

Abstract
Objective:
To conduct a clinicopathologic study to characterize clinical and neuropathologic features associated with cognitive impairment in participants with no neuritic amyloid plaques (primary age-related tauopathy [PART] definite) and sparse neuritic plaques (amyloid sparse).
Methods:
Using the National Alzheimer's Coordinating Center database, we identified 377 individuals who were PART definite (n = 170) or amyloid sparse (n = 207), clinically examined within 1 year of death, and autopsied at 1 of 26 National Institute on Aging–funded Alzheimer's Disease Centers. Factors associated with the odds of being symptomatic (global Clinical Dementia Rating [CDR] score >0) were identified with multivariable logistic regression.
Results:
PART-definite participants less often had a high Braak neurofibrillary tangle stage V or VI (4%) compared to amyloid sparse participants (28%, p < 0.001). Of the PART-definite participants, 98 were symptomatic and 72 asymptomatic according to their global CDR scores. PART-definite participants were less often symptomatic (58%) compared with amyloid sparse participants (80%, p < 0.001). Within the PART-definite group, independent predictors of symptomatic status included depression (adjusted odds ratio [aOR] 4.20, 95% confidence interval [CI] 2.15–8.19), Braak stage (aOR 1.42, 95% CI 1.04–1.95), and history of stroke (aOR 8.09, 95% CI 2.63–24.82). Within the amyloid sparse group, independent predictors of symptomatic status included education (aOR 0.80, 95% CI 0.65–0.99), Braak stage (aOR 1.91, 95% CI 1.07–3.43), and amyloid angiopathy (aOR 2.75, 95% CI 1.14–6.64).
Conclusions:
These findings support the hypothesis that participants with PART have an amyloid-independent dementing Alzheimer disease–like temporal lobe tauopathy.
Neurofibrillary tangles (NFTs) composed of the tau protein occur in a broad range of neurodegenerative diseases called tauopathies and in Alzheimer disease (AD) seem to develop secondarily to β-amyloid that accumulates as plaques.1,2 NFTs also develop independently of amyloid plaques in primary tauopathies such as frontotemporal dementia with tau gene mutation and progressive supranuclear palsy.3 In AD, cross-sectional autopsy studies suggest that NFTs develop in the medial temporal lobe early and then progress to neocortical regions.4 Occasional NFTs in the medial temporal lobe are ubiquitous in brain aging, and some patients with dementia without amyloid plaques, tau mutations, or other classifiable tauopathies display severe end-stage tauopathy consistent with Braak stage IV of less.5–8 Consequently, recent consensus criteria classified individuals with this spectrum of tau pathology as having primary age-related tauopathy (PART).9
PART dementia is frequently misdiagnosed as AD.10 PART NFTs affect brain regions that also are affected in early to moderate-stage AD (e.g., the medial temporal lobe) and in all other respects are indistinguishable from AD NFTs.11 However, individuals with PART usually display milder cognitive symptoms than patients with AD.9 Some evidence suggests that subjective memory complaints are common in PART12 and that PART dementia is more common in women and the oldest old.13 Our study aimed to investigate the clinical and neuropathologic features associated with PART to better characterize PART and to facilitate differentiation from other tauopathies, especially AD.
METHODS
Participants.
We used cross-sectional data from the National Alzheimer's Coordinating Center's (NACC’s) Uniform Data Set (UDS) and Neuropathology Data Set, data originating from 26 past and present US Alzheimer's Disease Centers (ADCs). Since 2005, ADCs have collected demographic, neuropsychological, clinical, and diagnostic data on those with normal cognition, mild cognitive impairment (MCI), and dementia. ADC participants are enrolled from various sources including clinic samples, other existing studies, and participant referrals and thus are considered a clinical case series. The UDS and Neuropathology Data Set data are described in detail elsewhere.14–16 We used data collected between September 2005 and December 2015.
Standard protocol approvals, registration, and patient consents.
All participants provided written informed consent at each ADC, and the University of Washington's Institutional Review Board approved the use of NACC data for research.
Inclusion and exclusion criteria.
The sample was restricted to those with a neuropathologic examination and a UDS visit ≤1 year before death (figure e-1 at Neurology.org). PART-definite participants had no neuritic plaques (NPs), and amyloid sparse (AS) participants had sparse NPs. It is debatable whether participants with sparse NPs should be considered PART possible or as having mild AD; thus, we have chosen AS, a neutral descriptive term. Braak stage 0 cases were assumed to have very mild PART, which is ubiquitous in aging.5
Given the scant studies characterizing PART to date, we focused on PART in its purest form and excluded 483 participants with clinical or neuropathologic Lewy body disease, including Parkinson disease; tau pathologies, including corticobasal degeneration, Picks disease, progressive supranuclear palsy, and other frontotemporal lobar degeneration (FTLD) tau-positive neuropathologies; other neurodegenerative disease, including Huntington disease, clinical or neuropathologic prion disease, and tau-negative FTLD (e.g., FTLD with TDP-43 inclusions); and other diseases, including normal-pressure hydrocephalus, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, and clinically diagnosed syndromes (e.g., primary progressive aphasia) frequently associated with FTLD.
