

Abstract
T cells undergo homeostatic expansion and acquire an activated phenotype in lymphopenic microenvironments. Restoration of normal lymphocyte numbers typically re-establishes normal homeostasis, and proinflammatory cytokine production returns to baseline. Mice deficient in guanine nucleotide exchange factor RasGRP1 exhibit dysregulated homeostatic expansion, which manifests as lymphoproliferative disease with autoantibody production. Our previous work revealed that autoreactive B cells lacking RasGRP1 break tolerance early during development, as well as during germinal center responses, suggesting that T cell–independent and T cell–dependent mechanisms are responsible. Examination of whether a particular T cell subset is involved in the breach of B cell tolerance revealed increased Th17 cells in Rasgrp1-deficient mice relative to control mice. Rasgrp1-deficient mice lacking IL-17R had fewer germinal centers, and germinal centers that formed contained fewer autoreactive B cells, suggesting that IL-17 signaling is required for a break in B cell tolerance in germinal centers. Interestingly, a fraction of Th17 cells from Rasgrp1-deficient mice were CXCR5+ and upregulated levels of CD278 coordinate with their appearance in germinal centers, all attributes of T follicular helper cells (Tfh17). To determine whether CD278–CD275 interactions were required for the development of Tfh17 cells and for autoantibody, Rasgrp1-deficient mice were crossed with CD275-deficient mice. Surprisingly, mice deficient in RasGRP1 and CD275 formed Tfh17 cells and germinal centers and produced similar titers of autoantibodies as mice deficient in only RasGRP1. Therefore, these studies suggest that requirements for Tfh cell development change in lymphopenia-associated autoimmune settings.
As central regulators of immunity, Th cell subsets dictate immunity to foreign Ags and self-Ags. In autoimmunity, altered frequency and function of Th1, Th17, and T follicular helper (Tfh) subsets are linked to proinflammatory autoimmune conditions, such as multiple sclerosis (1), Crohn’s disease (2), and systemic lupus erythematosus (SLE) (3). Targeted therapy disrupting T cell activation, costimulation and cytokines, or general T cell ablation generally ameliorates autoimmunity, thereby implicating particular Th subsets (4–10), although proof of their self-reactivity is often inferred rather than formally proven.
Tfh cells are defined by their location in germinal centers (GCs) resulting from upregulation of CXCR5 (11, 12), leading to migration toward ligand CXCL13 produced by follicular dendritic cells residing in B cell follicles (13, 14). In addition, Tfh cells exhibit increased levels of Bcl6 that are induced upon ICOS/CD278-ICOSL/CD275 interactions provided by both dendritic cells and B cells (15–17). CD275, CD278, and Bcl6 are each required for Tfh development, GCs, and Ab production (18, 19). Further, in autoimmune mice, such as the MRL-lpr strain, blockade or genetic deficiency in CD278 leads to amelioration of pathology, suggesting that this strategy may prevent development of autoreactive Tfh cells (20, 21).
In both humans and in mouse, deficiency in RasGRP1 predisposes to autoimmunity (22–25). For example, defective splicing of Rasgrp1 transcripts and single nucleotide polymorphisms of Rasgrp1 are associated with development of SLE and type I diabetes in humans, respectively (23, 26). Rasgrp1−/− mice develop a lymphoproliferative disorder that includes the development of spontaneous GCs and anti-nuclear Abs (ANAs) (27). Importantly, Rasgrp1-deficient nude mice do not exhibit splenomegaly and fail to produce ANAs, demonstrating that autoimmune features are T cell dependent (28). RasGRP1 is a guanine exchange factor (GEF) that is responsible, in part, for the activation of the Ras signal-transduction pathway downstream of AgRs in B and T cells (22, 29). In T cells, RasGRP1 is the predominant GEF mediating signals following AgR (TCR) engagement, and thymocytes deficient in Rasgrp1 are less able to survive selection as the result of impaired TCR signaling (30). The resultant block in T cell development at the CD4+CD8+ stage leads to T lymphopenia in the periphery (31, 32). Rasgrp1−/− CD4 cells that make it to the periphery are functionally impaired when strong signals are given via the TCR (33). Despite this functional impairment in vitro, Rasgrp1−/− CD4 T cells are activated in vivo as a consequence of homeostatic expansion (32). Paradoxically, the frequency and activity of regulatory T cells (Tregs) are reportedly increased in Rasgrp1−/− mice (34).
Ras pathway signaling operates upstream of ERK, and recent studies suggest that inhibiting ERK signaling skews Th cell differentiation toward the Th17 subset (35). Th17 cells are implicated in numerous autoimmune models as a pathogenic subset critical for disease. IL-17, the cytokine produced by Th17 cells, mediates class-switch recombination to all IgG subclasses, and Tfh-associated cytokine IL-21 can further enhance class-switch recombination to IgG1 and IgG2b (36). This contrasts with IFN-γ produced by Th1 cells that promotes IgG2a responses and suppresses IgG2b and IgG3 responses (37). Because Rasgrp1−/− mice show elevated serum Ab titers for all IgG subclasses, we speculated that Th17 cells have a role in regulating autoimmune responses.
Both the frequency and number of Th17 cells were increased in Rasgrp1−/− mice; further, these RORγt+ cells upregulated CXCR5 and CD278/ICOS and localized to GCs, consistent with a Tfh cell phenotype. To address whether Tfh17 cells in lymphopenic Rasgrp1-deficient mice were important for autoimmunity and, further, required CD278–CD275 interactions similar to nonlymphopenic mice, we assessed Tfh17 cells in mice deficient in both RasGRP1 and IL-17RA, as well as in mice deficient in both RasGRP1 and CD275 (ICOSL). The frequencies of autoreactive B cells, spontaneous GCs, and autoreactive B cells within spontaneous GCs were significantly reduced in Rasgrp1-deficient mice also lacking IL-17RA compared with autoimmune-prone mice lacking only RasGRP1. However, although CD275–CD278 interactions are typically important for GCs and Tfh cells, Rasgrp1-deficient mice lacking CD275 developed frequent GCs, Tfh17 cells, and autoantibody. We conclude that lymphopenia-driven autoimmunity and the development of Tfh17 cells in Rasgrp1-deficient mice occur independently of CD275.
Materials and Methods
Mice
Mice were housed at the University of South Alabama in an AALAC International–accredited specific pathogen–free facility. Rasgrp1-deficient (Rasgrp1−/−) (38) and littermate control Rasgrp1-sufficient heterozygous (Rasgrp1+/−) mice were maintained on a C57BL/6 background (>10 generations backcrossed). Importantly, no phenotypic or functional differences were measured between mice with one or two functional Rasgrp1 alleles. Mice lacking IL-17RA (C57BL/6 background) were crossed with Rasgrp1 strains harboring the BCR knock-in transgene 564Igi (39). The 564Igi BCR recognizes multiple self-Ags (40, 41), and B cells expressing this transgene can be readily identified using anti-idiotypic Ab (39). CD275/B7-H2/ICOSL–deficient mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Maintenance of breeding colonies and all procedures involving mice were performed according to protocols approved by the University of South Alabama Institutional Animal Care and Use Committee.
Flow cytometric analysis and Abs
Single-cell suspensions of splenic mononuclear cells (MNCs) were isolated by density gradient centrifugation using Lympholyte M (Cedarlane Laboratories, Burlington, NC). For intracellular cytokine staining of T cells, total splenocytes were incubated with PMA and ionomycin for 2 h at 37°C with 5% CO2, and GolgiStop and GolgiPlug (BD Biosciences, San Jose, CA) were added for an additional 3 h. Following staining with surface markers, splenocytes were permeabilized and fixed using the Foxp3 staining protocol (eBioscience, San Diego, CA). Intracellular staining for cytokines was subsequently performed. Abs used for the analysis of T cells included CD3ε (145-2C11), CD4 (GK1.5), CD8α (53-6.7), CD25 (PC61), CD44 (IM7), CD62L (MEL-14), CD69 (H1.2F3), CXCR5/CD185 (SPRCL5), CCR7/CD197 (4B12), ICOS/CD278 (7E.17G9), PD-1/CD279 (J43), Bcl6 (IG191E/A8, K112-91), IFN-γ (XMG1.2), IL-2 (JES6-5H4), IL-4 (11B11), IL-17A (eBio17B7), IL-21 (FFA21 or BL25168), and Foxp3 (FJK-16s). Combinations of these Abs conjugated to fluorophores FITC, PE, PE-Cy7, PE–Texas Red, PerCP-Cy5.5, allophycocyanin/eFluor 660, allophycocyanin-Cy7, and Pacific Blue/V450 were used (BD Biosciences, eBioscience, and BioLegend, San Diego, CA). Anti-idiotype Ab (B6.256) was used to identify 564Igi autoreactive B cells. Cells were analyzed by a FACSCanto II and sorted using a multilaser FACSAria II SORP housed in the University of South Alabama College of Medicine Flow Cytometry Laboratory. Data were analyzed with FlowJo software (TreeStar, Ashland, OR).
Immunofluorescent analysis of splenic sections
Five-micron cryosections of OCT-preserved (Tissue-Tek, Torrance, CA) spleens were prepared by placing trays onto a block of dry ice. Frozen tissues were stored at −80°C; 5-μm sections were placed onto Superfrost/Plus microscope slides (Fisher Scientific, Pittsburgh, PA) using a Shandon FE/FSE Cryotome (Thermo Scientific, Waltham, MA). After rehydration with PBS, sections were incubated with anti-CD16/CD32 (2.4G2; Bio X Cell, West Lebanon, NH) before immunostaining to resolve T cells (using anti-CD4 Ab), B cells (anti-CD45R), and GCs (PNA-FITC) or to resolve Th17 cell (PE-conjugated anti-mouse IL-17A) localization counterstained with FITC-conjugated anti-mouse CD4, PE-Cy7–conjugated anti-mouse CD45R, and allophycocyanin-conjugated GL7. Images were acquired using a Nikon A1R confocal microscope (University of South Alabama Microscope Core Facility) and analyzed with Nikon Elements Software (Nikon Instruments, Melville, NY).
