

Abstract
A number of recent studies have highlighted that splicing is frequently altered in cancer and that mutations affecting splicing of key cancer-associated genes as well as mutations and copy-number changes affecting spliceosomal proteins themselves are enriched in cancer. In parallel, there is also accumulating evidence that several molecular subtypes of cancer are highly dependent on wildtype splicing function for cell survival. These findings have resulted in a growing interest in targeting splicing catalysis, splicing regulatory proteins, and/or individual key altered splicing events in the treatment of cancer. In this review we present strategies that exist and are in development to target altered dependency on the spliceosome as well as aberrant splicing in cancer. These include drugs to target global splicing in cancer subtypes which are preferentially dependent on wildtype splicing for survival, methods to alter post-translational modifications of splice regulatory proteins, and strategies to modulate pathologic splicing events and protein/RNA interactions in cancer.
Introduction
The majority of genes in the human genome consist of multiple exons interspersed with introns that undergo splicing to form mature mRNA and protein products1,2. While alternative splicing (AS) provides cells a means to diversify the proteome, recent studies have revealed multiple ways by which splicing is pathologically altered to promote the initiation and/or maintenance of cancer. These include mutations affecting splicing regulatory sequences of critical cancer-associated genes3,4 as well as mutations5-12 and gene expression alterations13-15 affecting core and/or accessory components of the spliceosome complex (Figure 1A). Consistent with this, systematic transcriptomic analyses across cancer types have revealed widespread alterations in alternative as well as constitutive splicing relative to normal tissue counterparts3,4,16-18. These findings highlight the possibility that manipulation of splicing might provide therapeutic benefit in cancer. Splicing requires multiple protein/protein and protein/RNA interactions and is directed by a number of trans-acting proteins, which themselves are subjected to regulation by post-translational modifications and protein/RNA interactions for normal function. This multitude of interactions and regulatory steps provide a wide array of means to manipulate the splicing cascade for therapeutic purposes. In this review, we highlight the current and developing strategies to target aberrant splicing events as well as enhanced dependency on the spliceosome in cancer.
Figure 1. Diverse mechanisms by which alterations in splicing promote cancer and treatment resistance.
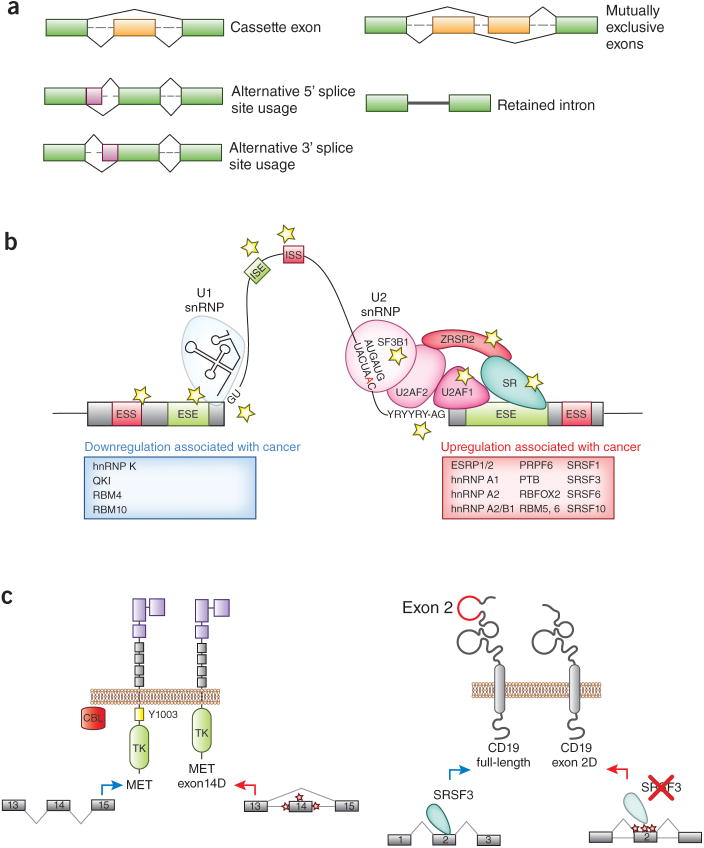
(A) The cis-regulatory sequences and trans-acting splicing factors affected by mutations in cancer are indicated by stars in the figure. Additional splicing regulatory factors whose upregulation have been shown to promote tumorigenesis are shown in the red box while those that have been demonstrated to function as tumor suppressors are shown in the blue box. (B) Examples of therapeutically important splicing alterations that promote cancer and/or resistance to cancer therapy. On the left is an example of mutations affecting splicing of MET exon 14 to promote a specific isoform of MET which lacks the juxtamembrane domain. Mutations affecting intronic sequences at the 5′ or 3′ splice sites of MET result in a form of MET which lacks the residue within exon 14 required for CBL-mediated downregulation. Expression of MET exon 14Δ drives lung adenocarcinomas and sensitizes them to MET inhibitors. On the right is a recently described event affecting expression of CD19. Acquired mutations affecting exon 2 of CD19 result in a stable form of CD19 lacking exon 2 (“CD19 exon 2Δ”) which is not recognized by anti-CD19 chimeric antigen receptor T-cells. Moreover, SRSF3 promotes inclusion of exon 2 and downregulation of SRSF3 expression has similarly been suggested to result in expression of CD19 exon 2Δ.
