presence of an mRNP complex on the extended 3′UTR which in mammalian cells is represented by an exon junction complex (EJC) downstream of the PTC. The terminating ribosome and the EJC are thought to be bridged by UPF2 that, according to this model, interacts with UPF1 at the termination site and EJC‐bound UPF3B, leading to the formation of a decay inducing complex that remodels the 3′ mRNP and recruits mRNA decay enzymes.
The faux 3′UTR model posits that NMD can be induced by an aberrant 3′UTR mRNP characterized by the absence of at least one termination‐enhancing factor that is associated with a normal 3′UTR (Amrani et al, 2004). Consequently, termination at a PTC is delayed and inefficient. Such an aberrant 3′UTR mRNP can be caused by inappropriate spacing between the termination codon and the poly(A) tail, preventing the termination‐promoting interaction between eRF3a and poly(A) binding protein, and instead allowing the recruitment of UPF1.
Both models converge on the central NMD effector UPF1 that interacts with the release factors (eRFs) at the terminating ribosome. For yeast NMD, all three UPF proteins are essential, whereas in higher eukaryotes, UPF2‐independent, UPF3B‐independent and EJC‐independent NMD branches have been described (Gehring et al, 2005; Bühler et al, 2006; Chan et al, 2007). How UPF2 and UPF3 are recruited to the termination site in EJC‐independent NMD is unknown. UPF2 and UPF3B are thought to stimulate the phosphorylation of UPF1 and to activate UPF1's ATPase and helicase functions that are necessary to remodel the 3′UTR mRNP and to recruit mRNA degradation enzymes (Kashima et al, 2006; Chamieh et al, 2008; Ivanov et al, 2008; Fiorini et al, 2015).
Although the necessity of an interaction between the UPF proteins and the translation termination apparatus is generally accepted, the sequence and timing of NMD factor recruitment to the termination site have not been addressed experimentally. The hypothesis that UPF1 is specifically recruited to aberrant termination events as an anchor point for the assembly of an NMD‐mRNP has been challenged by the finding that UPF1 is bound along the entire length of transcripts and that this binding occurs in a translation‐independent fashion (Hogg & Goff, 2010; Hurt et al, 2013; Zünd et al, 2013).
Translation termination, whether regular or aberrant, needs to recycle ribosomes to avoid deleterious consequences for the translation apparatus (Graille & Seraphin, 2012; Lykke‐Andersen & Bennett, 2014). In ribosome recycling, the ATPase ABCE1/Rli1 is needed for the ultimate dissociation of post‐termination ribosomes from the mRNA (Dever & Green, 2012; Franckenberg et al, 2012; Graille & Seraphin, 2012; Jackson et al, 2012). In yeast and human cells, depletion of the UPF proteins induces readthrough at PTCs in vivo as well as delayed termination in vitro (Amrani et al, 2004; Peixeiro et al, 2012). A recent attempt to reconcile all available data into a new NMD model posits that UPF1, UPF2 and UPF3 have roles in early and late phases of premature termination (He & Jacobson, 2015). Accordingly, UPF1's initially weak association with elongating ribosomes is proposed to be stabilized by UPF2 and UPF3 when a ribosome terminates prematurely, stimulating the initially delayed termination at a PTC by either recruiting the release factors or by enhancing peptide release. Subsequently, UPF2 and UPF3 promote ATP hydrolysis by UPF1 to fuel the dissociation of post‐terminating ribosomal complexes. UPF1, still bound to the 40S ribosomal subunit, then recruits mRNA decay enzymes to initiate mRNA degradation.
To shed light on these critical aspects of translation termination in an NMD context, we adopted an approach that combines a fully reconstituted in vitro translation termination system with in vitro and in vivo interaction studies to decipher the UPF‐eRF interactome in translation termination. We find that UPF3B interacts with eRF3a and forms a trimeric complex with both eRF3a and eRF1. Moreover, UPF3B binds to RNA, the ribosome and to UPF1. Unexpectedly, UPF1 plays no discernible functional role in this context, suggesting that it acts downstream to promote NMD. Importantly, UPF3B delays translation termination when release factors are limiting and dissolves post‐termination complexes after peptidyl‐tRNA hydrolysis.
Results
Validation of the experimental system
During termination at a PTC, the UPF1‐eRF interaction is thought to impede translation termination (Kashima et al, 2006; Ivanov et al, 2008). Here, we analyse whether UPF1 alone or together with UPF2 and/or UPF3B affects the efficiency of mammalian translation termination in vitro.
We produced full‐length eRF1, eRF3a, UPF1 and UPF3B. Because both the N‐ and C‐termini of purified full‐length UPF2 are unstable when expressed in Escherichia coli or insect cells, we produced a stable UPF2 variant (UPF2L) comprising amino acids (aa) 121–1227 (Fig EV1A). UPF2L contains the UPF1‐ and UPF3B‐binding domains and has the same activities as full‐length UPF2 (Chamieh et al, 2008; Chakrabarti et al, 2011). Ribosomal pre‐termination complexes (translating ribosomes stalled at a stop codon; pre‐TCs) were assembled on a model mRNA using ribosomal subunits, aminoacylated tRNAs, and purified initiation and elongation factors. The model mRNA (MVHC‐STOP) contained the β‐globin 5′UTR and a short open reading frame encoding a MVHC tetrapeptide followed by a UAA stop codon and a 3′UTR of ~400 nt (Fig 1A) (Alkalaeva et al, 2006; Fan‐Minogue et al, 2008). The formation of defined ribosomal complexes was analysed by sucrose density‐gradient (SDG) centrifugation and primer extension inhibition (toeprinting). Pre‐TCs containing the peptidyl‐tRNACys in the ribosomal P‐site and the stop codon in the A site are characterized by specific toeprints at position +16 nt 3′ to the U of the UGC (Cys) codon (Fig 1B, lane 1; Fig EV1B).
Figure EV1. Validation of the experimental system.
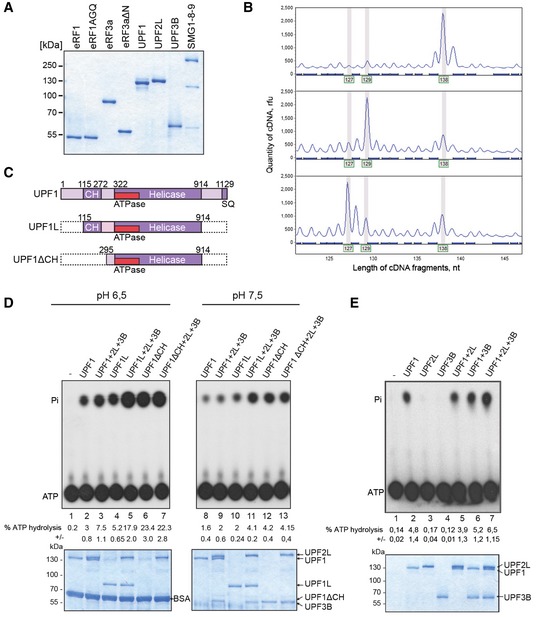
- SDS–PAGE analysis of purified recombinant proteins used in the toeprinting experiments as indicated.
- Toeprint analysis of translation complexes prepared by the reconstituted in vitro translation system. Above: 80S initiation complex; middle: pre‐termination complex (pre‐TC); below: termination complex formed in presence of eRF1, eRF3a and GTP. Peaks at 138 nt indicate the position of the 80S initiation complex on mRNA, peaks at 129 nt indicate the position of pre‐TC and peaks at 127 nt correspond to the termination complex (post‐TC). Rfu—relative fluorescence units.
- Schematic representation of UPF1 variants used in (D).
- Thin layer chromatography (TLC) analysis of the ATPase activity of UPF1 variants in the absence or presence of UPF2L and/or UPF3B at 30°C in MES buffer (pH 6.5, lanes 1–7) or translation buffer (pH 7.5, lanes 8–13), respectively. 1.5 μl of the samples was spotted on the TLC plates, and the residual 18.5 μl was analysed on SDS–PAGE gels for loading control (lower panels). The positions of γ32P‐ATP and γ32Pi are indicated. % ATP hydrolysis in (D and E) was calculated using a phosphoimager and displays the means ± SEM of four independent experiments.
- ATP hydrolysis experiment as in (D) at 37°C in translation buffer.
Source data are available online for this figure.
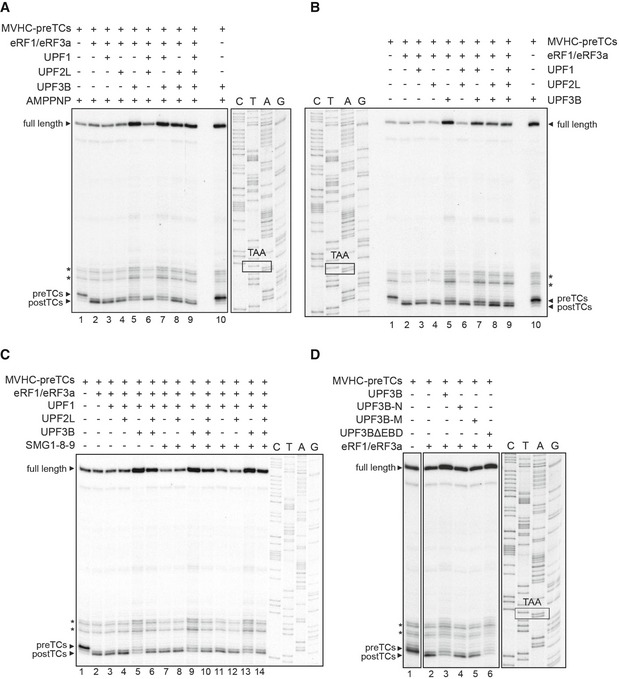
Figure 1. UPF3B delays translation termination in vitro .

- Scheme of the MVHC‐STOP mRNA.
- Toeprinting analysis of ribosomal complexes obtained by incubating pre‐TCs assembled on MVHC‐STOP mRNA (MVHC‐pre‐TCs) with UPF1, UPF2L, UPF3B or BSA at 1 mM free Mg2+ followed by termination with limiting amounts of eRF1 and eRF3a. The positions of pre‐TCs, post‐TCs and full‐length cDNA are indicated. In all toeprinting analyses the box marked TAA indicates the stop codon in the cDNA sequence corresponding to the MVHC‐STOP mRNA. Asterisks mark initiation and elongation complexes in all toeprinting analyses. Representative of five independent experiments. Lanes C, T, A, G indicates the sequence of the cDNA derived from the MVHC‐STOP mRNA using the same primer as the toeprint experiment. The box ‘TAA’ indicates the position of the stop codon.
- Kinetics of [35S]‐peptidyl‐tRNA hydrolysis in the presence of eRF1 and eRF3a (black circles) or eRF1, eRF3a and UPF3B (white triangles). Termination reactions were assembled as in (B). A value equal to 1 corresponds to the maximum value for peptide release triggered by eRF1 and eRF3a. Data points show the mean of three experiments ± SEM.
- UPF1 in vitro phosphorylation by SMG1‐8‐9 in the presence of UPF2L and/or UPF3B and in the presence (lanes 7–12) or absence (lanes 1–6) of the eRFs. In lanes 13–16 UPF2L and/or UPF3B were added after UPF1 phosphorylation. Samples were analysed by SDS–PAGE, Coomassie‐stained to control for equal loading (lower panel) and autoradiographed (upper panel). SMG1 autophosphorylation (P‐SMG1) confirms equal SMG1‐activity in all samples. UPF1 is represented by the lower and UPF2L by the upper of the two closely migrating bands between 125 and 130 kDa in the Coomassie‐stained gel. Representative of two independent experiments.
- Toeprinting analysis of ribosomal complexes obtained by incubating pre‐TCs with UPF1, UPF2L, UPF3B, SMG1‐8‐9 or BSA at 1 mM free Mg2+ as indicated followed by translation termination by eRF1 and eRF3a. See also Fig EV2. Representative of three independent experiments.
Source data are available online for this figure.
During translation termination, eRF1 in complex with eRF3a binds to the stop codon (Brown et al, 2015) inducing conformational rearrangements. The formation of such post‐termination complexes (post‐TCs) is manifested by a +1–2 nt forward shift relative to the pre‐TC toeprint (Fig 1B, lane 2; Fig EV1B) (Shirokikh et al, 2010). These shifted toeprint bands arise from mRNA compaction in the mRNA channel of the 40S subunit upon binding of the eRFs to the stop codon in the ribosomal A site (Brown et al, 2015; Matheisl et al, 2015; Ivanov et al, 2016). GTP hydrolysis by eRF3a leads to accommodation of the GGQ motif of eRF1 in the peptidyl transferase centre of the large ribosomal subunit, resulting in rapid peptide release. eRF1 (together with eRF3a or after dissociation of eRF3a) remains associated with post‐TCs, thus maintaining the +1–2 nt shift of their toeprint (Alkalaeva et al, 2006).