Asymptomatic and symptomatic PART.
The Clinical Dementia Rating (CDR) scale, a global measure of dementia severity,17 was completed by a clinician at each UDS visit on the basis of informant interview and examination (range 0 [no impairment] to 3 [greatest impairment]). Participants with global CDR score = 0 at their last visit were categorized as asymptomatic and those with CDR score >0 were categorized as symptomatic.
Neuropathology.
ADCs use a standardized Neuropathology Form to collect data on UDS participants who died and consented to autopsy and neuropathologic examination. The neuropathology data were used to assess PART-definite and AS pathology (defined further above) and the presence of diffuse plaques, originally ascertained as none, sparse, moderate, or frequent. For our regression analyses, diffuse plaques were defined as any (sparse, moderate, frequent) vs none. In addition, we described the following cerebrovascular disease findings: lacune/infarct, microinfarct, hemorrhage/microbleed, atherosclerosis of the circle of Willis, arteriolosclerosis, subcortical arteriosclerotic leukoencephalopathy, medial temporal lobe/hippocampal sclerosis (HS), amyloid angiopathy, and cortical laminar necrosis. The Neuropathology Form collects whether atherosclerosis of the circle of Willis, arteriolosclerosis, and amyloid angiopathy were mild, moderate, severe, or not present, and in our regression analyses, we defined these as any (mild, moderate, or severe) vs none.
Demographic, clinical, and genetic variables.
The demographic characteristics included age at last visit (years), sex, education (years), and race (nonwhite, white). A clinical diagnosis of normal cognition, MCI/impaired not MCI, or dementia was based on the participant's medical history, neurologic examination, and neuropsychological testing. Participants and their coparticipants reported history of stroke, hypertension, diabetes mellitus, and traumatic brain injury (TBI), and these variables were dichotomized as yes (recent [active/treated in last year] or remote) or no (absent). Depression included clinically diagnosed or self- or coparticipant-reported depression ≤2 years preceding the visit. Participants and their coparticipants reported if a first-degree family member had cognitive impairment (including dementia). APOE genotype was dichotomized into no ε4 alleles or ≥1 ε4 alleles (9.5% missing APOE data).
Statistical analysis.
We described the PART-definite and AS groups (separately) by their global CDR score, presence of NP (none or sparse), and Braak stage (0–VI). We then used descriptive statistics (mean and SD, frequency and percent) to describe the symptomatic and asymptomatic PART-definite and AS groups (separately) by demographics, presence of ≥1 APOE ε4 allele, family history of cognitive impairment, medical history, and other neuropathology (diffuse plaques and cerebrovascular pathology). Differences between symptomatic and asymptomatic participants were tested with χ2 tests or exact χ2 tests if any cell size included <5 participants.
Main analyses.
We used logistic regression models with generalized estimating equations to examine whether being symptomatic (vs asymptomatic) was associated with age (continuous measure); education (continuous measure); sex; race; depression; stroke or hypertension; diabetes; TBI; family history of cognitive impairment; ≥1 APOE ε4 allele; Braak stage (0 through VI); presence of any diffuse plaques; or presence of cerebrovascular disease at autopsy (e.g., infarct/lacune). Generalized estimating equations accounted for clustering by center. The unadjusted and adjusted logistic regression analyses were conducted separately for the PART-definite and AS groups. The unadjusted results informed which variables were included in the multivariable models (age was automatically included) on the basis of significance at an α level of 0.10 observed in either the PART-definite or AS group. We ran 2 separate multivariable logistic regression models, 1 for the PART-definite and 1 for the AS group. Statistical significance was determined with an α level of 0.05.
Sensitivity analyses.
