

Abstract
Organocatalyzed atom transfer radical polymerization (O-ATRP) has emerged as a metal-free variant of historically transition-metal reliant atom transfer radical polymerization. Strongly reducing organic photoredox catalysts have proven capable of mediating O-ATRP. To date, operation of photoinduced O-ATRP has been demonstrated in batch reactions. However, continuous flow approaches can provide efficient irradiation reaction conditions and thus enable increased polymerization performance. Herein, the adaptation of O-ATRP to a continuous flow approach has been performed with multiple visible-light absorbing photoredox catalysts. Using continuous flow conditions, improved polymerization results were achieved, consisting of narrow molecular weight distributions as low as 1.05 and quantitative initiator efficiencies. This system demonstrated success with 0.01% photocatalyst loadings and a diverse methacrylate monomer scope. Additionally, successful chain-extension polymerizations using 0.01 mol % photocatalyst loadings reveal continuous flow O-ATRP to be a robust and versatile method of polymerization.
Graphical abstract
Introduction
Photoinduced controlled radical polymerizations (CRPs) have become powerful tools for polymer synthesis to yield polymers with targeted molecular weights (MWs), low molecular weight dispersities (Đ), and controlled compositions with the added potential for spatial and temporal control.1–5 In the past two decades atom transfer radical polymerization (ATRP) has emerged as the most widely applied CRP.6,7 The popularity of ATRP is largely due to use of readily available reagents, robust implementation, and functional group tolerance. The ability of ATRP to preserve a functional chain-end group makes this methodology well-suited for the synthesis of advanced architectures, such as block copolymers.8 In ATRP, control over the polymerization relies on strict regulation over the equilibrium between a dormant alkyl halide and an active propagating radical species in order to maintain a low radical concentration and minimize undesirable bimolecular termination reactions.9 Traditionally, ATRP has employed transition metal catalysts to mediate this equilibrium, which ultimately contaminates the polymer product and can restrict the application potential of the polymer, especially in electronic applications. Significant advancements have enabled the use of lower levels of transition metal catalysts10–13 and increased ability for polymer purification,14–16 while recently organocatalyzed ATRP (O-ATRP) has risen as an approach to entirely eliminate metal contamination of the polymer product.17–22
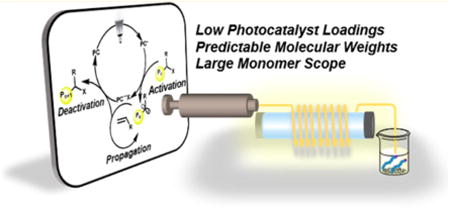
A proposed mechanism for O-ATRP proceeds through photoexcitation of the ground state photoredox catalyst (PC) to generate the singlet excited state (1PC*) and subsequent intersystem crossing to a highly reducing triplet excited state (3PC*) (Figure 1A). The 3PC* activates an alkyl halide through reduction to simultaneously form the PC radical cation halide anion ion pair (2PC•+X−) and the active carbon-centered radical species for polymerization propagation. The 2PC*+X− species deactivates the growing polymer chain via an oxidative event, generating a halide end-capped polymer. For CRPs, control over polymerization relies on a faster rate of deactivation than rate of activation or monomer propagation. Furthermore, the irradiation of the PC plays an important role in the efficiency of PC photoexcitation and thus relative concentrations of every PC species in the polymerization reaction, which culminates in control over the polymerization. As such, we hypothesized developing O-ATRP in continuous flow would enhance irradiation efficiency and facilitate the synthesis of well-defined polymers.
Figure 1.
(A) Proposed mechanism for O-ATRP proceeding through an oxidative quenching pathway. (B) Visible-light-absorbing PCs used in this study include N,N-diaryl phenazines (1 and 2), perylene (3), and N-aryl phenoxazines (4). (C) Photomediated flow reactors offer significant advantages to batch systems.
In 2014, perylene17 and N-aryl phenothiazines18 were presented as PCs able to orchestrate O-ATRP through an oxidative quenching pathway using visible and UV irradiation, respectively. Recently, our group has revealed N,N-diaryl dihydrophenazines19 and N-aryl phenoxazines,20 as strongly reducing visible-light-absorbing PCs efficient for O-ATRP. Mechanistic studies of O-ATRP mediated by N-aryl-phenothiazine catalysts highlighted the necessity for fast activation and deactivation for controlled polymerizations.23 Further studies have shown the importance of photoinduced charge transfer states of the PC and solvent stabilization of the resulting 2PC•+X− ion pair in N,N-diaryldihydrophenazine catalysts for the synthesis of well-defined polymers using O-ATRP.24 These studies reveal the free energy of 2PC•+X−, which is influenced by solvent polarity, to play a key role in efficient deactivation and to achieve control over the polymerization.