The spliceosome and splicing regulation
Splicing is accomplished by a megadalton complex of RNA and proteins consisting of 5 small nuclear ribonucleoprotein particles (snRNPs; U1, U2, U4, U5, and U6 snRNPs) and more than 200 proteins. Several recent studies have elucidated the structure of numerous components of the spliceosome at unprecedented, near-angstrom resolution19-22. Basic mechanisms of normal RNA splicing have been reviewed in detail recently23-26 and require cis-regulatory elements and trans factors that bind to these elements to promote splicing and/or recruitment of the spliceosome. As described below, each of these elements may be subjected to various forms of dysregulation as a means to promote tumorigenesis (Figure 1A). The key cis elements necessary for splicing are located at intron/exon junctions at the 5′ and 3′ splice sites and the branch point. Additional sequences within exons (exonic splicing enhancers (ESEs) and exonic splicing silencers (ESSs)) and introns (intronic splicing enhancers (ISEs) and intronic splicing silencers (ISSs)) are critical for the correct recognition of exons and modulation of splicing outcome. These additional regulatory sequences are recognized by splice regulatory proteins which may be differentially expressed in tissues to regulate tissue-specific splicing patterns. Many of these auxiliary splicing regulatory proteins are members of the serine/arginine-rich (SR)27 and heterogeneous nuclear ribonucleoprotein (hnRNP)28 families but numerous additional proteins outside of these 2 families have also been shown to promote or repress splicing.
Splicing alterations in cancer
Systematic genomic analyses of the transcriptional characteristics of cancer as well as the coding and noncoding mutations present across cancers have repeatedly identified alterations affecting splicing in diverse cancer types. Prior to the public deposition of high-depth mRNA sequencing (RNA-seq) analyses from a wide variety of cancer types, analysis of cancer transcriptomes using expressed sequence tag libraries suggested that cancer cells exhibit “noisier” splicing than their normal tissue counterparts.17—that is cancer cells express an elevated rate of transcripts containing premature termination codons (PTCs), consistent with an increased rate of mis-splicing relative to normal tissues. Analyzing this data based on dividing genes into oncogenes versus tumor suppressors genes (TSGs) revealed a far greater frequency of PTCs in transcripts encoding TSGs compared with oncogenes, suggesting that this process is non-random17.
More recent RNA-seq analyses of tumor tissues compared with neighboring normal tissues from the same individuals have further elucidated global, cancer-associated splicing features. Analysis of RNA-seq data across 16 distinct cancer types from The Cancer Genome Atlas (TCGA) revealed that common RNA processing differences exist between cancer cells and their normal tissue counterparts18. Interestingly, almost all cancer types exhibited abnormalities in intron retention (IR) specifically, far more commonly than alterations in other types of splicing events including cassette exon splicing or 5′ or 3′ splice site recognition. Increased IR was observed to affect both constitutive as well as alternatively spliced introns18. While an overriding molecular explanation for alterations in IR across cancers was not evident from this work, several specific molecular alterations have been associated with increased IR amongst cancer types. For example, an association between loss of function mutations in SETD2 and increased IR have been observed in clear cell renal cell carcinoma and presumed to be associated with a loss of histone H3 lysine 36 trimethylation at target exons following SETD2 loss29.