The fledgling understanding of PART and growing use of the term prompted a number of sensitivity analyses to ascertain the extent to which adjusting our definition of PART influenced our findings. While PART is generally considered a temporal lobe–predominant pathology, the challenges in operationalizing the Braak staging system18 and theoretical possibility of comorbid/nascent amyloid-driven tauopathy suggested that we explore the full spectrum of tau pathology in our sample. The sensitivity analyses used the same regression models as the main analysis. The first sensitivity analysis combined the PART-definite and AS participants, while the second kept the PART-definite and AS groups separate but excluded those with HS. None of the asymptomatic participants with PART had HS; thus, the regression model would not run with HS included as a covariate. Excluding those with HS ensured that the symptomatic and asymptomatic groups were comparable with respect to that pathology. The third sensitivity analysis excluded participants with Braak stage V to VI and combined the PART-definite and AS groups. The fourth sensitivity analysis replaced the comparison of CDR score >0 vs 0 with a comparison of those with a clinical diagnosis of cognitive impairment (MCI/impaired not MCI, dementia) vs no clinical diagnosis of cognitive impairment. The final sensitivity analyses included the 483 participants with comorbidities who were previously excluded, and the PART-definite and AS multivariable models were run including variables found to be statistically significant at p < 0.10 in updated unadjusted analyses and included tau pathologies, Lewy body dementia pathologies, and other neurodegenerative disease as covariates.
RESULTS