T cell isolation and stimulation assays
Splenic CD4 T cells were isolated using a MACS CD4 T Cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) and cultured using RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamine, 100 IU penicillin, and 0.1 mg/ml streptomycin (Invitrogen, Grand Island, NY) and 2 mM 2-ME. Cells were activated using plate-bound anti-CD3 (2C11, 5 μg/ml) in the presence or absence of mitomycin C–treated T cell–depleted splenocytes. After 48–72 h of culture, cells and supernatants were collected and analyzed.
ELISAs
To measure cytokines, culture supernatants were analyzed for IFN-γ and IL-17 by sandwich ELISA using anti-cytokine Abs (R&D Systems, Minneapolis, MN). Biotinylated anti-cytokine Ab and streptavidin HRP were used for cytokine detection. HRP was visualized using 2, 2′-azino-bis-(3-benzthiazoline-6-sulfonic acid), and absorbance signals two times above background (C57BL/6 sera) were used as a threshold. Standard curves were generated using recombinant cytokines, and linear regression was applied to quantitate levels of IFN-γ and IL-17 produced by activated T cells.
For measurement of 564Ig-derived autoantibody titers, ELISA plates (Thermo Scientific) were coated with Abs to IgM or IgG2b/2c/3 (Southern Biotechnology Associates, Birmingham, AL). After blocking plates with 5% dry milk (Carnation) in PBS, serial dilutions of serum were incubated, and plates were washed in PBS containing 0.05% Tween 20. Sandwich was completed by addition of biotinylated anti-idiotypic Ab (clone B6-256), followed by streptavidin-HRP. Color detection was performed by addition of ABTS, and absorbance was measured. Absorbance signals two times above background (C57BL/6 sera) were used to determine 564Ig Ab titers.
ANA assays
ANA assays were performed, according to the manufacturer’s protocol, using mitotic HEp-2 cells stabilized on microscope slides (Antibodies, Davis, CA).
Statistical analysis
Statistical comparisons reported were generated using GraphPad Prism 6 software. One-way ANOVA was used to compare more than two means, with Tukey’s multiple comparisons, as indicated in the figure legends. The unpaired Student t test was used to compare two means, and Welch’s correction was applied when variances were unequal, as determined by the F test.
Results
IL-17–producing CD4+ cells in Rasgrp1-deficient mice
Previous work demonstrated that Rasgrp1−/− mice produce increased autoantibodies of all IgG isotypes, as well as autoreactive B cells, within GCs (27). To examine T cell subsets that may contribute to autoantibody production and GCs, splenic CD4 T cells were isolated and stimulated in vitro with anti-CD3 alone and in the presence of APCs, and supernatants were analyzed for IFN-γ and IL-17A by ELISA (Fig. 1). Consistent with previous studies, CD4 T cells from Rasgrp1−/− mice and age-matched littermate control mice produced similar amounts of IFN-γ (Fig. 1A, 4.5 ± 2.2 ng/ml and 2.7 ± 1.0 ng/ml, respectively). In contrast to IFN-γ production, anti-CD3–stimulated CD4 T cells from Rasgrp1−/− mice produced >10-fold more IL-17A than did Rasgrp1-sufficient CD4 T cells (Fig. 1B, 8.0 ± 3.1 versus 0.5 ± 0.6 in the presence of APCs, p ≤ 0.02). These data suggest that Th17 cells are present in Rasgrp1−/− mice and that they can be activated via TCR engagement.
FIGURE 1.
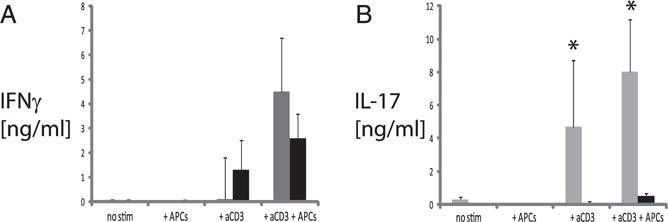
IL-17 production from stimulated CD4+ T cells from Rasgrp1-deficient mice. Isolated CD4+ T cells were stimulated for 48–72 h with anti-CD3 in the presence or absence of autologous mitomycin C–treated APCs. Culture supernatants containing control (black bars) and Rasgrp1−/− (gray bars) CD4 T cells from 10-wk-old mice were collected and analyzed by ELISA for IFN-γ (A) and IL-17 (B). Cultures containing unstimulated CD4 T cells and mitomycin C–treated APCs alone were used as negative controls. Data are mean ± SE and summarize the results from four independent experiments. *p ≤ 0.02, t test.
To determine the relative frequency and cell numbers of Th cell subsets, splenocytes from Rasgrp1−/− and littermate control mice were stained for intracellular cytokines and analyzed by flow cytometry (Fig. 2). Consistent with previous work, the frequency and number of Th1 cells (CD4+ IFNγ+ IL-17A−) was increased in Rasgrp1−/− mice relative to age-matched littermate control mice. The frequency of Th1 cells increased with age in spleens of Rasgrp1−/− and control mice, although the frequency and the number of Th1 cells remained significantly elevated in Rasgrp1−/− mice. Similarly, the frequency and number of Th17 cells (CD4+ IFNγ− IL-17A+) were elevated in Rasgrp1−/− mice compared with age-matched littermate control mice. Importantly, although the frequency of Th17 cells in Rasgrp1-deficient mice varied with age, the number of Th17 cells increased with age (Fig. 2). Interestingly, a minor population of T cells producing both IFN-γ and IL-17A was also elevated in Rasgrp1−/− mice relative to littermate controls. No IL-4+ staining was observed within splenic CD4+ T cells from Rasgrp1−/− or control mice. In sum, these analyses reveal that Rasgrp1−/− mice develop Th17 cells, and this subset exhibits marked increases in the frequency and number compared with nonautoimmune littermate control mice.
FIGURE 2.
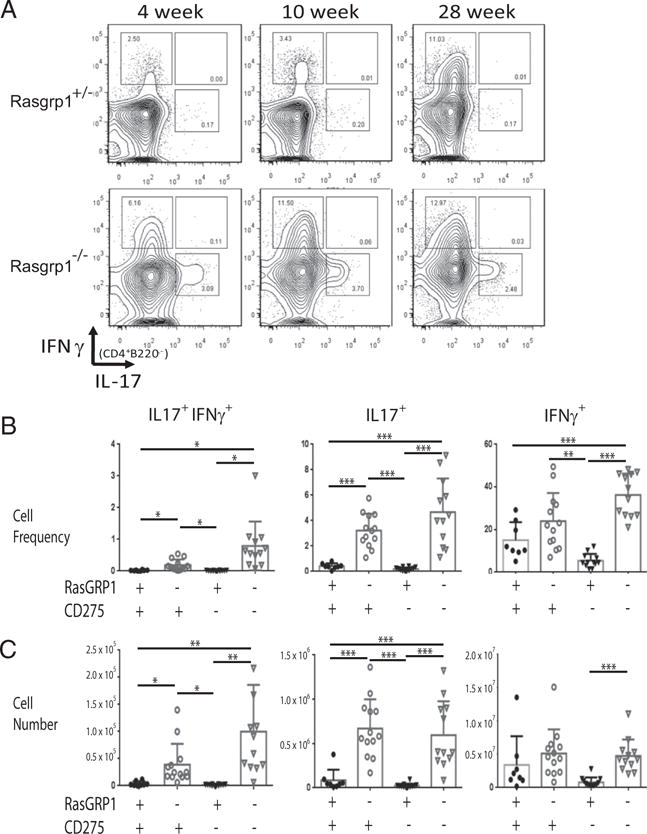
Frequency and number of IFN-γ– and IL-17–producing CD4 T cells. (A) Splenic MNCs were gated on CD4 T cells, and the frequency of IFN-γ and IL-17 production was determined by intracellular staining. Shown are FACS plots for 4-week-old (left panels), 10-week-old (middle panels), and 28-wk-old (right panels) Rasgrp1+/− littermate control (upper panels) and Rasgrp1−/− (lower panels) mice. Data are a representative from one of five independent experiments. Summary of the frequencies (B) and numbers (C) of IFN-γ+ IL-17+ (left panels), Th17 (middle panels), and Th1 (right panels) CD4 T cell subsets from individual 10- to 12-wk-old Rasgpr1+/− (●), Rasgrp1−/− (○), ICOSL/CD275−/− (▼), and DKO (∇) mice. Each symbol represents an individual mouse (one-way ANOVA with Tukey’s multiple-comparison test, *p ≤ 0.01, **p ≤ 0.001, ***p ≤ 0.0001).
Breach of GC B cell tolerance is IL-17R dependent
Autoreactive B cells break tolerance at multiple checkpoints in the absence of RasGRP1 (27). To measure the contribution of IL-17A production to the break in B cell tolerance within GCs, Rasgrp1-deficient mice containing the knock-in BCR transgene 564Igi were crossed with mice lacking IL-17RA. As previously reported (27), the frequency and number of autoreactive B cells were increased Rasgrp1-deficient mice compared with Rasgrp1-sufficient mice (Fig. 3A–C). Although the frequency and number of 564Igi B cells did not differ in spleens of Rasgrp1-sufficient mice with and without IL-17RA, the frequency and number of 564Igi B cells in autoimmune-prone Rasgrp1-deficient mice was significantly increased compared with autoimmune-prone mice lacking IL-17RA. Thus, for example, although Rasgrp1-deficient mice had a mean frequency of 38.0% and 4.7 × 107 564Igi splenic B cells, mice lacking RasGRP1 and IL-17RA had means of 22.8% and 1.1 × 106 564Igi B cells. Therefore, the increase in 564Igi autoreactive B cells in Rasgrp1-deficient mice is partially dependent on IL-17RA.
FIGURE 3.