Integration of RNA-seq analyses of cancer samples with whole exome and whole genome sequencing data from the same patients has provided further insights into the effects of mutations in cancer on RNA splicing. In these analyses, somatic, single nucleotide variants (SNVs) affecting splicing were associated with IR more commonly than cassette exon splicing or other categories of AS3,4. Moreover, SNVs causing IR were enriched in TSGs over oncogenes, potentially due to the fact that the majority of IR events were expected to result in generation of a PTC whereas only ∼50% of cassette exon splicing events would result in generation of a PTC4. This is consistent with several well-described and clinically important mutations activating proto-oncogenes by altering their splicing, including MET exon 14 splicing mutations in lung cancer30-32 (Figure 1B) and mutations activating a cryptic splice site to result in an aberrantly active form of NOTCH1 in chronic lymphocytic leukemia33. It is important to note that while mutations at intronic splicing donor or acceptor sites are commonly recognized as deleterious in cancer mutational studies, a substantial proportion of somatic mutations affecting splicing may occur as synonymous mutations within exons affecting ESE or ESS sequences3 (Figure 1A). Although synonymous exonic mutations might alter gene function in a variety of ways beyond affecting splicing, work by Supek et al. suggests that synonymous exonic mutations are enriched within oncogenes compared with TSGs and tend to cluster within 30 nucleotides of an exon boundary3. Such cancer-associated synonymous mutations close to exon boundaries appear to preferentially result in the gain of ESE motifs and loss of ESS motifs, a situation not frequently seen with synonymous mutations in TSGs3.
In parallel to the above work, numerous studies have now highlighted that splicing factors themselves are recurrently affected by somatic mutations in cancer (Figure 1A). The known mechanistic consequences of these mutations have been reviewed recently and it appears that these mutations, which predominantly affect SF3B15-7,9,10,12, U2AF15,11,34, SRSF25, and ZRSR25, confer an alteration of RNA splicing preference, distinct from those seen with loss of the wildtype protein35-37. Moreover, these mutations occur exclusively as heterozygous point mutations at restricted residues and, in disease where mutations in each of these genes are common, occur in a mutually exclusive manner with one another5. The mutual exclusivity of mutations in splicing factors along with their consistent heterozygous state, potentially suggests a requirement for wildtype splicing in the presence of a mutation in a splicing factor, a possibility that requires formal evaluation.
In addition to mutations altering splicing through changing the function of trans-acting splicing factors or altering splice sites of cancer-associated genes in cis, a series of studies have also identified numerous examples where altered expression of splicing regulatory proteins, even in the absence of mutation, themselves promote oncogenesis (Figure 1A and reviewed recently38). In addition, an elegant example where an alteration in splicing regulatory protein expression impacts response to cancer therapy was identified recently with the finding that alteration in SRSF3 expression in B-acute lymphoblastic leukemia (B-ALL) cells influences splicing of CD19, which, in turn, results in impaired recognition by anti-CD19 chimeric antigen receptor (CAR) T-cells39 (Figure 1B). Studying the mechanistic details by which alterations in splicing proteins contribute to cancer pathogenesis and/or treatment resistance will therefore be important given the potential methodologies described below to therapeutically target these events and/or their downstream consequences (Figure 2).
Figure 2. Methods by which splicing may be modulated for cancer therapy.
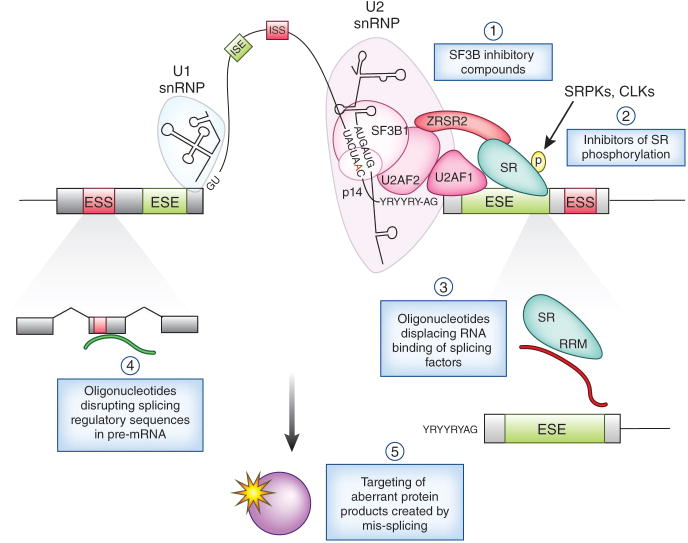
These include (1) use of a series of compounds that inhibit early spliceosome assembly by inhibiting SF3B1 and (2) inhibition of phosphorylation of Serine/Arginine-rich (SR) proteins through inhibition of CLKs (CDC2-like kinases) and/or SRPKs (SR protein kinases). In addition, use of oligonucleotides to (3) target the RNA binding activity of splicing regulatory proteins mutated or overexpressed in cancer, or (4) directly alter splicing of individual downstream mRNA's critical for cancer pathogenesis, may provide more selective tumor targeting. Alteration of specific events may be achieved by oligonucleotides that alter splicing by promoting or impairing the use of key splicing regulatory sequences (such as 5′/3′ splice sites, exonic splicing silencers (ESSs), exonic splicing enhancers (ESEs), intronic splicing enhancers (ISEs), and intronic splicing silencers (ISSs)). In addition, identification of novel proteins stably produced by aberrant splicing in cancer may result in therapeutic strategies to target these downstream pathologic products and pathways (as depicted in (5)). Additional therapeutic strategies that have been shown to affect splicing only in cell-free in vitro assays to date are not shown above. Inhibition of U2 snRNP by inhibition of SF3B1 function or methylation of Sm proteins is shown in more detail in Figure 3.