We identified 377 participants (mean age 85.9 years, SD = 10.0 years) meeting our criteria (table e-1 and figure e-1). The sample consisted of 98 symptomatic PART-definite, 72 asymptomatic PART-definite, 165 symptomatic AS, and 42 asymptomatic AS participants. PART definite overlaps with the “not AD neuropathologic change” and AS with the “low AD neuropathologic change” categories set forth by the National Institute on Aging (NIA)–Alzheimer's Association guidelines for AD neuropathologic assessment.19 PART-definite participants were less often symptomatic (58%) than AS participants (80%) (χ2 p < 0.001). Although the majority of the PART-definite and AS participants had a CDR score ≤ 1, more of the AS group (39.1%) had a CDR score >1 than the PART-definite group (18.2%) (figure 1, A and B and table e-1). Only 4.1% of the PART-definite group had Braak stage V to VI, compared to 28.0% of the AS group (χ2 p < 0.001) (figure 1, A and B and table e-1). There was no change in the proportion who were symptomatic with increasing age in the PART-definite and AS groups (figure 1, C and D).
Figure 1. NFT stage and age by CDR and NP scores.

CDR = Clinical Dementia Rating; NP = neuritic plaques; NFT = neurofibrillary tangle.
A higher percentage of the symptomatic than asymptomatic participants in the PART definite and AS groups were depressed and had ≥1 APOE ε4 allele (table 1). The distribution of participants by age, education, race, family history of cognitive impairment, and history of diabetes mellitus, hypertension, and TBI was fairly similar across the symptomatic and asymptomatic PART-definite and AS groups. In the PART-definite group, the symptomatic participants more often had a history of stroke.
Table 1.
Clinical and demographic characteristics of participants with PART at autopsy
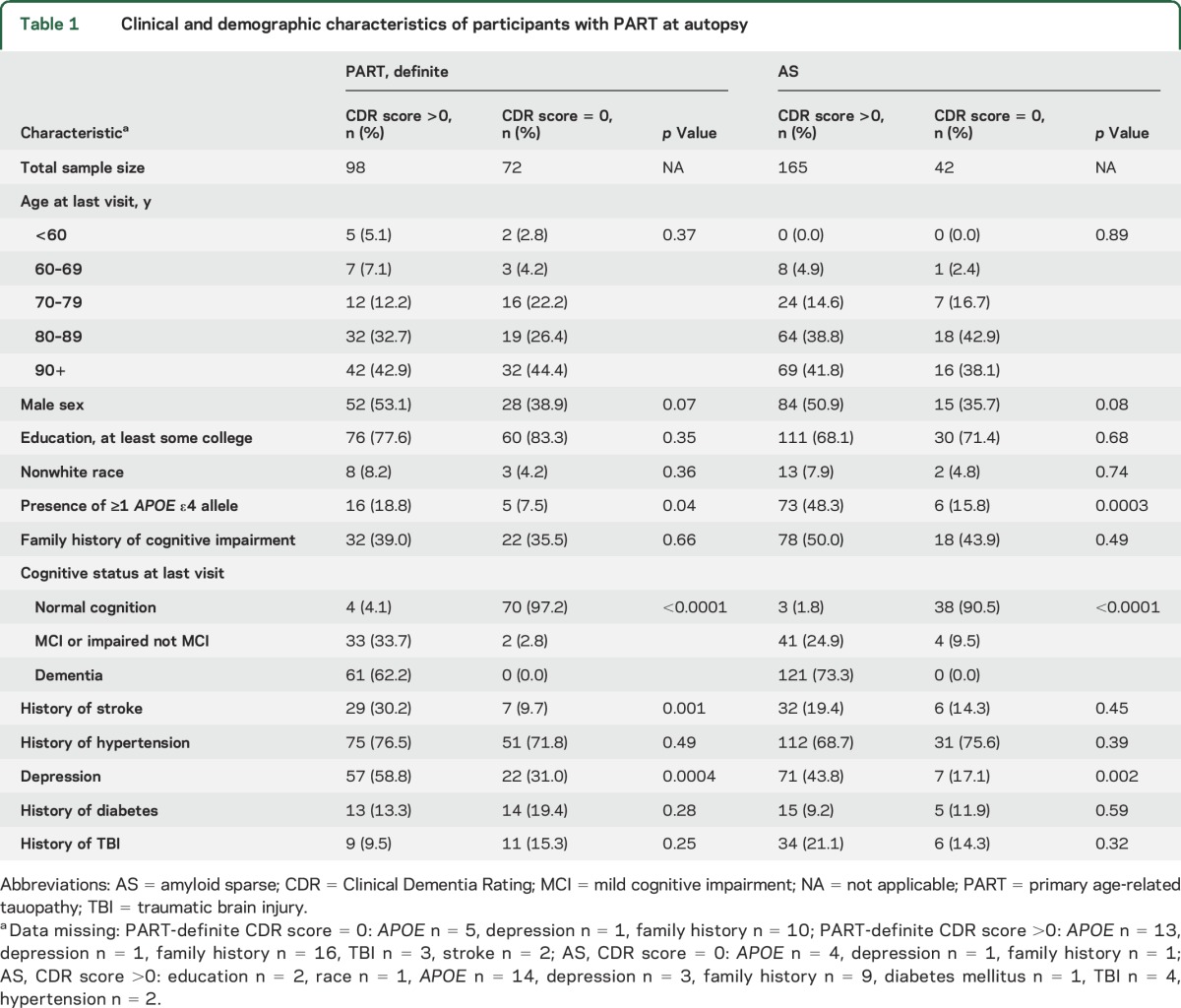