To confirm the catalytic activity of purified UPF1, UPF2L and UPF3B, we performed ATP hydrolysis experiments. In previous studies (Chamieh et al, 2008; Chakrabarti et al, 2011), maximal ATP hydrolysis by UPF1 was achieved at low pH (6–6.5) using N‐ and C‐terminally truncated UPF1 fragments UPF1L and UPF1ΔCH (Fig EV1C). Using these UPF1 isoforms as controls, we first tested ATP hydrolysis by UPF1 in end point experiments at pH 6.5 (Fig EV1D). UPF1ΔCH (aa 295–914) lacks the CH domain and exhibits high ATPase activity that cannot be further stimulated (Fig EV1D, lanes 6, 7), whereas UPF1L (aa 115–914) contains the CH domain and exhibits similar ATPase activity when stimulated by UPF2 and UPF3B (Fig EV1D, lanes 4, 5) (Chamieh et al, 2008). In full‐length UPF1 (UPF1), the C‐terminus contributes to maintaining UPF1 in an inactive state (Fiorini et al, 2013). Accordingly, UPF1 exhibited only modest ATPase activity at 30°C and pH 6.5 which was doubled in the presence of UPF2L/3B (Fig EV1D, lanes 2 and 3). These results using our recombinant UPF1 isoforms at pH 6.5 fully conformed to previous findings (Chamieh et al, 2008; Chakrabarti et al, 2011; Fiorini et al, 2013). At physiological pH 7.5, the activity of all proteins was lower, but followed a similar pattern (Fig EV1D, lanes 8–13). Under in vitro translation conditions at pH 7.5 and 37°C (Fig EV1E), neither UPF2L (lane 5) nor UPF3B (lane 6) had a significant effect on the ATPase function of UPF1. Taken together, these data show that our UPF proteins are catalytically active.
UPF3B delays inefficient translation termination in a fully reconstituted translation termination system
Termination at a PTC contrasts with termination at a NTC by being slowed and less efficient. This kinetic difference is thought to be either caused by the absence of the termination‐stimulating protein PABPC1 and/or by inefficient recruitment of the eRFs in the presence of UPF1 (Amrani et al, 2004; Peixeiro et al, 2012; He & Jacobson, 2015; Ivanov et al, 2016). To mimic this situation in vitro and to avoid missing relevant modulatory effects of the UPF proteins, we used limiting concentrations of eRFs for our termination experiments as judged by the retention of a faint, but discernible pre‐TC toeprint in addition to the appearance of post‐TC signals after termination (Figs 1B and E, EV2A and D, lanes 2).
Figure EV2. Validation of the termination‐delaying effect of UPF3B.

- Toeprinting analysis of ribosomal complexes obtained by incubating MVHC‐pre‐TCs at 1 mM free Mg2+ with decreasing amounts of eRFs. Representative example for the titration of eRF1 and eRF3a to identify concentrations slowing down the pre‐TC to post‐TC transition. The amount used for the sample in lane 5 was chosen for further experiments with this batch of pre‐TCs.
- Toeprinting analysis of ribosomal complexes obtained by incubating MVHC‐pre‐TCs with UPF3B, eIF4B, IRP1, SXL or BSA at 1 mM free Mg2+ and 1 mM ATP followed by termination with limiting amounts of eRF1 and eRF3a.
- Toeprinting analysis of ribosomal complexes obtained as in (B) by incubating MVHC‐pre‐TCs with UPF3B, UPF3B‐N, UPF3B‐M, UPF3BΔEBD or BSA.
- Toeprinting analysis of ribosomal complexes obtained as in Fig 1B in the presence of 1 mM ATP or AMPPNP, respectively.
To test whether UPF proteins affect the efficiency of translation termination, equal amounts of pre‐TCs that had been assembled on MVHC‐STOP mRNA and purified by SDG centrifugation were incubated with UPF1, UPF2L, UPF3B or combinations of these proteins as indicated (Fig 1B, lanes 3–9). UPF proteins were added in excess to saturate their interaction with the release factors, the mRNA and pre‐TCs. In fact, their local concentration, for example associated with the 3′UTR of natural NMD substrates, is impossible to estimate and might be in excess of terminating ribosomes (Zünd & Mühlemann, 2013; Hauer et al, 2016). As controls, pre‐TCs were either left untreated (lane 1) or the UPF proteins were replaced by BSA (lane 2). Subsequently, limiting amounts of eRFs were added to the reactions (except in lane 1), and translation termination was allowed to proceed for 5 min, followed by toeprinting analysis.
Neither UPF1 nor UPF2L individually (Fig 1B, lanes 3, 4) or together (lane 6) affected the intensity of pre‐ or post‐TC bands, compared to the control sample (lane 2). These findings indicate that when the eRFs are limiting, neither UPF1 nor UPF2L has a direct effect on translation termination in vitro.
By contrast, UPF3B (lane 5) substantially reduced the pre‐TC to post‐TC transformation rate to about 40% of the rate observed in the control reaction (lane 2) as estimated by calculating the ratio between the pre‐TC and post‐TC signal intensities using a phosphoimager. Notably, the addition of UPF1 to UPF3B resulted in a similar delay of termination (lane 7) and did not have an additive, synergistic or reversing effect. Addition of UPF2L abolished the effect of UPF3B on translation termination (lane 7, 8) confirming that the termination delay is specifically caused by UPF3B and indicating that binding to UPF2L may prevent UPF3B from interfering with the termination reaction. We observed that the toeprint signals corresponding to the full‐length RNA and to the termination complexes as well as to the traces of initiating and elongating ribosomes present in pre‐TC preparations were always stronger in the presence of UPF3B than in reactions without UPF3B. Therefore, we performed toeprinting of pre‐TCs that had been incubated with UPF3B but without eRFs. We found that here, too, all toeprint signals were stronger in the presence of UPF3B (Fig 1B, lanes 1 and 10), which is likely caused by more efficient recovery of ribosomal complexes and RNA in the reverse transcription reaction and the subsequent purification steps following the in vitro translation termination reaction. UPF3B has a basic pI of 9.5 and contains an RNA recognition motif (RRM). To exclude that the inhibitory effect of UPF3B on the pre‐TC‐post‐TC transition is due to unspecific binding to ribosomes and/or RNA, we tested other proteins with similar biochemical properties. Neither eIF4B (RNA‐ and ribosome‐binding), nor IRP1 (RNA‐binding), or SXL (RNA‐binding, pI 9.5) had an influence on in vitro translation termination (Fig EV2B). Likewise, we tested truncated versions of UPF3B for their capacity to delay translation termination. UPF3B‐N (aa 42–217, pI 8.0), comprising the RRM domain (Kadlec et al, 2004), and UPF3B‐M (aa 147–419, pI 9.7) comprising the middle domain had no influence on translation termination. In contrast, a UPF3B variant lacking the EJC binding motif (EBD; aa 421–434) but retaining both the RRM and the middle domain (UPF3BΔEBD, Gehring et al, 2003) delayed the pre‐TC‐post‐TC transition (Fig EV2C).
UPF3B reduces the efficiency of peptidyl‐tRNA hydrolysis at low concentrations of release factors
Toeprinting assays of termination reactions monitor stop codon recognition. To investigate whether UPF3B also affects peptidyl‐tRNA hydrolysis, pre‐TCs assembled on the MVHC‐STOP mRNA using 35S‐labelled initiator‐tRNA were incubated with or without UPF3B and with limiting amounts of eRFs. In comparison with the maximal rate of peptidyl‐tRNA hydrolysis achieved within the observed time window, peptide release efficiency was reduced in the presence of UPF3B by ~40–50% (Fig 1C). Both, the +1–2 nt toeprint shift can only occur when the eRFs bind to the stop codon in the ribosomal A site. Therefore, we conclude that UPF3B impairs stop codon recognition and peptidyl‐tRNA hydrolysis by the eRFs and thereby reduces termination efficiency.
Translation termination in vitro is independent of ATP binding or the ATPase activity of UPF1
ATP binding and hydrolysis by UPF1 are essential for NMD. We repeated the toeprinting experiment described above in the presence of either ATP (Fig EV2D, lanes 1–8) or its non‐hydrolyzable analogue AMPPNP (lanes 9–15). Under these conditions, neither UPF1 nor UPF2L individually (lanes 3, 4, 10, 11) or together (lanes 6, 13) affected the intensity of pre‐ or post‐TC signals compared to the control samples (lanes 2, 9). By contrast, UPF3B both alone and following addition of UPF1 reduced termination efficiency. These findings were independent of the presence of ATP or AMPPNP, indicating that neither the ATP binding nor the ATPase function of UPF1 influences the transition of pre‐TCs to post‐TCs.
Translation termination is independent of UPF1 phosphorylation and the presence of the SMG1‐8‐9 complex
According to current models, UPF1, the eRFs and the SMG1‐8‐9 complex form the termination‐stalling SURF complex (Kashima et al, 2006). UPF1 phosphorylation by SMG1 is thought to trigger UPF1's release from the eRFs (Kashima et al, 2006; Okada‐Katsuhata et al, 2012). Here, we explored whether in vitro phosphorylation of UPF1 affects translation termination. Maximal in vitro phosphorylation of UPF1 by SMG1 or SMG1‐8‐9 is achieved at pH 9.0, corresponding to the pH‐optimum of the kinase (Morita et al, 2007; Chakrabarti et al, 2014; Deniaud et al, 2015). We examined phosphorylation of UPF1 by SMG1‐8‐9 at physiological pH in the absence or presence of UPF2L, UPF3B and the eRFs (Fig 1D). UPF2L only slightly stimulates UPF1 phosphorylation (Fig 1D, compare lane 1 with lanes 4, 10) by SMG1‐8‐9. In contrast, UPF3B alone (lanes 5, 11) moderately and together with UPF2L (lanes 6, 12) strongly inhibits UPF1 phosphorylation by SMG1‐8‐9 irrespective of the presence of equimolar concentrations of the eRFs. We confirmed that our UPF2L or UPF3B preparations do not contain a phosphatase by co‐incubating the phosphorylated UPF1 (P‐UPF1) with the preparations of UPF2L and UPF3B for 15 min at 37°C (lanes 13–16), which did not affect the abundance of the phosphorylated UPF1.
We next investigated whether UPF1 phosphorylation or the presence of SMG1‐8‐9 per se affects in vitro translation termination. UPF1 alone or together with either UPF2L, UPF3B or both was incubated with SMG1‐8‐9 and ATP for 30 min and subsequently mixed with pre‐TCs for another 10 min at 37°C (Fig 1E, lanes 9–12) followed by termination with eRF1 and eRF3a and toeprint analysis. Reactions without SMG1‐8‐9 served as controls (lanes 3–8). We found that irrespective of the presence of either UPF2L or UPF2L and UPF3B, neither UPF1 phosphorylation nor the presence of SMG1‐8‐9 has a detectable influence on termination efficiency as judged by the rate of transformation of pre‐TCs to post‐TCs (compare lanes 3 and 9, lanes 6 and 10, lanes 8 and 12). The inhibitory effect of UPF3B on this transformation was independent of the presence of SMG1‐8‐9 and UPF1 (compare lanes 7 and 11).
UPF1 and UPF3B are part of release factor‐containing complexes in vivo
We next analysed the interaction of the UPF proteins with the termination complex in vivo. Based on co‐immunoprecipitation (co‐IP) experiments, human UPF1 has been suggested to interact with both eRF1 and eRF3a and thereby physically link the NMD apparatus with translation termination (Kashima et al, 2006; Ivanov et al, 2008; Singh et al, 2008). In yeast, all three Upf proteins were reported to bind to eRF3 (Sup35) (Wang et al, 2001). We thus transiently co‐transfected HeLa cells with FLAG‐tagged eRF1 or eRF3a and full‐length versions of V5‐tagged UPF1, UPF2 or UPF3B. Immunoprecipitations on FLAG‐antibody beads were performed in the presence of RNase A to ensure that interactions between the eRFs and the UPF proteins were not mediated by mRNA. Co‐IPs of FLAG‐eRF1 with V5‐eRF3a and of FLAG‐eRF3a with V5‐eRF1 served as positive controls and yielded strong eRF1‐eRF3a interactions (Fig 2A and B, lanes 2). Co‐IPs of FLAG‐eRFs with the EJC‐disassembly factor PYM (Gehring et al, 2009) served as specificity controls (Fig 2A and B, lanes 10).
Figure 2. In vivo interaction between release factors and UPF proteins.
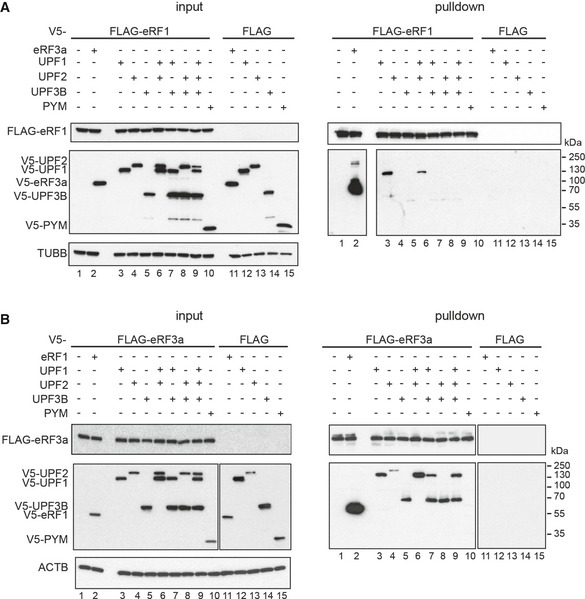
- Co‐immunoprecipitation from RNaseA‐treated lysates of HeLa cells transfected with FLAG‐eRF1 (lanes 1–10) or unfused FLAG (lanes 11–15) and V5‐eRF3a, V5‐UPF1, V5‐UPF2, V5‐UPF3B or V5‐PYM. Co‐precipitated proteins were detected using an anti‐V5 antibody. Lysate used for the immunoprecipitations was loaded in the input lanes (left). Re‐probing with anti‐TUBB antibody served as loading control.
- Co‐IP experiment as in (A) with FLAG‐eRF3a. Re‐probing with anti‐ACTB served as loading control.