As an alternative to traditional batch reactors, photomediated continuous flow reactors have materialized as an excellent approach in both small molecule25–28 and macromolecular29–31 syntheses to promote efficient and uniform irradiation conditions. According to the Beer–Lambert law, in a photomediated batch reaction the ability for photons to travel in the reaction medium decreases exponentially with increasing path length, equating to nonuniform irradiation and limiting the efficiency of the reaction. In particular, for photo-induced CRP reactions poor irradiation control leads to broad molecular weight distributions, slower reaction times, and limited scalability.26,33 In contrast to batch reactors, continuous flow offers a high surface-area-to-volume ratio of reactor to solution, allowing uniform irradiation, fast reaction times, efficient heat and mass transfer, reduction of batch-to-batch variations, and facile scalability (Figure 1C). 26,28,34 Considering reaction design, reaction parameters can be easily adjusted during polymerization by modulating flow rates, irradiation intensity, and reaction stoichiometry.35
To date, the scalability and irradiation efficiency of O-ATRP have been constrained by limitations inherent to batch reactor systems. Reports of continuous flow approaches have been presented for nitroxide-mediated polymerization (NMP),36,37 reversible addition–fragmentation chain transfer (RAFT),31,38 and metal-catalyzed ATRP.39,40 This method has been further extended to photoinduced RAFT,41–43 and photoinduced metal-catalyzed ATRP, 44,45 but photoinduced O-ATRP in continuous flow has not yet been reported. With that in mind, we hypothesized that the favorable characteristics of photomediated continuous flow systems, namely control over irradiation, would facilitate further study of the capabilities of recently developed organic PCs through heightened control over activation and deactivation, accessing enhanced results of polymerization. To that end, this work reports a study of the integration of visible-light-mediated O-ATRP into a continuous flow approach, improving and expanding the utility of the methodology while offering an energy-efficient method for producing polymers on scale.
Experimental Section
Continuous Flow Setup
A continuous flow tubular reactor system was designed from commercially available components consisting of a syringe pump, stainless steel syringe, Halar tubing, and a fluorescent tubular light bulb (see Supporting Information for full details). Halar tubing was used for the flow reactor based on studies optimizing a continuous flow reactor system using photo-induced ATRP catalyzed by fac-Ir(ppy)3.44
General Polymerization Procedure
A vial was charged with PC, brought inside a glovebox, and loaded under nonphotoactive lighting with N,N-dimethylacetamide (DMA), methyl methacrylate (MMA), and alkyl bromide initiator while stirring. Once the PC was fully dissolved, the reaction mixture was loaded into a stainless steel syringe to prevent undesired irradiation exposure during polymerization. The syringe was fitted with the first section of tubing (Figure S1), then brought outside of the glovebox, and quickly attached to the next section of tubing reactor. Once the syringe pump was started with the desired flow rate, the system was allowed to reach steady-state conditions by loading the reactor with the reaction mixture and running for 1.5 residence times. Residence time is defined as the amount of time the polymerization solution is exposed to irradiation conditions (see Supporting Information for a detailed explanation). The polymer product was collected and quenched by placing the third section of tubing into a 20 mL scintillation vial containing deuterated chloroform with 50 ppm BHT. The tubing reactor was flushed with DMA after polymerization was complete. The system was allowed to reach steady-state temperature conditions by turning on the light for 1 h before polymerization. The temperature of the system was monitored continuously during polymerization and remained at constant 60 °C, unless otherwise noted.
Polymer Characterization
Monomer conversion was determined by 1H NMR analysis immediately following collection by comparison of the integrations of the methyl ester monomer peak at 3.73 ppm to the corresponding methyl ester polymer peak at 3.57 ppm. The volatiles were removed from the remainder of the collected sample, and then the sample was dissolved in THF for molecular weight analysis using gel permeation chromatography (GPC) coupled to multiangle light scattering.
Results and Discussion
Synthesis of PMMA Using Visible-Light-Absorbing PCs
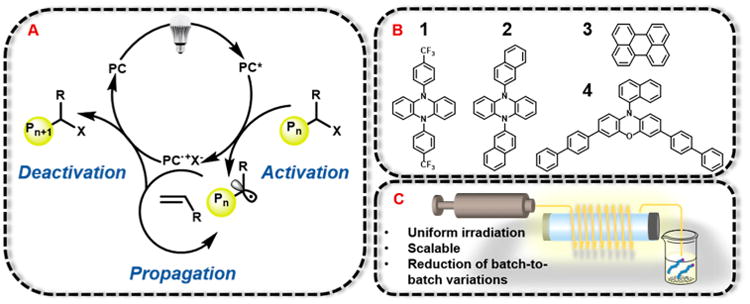
To examine the performance of O-ATRP using a continuous flow reactor, we investigated multiple previously reported PCs studied in a batch reactor system for the polymerization of methyl methacrylate (MMA). The PCs used in this study were selected due to absorption profiles in the visible light regime and familial diversity (Figure 1B). For a direct comparison of polymerization trends, similar reaction conditions were used to those reported in batch, including reaction stoichiometry and choice of alkyl bromide initiator. In batch conditions using broad spectrum white LEDs for irradiation for PC 1 and 2, Đ typically ranged from 1.10 to 1.18 and 1.03, respectively, with initiator efficiency (I*)46 values reaching 66% and 46%.19 For PC 3, batch conditions gave Đ of 1.3–1.8 but low I* (14–22%) and a lack of control over MW as polymerization proceeded.17 In the case of PC 4, in typical batch reactions Đ of 1.25–1.17 and I* of ∼100% were reported.24 A summary of key photophysical and redox properties of these PCs can be found in Table 1.