Methods to target splicing in cancer
Inhibition of the core spliceosome as cancer therapy
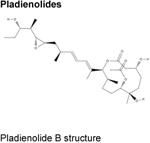
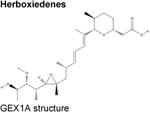
Over the last two decades, multiple natural products derived from bacteria and their analogues have been identified to bind to the SF3B component of U2 snRNP and interrupt early stages of spliceosome assembly. These compounds, which include FR901463, FR901464, and FR901465 (from Pseudomonas sp. 2663)40,41 and unrelated, structurally distinct compounds derived from Streptomyces (herboxiedenes (from Streptomyces sp. A7847)42 and pladienolides (from Streptomyces platensis Mer-11107)43,44) (Table), were originally identified as having potent cytotoxicity and resulting in cell cycle arrest in the G1 and G2/M phases of the cell cycle (reviewed previously45,46). Although these earliest natural compounds showed promising anti-cancer properties in various in vitro and in vivo studies with half-maximal inhibitory concentrations (IC50s) in the low nanomolar ranges, they were chemically unstable and thus unsuitable for therapeutic purposes. Total synthesis47-51 and further analogue efforts resulted in the successful development of additional compounds with improved stability, most notably E710752 (an analogue of pladienolide B), spliceostatin A (SSA; from FR901464)53 and the sudemycins54 (Table).
Table. Small molecules described to directly alter splicing and/or post-translational modifications of spliceosomal proteins in mammalian cells.
Compounds described to inhibit splicing only in non-mammalian cells108 or purely based on cell-free assays and/or with indirect effects on splicing109-111 are not described in this table. In addition, the thailanstatins (Thailanstatins A-C112) and FD-895113 are SF3B1 inhibitory compounds that are not shown below.
| Class | Compound | Target/Mechanism of Action (Note on specificity for substrate(s)) |
Effects on splicing and other phenotypes in mammalian cells |
Preclinical evaluation in context of cancer |
|---|---|---|---|---|
|
Pladienolides A-G43,44,114 |
|
Of 7 naturally occurring pladienolides, pladienolide B has the greatest in vivo activity. 43,44,114 | |
| E7107 (synthetic urethane derivative of pladienolideB)52,70 | Entered phase I clinical trials in 2006 but clinical development suspended due to toxicity68,69. | |||
|
Herboxiedene42 (GEX1A and other GEX family members), 6-norherboxidiene47 | |||
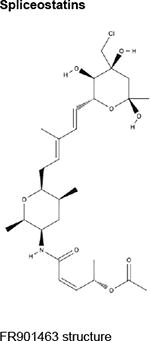
|
FR901463, FR901464, FR90146540,41 Meamycin49 Spliceostatin A53 (methylated derivative of FR901464) |
All demonstrate nanomolar antiproliferative activity against a wide array of cancer cell lines in vitro. | ||
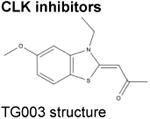
|
TG00390 | Inhibits CLK1, CLK2, and CLK4 through competitive antagonism of ATP binding. |
|
|
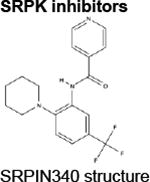
|
SRPIN340116 | ATP-competitive inhibitor of SRPK1 and SRPK2 (does not affect any other Ser/Thr kinases until 10μM)116 |
|
Preclinical studies limited to in vitro analyses of leukemia and lymphoma cell lines.117 |
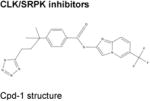
|
Cpd-1, Cpd-2, Cpd-391 | Cpd-1: inhibits SRPK1, SRPK2, CLK1, and CLK2.Cpd-3: inhibits CLK1 and CLK2 ≫ SRPK1, SRPK2. |
|
Preclinical studies limited to in vitro studies in MDA-MB-468 breast cancer cells.91 |
CLK (CDC2-like kinases); SRPIN340 (N-(2-(piperidin-1-yl)-5-(trifluoromethyl)phenyl); SRPK (Serine/Arginine protein kinases).