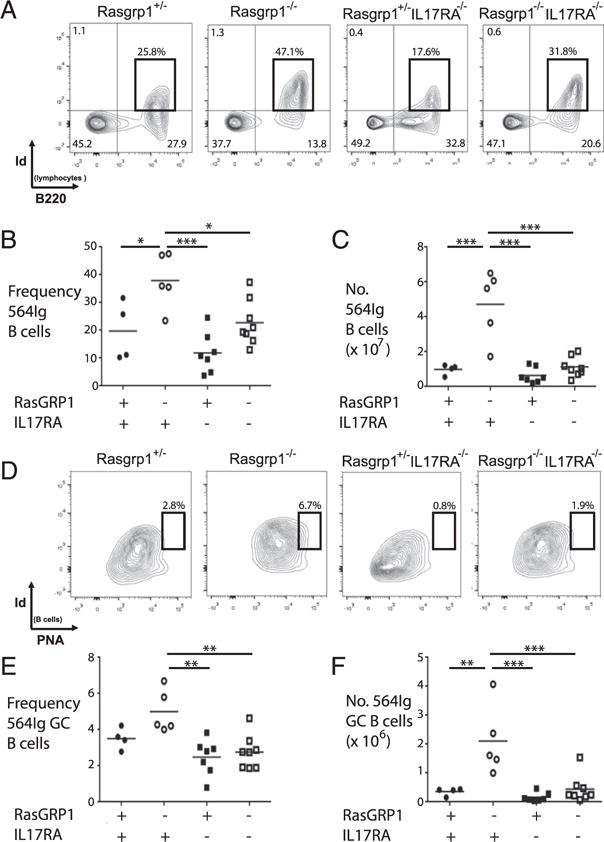
Frequency and number of self-reactive 564Igi B cells in GCs of Rasgrp1-deficient mice are IL-17R dependent. (A) Representative FACS plots from spleens of 12- to 16-wk-old sufficient (far left panel) and Rasgrp1-deficient (near left panel) mice with and without IL-17R (near right and far right panels, respectively). Self-reactive 564Igi B cells were identified with an anti-idiotypic Ab, and gates were set by comparing to minus one controls (i.e., no anti-idiotypic Ab added, data not shown). Summary of the frequencies (B) and numbers (C) of 564Igi B cells from spleens of individual Rasgpr1+/− (●), Rasgrp1−/− (○), IL17R−/− (■), and Rasgrp1−/− IL17R−/− (□) mice. (D) Representative FACS plots to identify 564Igi-derived germinal center B cells in the indicated strains of mice. Frequency (E) and number (F) of 564Igi B cells localizing to GCs (one-way ANOVA with Tukey’s multiple comparison test, *p ≤ 0.03, **p ≤ 0.003, ***p ≤ 0.0003).
IL-17RA signaling can promote B cell retention in GCs (42, 43). That Th17 cells are increased in Rasgrp1-deficient mice suggested that they may provide IL-17A to autoreactive B cells within GCs and, thereby, potentiate autoantibody production. Therefore, the frequency and number of 564Igi B cells within GCs was determined using flow cytometry (Fig. 3D, 3E). As previously reported (27), the frequency and number of autoreactive GC B cells were increased in Rasgrp1-deficient mice (5.0 ± 0.5%, 2.1 ± 0.5 × 106, mean ± SEM) compared with Rasgrp1-sufficient mice (3.5 ± 0.3%, 3.6 ± 0.7 × 105, Fig. 3D, 3E). Although the absence of IL-17RA did not significantly alter the frequency or number of auto-reactive GC B cells in the presence of RasGRP1, the frequency and number of 564Igi GC B cells were reduced in mice lacking RasGRP1 and IL-17RA (2.8 ± 0.3%, 4.4 ± 1.7 × 105) compared with Rasgrp1-deficient mice. These data are consistent with a model whereby IL-17–producing CD4 T cells in GCs (i.e., Tfh17) provide IL-17 to autoreactive B cells within GCs, thereby facilitating their survival, selection, and differentiation.
Th17 cells localize to GCs in Rasgrp1−/− mice
Tfh17 cells characteristically display a CXCR5+ PD1+ ICOS/CD278+ phenotype. To determine whether some Th17 cells observed in Rasgrp1−/− mice fulfill Tfh17 designation and localize to GCs, T cells were analyzed for Tfh markers and for their lo-calization in spleen. Compared with spleens from littermate control mice, 10-wk-old Rasgrp1−/− mice exhibited increases in the frequency and number of CXCR5+ CD278+ CD4 T cells (Fig. 4A–C). Notably, PD1 levels were increased on T cells from spleens of young Rasgrp1−/− mice that lacked GCs (presumably due to lymphopenia-associated activation; data not shown); therefore, PD1 was not used to distinguish Tfh cells. In contrast, CD278 levels on T cells from control and young Rasgrp1−/− mice were low. Although CD278 levels remained low on T cells from littermate control mice, CD278 levels increased markedly with age in Rasgrp1−/− mice (Fig. 4C, 4D). The increased levels of T cell CD278 correlated directly with the appearance of GCs in Rasgrp1−/− mice (27).
FIGURE 4.
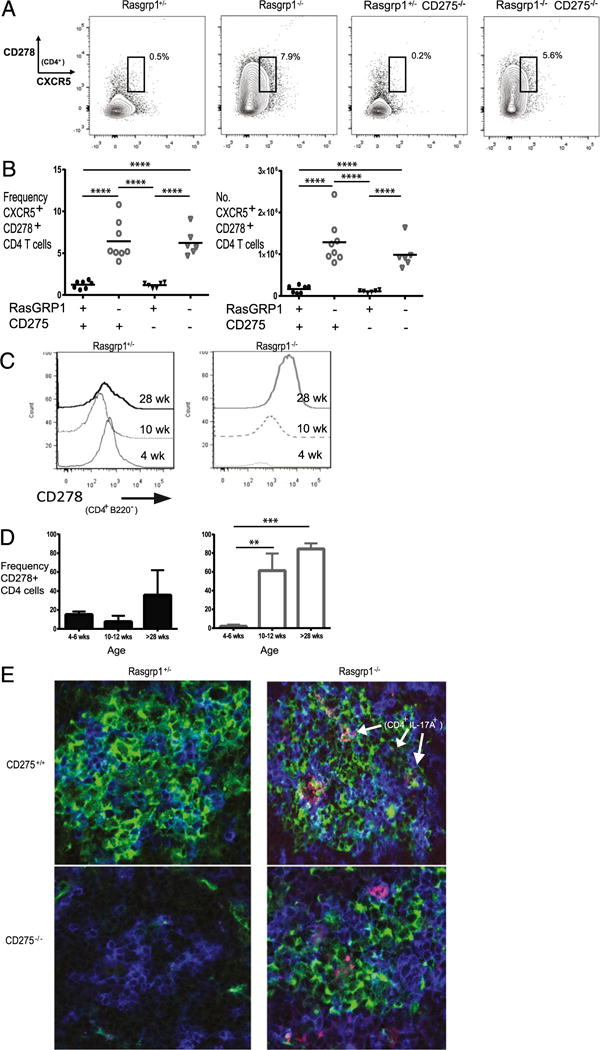
CD278 levels and localization of IL-17–producing CD4 T cells in spleens of Rasgrp1−/− mice. (A) Frequency of CXCR5+ CD278+ CD4 T cells in representative Rasgrp1+/− littermate control and Rasgrp1−/− mice, as well as in both sets of mice lacking CD275. (B) Summary of the frequency (left panel) and number (right panel) of CXCR5+ CD278+ CD4 T cells for all groups of mice; each individual symbol represents the value for a single mouse (one-way ANOVA with Tukey’s multiple comparison test, p < 0.0001). (C) Representative graphs of CD278 levels on CD4 T cells from spleens of 4-, 10-, and 28-wk-old Rasgrp1+/− littermate control mice (left panel) and Rasgrp1−/− mice (right panel). (D) Summary of age-related changes from multiple mice (**p < 0.01, ***p < 0.001, t test). (E) Representative splenic sections with GCs from 12-wk-old littermate control mice (upper left panel), Rasgrp1−/− mice (upper right panel), CD275−/− mice (lower left panel), and Rasgrp1−/− CD275−/− mice (DKO, lower right) were examined by confocal microscopy for GCs (PNA, green), CD4 (blue), and IL-17 (red) (original magnification ×600). Note that no GC was detected in the section from the CD275−/− mouse.
To directly examine localization of IL-17A+ CD4+ T cells, splenic sections were examined by immunofluorescent staining and confocal microscopy. GC staining in littermate control mice was infrequent, as was colocalized anti–IL-17A and anti-CD4 staining (Fig. 4E, left panels). In agreement with our earlier results, GCs were readily apparent in Rasgrp1−/− mice older than 2 mo of age. Further, IL-17A+ CD4 cells frequently localized within regions staining with PNA (Fig. 4E, right panels), indicating that at least some Th17 cells reside within GCs of Rasgrp1−/− mice. Based on these analyses, we conclude that Rasgrp1-deficient have increased Tfh17 cells.
Bcl6 is essential for Tfh development, whereas RORγt is the canonical transcription factor for Th17 cells. To further explore the phenotype of Tfh17 cells that apparently develop spontane ously in Rasgrp1−/− mice, Bcl6 and RORγt levels were examined in IL17A+ and CXCR5+ IL17A+ CD4 T cells (Fig. 5). Consistent with the canonical RORγt defining Th17 cells, Th17 cells from Rasgrp1-sufficient and -deficient mice stained positive for RORγt, whereas a smaller fraction was positive for RORγt and CXCR5 (Fig. 5A). Compared with Rasgrp1-sufficient mice that generated a low frequency (0.7 ± 0.4 [mean ± SE]) and number (3,913 ± 3,017) of RORγt+ CXCR5+ Th17 cells, spleens from Rasgrp1-deficient mice had substantial increases (5.1 ± 0.9 and 55,648 ± 23,476, respectively, Fig. 5A–C). In general, although Th17 cells from all mice stained positive for Bcl6 relative to the isotype control (data not shown), the levels of Bcl6 were elevated in Th17 cells from mice lacking RasGRP1 (Fig. 5D–F). In addition, a small fraction of Th17 cells from autoimmune-prone Rasgrp1-deficient mice was positive for Bcl6 and CXCR5, although levels of Bcl6 in CXCR5+ Th17 cells were lower compared with the CXCR5neg Th17 subset. Notably, few CXCR5+ Bcl6+ Th17 cells were observed in Rasgrp1-sufficient control mice. Taken together, these analyses suggest that CXCR5+ Th17 (i.e., Tfh17) cells are positive for canonical RORγt and Bcl6, although levels of Bcl6 were relatively low.
FIGURE 5.