Table 1. Summary of Redox and Photophysical Properties for PCs 1–4a.
The results for polymerizations using PCs 1, 2, 3, and 4 in continuous flow all showed similar trends of enhanced polymerization performance in comparison to batch reactor conditions, as demonstrated by improved predictability in MW and lowered Đ (Figure 2). When employing 1, Đ ranged from 1.10 to 1.18 over the course of polymerization, reaching 49% conversion after a 2 h residence time. In the case of 2, Đ ranged from 1.05 to 1.14 while reaching 64% conversion after a 1.5 h residence time. Using PC 3 showed distinct improvement over batch reactions and was able to produce PMMA with a Đ as low as 1.10. Although inferior to the N,N-diaryl dihydrophenazine and N-aryl phenoxazine catalysts, 3 still exhibited a linear growth in MW, a marked change in comparison to reported batch reaction results. The performance of PC 4 in continuous flow is highlighted by near 100% I*, a predictable increase in MW, and Đ ≤ 1.20 (Figure 2D).
Figure 2.
Plots of Mn (black diamonds) and Đ (red triangles) with respect to monomer conversion during the polymerization of MMA catalyzed by 1 (A), 2 (B), 3 (C), or 4 (D) performed in continuous flow at steady-state conditions and under different residence times. Shown is the theoretical evolution of Mn (dashed line) with respect to monomer conversion. Conditions for all plots: [1000]:[10]:[1] of [MMA]:[initiator]:[PC]; 3.73 mM PC; initiators are ethyl α-bromophenylacetate (EBP) when using 1–3 and diethyl 2-bromo-2-methylmalonate (DBMM) when using 4; 2:3 MMA to DMA by volume; 160 cm of tubing reactor, irradiated by 3000 K fluorescent light. See Supporting Information for flow rates and residence times used.
Most notably, for N,N-diaryldihydrophenazine catalysts 1 and 2, at low monomer conversion continuous flow demonstrated superior control in comparison to batch reactors, with experimentally measured MWs near theoretically predicted MWs resulting in nearly quantitative I*. These results can be explained through analysis of the proposed mechanism of O-ATRP in the context of continuous flow. In the O-ATRP mechanism, the rate of deactivation must be higher than activation in order to maintain control over the propagating radical species.23 This phenomenon is facilitated by the concentration, availability, and oxidation potential of the 2PC•+X− deactivator species. Low concentrations of 2PC•+X− hinder effective deactivation, limiting control over propagation via reversible end-capping of the active polymer chain. However, in a photoflow reactor a higher surface-to-volume ratio allows for more efficient irradiation and PC photo-excitation, subsequent activation, and therefore an indirect increase in deactivator concentrations. These conditions allow for control over propagation and result in enhanced control over MW at low polymer conversions for 1 and 2.
In total, these results demonstrate that a continuous flow approach can provide significant improvements in polymerization metrics using a variety of PCs, especially in regards to results at low monomer conversions. In a continuous-flow system, the maximum I* was reached at lower monomer conversions than in batch. This can be attributed to uniform irradiation resulting in higher concentrations of 3PC* and subsequent fast activation of the alkyl halide initiator. Because of low Đ and consistently high I* values near 100%, 4 was chosen as PC for additional study of continuous flow techniques using O-ATRP.
The effect of relative solvent volume on results of polymerization was explored using PC 4 (Table S7). Polymerization results did not vary significantly with varying reaction solution concentrations. The conditions tested included 1:1, 2:3, 1:2, and 1:3 ratios of MMA to DMA by volume. For all conditions, Đ remained relatively low, typically below 1.25, and MW predictability was conserved with I* near 100%. The most concentrated solution tested, a 1:1 ratio of MMA:DMA, exhibited the fastest reaction rate; however, remarkably increased viscosity upon reaching conversions above 70% posed issues with the continuous flow system. For fast reaction times and lower viscosities, a ratio of 2:3 MMA:DMA by volume was chosen for subsequent polymerizations.
As photoflow reactors facilitate efficient and homogeneous irradiation of the reaction solution due to increased surface-to-volume area of the reactor,32 we hypothesized sufficient concentrations of excited state 3PC* for successful polymerizations could be achieved using decreased PC loadings. To test this hypothesis, O-ATRP of MMA was performed using varying concentrations of 4. For mol % levels spanning 0.1%–0.01%, all loadings were well-controlled above 40% conversion (Table 2, runs 1-4). However, for 0.04–0.01% (runs 2–4), early stages of polymerization (<40% conversion) showed slightly higher MW polymers in comparison to theoretical MWs (Figure S4). This is likely a consequence of an insufficient concentration of critical deactivating 2PC*+X− species at low monomer conversions. Lowering the PC loading to 0.005% (Table 2, run 5) resulted in a loss of control over MW growth. A control experiment with no PC in the system resulted in low monomer conversions and Đ ≥ 1.85 (Table S4). To continue a comparison of general trends in batch reaction systems and continuous flow, PC loadings of 0.1% were employed in further studies.