While the main amyloid-related neuropathologic variable used for participant stratification was neuritic amyloid plaque burden, we also considered diffuse amyloid deposits. Diffuse plaques were absent in 68% of the PART-definite group but <2% of the AS group (table 2). Sparse diffuse plaques were present in 20% of the PART-definite and 66% of the AS groups. The percent with moderate or frequent diffuse plaques was similar between the symptomatic and asymptomatic participants within the PART-definite and AS groups.
Table 2.
Neuropathologic characteristics of participants with PART at autopsy

The symptomatic and asymptomatic individuals in the PART-definite and AS groups were similar with respect to the percent with lacune or infarct, microinfarct, hemorrhage or microbleed, atherosclerosis, arteriolosclerosis, cortical laminar necrosis, and subcortical arteriosclerotic leukoencephalopathy (table 2). In the AS but not the PART-definite group, symptomatic participants more often had amyloid angiopathy than asymptomatic participants. HS was present in ≈12% of symptomatic PART-definite and AS participants, respectively, but was absent in the asymptomatic PART-definite and AS participants.
Main analyses.
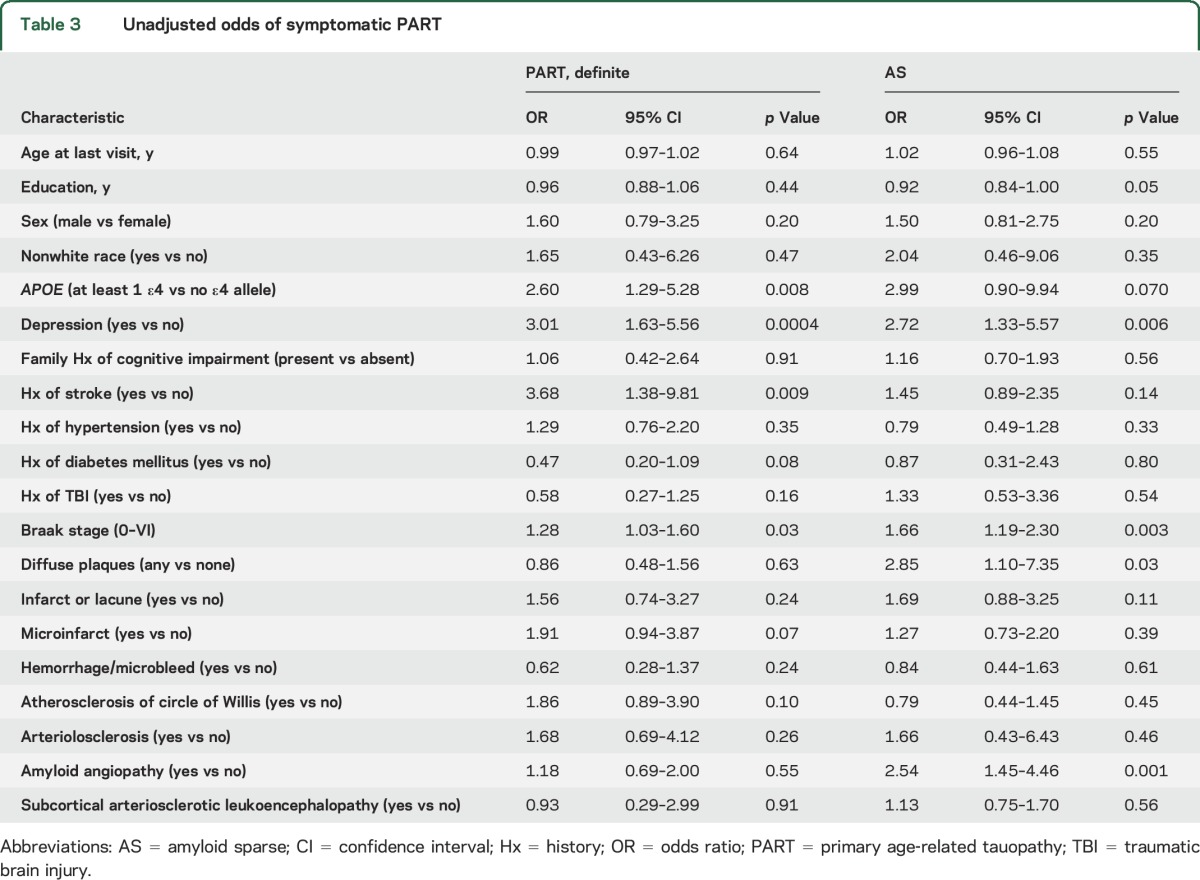
In the unadjusted analyses (table 3), depression, history of stroke, ≥1 APOE ε4 alleles, and Braak stage were associated with being symptomatic in the PART-definite group. Depression, Braak stage, diffuse plaques, and amyloid angiopathy were associated with being symptomatic in the AS group.
Table 3.
Unadjusted odds of symptomatic PART
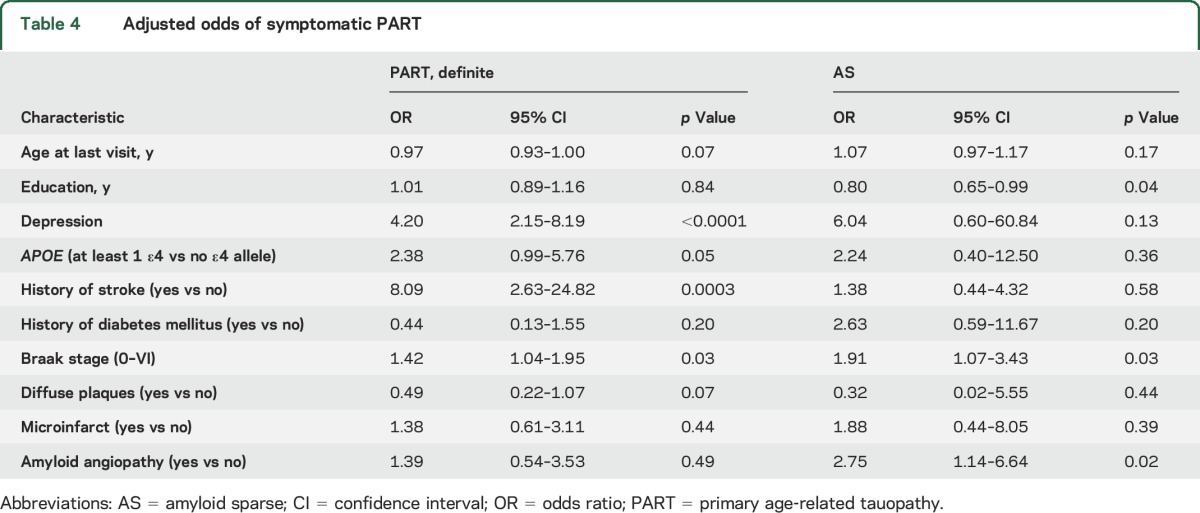
In the adjusted analyses (table 4), increasing Braak stage was associated with an increased odds of being symptomatic in the PART-definite and AS groups. In the PART-definite group, history of stroke and depression were associated with an increased odds of being symptomatic. In the AS group, being symptomatic was additionally associated with lower education and amyloid angiopathy.