Using FLAG‐eRF1 as bait, UPF1 (Fig 2A, lane 3), but not UPF2 (lanes 4, 6, 8, 9) or UPF3B (lane 5, 7–9), was co‐immunoprecipitated with eRF1. Importantly, co‐transfection of UPF3B and UPF1 prevented the formation of a complex containing eRF1 and UPF1 (lane 7) indicating that UPF3B either directly or indirectly competes with eRF1 for UPF1 binding.
Using FLAG‐eRF3a as bait, only little UPF2 (Fig 2B, lane 4) but considerably more UPF1 and UPF3B (lanes 3 and 5) were co‐immunoprecipitated. Interestingly, we found UPF3B in FLAG‐eRF3a immunoprecipitates together with UPF1 (lanes 7 and 9), indicating that UPF1 and UPF3B can bind to eRF3a complexes both, individually and together, but that these proteins do not compete for eRF3a‐binding. Surprisingly, UPF2 could not be detected in these complexes as well as in complexes containing eRF3a and UPF3B (lanes 6, 8 and 9). We conclude that UPF2 does not partake in complexes containing eRF3a together with UPF1, UPF3B or both.
UPF3B directly interacts with eRF3a in a magnesium‐sensitive manner forming a ternary complex with eRF1
Co‐IP experiments do not reveal whether the interactions identified are direct or indirect. Therefore, we performed in vitro pull‐down assays to analyse whether purified UPF proteins and release factors interact directly.
We incubated reaction mixtures containing His‐tagged UPF1, UPF2L or UPF3B and one or both untagged eRF(s) (Fig 3A) with Ni‐NTA beads, washed extensively and eluted the bound proteins with imidazole. We found that neither eRF1 nor eRF3a individually, nor the eRF1‐eRF3a complex, bound to UPF1 (Fig 3B, lanes 5–7), or to UPF2L (Fig 3C, lanes 5–7) above background (Fig 3B and C, lanes 1, 2, 4; Fig EV3A and C, lanes 3). UPF2 has recently been reported to directly interact with eRF3a (Lopez‐Perrote et al, 2016). However, under the conditions tested, UPF2L did not bind to eRF3a, although it comprises the part that was reported to interact with eRF3a. In contrast, eRF3a and, to a lesser extent, eRF1 co‐eluted with UPF3B individually (Fig 3D, lanes 5, 6) as well as simultaneously (lane 7) indicating that UPF3B directly interacts with both release factors.
Figure 3. UPF3B forms a complex with eRF3a and eRF1.
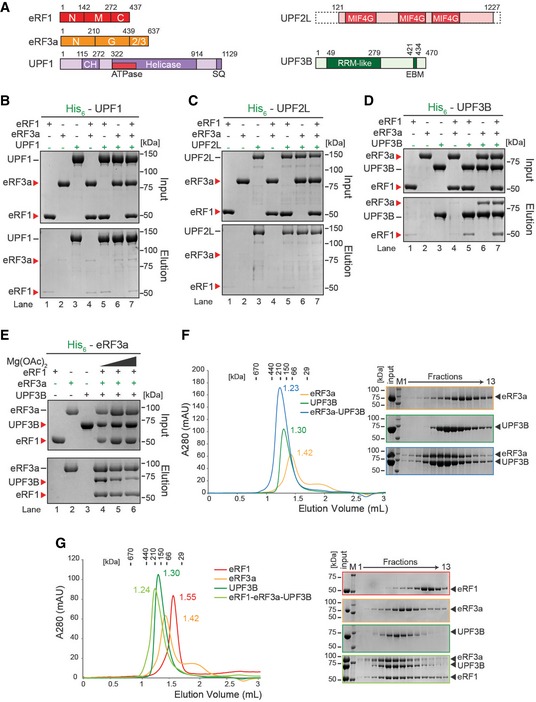
- Schematic representation of eRFs, UPF1, UPF2L and UPF3B proteins. Domains of known function and structural motifs are indicated. N, M, C, G, CH stand for N‐terminal, middle, C‐terminal, GTP‐binding and cysteine–histidine‐rich domain, respectively. 2/3: domains 2 and 3. MIF4G: middle fragment of eIF4G, RRM: RNA recognition motif, EBM: EJC‐binding motif.
- In vitro pulldown of eRF1 and/or eRF3a with His‐UPF1. Protein mixtures before loading onto the beads (input) or after elution (eluate) were separated by SDS–PAGE.
- Pulldown as in (B), with His‐UPF2L as bait.
- Pulldown as in (B), with His‐UPF3B as bait.
- Pulldown of eRF1, UPF3B, or both with His‐eRF3a in buffer containing 0 (lane 4), 2.5 (lane 5) or 5 mM (lane 6) Mg2+.
- Left: SEC elution profile of eRF3a (yellow), UPF3B (green), or both (blue). The elution volume (in ml) is indicated for each experiment. Column calibration was performed with globular proteins (shown above). Right: SDS–PAGE analysis of eluate fractions. M: protein molecular weight standards (kDa).
- SEC elution profile and SDS–PAGE analysis as in (F) of eRF1 (red), eRF3a (orange), UPF3B (dark green) or all three (light green). See also Fig EV3.
Figure EV3. Validation of UPF3B's complex formation with eRF1 and eRF3a.
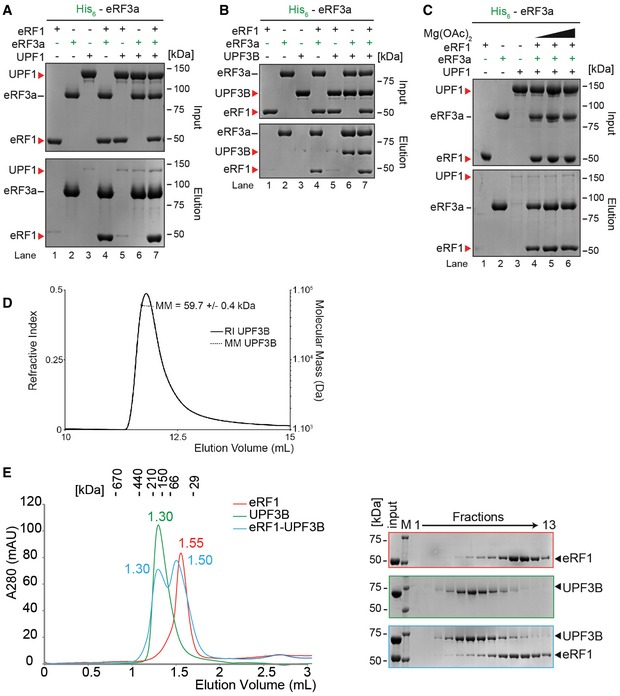
- In vitro pulldown of eRF1 and/or UPF1 with His‐eRF3a. Protein mixtures before loading onto the beads (input) or after elution (eluate) were separated by SDS–PAGE.
- Pull‐down experiment as in (A) with eRF1, UPF3B and His‐eRF3a.
- Pull‐down experiment as in (B) with eRF1, UPF1 and His‐eRF3a in buffer containing 0, 2.5 or 5 mM Mg2+ (lanes 4, 5, or 6 respectively).
- Molecular mass of UPF3B determined by size‐exclusion chromatography using a Superdex 200 column combined with detection by multiangle laser light scattering and refractometry (SEC‐MALLS‐RI). The SEC elution profiles as monitored by refractometry (RI) are represented for UPF3B. The molecular mass (MM) of UPF3B calculated from light scattering and refractometry data is indicated.
- Left: SEC elution profile of eRF1 (red), UPF3B (green) or both (blue). The elution volume (in ml) is indicated for each experiment. Calibration of the column was performed with globular proteins (shown above). Right: SDS–PAGE analysis of eluate fractions. M: protein molecular weight standards (kDa).
Reciprocal control experiments using His‐eRF3a as a bait for both UPF1 and UPF3B (Fig EV3) corroborated the eRF3a‐UPF3B interaction (Fig EV3B) and confirmed that UPF1 does not co‐elute with eRF3a irrespective of the presence of eRF1 (Fig EV3A).
Translation is modulated by the Mg2+ concentration both in vivo and in vitro. In our in vitro translation termination assays (Fig 1), we used 1 mM free Mg2+ which corresponds to the physiological intracellular level of unbound Mg2+ (Veloso et al, 1973; MacDermott, 1990). We explored the impact of Mg2+ on the UPF3B‐eRF interaction. At physiological [Mg2+], a substantial amount of UPF3B bound to eRF3a, whereas at > 5 mM Mg2+, the interaction between UPF3B and eRF3a was considerably weaker (Fig 3E, lanes 4–6). Notably, UPF1 did not directly interact with the eRFs at all Mg2+ concentrations tested (Fig EV3C).
To corroborate UPF3B‐eRF complex formation by an independent, established biophysical method, we incubated UPF3B with combinations of eRF1 and eRF3a and resolved the protein mixtures by size‐exclusion chromatography (SEC) under physiological buffer conditions. Co‐incubation of equimolar amounts of eRF3a and UPF3B resulted in a complex eluting at a higher apparent molecular weight than the individual proteins (Fig 3F), corroborating a direct interaction between eRF3a and UPF3B. Because UPF3B alone eluted at a higher apparent molecular weight than expected (Fig 3F), indicating possible oligomerization or a deviation from the globular shape, we subjected UPF3B to SEC coupled to online detection by Multi‐Angle Laser Light Scattering (SEC‐MALLS) and refractometry index measurements. The determined weight‐averaged molecular mass confirmed that UPF3B is monomeric in solution. This suggests a non‐globular shape of UPF3B (Fig EV3D).
In contrast, after co‐incubation of eRF1 and UPF3B, the proteins eluted in two peaks (Fig EV3E). The first peak eluted at the same volume as UPF3B when analysed individually and thus corresponds to UPF3B. The second peak eluted at a higher apparent molecular weight than eRF1 alone (1.55 ml vs. 1.50 ml). Accordingly, in the SDS–PAGE analysis, a slight shift of the eRF1 containing fractions can be observed in the gel analysing co‐migration of UPF3B and eRF1 in SEC as compared to the gel analysing the eRF1‐SEC fractions (Fig EV3E) suggesting a very weak interaction between eRF1 and UPF3B.
When UPF3B was mixed with both eRF1 and eRF3a, a single peak containing all three proteins eluted at a higher apparent molecular weight than each individual protein (Fig 3G) demonstrating that UPF3B, eRF1 and eRF3a can form a stable trimeric complex. The complex is likely stabilized by eRF3a, which can bind both UPF3B and eRF1. These findings suggest that the effect of UPF3B on translation termination can be fully or partially mediated by a direct interaction of UPF3B with either eRF3a or the eRF1‐eRF3a complex.
UPF3B and UPF1 interact directly
In EJC‐dependent NMD, UPF2 is thought to bridge the termination complex and the EJC by simultaneously binding to UPF1 at the termination site and UPF3B at the EJC (Kashima et al, 2006; Chamieh et al, 2008). However, UPF3B but not UPF1 binds to eRF3a in vitro and UPF2, in contrast to UPF1 and UPF3B, is excluded from eRF3a‐bound complexes in vivo (Fig 2 and 3). Although earlier in vitro binding studies using a truncated UPF1 variant revealed no direct interaction (Chamieh et al, 2008), we tested full‐length UPF1 binding to UPF3B. We incubated His‐UPF3B with UPF1 either in the presence or in the absence of eRF3a and found that UPF1 directly interacts with UPF3B (Fig 4A, lane 6). Binding of eRF3a to UPF3B was not affected by UPF1, suggesting that the two proteins can bind to UPF3B independently (lanes 5 and 7). In SEC analysis, a single peak containing both UPF1 and UPF3B eluted earlier than UPF1 and UPF3B alone, confirming the formation of a UPF1‐UPF3B complex. SDS–PAGE analysis revealed that the elution profile of UPF1 and UPF3B within this peak was not fully symmetric, indicating that the UPF1‐UPF3B complex partly dissociates during SEC (Fig 4B). Both protein preparations were virtually RNA‐free as indicated by their OD 260 nm/280 nm ratios of ~0.5 (Raynal et al, 2014). Therefore, the interaction between UPF1 and UPF3B is not mediated by RNA contaminations of the recombinant proteins.
Figure 4. UPF3B can directly interact with UPF1.
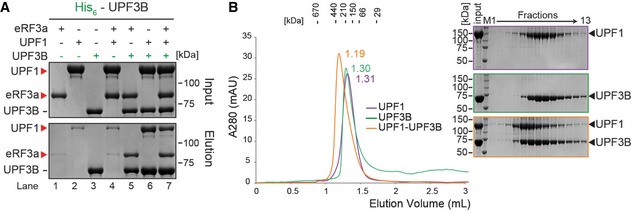
- In vitro pulldown of eRF3a, UPF1 or both with His‐UPF3B. Protein mixtures before loading onto the beads (input) or after elution (eluate) were separated by SDS–PAGE. Representative of four independent experiments.
- Left: SEC elution profile of UPF1 (purple), UPF3B (green) or both (orange). Right: SDS–PAGE analysis of eluate fractions. Representative of two independent experiments. Since the experiments described in Figs 3F and 4B were performed in parallel, the same UPF3B SEC elution profile (green) and the corresponding SDS–PAGE analysis served as control for both experiments. More UPF3B SEC experiments are depicted in Figs 3G, 6B and EV3E.
Source data are available online for this figure.