Table 2. Results of the O-ATRP of MMA Investigating PC 4 Loadinga.
| run. no. | [PC 4] (mol %) | conv (%)b | Mn (kDa)c | Đ (Mw/Mn)c | theor Mn (kDa)d | I* (%)e |
|---|---|---|---|---|---|---|
| 1 | 0.1 | 61 | 6.4 | 1.23 | 6.11 | 95 |
| 2 | 0.04 | 69 | 6.9 | 1.22 | 6.86 | 100 |
| 3 | 0.02 | 68 | 6.2 | 1.26 | 6.82 | 110 |
| 4 | 0.01 | 71 | 7.9 | 1.27 | 7.09 | 90 |
| 5 | 0.005 | 72 | 6.7 | 1.60 | 7.25 | 108 |
[MMA]:[DBMM]:[4] = [1000:[10]:[X]; 2:3 of MMA to DMA by volume and 160 cm tubing reactor irradiated by 3000 K fluorescent lamp. Results shown were achieved with a 90 min residence time at a flow rate of 8.11 μL/min.
Determined by 1H NMR.
Measured using GPC.
Calculated by (Conv × [Mon]/[Init] × Mw,Mon)/1000.
Initiator efficiency (I*) calculated by (Theor Mn/Calcd Mn) × 100.
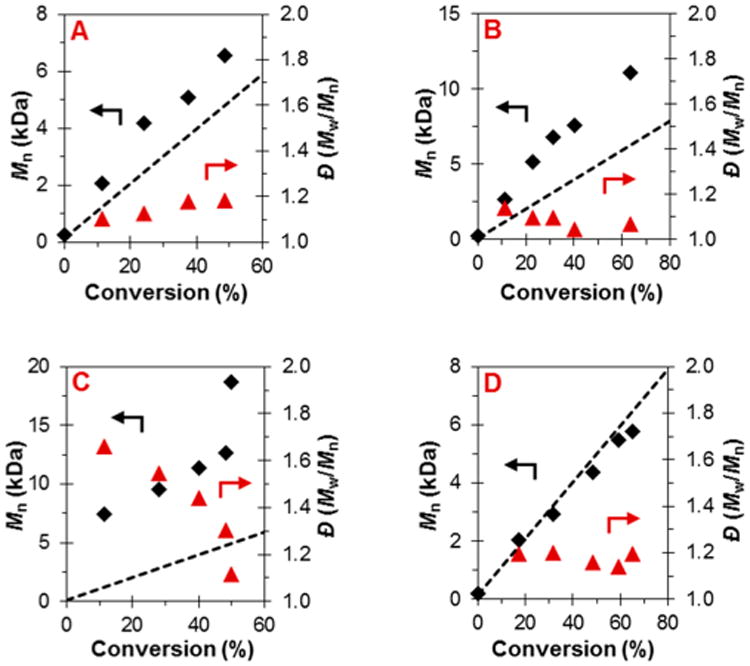
To validate that this system can be used to synthesize polymers with targeted MWs with a high level of control, the O-ATRP of MMA was performed with the goal of synthesizing PMMA with a range of MWs (Figure 3). These data showed well-controlled polymerizations through conversions above 80%, first-order kinetic behavior, nearly constant and relatively low Đ, and a linear increase in MW paired with quantitative I*.
Figure 3.
Results of the polymerization of MMA catalyzed by 4 performed in continuous flow at steady-state conditions and under different residence times: (A) first-order kinetic plot; (B) plot of the molecular weight (◆) in comparison to theoretical molecular weight (dashed line) with respect to monomer conversion and dispersity (▲); (C) GPC traces of the polymers corresponding to points shown in (A) and (B), color coded. Conditions used are [MMA]:[DBMM]:[4] = [1000]:[10]:[1]; 3.73 mM of PC, 2:3 MMA:DMA by volume, irradiated by 3000 K fluorescent lamp. See Supporting Information for flow rates and residence time used.
To synthesize PMMA with tunable MWs in continuous flow, concentrations of either monomer or initiator were adjusted while maintaining a constant [4] (Table 3). Through altering the stoichiometry, predictable MWs and relatively low Đ were obtained for low-MW polymers. However, when targeting high monomer-to-initiator loadings, Đ increased above 1.5 and I* exceeded 100%, showing a loss of control over the polymerization. This loss of control in continuous flow at high MW may be attributed to increased solution viscosities provided by high-MW polymers, which can cause increased shear rates within the reactor and poor diffusional mixing of the solution.47 As such, even at uniform flow rates polymer chains become elongated and move through the reactor at varying rates, leading to broadened residence time distributions that likely contribute to a higher Đ.35
Table 3. Results of O-ATRP of MMA in Continuous Flow for the Synthesis of PMMA with Tunable Molecular Weightsa.