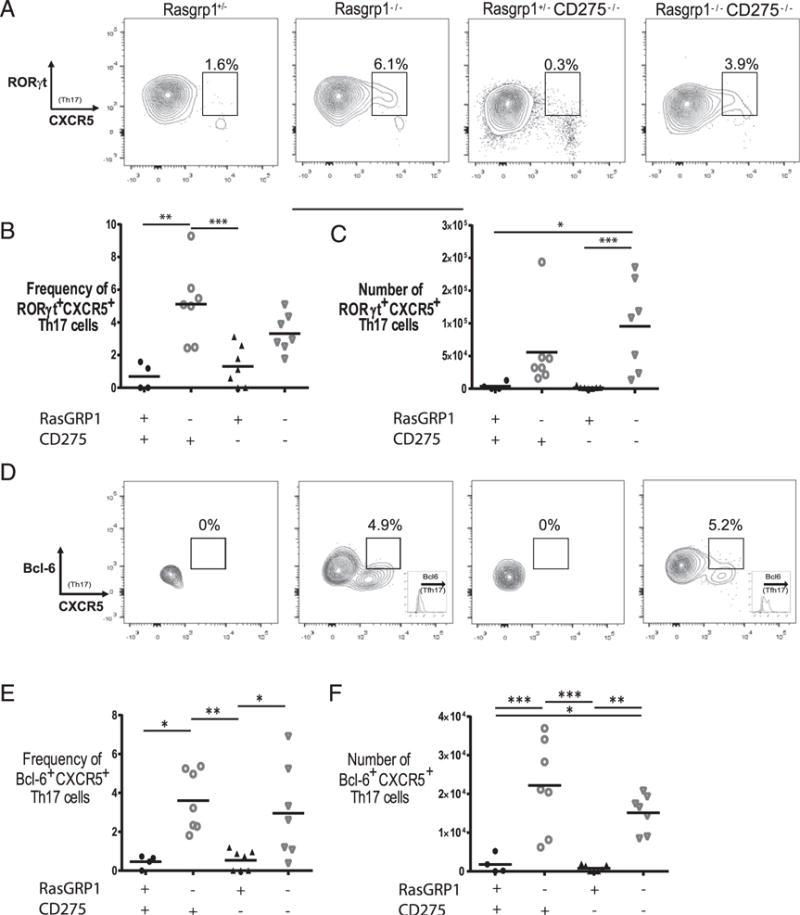
RORγt and Bcl6 levels in Th17 cells. Splenic MNCs were gated on Th17 cells, and the levels of RORγt (A) and Bcl6 (D) were plotted against CXCR5. Scatter plots of the cell frequency and number of RORγt+ CXCR5+ (B and C) or Bcl6+ CXCR5+ (E and F) Th17 cells from individual 12-to 16-wk-old Rasgpr1+/− (●), Rasgrp1−/− (○), ICOSL/CD275−/− (▴), and DKO (∇) mice (one-way ANOVA for all, except RORγt number [p = 0.007] with Tukey’s multiple-comparison test, *p ≤ 0.01, **p ≤ 0.001, ***p ≤ 0.0001).
Role of CD278 in Tfh17 cells
CD278-CD275/B7-H2/ICOSL interactions are required for the development of Tfh cells, GCs, and Ab production in wild-type mice (17, 44, 45). Increased CD278 levels on CD4 T cells from Rasgrp1−/− mice, concomitant with the appearance of GCs previously shown to harbor autoreactive B cells, suggested that CD278-CD275 may also be important for regulating Tfh cell development in this lymphoproliferative mouse strain. To test this hypothesis, 12- to 16-wk-old mice deficient in RasGRP1 and CD275 (double knockout [DKO]) were compared with single Rasgrp1- and CD275-deficient and Rasgrp1-heterozygous litter-mate control mice. The frequency and number of Th1, Th17, and CD4 T cells producing IFN-γ and IL-17 were minimal in litter-mate control and CD275-deficient mice (Fig. 2B, 2C). In comparison, the frequency and number of Th17-expressing, as well as IL-17 and IFN-γ–coexpressing, CD4 populations were elevated in Rasgrp1−/− and DKO mice. Therefore, disrupting CD278–CD275 interactions had no effect on the development of T effector subset populations in the absence of RasGRP1.
ICOS/CD278 is induced upon stimulation of CD4 T cells, and further CD275–CD278 interactions mediate upregulation of CXCR5 and Bcl6. As expected, CD4 T cells from naive control mice and control mice deficient in CD275 had minimal levels of CXCR5 and CD278 (Fig. 4A, 4B). Surprisingly, CD4 T cells from Rasgrp1-deficient and DKO mice exhibited an increased frequency of CXCR5+ CD278+ cells. To further address whether Th17 cells from DKO mice were Tfh like, Bcl6 and RORγt levels were measured (Fig. 5). Although Th17 cells from all groups of mice were largely RORγt+, few Th17 cells from control animals with and without CD275 stained positive for CXCR5. In addition, very few Th17 cells from control mice were positive for Bcl6. In contrast, a subpopulation of Th17 cells from Rasgrp1-deficient and DKO mice was double positive for Bcl6 and CXCR5, although, as noted previously, the levels of Bcl6 were lower in the CXCR5+ subset. Collectively, these data suggest that Tfh17-like cells in Rasgrp1-deficient mice express RORγt and low levels of Bcl6 in a CD275-independent manner.
To determine whether the absence of CD275 affected the development of splenomegaly, a feature linked to autoimmunity in Rasgrp1-deficient mice, splenic mass was measured in 10- to 12-wk-old mice. Consistent with earlier work, mice lacking RasGRP1 developed marked splenomegaly compared with control mice (Fig. 6A). Interestingly, splenic mass was comparable between Rasgrp1-deficient mice in the presence and absence of CD275, indicating that splenomegaly was CD275 independent.
FIGURE 6.
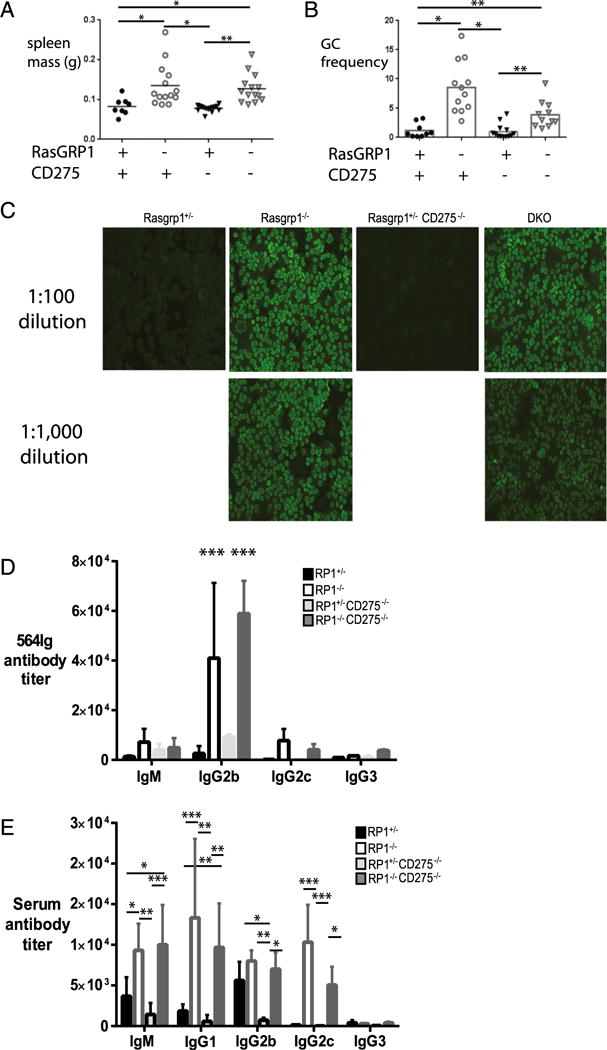
Autoimmunity in Rasgrp1-deficient mice is CD275 independent. (A) Splenic mass was measured in individual 12-to 16-wk-old Rasgpr1+/− (●), Rasgrp1−/− (○), ICOSL/CD275−/− (▼), and DKO (∇) mice (one-way ANOVA with Tukey’s multiple-comparison test, *p ≤ 0.03, **p ≤ 0.007, ***p = 0.0003). (B) Summary of the frequency of PNA+ CD95+ GC B cells in individual mice (one-way ANOVA, **p < 0.001, ***p < 0.0001). (C) Hep-2 ANA staining from representative Rasgpr1+/− mice (far left panel), Rasgrp1−/− mice (near left panels), ICOSL/CD275−/− mice (near right panel), and DKO mice (far right panels) mice at 1:100 (upper panels) and 1:1000 (lower panels) dilutions of serum (original magnification ×200). (D) Titers of 564Ig-derived serum autoantibodies were measured in Rasgpr1+/− (black bars), Rasgrp1−/− (white bars), ICOSL/CD275−/− (light gray bars), and DKO (dark gray bars) 564Igi-transgenic mice, as previously described (27). Data are mean ± SEM from five or six mice per genotype (two-way ANOVA with Tukey’s multiple-comparison test, ***p < 0.0001). (E) Total serum Ig titers in Rasgrp1-deficient mice. Titers of serum Abs in Rasgpr1+/−, Rasgrp1−/−, ICOSL/CD275−/−, and DKO mice were measured using ELISA. Data are mean ± SEM from five or six mice per genotype (two-way ANOVA with Tukey’s multiple-comparison test, *p < 0.04, **p < 0.001, ***p < 0.0003).
To understand whether the absence of CD275 affected the development of GCs, spleens of mice were examined using flow cytometry and immunohistology. As expected, the frequency of GC B cells, identified as PNAhi CD95+ B cells using flow cytometry, was low in Rasgrp1 heterozygous and CD275-deficient mice (Fig. 6B). In contrast, the frequency of GC B cells was signifi-cantly elevated in Rasgrp1−/− and DKO mice. These results were consistent with immunohistological analysis of splenic sections showing an increased frequency of GCs in Rasgrp1−/− mice with and without CD275 (data not shown). In addition, CD4+ IL-17+ T cells were detectable in splenic GCs from Rasgrp1−/− and DKO mice (Fig. 4E), suggesting that GC and Tfh cell development are not impaired in the absence of CD275.