To investigate whether RNA interferes with the formation of the UPF1‐UPF3B complex, we incubated UPF3B, or UPF1 and UPF3B with a threefold excess of a 24 nt RNA oligomer which is too small for a concomitant binding of both proteins (Fig EV4). In SEC analysis, the UPF3B‐UPF1 complex eluted earlier than UPF1 or UPF3B from the SEC column (1.18 ml vs. 1.31 or 1.3 ml, respectively) indicating that the oligomer did not compete with UPF1 for UPF3B binding or vice versa (Fig EV4A). The OD 260 nm/280 nm ratio of 0.76 in the UPF1‐UPF3B complex peak and the tailing of the peak (Fig EV4B, 3rd panel) suggest that a UPF1‐UPF3B‐RNA complex could transiently form since the complex partly dissociates during the SEC experiment.
Figure EV4. UPF1‐UPF3B complex formation is not prevented by RNA .
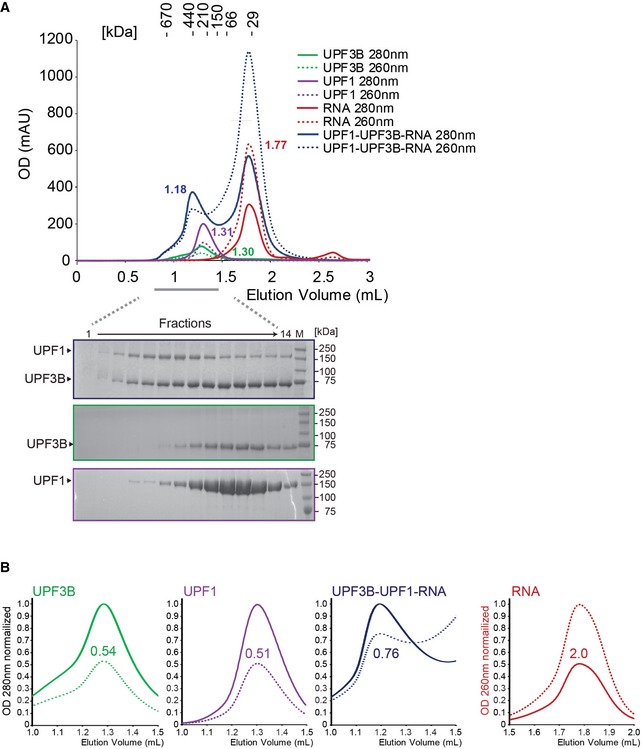
- SEC elution profile of UPF3B (green), UPF1 (purple), RNA (red) and a mix of UPF3B, UPF1 and a threefold excess of RNA (blue). Below: SDS–PAGE analysis of eluate fractions.
- Analysis of SEC peak fractions. Peaks representing UPF3B (green) and UPF1 (purple), elute with an OD 260 nm/280 nm ratio of 0.54 and 0.51, respectively, whereas the RNA oligomer (red) elutes with an OD 260 nm/280 nm ratio of 2.0. The peak containing the UPF3B‐UPF1 complex after incubation of UPF1, UPF3B and RNA (blue) has a higher OD 260 nm/280 nm ratio of 0.76 due to the presence of RNA in this peak.
Source data are available online for this figure.
The eRF3a‐UPF3B interaction requires the N‐terminus of eRF3a in vitro and in vivo
The N‐terminus of eRF3a is not required for the function of eRF3a in translation termination (Ter‐Avanesyan et al, 1993). We explored whether an eRF3a variant lacking the first 138 aa (eRF3aΔN) (Fig 5A) can bind UPF3B and found that in contrast to eRF3a (Fig 5B, lane 4), eRF3aΔN was not co‐eluted with His‐UPF3B (lane 5). In the reciprocal experiment using His‐eRF3a or His‐eRF3aΔN as bait, UPF3B co‐eluted with eRF3a (Fig 5C, lane 4) but not with eRF3aΔN (lane 5).
Figure 5. UPF3B interacts with the N‐terminus of eRF3a.
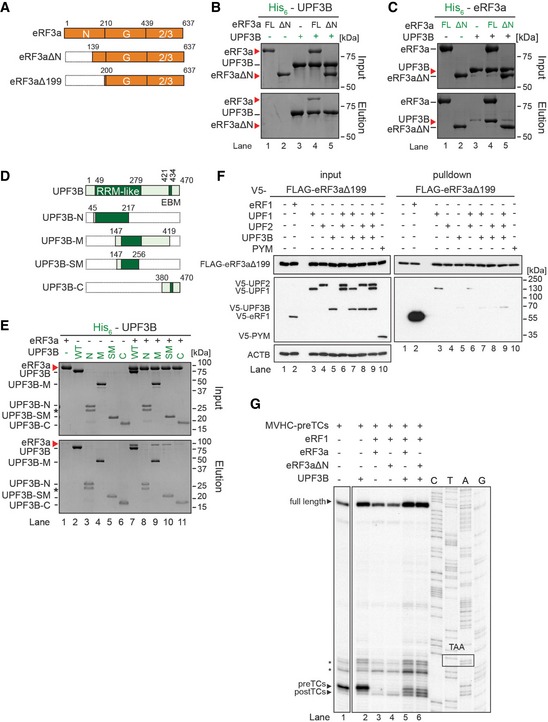
- Schematic representation of eRF3a constructs used for this Figure. eRF3aΔN, eRF3aΔN199: eRF3a variants lacking amino acids 1–100 and 1–199, respectively.
- In vitro pulldown as in Fig 3 of eRF3a (FL) or eRF3aΔN (ΔN) with His‐UPF3B.
- Pulldown of UPF3B with His‐eRF3a (FL) or His‐eRF3aΔN.
- Schematic representation of UPF3B constructs used in panel (E).
- Pulldown as in Fig 3 of eRF3a with His‐UPF3B variants. Asterisks mark the presence of a degradation product in the UPF3B‐N sample.
- Co‐IP experiment as in Fig 2 with FLAG‐eRF3aΔ199 and V5‐UPF1, V5‐UPF2L, V5‐UPF3B or V5‐PYM.
- Toeprinting analysis of ribosomal complexes obtained by incubating MVHC‐pre‐TCs with BSA or with UPF3B at 1 mM free Mg2+ as indicated. Termination was completed with limiting amounts of eRF1 and either eRF3a (lanes 3, 5) or eRF3aΔN (lanes 4, 6), respectively. Asterisks mark initiation and elongation complexes.
We further characterized the eRF3a‐UPF3B interaction using truncated versions of His‐UPF3B comprising the RRM domain (aa 42–217, UPF3B‐N) (Kadlec et al, 2004), the middle domain (aa 147–419, UPF3B‐M), part of the middle domain (aa 147–256, UPF3B‐SM) or the extended EJC‐binding motif (EBM) (aa 380–470, UPF3B‐C) (Fig 5D). UPF3B‐N and UPF3B‐C did not bind eRF3a, indicating that neither the RRM domain which binds UPF2 nor the EBM is sufficient to interact with eRF3a (Fig 5E, lanes 8, 11). In contrast, UPF3B‐M and UPF3B‐SM, comprising the hitherto uncharacterized middle domain of UPF3B, bound to eRF3a, albeit less efficiently than the full‐length protein (lanes 7, 9, 10). The protein–protein contact between eRF3a and UPF3B is thus established by binding between the eRF3a N‐terminus and the middle domain of UPF3B. Notably, most UPF3B mutations linked to neurodevelopmental disorders are located in this region (Alrahbeni et al, 2015).
To examine whether the interaction between UPF3B and N‐terminally truncated eRF3a was also impaired in vivo, we transiently co‐transfected HeLa cells with plasmids encoding a FLAG‐tagged version of eRF3a lacking the first 199 aa (FLAG‐eRF3aΔ199) and with V5‐eRF1 or V5‐UPF1, V5‐UPF2, or V5‐UPF3B, either individually or simultaneously (Fig 5F) and immunoprecipitated on FLAG‐antibody beads after digestion of the lysates with RNase A. We found that UPF1 still co‐precipitated with eRF3aΔ199 when it was co‐transfected individually or with UPF2 (Fig 5F, lanes 3, 6). In contrast, only trace amounts of UPF3B were found in FLAG‐eRF3aΔ199 complexes (lanes 5, 7–9), illustrating that the eRF3a N‐terminus is necessary for the interaction with UPF3B in vivo.
Next, we examined whether the inability of eRF3aΔN to interact with UPF3B affects the termination‐delaying function of UPF3B (Fig 5G). However, the pre‐ and post‐TC toeprints generated in the presence of UPF3B and eRF3a (lanes 3, 5) or eRF3aΔN (lanes 4, 6), respectively, were very similar.
We reasoned that the eRF3a(ΔN)‐eRF1 interaction is much stronger than the eRF3a‐UPF3B interaction. UPF3B's potential role in eRF3a recruitment or its inability to recruit eRF3aΔN could thus be bypassed by eRF1 and, therefore, cannot be mirrored by the in vitro translation system. Moreover, the effect of UPF3B in delaying translation termination may also involve direct binding to the ribosome, thus interfering with efficient stop codon recognition by the release factors. With an isoelectric point of 9.5, UPF3B is positively charged at physiological pH and may interact with negatively charged rRNAs or ribosomal proteins. The 24‐nt RNA oligomer used for the experiment described in Fig EV4 served to analyse in SEC the RNA‐binding capacity of full‐length UPF3B (Fig 6A). UPF3B incubated with RNA eluted earlier than UPF3B alone from the SEC column (1.22 ml vs. 1.30 ml) and had a higher OD260 nm signal, indicating the presence of nucleic acids in this peak. The second peak (~1.77 ml) contained unbound RNA oligomer. The majority of the RNA oligonucleotide shifted to the position of the UPF3B peak, demonstrating that full‐length UPF3B binds RNA, a finding that is consistent with recent iCLIP und RNA interactome data (Hauer et al, 2016).
Figure 6. UPF3B binds RNA and ribosomes.
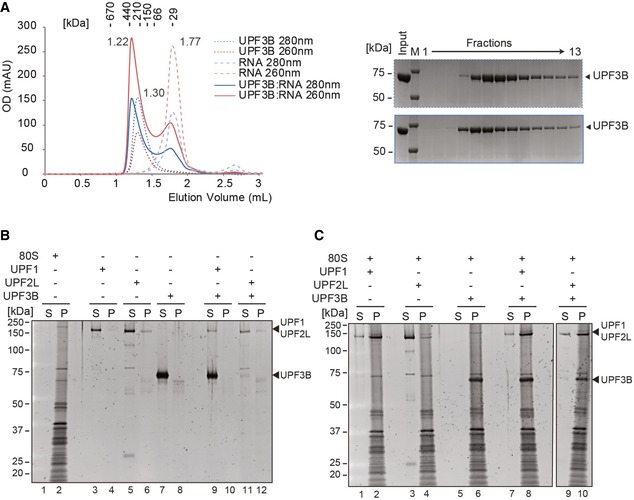
- Left panel: SEC elution profile of UPF3B (dotted lines), RNA (dashed lines) and the mix (solid lines). Optical density was recorded at 280 nm (blue) and at 260 nm (red). 40 μM of UPF3B or RNA oligonucleotide (24mer) or both was loaded onto a Superdex 200 column. The elution volumes are indicated next to the curves. Right panel: SDS–PAGE analysis of eluate fractions.
- Sucrose cushion co‐sedimentation analysis of either ribosomes (lanes 1, 2) or of UPF1, UPF2L or UPF3B or of combinations as indicated. After ultracentrifugation, the supernatant (S) and pellet (P) fractions were analysed by SDS–PAGE.
- Sucrose cushion co‐sedimentation analysis of UPF1, UPF2L or UPF3B as in (B) but in the presence of 80S ribosomes.
To explore the ability of UPF3B, UPF1 and UPF2L to interact with ribosomes, we performed co‐sedimentation assays (Fig 6C). Centrifugation without ribosomes served as controls (Fig 6B, lanes 3–12). UPF1 and UPF3B individually and simultaneously co‐sedimented with 80S ribosomes (Fig 6C, lanes 2, 6, 8), indicating that both proteins can bind independently to ribosomes. In contrast, the weak ribosome binding of UPF2L alone (lane 4) was considerably enhanced in the presence of UPF3B (lane 10). This finding indicates that UPF2L can be recruited to the ribosome by UPF3B and that the interaction with UPF2L on the ribosome may interfere with the function of UPF3B in termination.
UPF3B triggers the disassembly of post‐termination complexes
The ability of UPF3B to form complexes with UPF1 and the release factors suggests the existence of a previously unknown dynamic UPF‐eRF protein network. We reasoned that the influence of the UPF proteins on translation termination might differ from what we had observed with limiting amounts of the eRFs at equimolar and saturating amounts of eRFs and UPF proteins, allowing free interplay of all factors.
We thus complemented the experiments described in Figs 1B and E, and EV2 by experiments with saturating amounts of eRFs (Figs 7A and EV5). Addition of the eRFs induced the transformation of all pre‐TCs to post‐TCs as indicated by a shift of the toeprint by +1–2 nt relative to the pre‐TC toeprint (Figs 7A and EV5, lanes 2). Irrespective of the presence of ATP or AMPPNP, this pattern was essentially the same when pre‐TCs were pre‐incubated with either UPF1, UPF2L, both of these proteins (Figs 7A and EV5A and B, lanes 3, 4, and 6), with UPF2L and UPF3B (lanes 8), or with all three UPF proteins (lanes 9). We also investigated whether the phosphorylation of UPF1 by SMG1‐8‐9 either alone (Fig EV5C, lane 7), in presence of UPF2L and/or UPF3B (8–10), or before (lanes 11–14) addition of UPF2L and/or UPF3B influenced UPF1 function. We found that irrespective of the phosphorylation status, toeprints generated by termination with phosphorylated UPF1 (lanes 7–14) were indistinguishable from those generated with non‐phosphorylated UPF1 (lanes 3–6). Thus, independent of its functions in ATP binding or ATP hydrolysis or of its phosphorylation status, UPF1 has no impact on translation termination even when eRFs are present at saturating concentrations.