Eleven years after the initial description of these compounds, Kaida et al.53 and Kotake et al.52 utilized two separate target identification approaches to identify that FR901464, its methylated derivative, SSA, as well as E7107, all bind non-covalently to the SF3B component of U2 snRNP and result in impairment in pre-mRNA splicing in a dose- and time-dependent manner.53,55 Importantly, these effects were mimicked by RNAi-mediated silencing of SF3B1 at selected target genes53, a finding later confirmed at a broader level using splice-sensitive microarrays56. In addition, both studies identified that while exposure to these drugs resulted in nuclear accumulation of unspliced pre-mRNAs, some unspliced pre-mRNAs leaked out of the nucleus and underwent translation to generate stable, aberrant protein products52,53 (Figure 2 and 3). Interestingly, SSA53, E710752, and GEX1A57 all result in production of an aberrant truncated form of the cell cycle inhibitor p27 (encoded by CDKN1B), which retains the cell cycle inhibitory properties of p27 but is unusually stable due to truncation of the C-terminal domain necessary for its normal proteolytic degradation. Despite the convergent effects of FR901464 and the structurally distinct pladienolide compounds revealed in these two studies, the exact component of the SF3B complex targeted by these drugs was not fully resolved. SF3B is a 450kDa complex that comprises 7 subunits: SF3B1, SF3B2, SF3B3, SF3B4, SF3B5, SF3B14, and PHF5A (Figure 3). Initially, SF3B1 was identified as the most enriched protein binding to SSA53 while SF3B3 was initially suggested to be the main target of E710752. Interestingly, a functional genetic study performed 4 years later by Yokoi et al. clarified that SF3B1 is the exact target of both classes of compounds58, data which has confirmed that all of the cellular effects of these compounds can be attributed to binding to SF3B1. These findings were based on the characterization of 2 separate human colorectal cancer cell lines which acquired resistance to pladienolide B following long-term exposure to the drug. RNA-seq analysis of pladienolide-resistant versus parental, sensitive cells, revealed that resistant cells across both cell lines acquired the same point mutation in SF3B1 (SF3B1R1074H)58. Subsequent functional validation confirmed that this specific mutation confers resistance by reducing the binding affinity of each compound to SF3B1, abolishing inhibitory effects on pre-mRNA splicing. This same exact point mutation in SF3B1 also confers resistance to the sudemycins, providing further evidence that SF3B1 is the definitive target of these 2 major classes of spliceosomal inhibitory compounds58.
Figure 3. Pharmacologic methods to disrupt core spliceosome function.