T cell–dependent Ab responses in wild-type mice require CD278–CD275 interactions (44). In addition, disrupting these interactions through administration of blocking Ab regulates autoantibody levels in certain autoimmune murine models (4). Therefore, autoantibody production was also measured in Rasgrp1−/− mice with and without CD275. As expected, littermate control and CD275-deficient mice produced minimal ANAs (Fig. 6C). In contrast, ANAs were readily detectable in Rasgrp1−/− mice and could be detected at high dilutions of serum, indicating that autoantibody production was elevated. DKO mice also produced ANA titers comparable with those from Rasgrp1−/− mice. To understand whether autoantibody production from a known population of autoreactive B cells participating in GC responses of Rasgrp1−/− mice was regulated by CD275, CD275-deficient mice were crossed with 564Igi-transgenic mice, and serum 564Ig autoantibody levels were measured. Similar to the ANA results, the level of 564Ig-derived IgG2b subclass Ab was elevated in serum from Rasgrp1−/− and DKO mice compared with controls (Fig. 6D). Similarly, total serum levels of several classes of Ab were elevated comparably in Rasgrp1-deficient mice with and without CD275 (Fig. 6E). These data suggest that autoantibody production in Rasgrp1−/− mice occurs independently of CD275.
In addition to IL-17A, production of IL-21 is implicated in Tfh cell–mediated autoimmunity (46, 47). To determine whether IL-21 production by Tfh17 cells may contribute to the autoimmune phenotypes exhibited in Rasgrp1-deficient and Rasgrp1- CD275–DKO animals, the frequency and number of IL-21–producing Tfh17 cells were measured by flow cytometry (Fig. 7). Consistent with a potential role in mediating autoimmunity, the frequency and number of CD4+ IL17A+ CXCR5+ cells staining positive for IL-21 were increased in Rasgrp1-deficient mice relative to Rasgrp1-sufficient control mice (frequency, 0.8 ± 0.4 versus 4.1 ± 0.8; number, 427 ± 235 versus 21,987 ± 5,269 [mean ± SEM], Rasgrp1-sufficient versus Rasgrp1-deficient). Interestingly, the frequency and number of Tfh17 cells making IL-21 in mice lacking RasGRP1 and CD275 did not differ statistically from those of control mice (0.8 ± 0.3 and 2404 ± 805). Therefore, although production of IL-21 by Tfh17 cells may contribute to increased autoantibody titers in Rasgrp1-deficient mice, it does not seem to explain the increased autoantibody titers in mice lacking RasGRP1 and CD275.
FIGURE 7.
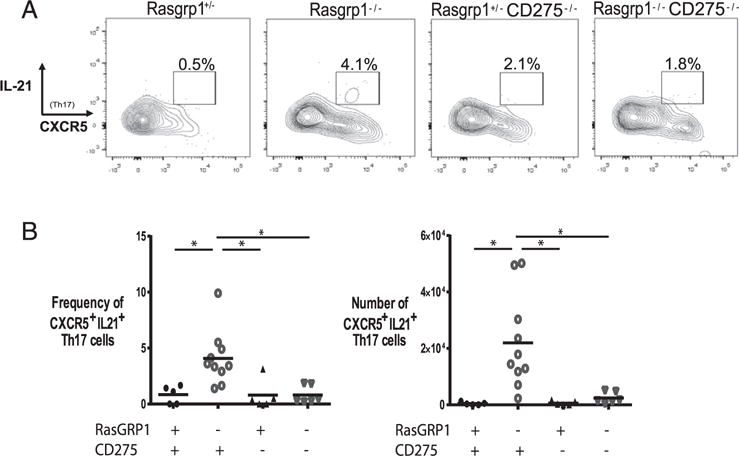
Generation of IL-21–producing Tfh17 cells is CD275 dependent. (A) Splenic MNCs were gated on Th17 cells, and levels of IL-21 were plotted against CXCR5. (B) Scatter plots of cell frequency (left panel) and number (right panel) of IL-21+ CXCR5+ Th17 cells from individual 12- to 16-wk-old Rasgpr1+/− (●), Rasgrp1−/− (○), ICOSL/CD275−/− (▴), and DKO (∇) mice (one-way ANOVA with Tukey’s multiple-comparison test, *p < 0.01).
CD278-CD275 is reportedly important for Treg survival and expansion (48). To understand whether the absence of CD275 may indirectly affect Th17 frequency and number by altering the development of Tregs, Treg frequency and number were quantified by measuring CD4+ CD25+ Foxp3+ cells (Fig. 8). Consistent with previous studies reporting that CD278-CD275 is required for Treg development and function, the frequency of Tregs was slightly reduced in CD275-deficient mice compared with CD275-sufficient mice. In contrast, the frequency and number of Tregs were similar between Rasgrp1−/− and DKO mice, suggesting that the absence of CD275 does not affect the development of Tregs in Rasgrp1−/− mice. The frequency and number of Foxp3+ CD25+ regulatory cells were also similar within gated Th1, Th17, and Tfh cell populations among all strains (data not shown), suggesting that differences in Tregs within GCs do not explain autoimmunity in Rasgrp1−/− and Rasgrp1−/− CD275−/− mice.
FIGURE 8.
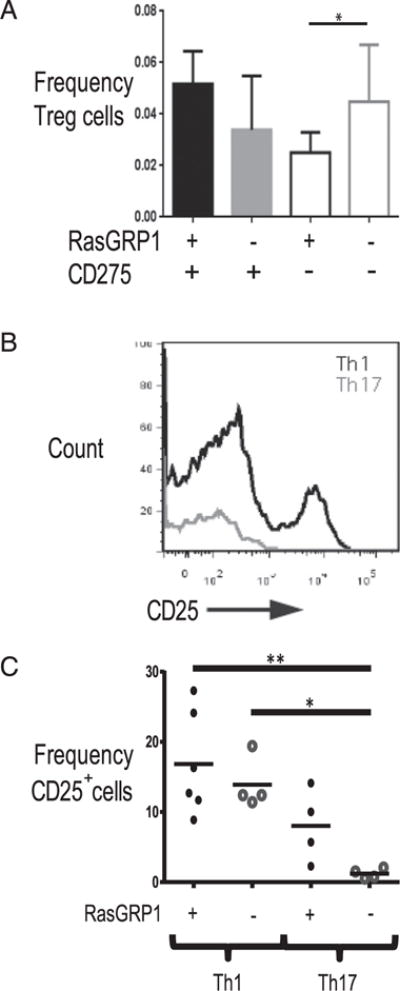
Frequency of Tregs in Rasgrp1-deficient mice with and without CD275 and CD25 levels on Th1 and Th17 cells. Tregs were identified as CD4+ CD25+ Foxp3+ cells. (A) Frequency of Tregs in 12- to 16-wk-old Rasgpr1+/− (filled black bar), Rasgrp1−/− (filled gray), ICOSL/CD275−/− (open black), and DKO (open gray) mice. Data are mean 6 SD from 5–10 mice per group (one-way ANOVA with Tukey’s multiple-comparison test, *p < 0.01). (B) CD25 levels on Th1 and Th17 cells from Rasgrp1−/− mice. (C) Summary of CD25-positive Th1 and Th17 cells from spleens of 12- to 16-wk-old control and Rasgrp1−/− mice (one-way ANOVA with Tukey’s multiple-comparison test, *p < 0.01, **p < 0.001).
Discussion
Early evidence from Rasgrp1-deficient mice indicated impaired T cell development, with subsequent T cell lymphopenia in the periphery (38). This leads to homeostatic expansion that was found to be defective in the absence of RasGRP1, causing clonal exhaustion of primarily Th1 cells (32). This observation, in combination with the finding that the frequency and activity of Tregs are also increased, led to the suggestion that autoimmunity was self-limiting in Rasgrp1-deficient mice (34). In contrast, we found that autoantibody levels of all IgG subclasses were elevated in Rasgrp1−/− mice, concomitant with the appearance of GCs containing autoreactive B cells (26). This autoimmune phenotype appears to be substantiated in other Rasgrp1 mutant strains of mice (33, 49). In this article, we demonstrate that the frequency and number of IL-17–producing CD4 T cells are increased in Rasgrp1−/− mice. These Th17 cells localize to GCs and resemble Tfh cells immunophenotypically. Importantly, autoreactive GC B cells are significantly reduced in mice lacking RasGRP1 and IL-17R, suggesting that Tfh17-like cells mediate a break in B cell tolerance within GCs. Surprisingly, the development and apparent function of Tfh17 cells, as well as the development of GCs and the production of autoantibody, occur independently of CD275 in Rasgrp1-deficient mice. We propose that this newly described Tfh17 cell population is uniquely regulated and mediates autoimmunity in lymphopenia-driven settings.
The frequency and number of Th17 cells were increased in Rasgrp1-deficient mice relative to Rasgrp1-sufficient littermate control animals, thereby accounting for increased IL-17A production by in vitro–stimulated splenic CD4 T cells from Rasgrp1-deficient mice (Fig. 1). In contrast, although the data for Th1 cells trend toward similar increases between Rasgrp1-deficient and Rasgrp1-sufficient animals, the differences were not statistically significant. Consistent with the Th1 cell–frequency data, IFN-γ production by in vitro–stimulated CD4 T cells also did not differ statistically. Interestingly, CD4 T cells in Rasgrp1-deficient mice exhibit an impaired IFN-γ response to challenge with Listeria monocytogenes, likely due to their propensity for clonal exhaustion (32). We did not test whether exhaustion of stimulated Rasgrp1-deficient Th1 cells (34) or impaired activation in the absence of the predominant GEF necessary for Ras signaling (33, 38, 49) contributes to IFN-γ responses of anti-CD3–stimulated Rasgrp1-deficient Th1 cells. However, phenotypic differences, such as CD25 levels (50), between Th1 and Th17 cells could potentially mediate differential sensitivity of Th subsets to clonal exhaustion. Indeed, we confirm that CD25 levels are significantly reduced on Th17 cells compared with Th1 cells in Rasgrp1−/− mice (Fig. 8B, 8C). Because IL-2 is critical for T cell expansion (17, 50–52), it follows that reduced CD25 levels presumably would reduce the ability of GC T cells to proliferate. Signaling via CD25 inhibits Th17 differentiation by a mechanism involving modulation of Stat3 and Stat5 binding to regulatory regions of the IL-17 promoter (50, 53, 54). Importantly, in contrast to IL-17, Rasgrp1-deficient CD4 T cells produce little IL-2 after stimulation with anti-CD3 in vitro (33). Therefore, reduced IL-2 may influence Th17 cell and Tfh17-like cell development in Rasgrp1-deficient mice. With regard to increased production of IL-17 by CD4 T cells from Rasgrp1−/− mice in vitro, Tfh cells are potent producers of cytokines compared with conventional effector T cells; therefore, the increase in IL-17 production in Rasgrp1−/− CD4 T cells can be most easily explained by the increased frequency of Tfh17 cells in deficient mice.