| run no. | [MMA]:[DBMM]:[4] | Conv. (%) | Mw (kDa) | Mn (kDa) | Đ (Mw/Mn) | Theo. Mn (kDa) | I* (%) |
|---|---|---|---|---|---|---|---|
| 1 | [1000]:[20]:[1] | 73 | 6.3 | 5.4 | 1.17 | 3.6 | 68 |
| 2 | [1000]:[15]:[1] | 48 | 3.7 | 3.0 | 1.24 | 3.2 | 108 |
| 3 | [1000]:[10]:[1] | 69 | 9.0 | 7.2 | 1.25 | 6.9 | 95 |
| 4 | [1000]:[5]:[1] | 76 | 25.2 | 19.3 | 1.30 | 15.3 | 79 |
| 5 | [1000]:[2]:[1] | 61 | 43.0 | 24.4 | 1.77 | 30.4 | 125 |
| 6 | [250]:[10]:[1] | 50 | 2.2 | 1.8 | 1.18 | 1.3 | 69 |
| 7 | [500]:[10]:[1] | 61 | 4.3 | 3.8 | 1.13 | 3.1 | 81 |
| 8 | [1500]:[10]:[1] | 85 | 18.9 | 13.6 | 1.39 | 12.7 | 94 |
| 9 | [2000]:[10]:[1] | 76 | 21.3 | 15.4 | 1.38 | 15.2 | 98 |
| 10 | [3000]:[10]:[1] | 79 | 29.9 | 20.5 | 1.46 | 23.6 | 115 |
| 11 | [4000]:[10]:[1] | 74 | 35.3 | 22.4 | 1.58 | 29.5 | 131 |
| 12 | [5000]:[10]:[1] | 65 | 37.0 | 22.2 | 1.67 | 32.6 | 147 |
[MMA]:[DBMM]:[4] = [X]:[Y]:[1]; 3.73 mM of 4, with 2:3 of MMA to DMA by volume and 160 cm tubing reactor irradiated by 3000 K fluorescent tubular lamp. Results were achieved using a 120 min residence time at a flow rate of 5.41 μL/min.
Application of O-ATRP for the Polymerization of Other Monomers
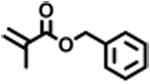
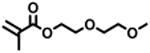
The expansion of this polymerization to a diverse group of methacrylate monomers using O-ATRP in continuous flow was investigated (Table 4). Polymerizations of benzyl methacrylate and ethyl methacrylate produced polymers with Đ's of 1.38 and 1.26, respectively, and linear increases in MW with monomer conversion (Table 4, runs 1 and 2). Synthesis of poly(2-ethylhexyl methacrylate) resulted in polymer with 98% I* and Đ of 1.40 (Table 4, run 3). The polymerizations of lauryl methacrylate and isodecyl methacrylate were well-controlled until reaching 60% and 66% conversion, respectively, although Đ's increased at higher monomer conversions (Table 3, runs 4 and 5). Applying this polymerization approach to diethylene glycol methyl ether methacrylate showed control over MW throughout polymerization with a polymer with Đ of 1.14 at 66% conversion (Table 4, run 7). Highlighting the robustness of O-ATRP in continuous flow, no additional optimization was necessary in order to accomplish a well-controlled polymerization of methacrylate monomers possessing varying functionalities.
Table 4. Monomer Scope of O-ATRP of Methacrylate Monomers Using Continuous Flow under Standard Conditionsa.
| run no. | Monomer | Res. Time (min) | Flow Rate (μL/min) | Conv. (%) | Mw (kDa) | Mn (kDa) | Đ (Mw/Mn) | Theo. Mn (kDa) | I* (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 |
|
90 | 8.11 | 82 | 20.7 | 15.0 | 1.38 | 14.4 | 96 |
| 2 |
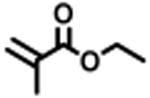
|
90 | 8.11 | 63 | 9.4 | 7.6 | 1.24 | 7.1 | 94 |
| 3 |
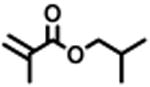
|
90 | 8.11 | 67 | 12.2 | 9.9 | 1.23 | 9.5 | 96 |
| 4 |
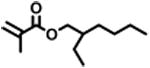
|
90 | 8.11 | 79 | 22.4 | 16.0 | 1.40 | 15.6 | 98 |
| 5 |
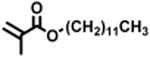
|
60 | 12.16 | 60 | 27.6 | 18.5 | 1.49 | 15.4 | 83 |
| 6 |
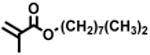
|
60 | 12.16 | 66 | 31.9 | 26.5 | 1.22 | 14.9 | 56 |
| 7 |
|
60 | 12.16 | 66 | 19.1 | 16.7 | 1.14 | 12.4 | 74 |
[Monomer]:[DBMM]:[PC 4] = [1000]:[10]:[1] 3.73 mM of 4, with 2:3 of MMA to DMA by volume and 160 cm tubing reactor irradiated by 3000 K fluorescent lamp.
Chain Extension of PMMA Synthesized in Continuous Flow
To further demonstrate the controlled nature of O-ATRP in a continuous flow system, chain-end group fidelity was validated by chain extension from a PMMA macroinitiator synthesized on large scale in continuous flow. The large-scale synthesis of the PMMA macroinitiator was carried out to obtain 3.3 g of purified PMMA macroinitiator in a 73% yield (see Supporting Information: Macroinitiator Synthesis and Chain-Extension Experiments). Low PC loadings were implemented for all steps (0.01 mol % of 4), and chain extensions were performed with isobutyl methacrylate or benzyl methacrylate. Macroinitiator chain-end group fidelity was confirmed through successful chain extension of macroinitiator to block copolymer, as shown by baseline-resolved shifts in GPC traces from the PMMA macroinitiator to higher molecular weight block copolymers (Figure 4B).