Figure 7. UPF3B dissociates post‐TCs.

- Toeprinting analysis of ribosomal complexes obtained by incubating MVHC‐pre‐TCs with UPF1, UPF2L, UPF3B, or BSA at 1 mM free Mg2+ and 1 mM ATP followed by termination with saturating amounts of eRF1 and eRF3a. Asterisks mark initiation and elongation complexes.
- Toeprinting analysis of ribosomal complexes obtained by incubating pre‐TCs as in (A) with UPF3B or BSA and combinations of eRF1, eRF1AGQ, eRF3a and puromycin in the presence of GTP or GMPPNP. Pre‐/post‐TC profiles of lanes 6–8 and 11–14 are enlarged to allow a better assessment. The gel on the left was exposed 2× longer than gel on the right. Note that puromycin‐treated pre‐TCs are relatively unstable at the low Mg2+ concentrations used (Skabkin et al, 2013).
- Mg2+ sensitivity of post‐TC dissociation by UPF3B. Toeprinting analysis of ribosomal complexes obtained by incubating MVHC‐pre‐TCs with BSA (lanes 1–6) or UPF3B (lanes 7–12) and at the indicated concentrations of free Mg2+. Termination was completed by adding eRF1 and eRF3a to the samples in lanes 4–9. Lanes 1–6 were exposed 2× longer than lanes 7–12. Asterisks mark initiation and elongation complexes.
Figure EV5. UPF3B's post‐TC‐dissolving activity is independent of ATP and SMG1‐8‐9 and requires both the RRM and the middle domain.
-
A, BToeprinting analysis of ribosomal complexes as in Fig 6A, but in the presence of 1 mM AMPPNP (A) or without adenosine nucleotide (B).
-
CImpact of UPF1‐phosphorylation on efficient translation termination and on ribosome dissociation by UPF3B. Toeprinting analysis of ribosomal complexes obtained by incubating pre‐TCs formed on MVHC‐STOP mRNA (MVHC‐pre‐TCs) with UPF1, UPF2L, UPF3B or BSA at 1 mM free Mg2+ and 1 mM ATP. In lanes 7–10, UPF1 was incubated with SMG1‐8‐9 and ATP for 30 min at 37°C either alone (lane 7) or in the presence of UPF2L, UPF3B, or both (lanes 8–10) before pre‐TCs were added to the mixture and again incubated for 10 min. In lanes 11–14, UPF1 was incubated with ATP and SMG1‐8‐9 for 30 min. Then, UPF2L and/or UPF3B were added for additional 15 min (lanes 12–14). Finally, MVHC‐pre‐TCs were added to the mixtures for 10 min followed by translation termination by eRF1 and eRF3a.
-
DToeprinting analysis of ribosomal complexes obtained by incubating MVHC‐pre‐TCs with UPF3B, UPF3B‐N, UPF3B‐M, UPF3BΔEBD or BSA at 1 mM free Mg2+ followed by termination with saturating amounts of eRF1 and eRF3a.
In termination reactions including UPF3B either alone or together with UPF1, a small amount of pre‐TCs was retained, suggesting that here, too, UPF3B delayed termination. However, most pre‐TCs were transformed to post‐TCs in the presence of either UPF3B alone or in the presence of both UPF1 and UPF3B when eRFs were not limiting (Figs 7A and EV5A and B, lanes 5, 7; Fig EV5C, lanes 5, 9, 13). Importantly, the post‐TC toeprints resulting from termination reactions in the presence of UPF3B (or of UPF1 and UPF3B; Figs 7A and EV5A and B, lanes 5 and 7) were considerably weaker and the readthrough full‐length signal clearly stronger than toeprints resulting from termination reactions without UPF3B (Figs 7A and EV5A and B, lanes 2–4, 6, 8, 9; Fig EV5C, lanes 3, 4, 7, 8, 11, 12) This finding indicates that UPF3B induced the release of post‐TCs from the mRNA. We thus conclude that UPF3B destabilizes post‐TCs when release factors are abundant. This activity is independent of UPF1 and is prevented by UPF2L (Figs 7A and EV5A and B, lanes 8, 9; Fig EV5C, lanes 6, 10, 14). The UPF2‐dependent inhibition of UPF3B's impact on termination confirms that both termination delay and ribosome dissociation are caused by UPF3B, and not by a potential low‐level contaminant escaping detection on Coomassie‐stained gels and which might be co‐purifying with recombinant UPF3B (Figs 1, 3, 4, 5, 6, and EV1, and EV3). We next included UPF3B‐N, UPF3B‐M, and UPF3BΔEBD into termination reactions with saturating amounts of eRFs. Only UPF3BΔEBD exerted a similar post‐TC dissociating activity as UPF3B, indicating that the EBD is dispensable for this function (Fig EV5D). Taken together with the results described in Fig EV2C and D this indicates that neither RNA/ribosome‐binding features nor a basic pI per se influence the pre‐TC‐post‐TC transition or the dissolution of post‐TC complexes and that neither the RRM nor the middle domain of UPF3B alone is sufficient to exert these functions.
The formation of post‐TCs in toeprinting assays reflects stop codon recognition by eRF1. To explore whether UPF3B performs its post‐TC‐dissociating activity before or after peptide release, we interfered with the termination reaction by adding GMPPNP, eRF1AGQ, or the peptide‐releasing reagent puromycin to the pre‐TCs, or by omitting eRF3a (Fig 7B). eRF1AGQ (Fig EV1A, lane 2) with a G183A mutation in the GGQ motif is inactive in peptide release, but recognizes stop codons, stimulates the GTPase activity of eRF3a, and together with eRF3a can induce the ribosomal rearrangements reflected by the +1–2 nt shift in the toeprint (Fig 7B, lanes 4, 5) (Frolova et al, 1998; Alkalaeva et al, 2006). Likewise, eRF3a supports stop codon recognition in the presence of GMPPNP (lane 3), but impairs peptidyl‐tRNA hydrolysis by eRF1. eRF1 alone can induce termination and ribosomal rearrangements as well as peptide release, but the reaction is considerably less efficient than in the presence of eRF3a and GTP (lane 6) (Alkalaeva et al, 2006). UPF3B efficiently dissociated post‐TCs generated in the presence of eRF1, eRF3a and GTP (Fig 7B, lane 10), but not ribosomal complexes that were deficient in peptide release either due to blocking the activity of eRF3a by GMPPNP (lane 11), the peptide hydrolysis defective eRF1AGQ (lane 12) or the absence of eRF3a (lane 14). When peptide release was enforced by the addition of puromycin, UPF3B effectively dissociated the resulting post‐TCs (lanes 13, 15), which was also reflected by a concomitant increase of the toeprint corresponding to the ribosome‐free full‐length mRNA. Notably, UPF3B was unable to dissociate pre‐TCs in the absence of eRFs (Fig 7B, lane 9) or residual pre‐TCs in termination reactions that were incubated with eRF1 or puromycin alone (lanes 15, 16 in main panel and enlargement). These data indicate that UPF3B dissociates post‐TCs after both GTP and peptidyl‐tRNA hydrolysis, but not pre‐TCs or post‐TCs before peptide hydrolysis. UPF3B also dissociates post‐TCs that have been generated in the absence of eRF3a (Fig 7B, compare lanes 7 and 15 in main panel and enlargement). Therefore, the eRF3a‐UPF3B interaction is not required for the function of UPF3B in ribosome dissociation.
The ability of UPF3B to promote the dissociation of post‐TCs is reminiscent of the energy‐free ribosome recycling activity mediated by eIF3, eIF1 and eIF1A (Pisarev et al, 2007a), which is apparent only at low Mg2+ concentrations. Furthermore, ribosomal inter‐subunit association is dynamic and more flexible at physiological rather than at higher Mg2+ concentrations (Shenvi et al, 2005). Therefore, we investigated the ability of UPF3B to dissociate post‐TCs at Mg2+ concentrations higher than 1 mM (Fig 7C). Importantly, we found that no post‐TC dissociation occurred at 2.5 or 5 mM Mg2+ in the presence of equimolar amounts of the eRFs and UPF3B (compare lanes 7/10, 8/11, and 9/12).
These findings suggest that UPF3B dissociates post‐TCs with flexible subunit association, possibly by accessing the ribosome subunit interface which is stabilized at higher Mg2+ concentrations.
Discussion
How translation termination at a premature termination codon differs from termination at a NTC has long been a matter of debate. All prevailing models from yeast to man ascribe a critical role to UPF1 not only in the mRNA degradation phase, but already in the translation termination phase of NMD. These hypotheses are founded on the interaction of UPF1 with eRF1 and eRF3a, which were identified in co‐IP experiments (Wang et al, 2001; Ivanov et al, 2008; Singh et al, 2008) (Fig 2). UPF1 is thought to recruit the eRFs to ribosomes that are stalled at a PTC in an early phase of termination and to promote ribosome disassembly in a late phase of termination via its ATPase function that is activated by UPF2 and UPF3 binding (reviewed in Brogna et al, 2016; Celik et al, 2015; He & Jacobson, 2015). However, it has not been possible to experimentally address the hypothetical functions of NMD factors in translation termination in cells and organisms, because no adequate in vivo termination assay is available to date. Deletion of the UPF genes in yeast leads to increased stop codon suppression (Wang et al, 2001; Keeling et al, 2004), whereas RNAi‐mediated depletion of UPF1 in human cells reduces stop codon readthrough (Ivanov et al, 2008). Yet, it is unclear whether these manipulations disturb or reflect direct interactions of UPF proteins with the translation termination machinery.
Here, we tested the functional interactions of key NMD factors in vitro using a fully reconstituted translation termination system that has been demonstrated to faithfully mirror all phases of eukaryotic translation (Alkalaeva et al, 2006; Pisarev et al, 2007b; Pisareva et al, 2008). Although this system cannot per se differentiate between termination at a NTC and a PTC, respectively, we simulated the situation at a PTC by combining terminating ribosomes and NMD factors as well as by omitting termination‐stimulating factors. In agreement with current models, we hypothesized that in such a system the central NMD factor UPF1 interacts with the eRFs and possibly the ribosome (Min et al, 2013), thereby delaying translation termination. In these models, UPF2 and UPF3B serve as activators of UPF1 functions. They support UPF1 phosphorylation by SMG1‐8‐9 which is thought to dissolve the UPF1‐eRF interaction, to release UPF1‐induced ribosomal stalling and to activate its RNP remodelling function in a post‐termination phase (Kashima et al, 2006; Ivanov et al, 2008). Accordingly, we hypothesized that the addition of UPF2, UPF3B, ATP and/or SMG1‐8‐9 would release the UPF1‐induced break and allow for efficient termination.
Surprisingly, we find that neither UPF1 per se nor its biochemical functions such as ATP binding, ATP–hydrolysis or its phosphorylation play a discernible role in early or late phases of translation termination. Furthermore, UPF1 does not appear to bind eRF1 and eRF3a directly, and the previously described interactions between UPF1 and the eRFs, found in co‐IP experiments, are thus likely to be indirect. Cumulatively, our data demonstrate that UPF1 remains inactive or functionally dispensable during translation termination and that the essential role of UPF1 in human NMD as well as the function of UPF2 may be exerted in the post‐termination phase of NMD. This conclusion is supported by findings that show UPF1 phosphorylation and its ATPase and helicase activities in metazoans to be important for its functions in 3′UTR mRNP remodelling and the recruitment of mRNA decay factors (Franks et al, 2010; Okada‐Katsuhata et al, 2012; Kurosaki et al, 2014; Fiorini et al, 2015). However, yeast Upf1p has recently been implicated in translation termination and ribosome release at PTCs (Serdar et al, 2016), an activity that required Upf1p's ATPase function as well as Upf2p and Upf3p. The higher complexity of metazoan NMD involving several NMD branches as well as a considerably larger number of factors, regulatory steps and feedback mechanisms as compared to yeast NMD may underlie these differences.
Unexpectedly, we discover that UPF3B exerts the bifunctional influence on translation termination that has hitherto been attributed to UPF1. When release factors are limiting and translation termination is inefficient, UPF3B further delays termination and inhibits peptide release. After release of the nascent peptide, UPF3B promotes the dissociation of post‐termination ribosomal complexes. Both activities are prevented by UPF2L, which is likely caused by interference with its function at the termination site. This dual function of UPF3B is in excellent accord with the observation that termination at PTCs is considerably slower than termination at NTCs (Amrani et al, 2004; Peixeiro et al, 2012) and that deletion of any of the UPF genes in yeast causes defects in ribosome release both in vitro and in vivo (Ghosh et al, 2010). It has been suggested that the kinetics of termination determines the discrimination between NTCs and PTCs in vivo (Hilleren & Parker, 1999; Zünd & Mühlemann, 2013). Slow termination defines aberrant termination events and triggers the recruitment of decay enzymes, whereas fast termination is promoted by the interaction of PABPC1 with eRF3a.
Because UPF3B interferes with translation termination, we assign a central role to UPF3B in a modified model for NMD. Mechanistically, we suggest that at a PTC in the absence of PABPC1, either EJC‐bound or free UPF3B binds to the terminating ribosome, interacts with the release factors and then delays termination by sterically impeding stop codon recognition and peptide release by eRF1. This hypothesis also provides a mechanistic rationale for the NMD‐enhancing effect of EJCs, which may increase the local concentration of UPF3B at the premature termination site.