Th17 and Tfh17 cells can produce IL-17A, as well as IL-21 (55), and both of these cytokines can influence GC B cell responses (56, 57). The importance of IL-17A in mediating autoimmunity in Rasgrp1-deficient mice was tested in mice lacking IL-17RA; a specific population of autoreactive B cells (564Igi transgenic) was reduced in GCs (Fig. 3). Notably, in nonautoimmune settings, IL-21 and CD275 are both required for the generation of Tfh cells (55). Tfh17 cells from Rasgrp1-deficient mice also produced IL-21, raising the possibility that IL-21 may contribute to anti-nuclear and 564Igi-derived autoantibody production. However, mice lacking RasGRP1 and CD275 generated similar titers of these autoantibodies, yet lacked the IL-21–producing Tfh17 population (Fig. 7). Although a more direct test of the importance of IL-21 is needed, we infer from our data that IL-21 production by Tfh17-like cells is not required for at least some autoantibodies produced in Rasgrp1-deficient mice. These results from Rasgrp1-deficient mice seemingly contrast with other autoimmune disease models, including MRL/lpr (20), sanroque (58, 59), and BXD2 (46) mice strains, in which CD275 and/or IL-21 potentiate Tfh cell development, GCs, and autoantibody production. Thus, Tfh17-like cell development in Rasgrp1-deficient mice may well involve a novel pathway.
Tfh17 cells are linked to several autoimmune disorders in mice and in humans. For example, an increase in Th17 cells was noted in peripheral blood from multiple cohorts of patients with SLE compared with healthy control samples (60–62). Th17 cells support class-switch recombination to multiple IgG subclasses in B cells (36); therefore, our observation that the frequency and number Tfh17 cells are increased in Rasgrp1−/− mice provides an explanation for increased autoantibodies of multiple IgG subclasses in deficient mice (Fig. 6) (27). Notably, Tfh17 cells in Rasgrp1−/− mice possess the conventional CD4+ CXCR5+ PD-1+ CD278+ immunophenotype, as well as express RORγt, although with lower than reported levels of Bcl6 (Figs. 4, 5). Previous work established that Bcl6 is necessary and sufficient for the development of Tfh cells (11, 18, 19). Bcl6 upregulation in T cells is potentiated by CD278–CD275 interactions, leading to upregulation of CXCR5 to potentiate migration of activated T cells into the B cell follicle. Therefore, it is surprising that the frequency and number of Tfh17 cells are comparable in Rasgrp1-deficient mice with and without CD275 (Figs. 4, 5). Under healthy physiologic settings, Tfh cell development is also dependent on Stat3 (63). Interestingly, Stat3 can be activated through IL-6 (64), and we observed elevated serum levels of IL-6 in Rasgrp1-deficient mice (∼5 pg/ml and undetectable levels in Rasgrp1−/− and Rasgrp1+/− mice, respectively; data not shown). In addition, IL-6 and IL-21 were shown to be required for optimal Tfh development in lymphocytic choriomeningitis virus–infected mice (65). We propose that the proinflammatory state of lymphopenic Rasgrp1-deficient mice can compensate, in part, for CD278–CD275 interactions. If true, this could hold important implications for understanding and treating lymphopenia-coupled autoimmune diseases.
In addition to the increased frequency and number of Th17 cells, Th cells producing IFN-γ and IL-17 are increased in Rasgrp1−/− mice (Fig. 2). Similar double-positive subsets were observed in other models of autoimmunity (66), in which it was suggested that this population represented the most pathogenic of autoreactive Th subsets as a result of their ability to produce multiple proinflammatory cytokines. Another possibility is that this population represents an intermediate between Th1 and Th17 cells (67, 68). Regardless of their origin, the increase in Rasgrp1-deficient mice is at least consistent with a potential role in autoimmunity. However, CD4 T cells producing IFN-γ and IL-17 did not meet the immunophenotypic definition of Tfh cells (data not shown). The increased numbers of Tfh17 and IFNγ+ IL-17+ Th cells are likely caused by defective homeostatic expansion in Rasgrp1−/− mice and are grossly reflected by the development of splenomegaly. In nonautoimmune-prone mice, homeostatic expansion is regulated by IL-7 (69–71). In mice developing lymphoproliferative syndrome, additional mechanisms override IL-7 regulation, leading to accumulation of CD4 T cells (72–75), as well as other lymphocytes. The root cause of defective homeostatic expansion in Rasgrp1-deficient mice is unknown.
CD4 T cells from Rasgrp1-deficient mice upregulate several markers associated with homeostatic activation and expansion of CD4 T cells similar to what is reported for wild-type T cells placed in lymphopenic environments (OX-40, PD-1, PD-L1, CD69, and CD80; data not shown). Unlike CD4 T cells from wild-type mice (76), restitution of normal CD4 T cell numbers in Rasgrp1−/− mice does not result in reduction of the above markers to levels found in quiescent cells. Interestingly, CD4 T cells from Rasgrp1−/− mice do not upregulate CD278 during lymphopenia. Instead, CD278 levels only increase concomitantly with the appearance of spontaneous GCs (Fig. 4). This led us to test whether CD278–CD275 interactions are required for the development of Tfh cells and GCs. Notably, we observed that the development of Tfh17 cells and GCs is CD275 independent (Figs. 4, 5). We speculate that defective homeostatic expansion in mice lacking RasGRP1 contributes to a novel Tfh17 population that can arise independently of CD278–CD275 interactions.
Whether these novel Tfh17 cells in Rasgrp1−/− mice are auto-reactive is unknown. We showed previously that GCs developing spontaneously (i.e., without manipulation) in Rasgrp1-deficient mice contain autoreactive B cells (27); therefore, the presence of Tfh17 cells may directly affect autoantibody production. In this study, we observed that autoantibody production is elevated in Rasgrp1−/− mice independently of CD275. IL-17 potentiates B cell survival and promotes class switch to multiple IgG subclasses (36); therefore, it is noteworthy that 564Ig-derived autoantibody of multiple IgG subclasses (IgG1 could not be tested as a result of the anti-idiotypic detection Ab also being IgG1) was increased in Rasgrp1-deficient mice independently of CD275 (Fig. 6). Increased autoantibody of multiple IgG subclasses is consistent with at least some autoantibody being T cell dependent. CD278-CD275 is thought to enhance synapses between Ag-specific GC B cells and Tfh cells, leading to increased survival, as well as differentiation of GC B cells into memory and plasma cells. Surprisingly, our observations using DKO mice indicate that CD278–CD275 interactions are not required for autoantibody production. These results contrast with those derived from other autoimmune murine models (20, 58).
Tregs are key components for peripheral tolerance and are dependent on CD278/CD275 for their function. The frequency and function of Tregs are reportedly increased in Rasgrp1-deficient mice (34). Our own studies corroborate the increased frequency of Tregs, although we find this difference only within the first several weeks of life. Further, our studies indicate that Tregs lacking RasGRP1 are comparable functionally to Tregs from control mice (data not shown). In the current study, although the lack of CD275 appeared to affect the frequency of Tregs in Rasgrp1-sufficient mice, there was no statistical change in the frequency of Tregs in Rasgrp1-deficient mice (Fig. 8). Although we cannot rule out functional differences, it appears that CD275–CD278 interactions may not be required for Treg development in lymphopenia-associated autoimmune-prone Rasgrp1-deficient mice. A separate Treg subset, called T follicular regulatory (Tfr) cells, locate to GCs and share many markers of Tfh cells, including CXCR5, PD-1, and CD278, although they can be distinguished from Tfh cells based on Foxp3, CTLA-4, and higher levels of CD25 (77). Tfr cells inhibit GC responses by limiting numbers of Tfh and GC B cells, and altered Tfh/Tfr balance was recently implicated in autoimmunity in BXD2 mice (46). Interestingly, the imbalance in autoimmune BXD2 mice was potentiated by IL-21 production. Although we observed an increased frequency of IL-21+ Tfh17 cells in Rasgrp1-deficient mice relative to littermate control mice (Fig. 7), no differences in the frequency of Foxp3+ CXCR5+ CD4 T cells were observed (data not shown). Moreover, Rasgrp1-deficient mice remained autoimmune prone in the absence of CD275, despite the reduced frequency of IL-21–producing Tfh17 cells. Collectively, these data appear inconsistent with Tfr cells having a profound role in regulating autoimmunity in the absence of RasGRP1.
Blocking CD278–CD275 interactions shows efficacy in several autoimmune settings (78–80). Given that autoantibody production in Rasgrp1-deficient mice occurs independently of CD275, it is tempting to speculate that there may be subsets of patients with autoimmune disease who are unresponsive to such therapy. This may apply more specifically to autoimmunity developing in lymphopenic individuals, as appears in the Rasgrp1-deficient mouse model. More work is needed to further define the requirements and functions of Tfh17 cells in Rasgrp1-deficient and other lymphopenia-associated autoimmune mouse models.
Acknowledgments
This work was supported by American Lung Association Biomedical Research Grant RG-349167 (to R.A.B.), as well as by start-up funds provided by the University of South Alabama College of Medicine (to R.A.B.).
Abbreviations used
- ANA
anti-nuclear Ab
- DKO
double knockout
- GC
germinal center
- GEF
guanine exchange factor
- MNC
mononuclear cell
- SLE
systemic lupus erythematosus
- Tfh
T follicular helper
- Tfr
T follicular regulatory
- Treg
regulatory T cell
Footnotes
ORCID: 0000-0001-5713-5369 (R.A.B.).