Table 4.
Adjusted odds of symptomatic PART
Sensitivity analyses.
Combining the PART-definite and AS participants into 1 group resulted in an adjusted association between depression, increasing Braak stage, and history of stroke and being symptomatic (table e-2). After participants with HS were removed, the same variables were associated with being symptomatic as found in the main analyses (table e-3). After those with Braak stage V to VI were excluded and the PART-definite and AS groups were combined, being symptomatic was associated with less education, depression, increasing Braak stage, and history of stroke (table e-4). Comparisons based on clinical diagnosis of cognitive impairment vs no cognitive impairment instead of based on CDR score resulted in similar findings, except that increasing age was now a significant predictor and Braak stage was no longer a statistically significant predictor of being symptomatic among PART-definite participants (table e-5). In the final sensitivity analysis, depression, increasing Braak stage, presence of tau pathologies, and presence of other neurodegenerative disease were associated with being symptomatic in the PART-definite group, and male sex, depression, increasing Braak stage, presence of other neurodegenerative disease, and presence of Lewy body disease were associated with being symptomatic in the AS group (table e-6).
DISCUSSION
Understanding the selective vulnerability of the medial temporal lobe to neurofibrillary degeneration has the potential to reveal factors that trigger neurodegeneration and to pave the way toward new methods to diagnose and treat patients with dementia at the earliest stages. While temporal lobe tangles are generally thought to be driven by amyloid toxicity, the possibility that an alternative pathway exists has been proposed9,20; however, this is controversial.21,22 Nevertheless, there is a growing interest in whether PART is best considered a component of the AD spectrum despite individuals having no or minimal amyloid but who nonetheless develop tauopathy.20 Here, by examining the clinical and neuropathologic features of such individuals, we made a number of observations that support the hypothesis that PART is a dementing illness that might represent such an alternative pathway.
The finding that the absence of NPs was associated with a restricted distribution of tau pathology (Braak NFT stage ≤IV) is consistent with previous reports and further supports the assertions that PART is predominantly a temporal lobe pathology and that isocortical spread of NFTs is highly correlated with the presence of amyloid pathology.9 This difference in regional distribution may reflect differences in pathologic drivers but might alternatively indicate an interaction whereby amyloid facilitates spread of tau pathology.23 Our PART sample had normal cognition, MCI, or dementia, as previously reported.9 We further found that Braak stage was associated with an increased odds of cognitive impairment in both the PART-definite and AS groups, before and after the exclusion of the Braak V to VI cases. While these findings are consistent with previous autopsy studies demonstrating that severe PART is correlated with cognitive impairment,24,25 more sensitive or quantitative measures of tangle burden might be necessary to demonstrate robust associations at milder NFT disease stages, which are difficult to accurately assess neuropathologically with the Braak system.18
AS participants were significantly more likely to be symptomatic. In addition to considering neuritic amyloid plaques, which were the basis for our PART-definite and AS groups, we looked at a number of other amyloid-related variables. The presence of an APOE ε4 allele, which is strongly associated with amyloid deposition, was associated with being symptomatic in the PART-definite group in our main unadjusted analysis. In addition, the presence of diffuse amyloid plaques was found to be associated with symptomatology in the unadjusted analysis, but only in the AS group. However, neither of these factors survived adjustment for multiple covariates. In contrast, amyloid angiopathy, which is associated with cognitive impairment independently of Alzheimer pathology,26 was strongly associated with cognitive impairment even after adjustment, but only in the AS group.
Curiously, symptomatic PART was associated with depressive symptoms in our adjusted main analysis and our sensitivity analyses. Notably, self-reported memory complaints have previously been shown to be associated with depression,27 and depression has been found to be a risk factor for Parkinson disease and vascular and AD dementia.28,29 Therefore, similar to other neurodegenerative diseases, depression may be a risk factor or early symptom of symptomatic PART. Alternatively, the underlying disease process involved in PART may cause depression.
Sex differences have become an increasingly important issue in AD research. While previous studies have suggested that PART dementia might be more common among women,13 our cohort had a higher number of symptomatic male participants. The reason for the inconsistency may be that our study combined individuals with MCI and dementia in the symptomatic group. In addition, the referenced study13 was not focused on comparing symptomatic and asymptomatic individuals with PART but compared individuals with dementia with PART to those with traditional “plaque and tangle” AD.