According to our model, UPF3B binds in the vicinity of the A site of the ribosome and assists in the recruitment of eRF1‐eRF3a during the initial slow phase of termination at a PTC (Fig 8, phase 1). UPF1 molecules bound to the 3′UTR nearby may interact with terminating ribosomes as well but remain inactive in the proceedings of termination. After peptide release (Fig 8, phase 2), UPF3B, possibly promoted by a conformational or positional change at the ribosome, contributes to the rescue of ribosomes stalled at a PTC and dissolves the post‐TC (Fig 7A and B, and Fig 8, phase 3). This phase is independent of eRF binding and may be promoted by interactions of UPF3B with the ribosome subunit interface. Because UPF2 is not detected in complexes that contain both UPF3B and eRF3a (Fig 2B), we propose that UPF2 is recruited to post‐TCs after the release of eRF3a and dissociation of the ribosome by UPF3B. Subsequently UPF1, supported by UPF2 and UPF3B and possibly other proteins, can engage in 3′UTR remodelling and the recruitment of decay enzymes triggering the decay phase of NMD (Fig 8, phase 4).
Figure 8. Model for early and late UPF3B function in translation termination.
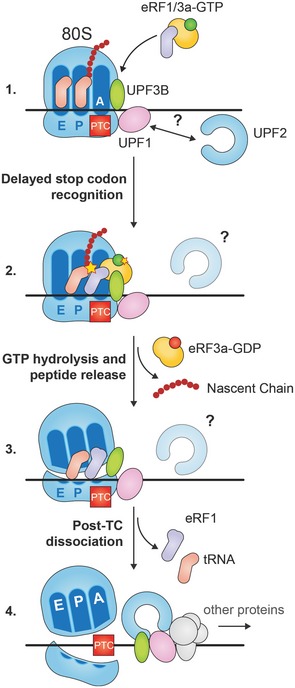
During termination at a PTC ribosome‐bound UPF3B interacts with the eRF1/eRF3a‐GTP complex impeding efficient stop codon recognition. UPF1 bound to the 3′UTR can contact the termination complex, but does not interfere with termination. After GTP hydrolysis and peptide release UPF3B destabilizes the post‐termination ribosomal complex leading to its dissociation. Subsequently, UPF3B, UPF1, UPF2 and other factors activate UPF1's ATPase and helicase functions to remodel the 3′UTR mRNP and attract decay enzymes.
Importantly, UPF3B can neither destabilize pre‐TCs nor post‐TCs, when either GTP hydrolysis by eRF3a or peptidyl‐tRNA hydrolysis is inhibited (Fig 7B). Ribosome recycling after proper or faulty translation termination is crucial for the protein synthesis machinery to avoid sequestration of essential components of the translation apparatus. In normal termination, no‐go decay (NGD) and non‐stop decay (NSD) post‐TCs or stalled ribosomes are dissociated by ABCE1 (reviewed in Graille & Seraphin, 2012; Lykke‐Andersen & Bennett, 2014). A specific mechanism for ribosome rescue in NMD has not yet been identified. UPF1 has been proposed to dissolve ribosomes stalled at a PTC, possibly because eRF3a is not able to leave the complex and therefore prevents the interaction of ABCE1 with eRF1 (Celik et al, 2015; Serdar et al, 2016). Here, we uncover that UPF3B dissociates post‐TCs and may, therefore, function as a dedicated NMD ribosome dissociation factor in metazoans. This conclusion is indirectly supported by the finding that directing UPF3B close to the 3′ end of the ORF stimulates translation of a reporter RNA in vivo (Kunz et al, 2006). Since UPF3B is not an ATPase, its activity is reminiscent of the energy‐free activity of initiation factors that can recycle post‐termination complexes only at a narrow range of low Mg2+ (Pisarev et al, 2007a, 2010). Dissociation of post‐TCs by eIF3 alone is relatively inefficient and is enhanced by eIF1, eIF1A and eIF3j. Similarly, UPF3B does not dissolve all post‐TCs even when present in large excess over the pre‐TCs (Fig 7). Hence, future work will determine whether UPF3B‐mediated ribosome dissociation is simply slower than ribosome release in normal termination as has been suggested (He & Jacobson, 2015), or whether it can be stimulated by other proteins reminiscent of the cooperation of initiation factors and ABCE1 (Pisarev et al, 2010).
Remaining questions concern the role of UPF3B both in NMD and in NMD‐related diseases. Loss‐of‐function mutations of the UPF3B gene result in X‐linked intellectual disability disorders. However, their underlying molecular mechanisms are unknown (Tarpey et al, 2007; Jolly et al, 2013). Most of these mutations impair NMD and have been mapped to the functionally uncharacterized middle domain of UPF3B (Alrahbeni et al, 2015). We show here that this domain mediates the interaction with eRF3a (Fig 6E). This finding will enable investigation of whether disruption of the UPF3B‐eRF3a interaction leads to the deregulation of genes that are required for normal neurodevelopment.
Upf3 is essential for NMD in yeast. For human NMD, both UPF2‐independent and UPF3B‐independent branches have been reported (Gehring et al, 2005; Chan et al, 2007). Our discovery that UPF1 can directly interact with UPF3B contributes to the understanding of the UPF2‐independent NMD branch, since UPF2 was assumed to bridge between UPF1 and UPF3B. However, if a major role of UPF3B in NMD is confined to translation termination, it remains to be investigated how ribosomes stalled at a PTC are recognized in the UPF3B‐independent branch. Notably, UPF3 exists in two paralogs in higher eukaryotes, UPF3A and UPF3B. UPF3A has hitherto been considered to be a “backup molecule” that can substitute for UPF3B in NMD (Chan et al, 2009). By contrast, UPF3A has recently been shown to also act as an antagonist of UPF3B and to function as a suppressor of NMD (Shum et al, 2016). This fundamental functional difference between the two proteins appears to root in the impairment of UPF3A's EJC‐binding domain (EBD). Swapping of UPF3A's weak EBD with UPF3B's strong EBD converts UPF3A into an NMD activator and UPF3B into an NMD suppressor (Shum et al, 2016). However, the EBD plays no role in the dual function of UPF3B described here. Interestingly, UPF3A and UPF3B not only differ in their NMD activity but also in their ability to stimulate translation (Kunz et al, 2006), a function that may be related to translation termination and ribosome dissociation at a PTC. Therefore, it will be interesting to probe if UPF3A can either substitute or antagonize the role of UPF3B in translation termination.
Materials and Methods
All experiments were performed between two and four times with comparable results using different batches of cells, of recombinant proteins, and/or pre‐TCs.
Plasmids
The pPROExHtb_eRF1 plasmid was generated by subcloning the PCR‐amplified gene encoding from pQE30_eRF1 (Frolova et al, 2000) into pPROExHtb (Life Technologies) using restriction enzymes NcoI and NotI. The plasmid pET21d_UPF2L (121‐1227) was generated by subcloning pPROExHtb_UPF2 (121‐1227) into pET21d (EMD Biosciences) using restriction enzymes NcoI and NotI. pFastBacHtb_eRF3a was generated by subcloning the NcoI/HindIII fragment encoding full‐size wild‐type eRF3a from pET15b‐eRF3a into pFastBacHtb (Life Technologies). Deletion constructs for pFast‐BacHtb_UPF3B or pFastBacHtb_eRF3a were engineered by Self‐SLIC, an insert‐free SLIC reaction (Li & Elledge, 2007). Plasmids encoding human UPF1, UPF2L, UPF3A and UPF3B were generated by subcloning NcoI/NotI digested fragments from the respective pCI Neo/pcDNA vectors (Promega) into pFastBacHtb. Plasmids pCIneo‐FLAG‐eRF1, pCIneo‐FLAG‐eRF3a, and eRF3a variant plasmids as well as pCIneo‐V5‐UPF1, pCIneo‐UPF2, and pCIneo‐UPF3B plasmids for eukaryotic expression have been described (Ivanov et al, 2008).
Protein production and purification
UPF1(115‐914) and UPF1(295‐914) were expressed from plasmids pET28‐UPF1(115‐914) and pET28a‐UPF1(295‐914) and purified as described (Chamieh et al, 2008). His‐tagged human eRF3a, UPF1, UPF2L and UPF3B were expressed using the Multibac expression system (Fitzgerald et al, 2006). His‐tagged eRF1 and UPF2L were expressed in the E. coli strain BL21‐Gold(DE3) (Life Technologies). Cells were lysed in buffer A (25 mM HEPES‐KOH, 10 mM imidazole, 300 mM KCl, 5 mM 2‐mercaptoethanol, 5% v/v glycerol, pH 7.5) supplemented with protease inhibitor cocktail (Roche) and 0.1% NP40 for the insect cell expressed proteins. Lysed cells were centrifuged at 30,000 × g, 30 min. The supernatant was subjected to Ni‐NTA affinity chromatography (QIAGEN). After removal of the His‐tag with tobacco etch virus (TEV) protease, proteins were further purified using a HiTrap QXL column (GE Healthcare), followed by cation exchange chromatography using a HiTrap SP/HP column (GE Healthcare) for UPF3B. UPF1, eRF1 and eRF3a were further purified by SEC using a Superdex 200 column (GE Healthcare) equilibrated with buffer B (25 mM HEPES‐KOH, 300 mM KCl, 5 mM dithiothreitol (DTT), 5% v/v glycerol, pH 7.5). The SMG1‐8‐9 complex was expressed in HEK‐293T cells and purified via its streptavidin‐binding tag and SEC as described (Deniaud et al, 2015). For in vitro termination, ATPase and in vitro phosphorylation assays all proteins used were diluted to 3 μM in the protein storage buffer (25 mM Tris, pH 7.5, 150 mM NaCl, 1 mM Mg(OAc)2, 1 μM ZnSO4, 1 mM DTT, 10% glycerol). Aliquots were stored at −80°C. As judged by an OD 260 nm/280 nm ratio of ~0.5, all recombinant proteins were free of nucleic acid contaminations.
Pull‐down assays to probe protein–protein interaction using purified proteins
All experiments were performed with 20 μM of each protein in the final reaction volume of 20 μl in buffer C (25 mM HEPES/KOH, 150 mM KCl, 10 mM imidazole, 0.05% v/v Tween‐20, 5 mM DTT, 5% v/v glycerol, pH 7.5), if not indicated otherwise. The protein mixtures were incubated for 1 h on ice; subsequently, 20 μl of Ni‐NTA agarose (QIAGEN) was added to the mix and incubated for 1 h on ice. The reaction mixtures were washed four times with 200 μl of buffer C, and proteins were eluted using buffer C supplemented with 200 mM imidazole. 8 μl of SDS loading dye was mixed with either 2 μl of input reactions or 20 μl for the eluted complexes. 5 μl of the input sample, and 10 μl of the elution sample was loaded onto a 10 or 12% SDS–PAGE gel.
Size‐exclusion chromatography
Size‐exclusion chromatography (SEC) was performed under physiological conditions similar to those used for the reconstitution of the UPF1‐UPF2‐UPF3‐EJC complex (Melero et al, 2012) and for the purification of the SMG1‐8‐9 complex (Deniaud et al, 2015). Briefly, for SEC, 40 μM of each protein was added in a final reaction volume of 60 μl in buffer D (25 mM HEPES/KOH, 200 mM KCl, 3 mM Mg(OAc)2, 0.05% v/v Tween‐20, 5 mM DTT, 5% v/v glycerol, pH 7.5). The protein mixtures were incubated for 1 h on ice before loading onto a Superdex 200 PC3.2/30 column (AEKTA micro system, GE Healthcare). 10 μl of each elution fraction was loaded onto a 10% SDS–PAGE gel. For the UPF3B‐RNA interaction experiment, 40 μM of a 24 nucleotide long RNA oligonucleotide (5′‐CCCUGAGCUGACGCAGCACCUGGG‐3′) was mixed with 40 μM UPF3B. For the UPF1‐UPF3B‐RNA interaction experiment, a threefold excess of the RNA oligomer was used.
Size‐exclusion chromatography coupled to multiple angle laser light scattering (SEC‐MALLS)
SEC‐MALLS of UPF3B was performed with a Superdex 200 increase column (GE Healthcare) equilibrated with 25 mM HEPES‐KOH, 300 mM KCl, 5 mM DTT, 5% v/v glycerol, pH 7.5. The column was calibrated with globular standard proteins. The experiments were performed at 20°C with a flowrate of 0.5 ml/min. A DAWN‐HELEOS II detector (Wyatt Technology Corp.) with a laser emitting at 690 nm was used for detection. The protein concentration was determined online by differential refractive index measurements, using an Optilab T‐rEX detector (Wyatt Technology Corp.) and a refractive index increment, dn/dc, of 0.185 ml/g. The weight‐averaged molar masses were calculated using the ASTRA software (Wyatt Technology Corp.).
Ribosome‐binding experiments
For co‐sedimentation experiments, 5 pmol of rabbit 80S ribosomes was mixed with a tenfold molar excess of UPF1, UPF2L and UPF3B in 20 μl of 25 mM HEPES‐KOH pH 7.5, 100 mM KOAc, 2.5 mM Mg(OAc)2, 2 mM DTT and incubated for 1 h on ice. Subsequently, the reaction mixtures were applied on a sucrose cushion (same buffer containing 250 mM KOAc and 750 mM sucrose) and spun for 3 h at 135,000 × g at 4°C using a TLA‐55 rotor (Beckman). Supernatant and pellet fractions were analysed by SDS–PAGE followed by SYPRO Ruby (Thermo Scientific) staining.