Figure 4.
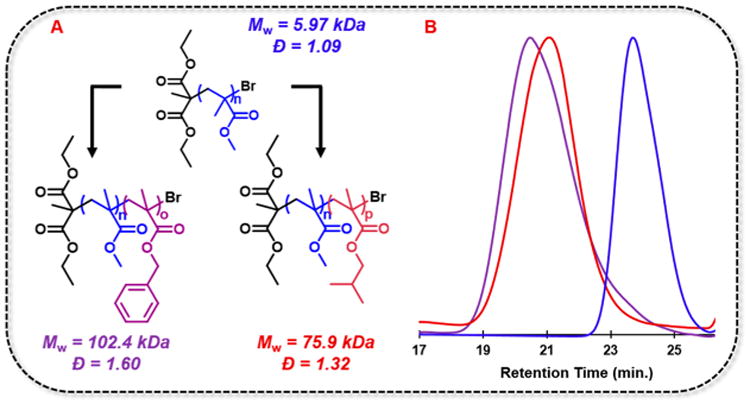
(A) Chain extension using O-ATRP from a PMMA macroinitiator extended with BzMA (purple) and iBuMA (red). (B) Gel permeation chromatography traces of the polymers are shown with corresponding color schemes.
Conclusions
Photoinduced O-ATRP performed in continuous flow has been established as a robust and efficient method of polymerization. For each photoredox catalyst tested, results show good control over all metrics of polymerization to produce polymers with relatively low Đ and high MW predictability. Control over the course of the polymerization was maintained when catalyst loadings were lowered to 0.01 mol %. Further, this technique has been successfully applied to the polymerization of a diverse scope of methacrylate monomers with no further optimization required and provides the capability for the scalable synthesis of well-defined block copolymers. The success of O-ATRP in this system is enhanced by the efficient irradiation characteristics offered by continuous flow.
Supplementary Material
Materials and methods, detailed polymerization procedures, optimization experiments, and supplemental data (PDF)
Acknowledgments
This work was supported by the University of Colorado Boulder and the Advanced Research Projects Agency-Energy (DE-AR0000683). Acknowledgement is made to the donors of The American Chemical Society Petroleum Research Fund for partial support of this research (56501-DNI7). Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award R35GM119702. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. R.M.P. is grateful for an award from the IBM Students for a Smarter Planet Challenge. The authors thank Antonio Garcia and Steven Sartor for technical assistance and Tracy French for photography.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macro-mol.6b02791.
Notes: The authors declare no competing financial interest.
References
- 1.Pan X, Tasdelen MA, Laun J, Junkers T, Yagci Y, Matyjaszewski K. Photomediated Controlled Radical Polymerization. Prog Polym Sci. 2016;62:73–125. [Google Scholar]
- 2.Corrigan N, Shanmugam S, Xu J, Boyer C. Photocatalysis in Organic and Polymer Synthesis. Chem Soc Rev. 2016;45:6165–6212. doi: 10.1039/c6cs00185h. [DOI] [PubMed] [Google Scholar]
- 3.Chatani S, Kloxin CJ, Bowman CN. The power of light in polymer science: photochemical processes to manipulate polymer formation, structure, and properties. Polym Chem. 2014;5:2187–2201. [Google Scholar]
- 4.Yagci Y, Jockusch S, Turro NJ. Photoinitiated Polymerization: Advances, Challenges, and Opportunities. Macromolecules. 2010;43:6245–6260. [Google Scholar]
- 5.Zhou yN, Guo JK, Li JJ, Luo ZH. Photoinduced Iron(III)-Mediated Atom Transfer Radical Polymerization with In Situ Generated Initiator: Mechanism and Kinetics Studies. Ind Eng Chem Res. 2016;55:10235–10242. [Google Scholar]
- 6.Patten TE, Xia J, Abernathy T, Matyjaszewski K. Polymers with Very Low Polydispersities from Atom Transfer Radical Polymerization. Science. 1996;272:866–868. doi: 10.1126/science.272.5263.866. [DOI] [PubMed] [Google Scholar]
- 7.Matyjaszewski K, Xia J. Atom transfer radical polymerization. Chem Rev. 2001;101:2921–2990. doi: 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- 8.Matyjaszewski K, Tsarevsky NV. Macromolecular engineering by atom transfer radical polymerization. J Am Chem Soc. 2014;136:6513–6533. doi: 10.1021/ja408069v. [DOI] [PubMed] [Google Scholar]
- 9.Matyjaszewski K. Controlled radical polymerization: State-of-the-Art in 2014. ACS Symp Ser. 2015;1187:1–17. [Google Scholar]
- 10.Konkolewicz D, Schröder K, Buback J, Bernhard S, Matyjaszewski K. Visible Light and Sunlight Photoinduced ATRP with ppm of Cu Catalyst. ACS Macro Lett. 2012;1:1219–1223. doi: 10.1021/mz300457e. [DOI] [PubMed] [Google Scholar]
- 11.Mosnaćek J, Ilčíkoviá M. Photochemically mediated Atom Transfer Radical Polymerization of Methyl Methacrylate Using ppm Amounts of Catalyst. Macromolecules. 2012;45:5859–5865. [Google Scholar]