While this study had some notable strengths, including the relatively large sample size and the rich clinical and neuropathologic data, there are a number of important limitations. First, it was based on a convenience sample, which limits its generalizability to other populations. Second, a larger sample size may have increased our ability to detect associations. In addition, clinically confirmed medical conditions would have been preferred over the self-reported conditions available in the UDS. With respect to neuropathologic data, Thal amyloid phase has recently been incorporated into consensus recommendations for the neuropathologic diagnosis of AD,30 but these data are not available for most participants in the NACC cohort. Nevertheless, recent studies suggest that Thal phase might not substantially contribute to predicting antemortem cognition compared with Consortium to Establish a Registry for Alzheimer's Disease NP scores and Braak NFT stages.31
Our findings provide evidence that PART is an amyloid-independent tauopathy that is associated with cognitive impairment. While APOE ε4 genotype and presence of diffuse plaques were not associated with symptomatic PART, increasing Braak stage and depression were consistently associated with being symptomatic. After controlling for the presence of depression and stroke and after excluding and alternatively controlling for comorbid pathologies, we found that increasing Braak stage in the absence of amyloid NPs was associated with being symptomatic, and these findings persisted in our various sensitivity analyses. Thus, our study supports the hypothesis that PART is a distinct (i.e., amyloid-independent) pathology associated with cognitive impairment. Future work will need to confirm our results in other representative and diverse cohorts and to examine more quantitative measures of tangle burden to further refine the understanding of PART pathology and its associated clinical and neuropathologic features.
Supplementary Material
ACKNOWLEDGMENT
The authors acknowledge the patients and families enrolled at the ADCs who contributed data to the UDS and the faculty and staff of the ADCs who conducted the evaluations and collected the data used in these analyses. The authors also thank the NIA, which provided support for the ADCs and NACC, as well as NACC staff (Duane Beekly, George Thomas, Mark Bollenbeck, Janene Hubbard, Mary Jacka, Joylee Wu, Elizabeth Robichaud, Nicole Barlow, Simone Wilk, Merilee Teylan, Kristen Schwabe-Fry, and Margaret Dean) who help in programming of the data submission systems, data management, research coordination, and administration, without whom this research would not be possible.
GLOSSARY
- AD
Alzheimer disease
- ADC
Alzheimer's Disease Centers
- AS
amyloid sparse
- CDR
Clinical Dementia Rating
- FTLD
frontotemporal lobar degeneration
- HS
hippocampal sclerosis
- MCI
mild cognitive impairment
- NACC
National Alzheimer's Coordinating Center
- NFT
neurofibrillary tangle
- NIA
National Institute on Aging
- NP
neuritic plaque
- PART
primary age-related tauopathy
- TBI
traumatic brain injury
- UDS
Uniform Data Set
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Besser helped with the study design, performed the analyses, and wrote the manuscript. Dr. Crary helped with the study design and wrote the manuscript. Dr. Mock helped with the study design and idea conception and edited the manuscript. Dr. Kukull supervised the study and helped with the study design.
STUDY FUNDING
The NACC database is funded by NIA/NIH grant U01 AG016976. NACC data are contributed by the NIA-funded ADCs: P30 AG019610 (principal investigator [PI] Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P50 AG047266 (PI Todd Golde, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P50 AG005134 (PI Bradley Hyman, MD, PhD), P50 AG016574 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Steven Ferris, PhD), P30 AG013854 (PI M. Marsel Mesulam, MD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P50 AG047366 (PI Victor Henderson, MD, MS), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P50 AG016570 (PI Marie-Francoise Chesselet, MD, PhD), P50 AG005131 (PI Douglas Galasko, MD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P50 AG005136 (PI Thomas Montine, MD, PhD), P50 AG033514 (PI Sanjay Asthana, MD, FRCP), P50 AG005681 (PI John Morris, MD), and P50 AG047270 (PI Stephen Strittmatter, MD, PhD). We also acknowledge the following funding sources: NIH grants R01 NS095252 (PI Dr. Crary) and R01AG054008 (PI Dr. Crary) and Alzheimer's Association NIRG-15-363188 (PI Dr. Crary).