ATPase assays
ATPase assays were performed in a total volume of 20 μl essentially as described (Chamieh et al, 2008). Briefly, 1.5 pmol UPF1 either alone or in presence of 3 pmol UPF2L and/or 3 pmol UPF3B was mixed with 4 μl 5× MES buffer (250 mM MES pH 6.5, 250 mM KOAc, 25 mM Mg(OAc)2, 10 mM DTT, 0.5 mg/ml BSA) or 5× translation buffer (100 mM Tris pH 7.5, 500 mM KCl, 12.5 mM MgCl2, 10 mM DTT, 1.25 mM spermidine), 2 μl Poly(U) RNA (Sigma, 2 mg/ml in H2O) and H2O to a final volume of 16 μl. When variable protein compositions were tested, the total volume of proteins added was adjusted to equal using protein storage buffer. The reaction was started by adding 3.9 μl 10 mM ATP and 0.1 μl γ32P‐ATP (Hartmann Analytic, 3,000 Ci/mmol). Reactions proceeded for 1 h at 30 or 37°C. 1.5 μl per sample was spotted on PEI cellulose TLC plates (Merck) that had been pre‐run in water. Plates were developed in 0.4 M LiCl, 0.8 M acetic acid, dried and visualized by autoradiography.
In vitro phosphorylation
1.2 pmol UPF1 either alone or in presence of various combinations of 2.4 pmol of UPF2L, UPF3B, eRF1/eRF3a and 40 fmol of SMG1‐8‐9 was mixed with 4 μl 5× translation buffer and H2O to a final volume of 16 μl. When variable protein compositions were tested, the total volume of proteins added was adjusted to equal using protein storage buffer. The reaction was started by adding 2.5 μl 10 mM ATP and 1.5 μl γ32P‐ATP and was allowed to proceed for 30 min at 37°C. Samples were separated by SDS–PAGE and visualized by autoradiography.
Cell culture and transfections
HeLa cells were grown in DMEM and transfected in 10‐cm plates using JetPrime transfection reagent (Polyplus), 1–4.5 μg of the test plasmids and 0.4 μg of a YFP plasmid. Empty pGEMG‐3z vector (Promega) was used to adjust total amounts of transfected DNA.
Co‐immunoprecipitation assays from transfected cells
Twenty‐four hours after transfection, cells were harvested in 400 μl/10‐cm plate of buffer E (20 mM Tris pH 7.5, 150 mM KCl, 0.1% NP40) supplemented with 0.3 mM MgCl2 and EDTA‐free complete (Sigma‐Aldrich) and lysed 30 min on ice. Magnetic M2 anti‐FLAG beads (Sigma‐Aldrich) were used to immunoprecipitate FLAG‐tagged complexes from RNaseA‐treated (30–40 μg/ml) cell lysates after 1 h incubation with the beads at 4°C. Beads were washed eight times with buffer E supplemented with 0.6 mM MgCl2. Tubes were changed before the last wash. FLAG complexes were eluted with 25 μl 0.1 M glycine (pH 3.0), added to 6 μl 5× loading buffer and neutralized with 1.5 μl 1 M Tris pH 7.5. 6 μl of the samples was loaded for anti‐FLAG detection and 20 μl for anti‐V5 detection, separated by SDS–PAGE and analysed by immunoblotting using anti‐FLAG and anti‐V5‐antibodies (Sigma‐Aldrich F7425 and V8137, respectively). Anti‐TUBB (Sigma‐Aldrich T5168) and anti‐ACTB (Sigma‐Aldrich A1978) antibodies were used for loading controls.
Pre‐termination complex assembly and purification
Pre‐termination complexes (pre‐TC) were assembled as described (Alkalaeva et al, 2006) with the following modifications: The translation reaction was performed in translation buffer D (20 mM Tris–HCl, pH 7.5, 50 mM KOAc, 1.3 mM free MgCl2, 2 mM DTT, 0.25 mM spermidine) supplemented with 200 U RNase inhibitor (RiboLock, Fermentas), 1 mM ATP, 0.2 mM GTP, 35 pmol of MVHC‐STOP mRNA, 35 pmol initiator‐tRNAi (acylated with [35S]‐methionine for peptide release assays), 75 μg total tRNA (acylated with individual amino acids), 50 pmol purified human ribosomal subunits (40S and 60S), 100 pmol eIF2, 50 pmol eIF3, 80 pmol eIF4GΔ, eIF4A, eIF4B, eIF1, eIF1A, eIF5, eIF5BΔ each, 200 pmol eEF1H and 50 pmol eEF2 in the volume of 500 μl. The reaction mix was incubated for 40 min at 37°C. Subsequently, the reaction mix was loaded onto a 10–30% w/w linear sucrose density gradient (SDG) prepared in buffer D with 5 mM MgCl2 and centrifuged for 115 min at 4°C at 255,000 × g using a Beckman SW60 rotor. The fractions corresponding to pre‐TC complexes according to optical density and the presence of [35S]‐methionine were combined and used for toe printing and peptide release assays.
In vitro translation and toeprinting analysis of pre‐ and post‐termination complexes
In vitro translation termination was performed essentially as described (Alkalaeva et al, 2006). Briefly, 0.1 pmol pre‐TCs that had been assembled on MVHC‐STOP mRNA and purified by SDG centrifugation as described under “Pre‐termination complex assembly and purification” was incubated for 10 min at 37°C with 3 pmol of UPF proteins or BSA in a total volume of 35 μl translation buffer E (10 mM Tris pH 7.5, 100 mM KCl, 1 mM free MgCl2, 2 mM DTT, 0.25 mM spermidine) supplemented with 0.5 mM GTP and 1 mM ATP or AMPPNP as indicated. Protein storage buffer was used to compensate for varying amounts of proteins. The amounts of eRF1 and eRF3a required to limit the termination rate were determined for each batch of pre‐TCs. eRF1 and eRF3a were added to the reaction mix, and termination was allowed to proceed for 5 min at 37°C. Ribosomal complexes were analysed by primer extension inhibition using AMV reverse transcriptase and a [32P]‐labelled primer (5′‐GCAATGAAAATAAATTTCC‐3′) complementary to a coding region of β‐globin mRNA (Pestova et al, 1996). For the experiment described in Fig 1E, 3 pmol of UPF1 was incubated in ATP‐supplemented translation buffer either alone or in the presence of equal amounts of UPF2L and/or UPF3B and with or without 50 fmol SMG1‐8‐9 as indicated for 30 min at 37°C. After the addition of pre‐TCs, the mixture was incubated for 10 min at 37°C. The termination reaction was initiated by the addition of eRF1 and eRF3a and allowed to proceed for 5 min.
Peptide release assays
The termination efficiency was determined as described (Alkalaeva et al, 2006) with the following modifications. Aliquots containing 0.01 pmol of [35S]‐methionine‐containing pre‐TCs were incubated for 10 min at 37°C with 0.3 pmol BSA or UPF3B in translation buffer E supplemented with 1 mM ATP and 0.5 mM GTP. The peptide release reaction was started by addition of termination‐rate limiting amounts eRF1 and eRF3a. Subsequently, the reaction was allowed to proceed for 0–60 s at 37°C. The ribosomal complexes were pelleted with ice‐cold 5% trichloroacetic acid and centrifuged for 30 min at 4°C and 14,000 × g. The amount of released [35S]‐methionine‐containing tetrapeptide was determined by scintillation counting of the supernatants using a Wallac 1504 liquid scintillation spectrometer.
Author contributions
GN‐Y, CS, AEK and MWH designed the study and wrote the manuscript with input from ER, BE and LY; GN‐Y, BA and KK performed toeprinting experiments, ATPase assays, in vitro phosphorylation and Co‐IPs; ER, BE, LY, AD and KH produced proteins; ER performed in vitro pulldown, co‐sedimentation and SEC (SEC‐MALLS) experiments; BE generated pre‐TCs and performed peptide release experiments.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We are much obliged to Tatyana Pestova for generously introducing KK to the reconstituted in vitro translation system and for providing the MVHC‐STOP plasmid. We thank Hervé Le Hir for the UPF1L and UPF1ΔCH expression plasmids and providing protocols for their production as well as for the ATPase assay, Ludmila Frolova for providing the plasmid pET15b‐eRF3a, and Stephen Cusack for plasmids pPROExHtb_UPF2 (121‐1227), pPROExHtb_UPF3B‐N, and the RNA oligonucleotide for RNA interaction assays. We thank Elena Alkalaeva for providing Met‐tRNA and the plasmids for initiation factors used for in vitro translation, Alain Miller (Calbiotech S.A., Mons, Belgium) for providing HeLa cytoplasmic extracts, Jan Medenbach for the gift of SXL, and Dmytro Dziuba for the gift of IRP1. AD acknowledges support by an EC‐EMBL CoFUNDS EIPOD fellowship; CS is supported by a European Research Council Starting Grant (ComplexNMD, 281331). The experimental work of MWH and AEK is supported by the DFG through the SFB 1036 grant and together with GN‐Y through the SPP1935 grant.
See also: O Mühlemann & ED Karousis (October 2017)
Contributor Information
Matthias W Hentze, Email: hentze@embl.de.
Christiane Schaffitzel, Email: christiane.berger-schaffitzel@bristol.ac.uk.
Andreas E Kulozik, Email: andreas.kulozik@med.uni-heidelberg.de.
References
- Alkalaeva EZ, Pisarev AV, Frolova LY, Kisselev LL, Pestova TV (2006) In vitro reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and eRF3. Cell 125: 1125–1136 [DOI] [PubMed] [Google Scholar]
- Alrahbeni T, Sartor F, Anderson J, Miedzybrodzka Z, McCaig C, Muller B (2015) Full UPF3B function is critical for neuronal differentiation of neural stem cells. Mol Brain 8: 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A (2004) A faux 3′‐UTR promotes aberrant termination and triggers nonsense‐mediated mRNA decay. Nature 432: 112–118 [DOI] [PubMed] [Google Scholar]
- Bhuvanagiri M, Schlitter AM, Hentze MW, Kulozik AE (2010) NMD: RNA biology meets human genetic medicine. Biochem J 430: 365–377 [DOI] [PubMed] [Google Scholar]
- Brogna S, McLeod T, Petric M (2016) The meaning of NMD: translate or perish. Trends Genet 32: 395–407 [DOI] [PubMed] [Google Scholar]
- Brown A, Shao S, Murray J, Hegde RS, Ramakrishnan V (2015) Structural basis for stop codon recognition in eukaryotes. Nature 524: 493–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bühler M, Steiner S, Mohn F, Paillusson A, Mühlemann O (2006) EJC‐independent degradation of nonsense immunoglobulin‐mu mRNA depends on 3′ UTR length. Nat Struct Mol Biol 13: 462–464 [DOI] [PubMed] [Google Scholar]
- Celik A, Kervestin S, Jacobson A (2015) NMD: at the crossroads between translation termination and ribosome recycling. Biochimie 114: 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Jayachandran U, Bonneau F, Fiorini F, Basquin C, Domcke S, Le Hir H, Conti E (2011) Molecular mechanisms for the RNA‐dependent ATPase activity of Upf1 and its regulation by Upf2. Mol Cell 41: 693–703 [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Bonneau F, Schussler S, Eppinger E, Conti E (2014) Phospho‐dependent and phospho‐independent interactions of the helicase UPF1 with the NMD factors SMG5‐SMG7 and SMG6. Nucleic Acids Res 42: 9447–9460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamieh H, Ballut L, Bonneau F, Le Hir H (2008) NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat Struct Mol Biol 15: 85–93 [DOI] [PubMed] [Google Scholar]
- Chan WK, Huang L, Gudikote JP, Chang YF, Imam JS, MacLean JA II, Wilkinson MF (2007) An alternative branch of the nonsense‐mediated decay pathway. EMBO J 26: 1820–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WK, Bhalla AD, Le Hir H, Nguyen LS, Huang L, Gecz J, Wilkinson MF (2009) A UPF3‐mediated regulatory switch that maintains RNA surveillance. Nat Struct Mol Biol 16: 747–753 [DOI] [PubMed] [Google Scholar]
- Deniaud A, Karuppasamy M, Bock T, Masiulis S, Huard K, Garzoni F, Kerschgens K, Hentze MW, Kulozik AE, Beck M, Neu‐Yilik G, Schaffitzel C (2015) A network of SMG‐8, SMG‐9 and SMG‐1 C‐terminal insertion domain regulates UPF1 substrate recruitment and phosphorylation. Nucleic Acids Res 43: 7600–7611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever TE, Green R (2012) The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb Perspect Biol 4: a013706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan‐Minogue H, Du M, Pisarev AV, Kallmeyer AK, Salas‐Marco J, Keeling KM, Thompson SR, Pestova TV, Bedwell DM (2008) Distinct eRF3 requirements suggest alternate eRF1 conformations mediate peptide release during eukaryotic translation termination. Mol Cell 30: 599–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatscher T, Boehm V, Gehring NH (2015) Mechanism, factors, and physiological role of nonsense‐mediated mRNA decay. Cell Mol Life Sci 72: 4523–4544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorini F, Boudvillain M, Le Hir H (2013) Tight intramolecular regulation of the human Upf1 helicase by its N‐ and C‐terminal domains. Nucleic Acids Res 41: 2404–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorini F, Bagchi D, Le Hir H, Croquette V (2015) Human Upf1 is a highly processive RNA helicase and translocase with RNP remodelling activities. Nat Commun 6: 7581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald DJ, Berger P, Schaffitzel C, Yamada K, Richmond TJ, Berger I (2006) Protein complex expression by using multigene baculoviral vectors. Nat Methods 3: 1021–1032 [DOI] [PubMed] [Google Scholar]
- Franckenberg S, Becker T, Beckmann R (2012) Structural view on recycling of archaeal and eukaryotic ribosomes after canonical termination and ribosome rescue. Curr Opin Struct Biol 22: 786–796 [DOI] [PubMed] [Google Scholar]