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, Naves R, Han M, Zhong F, Castellanos JG, et al. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med. 2010;16:406–412. doi: 10.1038/nm.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monteleone G, Monteleone I, Fina D, Vavassori P, Del Vecchio Blanco G, Caruso R, Tersigni R, Alessandroni L, Biancone L, Naccari GC, et al. Interleukin-21 enhances T-helper cell type I signaling and interferon-gamma production in Crohn’s disease. Gastroenterology. 2005;128:687–694. doi: 10.1053/j.gastro.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 3.Moulton VR, Tsokos GC. Abnormalities of T cell signaling in systemic lupus erythematosus. Arthritis Res Ther. 2011;13:207. doi: 10.1186/ar3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu YL, Metz DP, Chung J, Siu G, Zhang M. B7RP-1 blockade ameliorates autoimmunity through regulation of follicular helper T cells. J Immunol. 2009;182:1421–1428. doi: 10.4049/jimmunol.182.3.1421. [DOI] [PubMed] [Google Scholar]
- 5.Wang X, Huang W, Mihara M, Sinha J, Davidson A. Mechanism of action of combined short-term CTLA4Ig and anti-CD40 ligand in murine systemic lupus erythematosus. J Immunol. 2002;168:2046–2053. doi: 10.4049/jimmunol.168.4.2046. [DOI] [PubMed] [Google Scholar]
- 6.Huang W, Sinha J, Newman J, Reddy B, Budhai L, Furie R, Vaishnaw A, Davidson A. The effect of anti-CD40 ligand antibody on B cells in human systemic lupus erythematosus. Arthritis Rheum. 2002;46:1554–1562. doi: 10.1002/art.10273. [DOI] [PubMed] [Google Scholar]
- 7.Kyttaris VC, Zhang Z, Kuchroo VK, Oukka M, Tsokos GC. Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice. J Immunol. 2010;184:4605–4609. doi: 10.4049/jimmunol.0903595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu HY, Quintana FJ, Weiner HL. Nasal anti-CD3 antibody ameliorates lupus by inducing an IL-10-secreting CD4+ CD25− LAP+ regulatory T cell and is associated with down-regulation of IL-17+ CD4+ ICOS+ CXCR5+ follicular helper T cells. J Immunol. 2008;181:6038–6050. doi: 10.4049/jimmunol.181.9.6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu HY, Center EM, Tsokos GC, Weiner HL. Suppression of murine SLE by oral anti-CD3: inducible CD4+CD25−LAP+ regulatory T cells control the expansion of IL-17+ follicular helper T cells. Lupus. 2009;18:586–596. doi: 10.1177/0961203308100511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones JL, Thompson SA, Loh P, Davies JL, Tuohy OC, Curry AJ, Azzopardi L, Hill-Cawthorne G, Fahey MT, Compston A, Coles AJ. Human autoimmunity after lymphocyte depletion is caused by homeo-static T-cell proliferation. Proc Natl Acad Sci USA. 2013;110:20200–20205. doi: 10.1073/pnas.1313654110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, Srivastava M, Linterman M, Zheng L, Simpson N, et al. The transcriptional re-pressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity. 2009;31:457–468. doi: 10.1016/j.immuni.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 12.Haynes NM, Allen CD, Lesley R, Ansel KM, Killeen N, Cyster JG. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated sub-population. J Immunol. 2007;179:5099–5108. doi: 10.4049/jimmunol.179.8.5099. [DOI] [PubMed] [Google Scholar]
- 13.Debes GF, Arnold CN, Young AJ, Krautwald S, Lipp M, Hay JB, Butcher EC. Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat Immunol. 2005;6:889–894. doi: 10.1038/ni1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veerman KM, Williams MJ, Uchimura K, Singer MS, Merzaban JS, Naus S, Carlow DA, Owen P, Rivera-Nieves J, Rosen SD, Ziltener HJ. Interaction of the selectin ligand PSGL-1 with chemokines CCL21 and CCL19 facilitates efficient homing of T cells to secondary lymphoid organs. Nat Immunol. 2007;8:532–539. doi: 10.1038/ni1456. [DOI] [PubMed] [Google Scholar]
- 15.Goenka R, Barnett LG, Silver JS, O’Neill PJ, Hunter CA, Cancro MP, Laufer TM. Cutting edge: dendritic cell-restricted antigen presentation initiates the follicular helper T cell program but cannot complete ultimate effector differentiation. J Immunol. 2011;187:1091–1095. doi: 10.4049/jimmunol.1100853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu H, Li X, Liu D, Li J, Zhang X, Chen X, Hou S, Peng L, Xu C, Liu W, et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature. 2013;496:523–527. doi: 10.1038/nature12058. [DOI] [PubMed] [Google Scholar]
- 17.Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C, Crotty S. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34:932–946. doi: 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnston RJ, Poholek AC, Di Toro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, Crotty S. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang YH, Dong C. Bcl6 mediates the development of T follicular helper cells. Science. 2009;325:1001–1005. doi: 10.1126/science.1176676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, Flavell RA, Craft J. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med. 2008;205:2873–2886. doi: 10.1084/jem.20080840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Odegard JM, DiPlacido LD, Greenwald L, Kashgarian M, Kono DH, Dong C, Flavell RA, Craft J. ICOS controls effector function but not trafficking receptor expression of kidney-infiltrating effector T cells in mu-rine lupus. J Immunol. 2009;182:4076–4084. doi: 10.4049/jimmunol.0800758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coughlin JJ, Stang SL, Dower NA, Stone JC. RasGRP1 and RasGRP3 regulate B cell proliferation by facilitating B cell receptor-Ras sig-naling. J Immunol. 2005;175:7179–7184. doi: 10.4049/jimmunol.175.11.7179. [DOI] [PubMed] [Google Scholar]
- 23.Yasuda S, Stevens RL, Terada T, Takeda M, Hashimoto T, Fukae J, Horita T, Kataoka H, Atsumi T, Koike T. Defective expression of Ras guanyl nucleotide-releasing protein 1 in a subset of patients with systemic lupus erythematosus. J Immunol. 2007;179:4890–4900. doi: 10.4049/jimmunol.179.7.4890. [DOI] [PubMed] [Google Scholar]
- 24.Pan W, Zhu S, Yuan M, Cui H, Wang L, Luo X, Li J, Zhou H, Tang Y, Shen N. MicroRNA-21 and microRNA-148a contribute to DNA hypo-methylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol. 2010;184:6773–6781. doi: 10.4049/jimmunol.0904060. [DOI] [PubMed] [Google Scholar]
- 25.Sun C, Molineros JE, Looger LL, Zhou XJ, Kim K, Okada Y, Ma J, Qi YY, Kim-Howard X, Motghare P, et al. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat Genet. 2016;48:323–330. doi: 10.1038/ng.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qu HQ, Grant SF, Bradfield JP, Kim C, Frackelton E, Hakonarson H, Polychronakos C. Association of RASGRP1 with type 1 diabetes is revealed by combined follow-up of two genome-wide studies. J Med Genet. 2009;46:553–554. doi: 10.1136/jmg.2009.067140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bartlett A, Buhlmann JE, Stone J, Lim B, Barrington RA. Multiple checkpoint breach of B cell tolerance in Rasgrp1-deficient mice. J Immunol. 2013;191:3605–3613. doi: 10.4049/jimmunol.1202892. [DOI] [PubMed] [Google Scholar]
- 28.Coughlin JJ, Stang SL, Dower NA, Stone JC. The role of RasGRPs in regulation of lymphocyte proliferation. Immunol Lett. 2006;105:77–82. doi: 10.1016/j.imlet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 29.Stone JC. Regulation and Function of the RasGRP Family of Ras Activators in Blood Cells. Genes Cancer. 2011;2:320–334. doi: 10.1177/1947601911408082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alberola-Ila J, Hernández-Hoyos G. The Ras/MAPK cascade and the control of positive selection. Immunol Rev. 2003;191:79–96. doi: 10.1034/j.1600-065x.2003.00012.x. [DOI] [PubMed] [Google Scholar]
- 31.Priatel JJ, Teh SJ, Dower NA, Stone JC, Teh HS. RasGRP1 transduces low-grade TCR signals which are critical for T cell development, homeostasis, and differentiation. Immunity. 2002;17:617–627. doi: 10.1016/s1074-7613(02)00451-x. [DOI] [PubMed] [Google Scholar]
- 32.Priatel JJ, Chen X, Zenewicz LA, Shen H, Harder KW, Horwitz MS, Teh HS. Chronic immunodeficiency in mice lacking RasGRP1 results in CD4 T cell immune activation and exhaustion. J Immunol. 2007;179:2143–2152. doi: 10.4049/jimmunol.179.4.2143. [DOI] [PubMed] [Google Scholar]
- 33.Fuller DM, Zhu M, Song X, Ou-Yang CW, Sullivan SA, Stone JC, Zhang W. Regulation of RasGRP1 function in T cell development and activation by its unique tail domain. PLoS One. 2012;7:e38796. doi: 10.1371/journal.pone.0038796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X, Priatel JJ, Chow MT, Teh HS. Preferential development of CD4 and CD8 T regulatory cells in RasGRP1-deficient mice. J Immunol. 2008;180:5973–5982. doi: 10.4049/jimmunol.180.9.5973. [DOI] [PubMed] [Google Scholar]
- 35.Tan AH, Lam KP. Pharmacologic inhibition of MEK-ERK sig-naling enhances Th17 differentiation. J Immunol. 2010;184:1849–1857. doi: 10.4049/jimmunol.0901509. [DOI] [PubMed] [Google Scholar]
- 36.Mitsdoerffer M, Lee Y, Ja¨ger A, Kim HJ, Korn T, Kolls JK, Cantor H, Bettelli E, Kuchroo VK. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci USA. 2010;107:14292–14297. doi: 10.1073/pnas.1009234107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith KM, Pottage L, Thomas ER, Leishman AJ, Doig TN, Xu D, Liew FY, Garside P. Th1 and Th2 CD4+ T cells provide help for B cell clonal expansion and antibody synthesis in a similar manner in vivo. J Immunol. 2000;165:3136–3144. doi: 10.4049/jimmunol.165.6.3136. [DOI] [PubMed] [Google Scholar]
- 38.Dower NA, Stang SL, Bottorff DA, Ebinu JO, Dickie P, Ostergaard HL, Stone JC. RasGRP is essential for mouse thy-mocyte differentiation and TCR signaling. Nat Immunol. 2000;1:317–321. doi: 10.1038/79766. [DOI] [PubMed] [Google Scholar]