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Jack CR, Knopman DS, Weigand SD, et al. An operational approach to National Institute on Aging-Alzheimer's Association criteria for preclinical Alzheimer disease. Ann Neurol 2012;71:765–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Irwin DJ. Tauopathies as clinicopathological entities. Parkinsonism Relat Disord 2016;22(1 suppl):S29–S33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction. Eur Neurol 1993;33:403–408. [DOI] [PubMed] [Google Scholar]
- 5.Bouras C, Hof PR, Giannakopoulos P, Michel JP, Morrison JH. Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: a quantitative evaluation of a one-year autopsy population from a geriatric hospital. Cereb Cortex 1994;4:138–150. [DOI] [PubMed] [Google Scholar]
- 6.Ulrich J, Spillantini M, Goedert M, Dukas L, Staehelin H. Abundant neurofibrillary tangles without senile plaques in a subset of patients with senile dementia. Neurodegeneration 1992;1:257–284. [Google Scholar]
- 7.Santa-Maria I, Haggiagi A, Liu X, et al. The MAPT H1 haplotype is associated with tangle-predominant dementia. Acta Neuropathol 2012;124:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamada M. Senile dementia of the neurofibrillary tangle type (tangle-only dementia): neuropathological criteria and clinical guidelines for diagnosis. Neuropathology 2003;23:311–317. [DOI] [PubMed] [Google Scholar]
- 9.Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014;128:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beach TG, Monsell SE, Phillips LE, Kukull W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005-2010. J Neuropathol Exp Neurol 2012;71:266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jellinger KA, Bancher C. Senile dementia with tangles (tangle predominant form of senile dementia). Brain Pathol 1998;8:367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kryscio RJ, Abner EL, Jicha GA, et al. Self-reported memory complaints: a comparison of demented and unimpaired outcomes. J Prev Alzheimers Dis 2016;3:13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jellinger K, Attems J. Tangle dominant dementia: comparison with classical Alzheimer disease. Neuropathol Appl Neurobiol 2011;37:11. [Google Scholar]
- 14.Morris JC, Weintraub S, Chui HC, et al. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord 2006;20:210–216. [DOI] [PubMed] [Google Scholar]
- 15.Beekly DL, Ramos EM, Lee WW, et al. The National Alzheimer's Coordinating Center (NACC) database: the Uniform Data Set. Alzheimer Dis Assoc Disord 2007;21:249–258. [DOI] [PubMed] [Google Scholar]
- 16.Beekly DL, Ramos EM, van Belle G, et al. The National Alzheimer's Coordinating Center (NACC) database: an Alzheimer disease database. Alzheimer Dis Assoc Disord 2004;18:270–277. [PubMed] [Google Scholar]
- 17.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 18.Alafuzoff I, Arzberger T, Al-Sarraj S, et al. Staging of neurofibrillary pathology in Alzheimer's disease: a study of the Brainnet Europe Consortium. Brain Pathol 2008;18:484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crary JF. Primary age-related tauopathy and the amyloid cascade hypothesis: the exception that proves the rule? J Neurol Neuromedicine 2016;1:53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duyckaerts C, Braak H, Brion JP, et al. PART is part of Alzheimer disease. Acta Neuropathol 2015;129:749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braak H, Del Tredici K. Are cases with tau pathology occurring in the absence of Abeta deposits part of the AD-related pathological process? Acta Neuropathol 2014;128:767–772. [DOI] [PubMed] [Google Scholar]
- 23.Jack CR. PART and SNAP. Acta Neuropathol 2014;128:773–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikeda K, Akiyama H, Arai T, et al. Clinical aspects of “senile dementia of the tangle type”: a subset of dementia in the senium separable from late-onset Alzheimer's disease. Dement Geriatr Cogn Disord 1999;10:6–11. [DOI] [PubMed] [Google Scholar]
- 25.Nelson PT, Abner EL, Schmitt FA, et al. Brains with medial temporal lobe neurofibrillary tangles but no neuritic amyloid plaques are a diagnostic dilemma but may have pathogenetic aspects distinct from Alzheimer disease. J Neuropathol Exp Neurol 2009;68:774–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arvanitakis Z, Leurgans SE, Wang Z, Wilson RS, Bennett DA, Schneider JA. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol 2011;69:320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benito-Leon J, Mitchell AJ, Vega S, Bermejo-Pareja F. A population-based study of cognitive function in older people with subjective memory complaints. J Alzheimers Dis 2010;22:159–170. [DOI] [PubMed] [Google Scholar]
- 28.Diniz BS, Butters MA, Albert SM, Dew MA, Reynolds CF. Late-life depression and risk of vascular dementia and Alzheimer's disease: systematic review and meta-analysis of community-based cohort studies. Br J Psychiatry 2013;202:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leentjens AF. Parkinson disease: depression-risk factor or early symptom in Parkinson disease? Nat Rev Neurol 2015;11:432–433. [DOI] [PubMed] [Google Scholar]
- 30.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002;58:1791–1800. [DOI] [PubMed] [Google Scholar]
- 31.Serrano-Pozo A, Qian J, Muzikansky A, et al. Thal amyloid stages do not significantly impact the correlation between neuropathological change and cognition in the Alzheimer disease continuum. J Neuropathol Exp Neurol 2016;75:516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.