- Franks TM, Singh G, Lykke‐Andersen J (2010) Upf1 ATPase‐dependent mRNP disassembly is required for completion of nonsense‐ mediated mRNA decay. Cell 143: 938–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolova LY, Simonsen JL, Merkulova TI, Litvinov DY, Martensen PM, Rechinsky VO, Camonis JH, Kisselev LL, Justesen J (1998) Functional expression of eukaryotic polypeptide chain release factors 1 and 3 by means of baculovirus/insect cells and complex formation between the factors. Eur J Biochem 256: 36–44 [DOI] [PubMed] [Google Scholar]
- Frolova LY, Merkulova TI, Kisselev LL (2000) Translation termination in eukaryotes: polypeptide release factor eRF1 is composed of functionally and structurally distinct domains. RNA 6: 381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring NH, Neu‐Yilik G, Schell T, Hentze MW, Kulozik AE (2003) Y14 and hUpf3b form an NMD‐activating complex. Mol Cell 11: 939–949 [DOI] [PubMed] [Google Scholar]
- Gehring NH, Kunz JB, Neu‐Yilik G, Breit S, Viegas MH, Hentze MW, Kulozik AE (2005) Exon‐junction complex components specify distinct routes of nonsense‐mediated mRNA decay with differential cofactor requirements. Mol Cell 20: 65–75 [DOI] [PubMed] [Google Scholar]
- Gehring NH, Lamprinaki S, Kulozik AE, Hentze MW (2009) Disassembly of exon junction complexes by PYM. Cell 137: 536–548 [DOI] [PubMed] [Google Scholar]
- Ghosh S, Ganesan R, Amrani N, Jacobson A (2010) Translational competence of ribosomes released from a premature termination codon is modulated by NMD factors. RNA 16: 1832–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graille M, Seraphin B (2012) Surveillance pathways rescuing eukaryotic ribosomes lost in translation. Nat Rev Mol Cell Biol 13: 727–735 [DOI] [PubMed] [Google Scholar]
- Hauer C, Sieber J, Schwarzl T, Hollerer I, Curk T, Alleaume AM, Hentze MW, Kulozik AE (2016) Exon junction complexes show a distributional bias toward alternatively spliced mRNAs and against mRNAs coding for ribosomal proteins. Cell Rep 16: 1588–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Jacobson A (2015) Nonsense‐mediated mRNA decay: degradation of defective transcripts is only part of the story. Annu Rev Genet 49: 339–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilleren P, Parker R (1999) mRNA surveillance in eukaryotes: kinetic proofreading of proper translation termination as assessed by mRNP domain organization? RNA 5: 711–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg JR, Goff SP (2010) Upf1 senses 3′UTR length to potentiate mRNA decay. Cell 143: 379–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurt JA, Robertson AD, Burge CB (2013) Global analyses of UPF1 binding and function reveal expanded scope of nonsense‐mediated mRNA decay. Genome Res 23: 1636–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov PV, Gehring NH, Kunz JB, Hentze MW, Kulozik AE (2008) Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. EMBO J 27: 736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Mikhailova T, Eliseev B, Yeramala L, Sokolova E, Susorov D, Shuvalov A, Schaffitzel C, Alkalaeva E (2016) PABP enhances release factor recruitment and stop codon recognition during translation termination. Nucleic Acids Res 44: 7766–7776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CU, Pestova TV (2012) Termination and post‐termination events in eukaryotic translation. Adv Protein Chem Struct Biol 86: 45–93 [DOI] [PubMed] [Google Scholar]
- Jolly LA, Homan CC, Jacob R, Barry S, Gecz J (2013) The UPF3B gene, implicated in intellectual disability, autism, ADHD and childhood onset schizophrenia regulates neural progenitor cell behaviour and neuronal outgrowth. Hum Mol Genet 22: 4673–4687 [DOI] [PubMed] [Google Scholar]
- Kadlec J, Izaurralde E, Cusack S (2004) The structural basis for the interaction between nonsense‐mediated mRNA decay factors UPF2 and UPF3. Nat Struct Mol Biol 11: 330–337 [DOI] [PubMed] [Google Scholar]
- Karousis ED, Nasif S, Muhlemann O (2016) Nonsense‐mediated mRNA decay: novel mechanistic insights and biological impact. Wiley Interdiscip Rev RNA 7: 661–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima I, Yamashita A, Izumi N, Kataoka N, Morishita R, Hoshino S, Ohno M, Dreyfuss G, Ohno S (2006) Binding of a novel SMG‐1‐Upf1‐eRF1‐eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense‐mediated mRNA decay. Genes Dev 20: 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling KM, Lanier J, Du M, Salas‐Marco J, Gao L, Kaenjak‐Angeletti A, Bedwell DM (2004) Leaky termination at premature stop codons antagonizes nonsense‐mediated mRNA decay in S. cerevisiae . RNA 10: 691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz JB, Neu‐Yilik G, Hentze MW, Kulozik AE, Gehring NH (2006) Functions of hUpf3a and hUpf3b in nonsense‐mediated mRNA decay and translation. RNA 12: 1015–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T, Li W, Hoque M, Popp MW, Ermolenko DN, Tian B, Maquat LE (2014) A post‐translational regulatory switch on UPF1 controls targeted mRNA degradation. Genes Dev 28: 1900–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T, Maquat LE (2016) Nonsense‐mediated mRNA decay in humans at a glance. J Cell Sci 129: 461–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MZ, Elledge SJ (2007) Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods 4: 251–256 [DOI] [PubMed] [Google Scholar]
- Linder B, Fischer U, Gehring NH (2015) mRNA metabolism and neuronal disease. FEBS Lett 589: 1598–1606 [DOI] [PubMed] [Google Scholar]
- Lopez‐Perrote A, Castano R, Melero R, Zamarro T, Kurosawa H, Ohnishi T, Uchiyama A, Aoyagi K, Buchwald G, Kataoka N, Yamashita A, Llorca O (2016) Human nonsense‐mediated mRNA decay factor UPF2 interacts directly with eRF3 and the SURF complex. Nucleic Acids Res 44: 1909–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke‐Andersen J, Bennett EJ (2014) Protecting the proteome: eukaryotic cotranslational quality control pathways. J Cell Biol 204: 467–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke‐Andersen S, Jensen TH (2015) Nonsense‐mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol 16: 665–677 [DOI] [PubMed] [Google Scholar]
- MacDermott M (1990) The intracellular concentration of free magnesium in extensor digitorum longus muscles of the rat. Exp Physiol 75: 763–769 [DOI] [PubMed] [Google Scholar]
- Matheisl S, Berninghausen O, Becker T, Beckmann R (2015) Structure of a human translation termination complex. Nucleic Acids Res 43: 8615–8626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melero R, Buchwald G, Castano R, Raabe M, Gil D, Lazaro M, Urlaub H, Conti E, Llorca O (2012) The cryo‐EM structure of the UPF‐EJC complex shows UPF1 poised toward the RNA 3′ end. Nat Struct Mol Biol 19: 498–505, S1–2 [DOI] [PubMed] [Google Scholar]
- Mendell JT, Sharifi NA, Meyers JL, Martinez‐Murillo F, Dietz HC (2004) Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet 36: 1073–1078 [DOI] [PubMed] [Google Scholar]
- Min EE, Roy B, Amrani N, He F, Jacobson A (2013) Yeast Upf1 CH domain interacts with Rps26 of the 40S ribosomal subunit. RNA 19: 1105–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita T, Yamashita A, Kashima I, Ogata K, Ishiura S, Ohno S (2007) Distant N‐ and C‐terminal domains are required for intrinsic kinase activity of SMG‐1, a critical component of nonsense‐mediated mRNA decay. J Biol Chem 282: 7799–7808 [DOI] [PubMed] [Google Scholar]
- Nguyen LS, Wilkinson MF, Gecz J (2014) Nonsense‐mediated mRNA decay: inter‐individual variability and human disease. Neurosci Biobehav Rev 46(Pt 2): 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada‐Katsuhata Y, Yamashita A, Kutsuzawa K, Izumi N, Hirahara F, Ohno S (2012) N‐ and C‐terminal Upf1 phosphorylations create binding platforms for SMG‐6 and SMG‐5: SMG‐7 during NMD. Nucleic Acids Res 40: 1251–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottens F, Gehring NH (2016) Physiological and pathophysiological role of nonsense‐mediated mRNA decay. Pflugers Arch 468: 1013–1028 [DOI] [PubMed] [Google Scholar]
- Peixeiro I, Inacio A, Barbosa C, Silva AL, Liebhaber SA, Romao L (2012) Interaction of PABPC1 with the translation initiation complex is critical to the NMD resistance of AUG‐proximal nonsense mutations. Nucleic Acids Res 40: 1160–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova TV, Hellen CU, Shatsky IN (1996) Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol Cell Biol 16: 6859–6869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarev AV, Hellen CU, Pestova TV (2007a) Recycling of eukaryotic posttermination ribosomal complexes. Cell 131: 286–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarev AV, Unbehaun A, Hellen CU, Pestova TV (2007b) Assembly and analysis of eukaryotic translation initiation complexes. Methods Enzymol 430: 147–177 [DOI] [PubMed] [Google Scholar]
- Pisarev AV, Skabkin MA, Pisareva VP, Skabkina OV, Rakotondrafara AM, Hentze MW, Hellen CU, Pestova TV (2010) The role of ABCE1 in eukaryotic posttermination ribosomal recycling. Mol Cell 37: 196–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisareva VP, Pisarev AV, Komar AA, Hellen CU, Pestova TV (2008) Translation initiation on mammalian mRNAs with structured 5′UTRs requires DExH‐box protein DHX29. Cell 135: 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynal B, Lenormand P, Baron B, Hoos S, England P (2014) Quality assessment and optimization of purified protein samples: why and how? Microb Cell Fact 13: 180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweingruber C, Rufener SC, Zünd D, Yamashita A, Mühlemann O (2013) Nonsense‐mediated mRNA decay – mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim Biophys Acta 1829: 612–623 [DOI] [PubMed] [Google Scholar]
- Serdar LD, Whiteside DL, Baker KE (2016) ATP hydrolysis by UPF1 is required for efficient translation termination at premature stop codons. Nat Commun 7: 14021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenvi CL, Dong KC, Friedman EM, Hanson JA, Cate JH (2005) Accessibility of 18S rRNA in human 40S subunits and 80S ribosomes at physiological magnesium ion concentrations–implications for the study of ribosome dynamics. RNA 11: 1898–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirokikh NE, Alkalaeva EZ, Vassilenko KS, Afonina ZA, Alekhina OM, Kisselev LL, Spirin AS (2010) Quantitative analysis of ribosome‐mRNA complexes at different translation stages. Nucleic Acids Res 38: e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shum EY, Jones SH, Shao A, Dumdie J, Krause MD, Chan WK, Lou CH, Espinoza JL, Song HW, Phan MH, Ramaiah M, Huang L, McCarrey JR, Peterson KJ, De Rooij DG, Cook‐Andersen H, Wilkinson MF (2016) The antagonistic gene paralogs Upf3a and Upf3b govern nonsense‐mediated RNA decay. Cell 165: 382–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Rebbapragada I, Lykke‐Andersen J (2008) A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense‐mediated mRNA decay. PLoS Biol 6: e111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skabkin MA, Skabkina OV, Hellen CU, Pestova TV (2013) Reinitiation and other unconventional posttermination events during eukaryotic translation. Mol Cell 51: 249–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpey PS, Raymond FL, Nguyen LS, Rodriguez J, Hackett A, Vandeleur L, Smith R, Shoubridge C, Edkins S, Stevens C, O'Meara S, Tofts C, Barthorpe S, Buck G, Cole J, Halliday K, Hills K, Jones D, Mironenko T, Perry J et al (2007) Mutations in UPF3B, a member of the nonsense‐mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat Genet 39: 1127–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ter‐Avanesyan MD, Kushnirov VV, Dagkesamanskaya AR, Didichenko SA, Chernoff YO, Inge‐Vechtomov SG, Smirnov VN (1993) Deletion analysis of the SUP35 gene of the yeast Saccharomyces cerevisiae reveals two non‐overlapping functional regions in the encoded protein. Mol Microbiol 7: 683–692 [DOI] [PubMed] [Google Scholar]
- Veloso D, Guynn RW, Oskarsson M, Veech RL (1973) The concentrations of free and bound magnesium in rat tissues. Relative constancy of free Mg2+ concentrations. J Biol Chem 248: 4811–4819 [PubMed] [Google Scholar]
- Wang W, Czaplinski K, Rao Y, Peltz SW (2001) The role of Upf proteins in modulating the translation read‐through of nonsense‐containing transcripts. EMBO J 20: 880–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zünd D, Gruber AR, Zavolan M, Mühlemann O (2013) Translation‐dependent displacement of UPF1 from coding sequences causes its enrichment in 3′ UTRs. Nat Struct Mol Biol 20: 936–943 [DOI] [PubMed] [Google Scholar]
- Zünd D, Mühlemann O (2013) Recent transcriptome‐wide mapping of UPF1 binding sites reveals evidence for its recruitment to mRNA before translation. Translation (Austin) 1: e26977 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7