- 39.Berland R, Fernandez L, Kari E, Han JH, Lomakin I, Akira S, Wortis HH, Kearney JF, Ucci AA, Imanishi-Kari T. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity. 2006;25:429–440. doi: 10.1016/j.immuni.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 40.Gavalchin J, Datta SK. The NZB X SWR model of lupus nephritis. II Autoantibodies deposited in renal lesions show a distinctive and restricted idiotypic diversity. J Immunol. 1987;138:138–148. [PubMed] [Google Scholar]
- 41.Mohan C, Adams S, Stanik V, Datta SK. Nucleosome: a major immunogen for pathogenic autoantibody-inducing T cells of lupus. J Exp Med. 1993;177:1367–1381. doi: 10.1084/jem.177.5.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xie S, Li J, Wang JH, Wu Q, Yang P, Hsu HC, Smythies LE, Mountz JD. IL-17 activates the canonical NF-kappaB signaling pathway in autoimmune B cells of BXD2 mice to upregulate the expression of regulators of G-protein signaling 16. J Immunol. 2010;184:2289–2296. doi: 10.4049/jimmunol.0903133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus AL, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- 44.Tafuri A, Shahinian A, Bladt F, Yoshinaga SK, Jordana M, Wakeham A, Boucher LM, Bouchard D, Chan VS, Duncan G, et al. ICOS is essential for effective T-helper-cell responses. Nature. 2001;409:105–109. doi: 10.1038/35051113. [DOI] [PubMed] [Google Scholar]
- 45.Warnatz K, Bossaller L, Salzer U, Skrabl-Baumgartner A, Schwinger W, van der Burg M, van Dongen JJ, Orlowska-Volk M, Knoth R, Durandy A, et al. Human ICOS deficiency abrogates the germinal center reaction and provides a monogenic model for common variable immunodeficiency. Blood. 2006;107:3045–3052. doi: 10.1182/blood-2005-07-2955. [DOI] [PubMed] [Google Scholar]
- 46.Ding Y, Li J, Yang P, Luo B, Wu Q, Zajac AJ, Wildner O, Hsu HC, Mountz JD. Interleukin-21 promotes germinal center reaction by skewing the follicular regulatory T cell to follicular helper T cell balance in autoimmune BXD2 mice. Arthritis Rheumatol. 2014;66:2601–2612. doi: 10.1002/art.38735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med. 2012;209:1241–1253. doi: 10.1084/jem.20120994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y, Shen S, Gorentla BK, Gao J, Zhong XP. Murine regulatory T cells contain hyperproliferative and death-prone subsets with differential ICOS expression. J Immunol. 2012;188:1698–1707. doi: 10.4049/jimmunol.1102448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daley SR, Coakley KM, Hu DY, Randall KL, Jenne CN, Limnander A, Myers DR, Polakos NK, Enders A, Roots C, et al. Rasgrp1 mutation increases naive T-cell CD44 expression and drives mTOR-dependent accumulation of Helios+ T cells and autoantibodies. eLife. 2013;2:e01020. doi: 10.7554/eLife.01020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ballesteros-Tato A, León B, Graf BA, Moquin A, Adams PS, Lund FE, Randall TD. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity. 2012;36:847–856. doi: 10.1016/j.immuni.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science. 1976;193:1007–1008. doi: 10.1126/science.181845. [DOI] [PubMed] [Google Scholar]
- 52.Smith KA. Interleukin-2: inception, impact, and implications. Science. 1988;240:1169–1176. doi: 10.1126/science.3131876. [DOI] [PubMed] [Google Scholar]
- 53.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 54.Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med. 2012;209:243–250. doi: 10.1084/jem.20111174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, Wang YH, Watowich SS, Jetten AM, Tian Q, Dong C. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29:138–149. doi: 10.1016/j.immuni.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ozaki K, Spolski R, Ettinger R, Kim HP, Wang G, Qi CF, Hwu P, Shaffer DJ, Akilesh S, Roopenian DC, et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J Immunol. 2004;173:5361–5371. doi: 10.4049/jimmunol.173.9.5361. [DOI] [PubMed] [Google Scholar]
- 57.Spolski R, Leonard WJ. Interleukin-21: basic biology and implications for cancer and autoimmunity. Annu Rev Immunol. 2008;26:57–79. doi: 10.1146/annurev.immunol.26.021607.090316. [DOI] [PubMed] [Google Scholar]
- 58.Linterman MA, Rigby RJ, Wong R, Silva D, Withers D, Anderson G, Verma NK, Brink R, Hutloff A, Goodnow CC, Vinuesa CG. Roquin differentiates the specialized functions of duplicated T cell costimulatory receptor genes CD28 and ICOS. Immunity. 2009;30:228–241. doi: 10.1016/j.immuni.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 59.Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, Yu D, Domaschenz H, Whittle B, Lambe T, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature. 2005;435:452–458. doi: 10.1038/nature03555. [DOI] [PubMed] [Google Scholar]
- 60.Xing Q, Wang B, Su H, Cui J, Li J. Elevated Th17 cells are accompanied by FoxP3+ Treg cells decrease in patients with lupus nephritis. Rheumatol Int. 2012;32:949–958. doi: 10.1007/s00296-010-1771-0. [DOI] [PubMed] [Google Scholar]
- 61.Shah K, Lee WW, Lee SH, Kim SH, Kang SW, Craft J, Kang I. Dysregulated balance of Th17 and Th1 cells in systemic lupus eryth-ematosus. Arthritis Res Ther. 2010;12:R53. doi: 10.1186/ar2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dolff S, Quandt D, Wilde B, Feldkamp T, Hua F, Cai X, Specker C, Kribben A, Kallenberg CG, Witzke O. Increased expression of costimulatory markers CD134 and CD80 on interleukin-17 producing T cells in patients with systemic lupus erythematosus. Arthritis Res Ther. 2010;12:R150. doi: 10.1186/ar3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ray JP, Marshall HD, Laidlaw BJ, Staron MM, Kaech SM, Craft J. Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity. 2014;40:367–377. doi: 10.1016/j.immuni.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sengupta TK, Talbot ES, Scherle PA, Ivashkiv LB. Rapid inhibition of interleukin-6 signaling and Stat3 activation mediated by mitogen-activated protein kinases. Proc Natl Acad Sci USA. 1998;95:11107–11112. doi: 10.1073/pnas.95.19.11107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eto D, Lao C, DiToro D, Barnett B, Escobar TC, Kageyama R, Yusuf I, Crotty S. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS One. 2011;6:e17739. doi: 10.1371/journal.pone.0017739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Axtell RC, Xu L, Barnum SR, Raman C. CD5-CK2 binding/activation-deficient mice are resistant to experimental autoimmune encephalo-myelitis: protection is associated with diminished populations of IL-17-expressing T cells in the central nervous system. J Immunol. 2006;177:8542–8549. doi: 10.4049/jimmunol.177.12.8542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lexberg MH, Taubner A, Albrecht I, Lepenies I, Richter A, Kamradt T, Radbruch A, Chang HD. IFN-γ and IL-12 synergize to convert in vivo generated Th17 into Th1/Th17 cells. Eur J Immunol. 2010;40:3017–3027. doi: 10.1002/eji.201040539. [DOI] [PubMed] [Google Scholar]
- 68.Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W, Evans JG, Cimaz R, Bajaj-Elliott M, Wedderburn LR. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci USA. 2010;107:14751–14756. doi: 10.1073/pnas.1003852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schluns KS, Kieper WC, Jameson SC, Lefrançois L. Interleukin-7 mediates the homeostasis of naı¨ve and memory CD8 T cells in vivo. Nat Immunol. 2000;1:426–432. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 70.Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, Surh CD. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci USA. 2001;98:8732–8737. doi: 10.1073/pnas.161126098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fry TJ, Mackall CL. The many faces of IL-7: from lymphopoiesis to peripheral T cell maintenance. J Immunol. 2005;174:6571–6576. doi: 10.4049/jimmunol.174.11.6571. [DOI] [PubMed] [Google Scholar]
- 72.Gudmundsdottir H, Turka LA. A closer look at homeostatic proliferation of CD4+ T cells: costimulatory requirements and role in memory formation. J Immunol. 2001;167:3699–3707. doi: 10.4049/jimmunol.167.7.3699. [DOI] [PubMed] [Google Scholar]
- 73.Kieper WC, Troy A, Burghardt JT, Ramsey C, Lee JY, Jiang HQ, Dummer W, Shen H, Cebra JJ, Surh CD. Recent immune status determines the source of antigens that drive homeostatic T cell expansion. J Immunol. 2005;174:3158–3163. doi: 10.4049/jimmunol.174.6.3158. [DOI] [PubMed] [Google Scholar]
- 74.Min B, Yamane H, Hu-Li J, Paul WE. Spontaneous and ho-meostatic proliferation of CD4 T cells are regulated by different mechanisms. J Immunol. 2005;174:6039–6044. doi: 10.4049/jimmunol.174.10.6039. [DOI] [PubMed] [Google Scholar]
- 75.Prlic M, Blazar BR, Khoruts A, Zell T, Jameson SC. Homeo-static expansion occurs independently of costimulatory signals. J Immunol. 2001;167:5664–5668. doi: 10.4049/jimmunol.167.10.5664. [DOI] [PubMed] [Google Scholar]
- 76.Ernst B, Lee DS, Chang JM, Sprent J, Surh CD. The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity. 1999;11:173–181. doi: 10.1016/s1074-7613(00)80092-8. [DOI] [PubMed] [Google Scholar]
- 77.Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, Srivastava M, Divekar DP, Beaton L, Hogan JJ, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. 2011;17:975–982. doi: 10.1038/nm.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nurieva RI, Liu X, Dong C. Yin-Yang of costimulation: crucial controls of immune tolerance and function. Immunol Rev. 2009;229:88–100. doi: 10.1111/j.1600-065X.2009.00769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frey O, Meisel J, Hutloff A, Bonhagen K, Bruns L, Kroczek RA, Morawietz L, Kamradt T. Inducible costimulator (ICOS) blockade inhibits accumulation of polyfunctional T helper 1/T helper 17 cells and mitigates autoimmune arthritis. Ann Rheum Dis. 2010;69:1495–1501. doi: 10.1136/ard.2009.119164. [DOI] [PubMed] [Google Scholar]
- 80.Ansari MJ, Fiorina P, Dada S, Guleria I, Ueno T, Yuan X, Trikudanathan S, Smith RN, Freeman G, Sayegh MH. Role of ICOS pathway in autoimmune and alloimmune responses in NOD mice. Clin Immunol. 2008;126:140–147. doi: 10.1016/j.clim.2007.07.019. [DOI] [PubMed] [Google Scholar]