- 12.Jones GR, Whitfield R, Anastasaki A, Haddleton DM. Aqueous Copper(II) Photoinduced Polymerization of Acrylates: Low Copper Concentration and the Importance of Sodium Halide Salts. J Am Chem Soc. 2016;138:7346–7352. doi: 10.1021/jacs.6b02701. [DOI] [PubMed] [Google Scholar]
- 13.Zhou YN, Luo ZH. An Old Kinetic Method for a new Polymerization Mechanism: Toward Photochemically Mediated ATRP. AIChE J. 2015;61:1947–1958. [Google Scholar]
- 14.Faucher S, Okrutny P, Zhu S. Facile and Effective Purification of Polymers Produced by Atom Transfer Radical Polymerization via Simple Catalyst Precipitation and Microfiltration. Macromolecules. 2006;39:3–5. [Google Scholar]
- 15.Zhang H, Abeln CH, Fijten MWM, Schubert US. High-throughput Experimentation Applied to Atom Transfer Radical Polymerization: Automated Optimization of the Copper Catalysts Removal from Polymers. e-Polym. 2006;6:90–98. [Google Scholar]
- 16.Matyjaszewski K, Pintauer T, Gaynor S. Removal of Copper-Based Catalyst in Atom Transfer Radical Polymerization Using Ion Exchange Resins. Macromolecules. 2000;33:1476–1478. [Google Scholar]
- 17.Miyake GM, Theriot JC. Perylene as an Organic Photocatalyst for the Radical Polymerization of Functionalized Vinyl Monomers through Oxidative Quenching with Alkyl Bromides and Visible Light. Macromolecules. 2014;47:8255–8261. [Google Scholar]
- 18.Treat NJ, Sprafke H, Kramer JW, Clark PG, Barton BE, Read de Alaniz J, Fors BP, Hawker CJ. Metal-Free Atom Transfer Radical Polymerization. J Am Chem Soc. 2014;136:16096–16101. doi: 10.1021/ja510389m. [DOI] [PubMed] [Google Scholar]
- 19.Theriot JC, Lim CH, Yang H, Ryan MD, Musgrave CB, Miyake GM. Organocatalyzed Atom Transfer Radical Polymerization Driven by Visible Light. Science. 2016;352:1082. doi: 10.1126/science.aaf3935. [DOI] [PubMed] [Google Scholar]
- 20.Pearson RM, Lim CH, McCarthy BG, Musgrave CB, Miyake GM. Organocatalyzed Atom Transfer Radical Polymerization Using N-Aryl Phenoxazines as Photoredox Catalysts. J Am Chem Soc. 2016;138:11399–11407. doi: 10.1021/jacs.6b08068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allushi A, Jockusch S, Yilmaz G, Yagci Y. Photoinitiated Metal-Free Controlled/Living Radical Polymerization Using Poly-nuclear Aromatic Hydrocarbons. Macromolecules. 2016;49:7785–7792. [Google Scholar]
- 22.Huang Z, Gu Y, Liu X, Zhang L, Cheng Z, Zhu X. Metal-Free Atom Transfer Radical Polymerization of Methyl Methacrylate with ppm Level of Organic Photocatalyst. Macromol Rapid Commun. 2016 doi: 10.1002/marc.201600461. [DOI] [PubMed] [Google Scholar]
- 23.Pan X, Fang C, Fantin M, Malhotra N, So WY, Peteanu LA, Isse AA, Gennaro A, Liu P, Matyjaszewski K. Mechanism of Photoinduced Metal-Free Atom Transfer Radical Polymerization: Experimental and Computational Studies. J Am Chem Soc. 2016;138:2411–2425. doi: 10.1021/jacs.5b13455. [DOI] [PubMed] [Google Scholar]
- 24.Lim CH, Ryan MD, McCarthy BG, Theriot JC, Sartor SM, Damrauer NH, Musgrave CB, Miyake GM. Intramolecular Charge Transfer and Ion Pairing in N,N-Diaryl Dihydrophenazine Photoredox Catalyst for Efficient Organocatalyzed Atom Transfer Radical Polymerization. J Am Chem Soc. 2016;139:348–355. doi: 10.1021/jacs.6b11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tucker JW, Zhang Y, Jamison TF, Stephenson CRJ. Visible-Light Photoredox Catalysis in Flow. Angew Chem Int Ed. 2012;51:4144–4147. doi: 10.1002/anie.201200961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hook BDA, Dohle W, Hirst PR, Pickworth M, Berry MB, Booker-Milburn KI. A Practical Flow Reactor for Continuous Organic Photochemistry. J Org Chem. 2005;70:7558–7564. doi: 10.1021/jo050705p. [DOI] [PubMed] [Google Scholar]
- 27.Beatty JW, Douglas JJ, Miller R, McAtee RC, Cole KP, Stephenson CRJ. Photochemical Perfluoroalkylation with Pyridine N-Oxides: Mechanistic Insights and Performance on a Kilogram Scale. Chem. 2016;1:456–472. doi: 10.1016/j.chempr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garlets ZJ, Nguyen JD, Stephenson CRJ. The Development of Visible Light Photoredox Catalysis in Flow. Isr J Chem. 2014;54:351. doi: 10.1002/ijch.201300136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steinbacher JL, McQuade DT. Polymer Chemistry in Flow: New Polymers, Beads, Capsules, and Fibers. J Polym Sci Part A Polym Chem. 2006;44:6505–6533. [Google Scholar]
- 30.Natalello A, Morsbach J, Friedel A, Alkan A, Tonhauser C, Müller AHE, Frey H. Living Anionic Polymerization in Continuous Flow: Facilitated Synthesis of High-Molecular Weight Poly(2-vinylpyridine) and Polystyrene. Org Process Res Dev. 2014;18:1408–1412. [Google Scholar]
- 31.Diehl C, Laurino P, Azzouz N, Seeberger PH. Accelerated Continuous Flow RAFT Polymerization. Macromolecules. 2010;43:10311–10314. [Google Scholar]
- 32.Bou-Hamdan FR, Seeberger PH. Visible-light-mediated photochemistry: accelerating Ru(bpy)32+-catalyzed reactions in continuous flow. Chem Sci. 2012;3:1612–1616. [Google Scholar]
- 33.Lu H, Schmidt MA, Jensen KF. Photochemical Reactions and On-line UV detection in Microfabricated Reactors. Lab Chip. 2001;1:22–28. doi: 10.1039/b104037p. [DOI] [PubMed] [Google Scholar]
- 34.Elliott LD, Knowles JP, Koovits PJ, Maskill LG, Ralph MJ, Lejeune G, Edwards LJ, Robinson RI, Clemens IR, Cox B, Pascoe DD, Koch G, Eberle M, Berry MB, Booker-Milburn KI. Batch versus Flow Photochemistry: A Revealing Comparison of Yield and Productivity. Chem Eur J. 2014;20:15226–15232. doi: 10.1002/chem.201404347. [DOI] [PubMed] [Google Scholar]
- 35.Wu T, Mei Y, Cabral JT, Xu C, Beers KL. A New Synthetic Method for Controlled Polymerization Using a Microfluidic System. J Am Chem Soc. 2004;126:9880–9881. doi: 10.1021/ja048432n. [DOI] [PubMed] [Google Scholar]
- 36.Fukuyama T, Kajihara Y, Ryu I, Studer A. Nitroxide- Mediated Polymerization of Styrene, Butyl Acrylate, or Methyl Methacrylate by Microflow Reactor Technology. Synthesis. 2012;44:2555–2559. [Google Scholar]
- 37.Enright TE, Cunningham MF, Keoshkerian B. Nitroxide-Mediated Polymerization of Styrene in a Continuous Tubular Reactor. Macromol Rapid Commun. 2005;26:221–225. [Google Scholar]
- 38.Hornung CH, Guerrero-Sanchez C, Brasholz M, Saubern S, Chiefari J, Moad G, Rizzardo E, Thang SH. Controlled RAFT Polymerization in a Continuous Flow Microreactor. Org Process Res Dev. 2011;15:593–601. [Google Scholar]
- 39.Muller M, Cunningham MF, Hutchinson RA. Continuous Atom Transfer Radical Polymerization in a Tubular Reactor. Macromol React Eng. 2008;2:31–36. [Google Scholar]
- 40.Noda T, Grice AJ, Levere ME, Haddleton DM. Continuous process for ATRP: Synthesis of homo and block copolymers. Eur Polym J. 2007;43:2321–2330. [Google Scholar]
- 41.Chen M, Johnson JA. Improving photo-controlled living radical polymerization from trithiocarbonates through the use of continuous-flow techniques. Chem Commun. 2015;51:6742–6745. doi: 10.1039/c5cc01562f. [DOI] [PubMed] [Google Scholar]
- 42.Gardiner J, Hornung CH, Tsanaktsidis J, Guthrie D. Continuous flow photo-initiated RAFT polymerization using a tubular photochemical reactor. Eur Polym J. 2016;80:200–207. [Google Scholar]
- 43.Corrigan N, Rosli D, Jones JWJ, Xu J, Boyer C. Oxygen Tolerance in Living Radical Polymerization: Investigation of Mechanism and Implementation in Continuous Flow Polymerization. Macromolecules. 2016;49:6779–6789. [Google Scholar]
- 44.Melker A, Fors BP, Hawker CJ, Poelma JE. Continuous Flow Synthesis of Poly(methyl methacrylate) via a Light-Mediated Controlled Radical Polymerization. J Polym Sci Part A Polym Chem. 2015;53:2693–2698. [Google Scholar]
- 45.Wenn B, Conradi M, Carreiras AD, Haddleton DM, Junkers T. Photo-induced copper-mediated polymerization of methyl acrylate in continuous flow reactors. Polym Chem. 2014;5:3053–3060. [Google Scholar]
- 46.Initiator efficiency (I*) = theoretical number average molecular weight (Mn)/experimentally measured Mn × 100.
- 47.Parida D, Serra CA, Gómez RI, Garg DK, Hoarau Y, Bouquey M, Muller R. Atom Transfer Radical Polymerization in Continuous Microflow: Effect of Process Parameters. J Flow Chem. 2014;4:92–96. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and methods, detailed polymerization procedures, optimization experiments, and supplemental data (PDF)