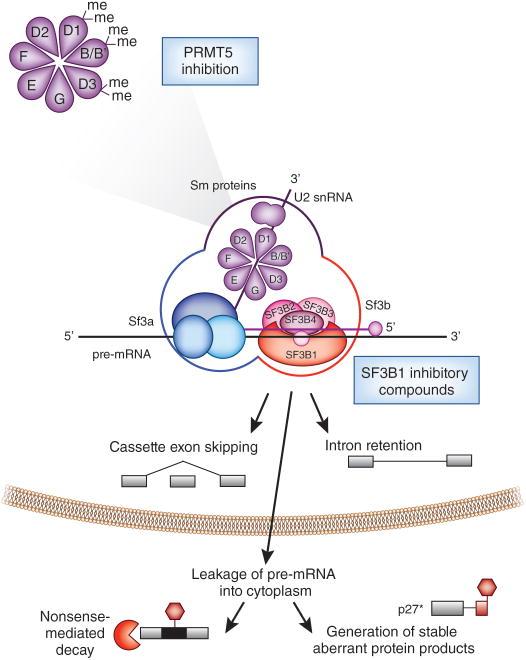
Current methods to directly inhibit spliceosome function include a series of compounds that bind to the SF3B component of U2 snRNP and inhibit early spliceosome assembly. While the precise biochemical interactions between SF3B1 inhibitors and U2 snRNP are not well understood, the effects of each of these drugs on cell toxicity is nearly completely abrogated by mutation of a single residue of SF3B1 (SF3B1 R1074H), suggesting that each of these compounds specifically functions through interactions with SF3B1. Inhibition of U2 snRNP function has been shown to result in widespread intron retention and cassette exon skipping in a time- and dose-dependent manner in a variety of cell types. While this inhibition of splicing results in an accumulation of pre-mRNAs in the nucleus, pharmacologic inhibition of U2 snRNP is also associated with leakage of pre-mRNA into the cytoplasm. Although most unspliced mRNAs are expected to become substrates for nonsense-mediated decay, a portion of these mRNAs may undergo translation to generate aberrant protein products which themselves may have cellular toxicity. For example, a functionally active form of the cell cycle inhibitory protein p27 (termed “p27*”) which lacks the C-terminal domains required for degradation is generated following exposure to several SF3B1 inhibitory compounds.
In addition to SF3B1 inhibitory compounds, recent data suggests that inhibition of Sm protein methylation through downregulation of PRMT5 (protein arginine methyltransferase-5) may also inhibit splicing. PRMT5 symmetrically methylates arginine residues of SmB/B′, SmD1, and SmD3.
The above data clearly suggest that SSA, pladienolides, and herboxiedenes, despite being structurally distinct, appear to interact with SF3B1 in the exact same manner. Although it is clear that these compounds influence pre-mRNA splicing by binding to the SF3B complex of the U2 snRNP and destabilizing its interaction with pre-mRNA (Figure 3), the precise mechanistic effects of these compounds on splicing has been reported differently across studies. In early spliceosome assembly, U2 snRNP is recruited to the branch point adenosine within introns via interactions between SF3B1 and U2AF65. ATP-dependent stabilization of the interaction between U2 snRNP and the branch point sequence (BPS) is a hallmark of complex A formation23,24,26. Folco et al. proposed that E7107 results in defective formation of the spliceosome at the step in which U2 snRNA binds pre-mRNA by abolishing binding of the branch point-binding region (BBR) of U2 snRNA to the BPS in the intron59. More specifically, E7107 was reported to block an ATP-dependent conformational change in U2 sRNP required to expose the BBR59. In contrast, Roybal et al. suggested that SSA interferes with splicing after U2 snRNP has stably integrated and at the transition of the pre-spliceosome to the B-complex60. However, more recent work by the same group has now revealed that the mechanistic effects of 3 main classes of SF3B1 inhibitory compounds are very likely to be similar. Each of these compounds inhibits cell-free in vitro splicing at 1 μM while addition of the inactive analogs of each compound restores splicing61. Moreover, the inactive analogs of each of these compounds can interfere with the function of the active versions of each compound in a totally interchangeable fashion, suggesting a shared mechanism of splicing inhibition amongst each of these compounds61.
Despite the mechanistic insights regarding the interaction of SF3B1 inhibitors with U2 snRNP, greater systematic efforts to characterize the effects of these compounds on the transcriptome are needed. For example, several studies have suggested that the main effects of these compounds on splicing is the generation of widespread IR53,62, yet numerous examples of alteration of other splicing events by SF3B1 inhibition have been identified and there are suggestions that the strength of the 3′ splice site is an important determinant of which splice sites are affected by these compounds56. Similarly, a more systematic effort to identify the effects of splicing inhibitory compounds on the proteome is needed. It is entirely possible that the cellular effects of these compounds can be attributed to the generation of aberrant protein products, beyond p27, which themselves might be toxic to cells and enlighten future drug development efforts.
Preclinical and clinical evaluation of splicing inhibition in cancer
The recent discovery of mutations in splicing factors in cancer has prompted interest to understand if cancer cells bearing mutations in splicing factors might have preferential sensitivity to compounds interrupting splicing. The rationale for these studies is the aforementioned fact that splicing factor mutations occur in a mutually exclusive manner with one another in cancer and consistently occur in a heterozygous state5, suggesting that cells will not tolerate further perturbations to normal splicing catalysis in the setting of expression of these mutations. Recent work from our group63 and others64 has revealed that the wildtype allele of a splicing factor is required in the setting of expression of a mutant allele. Moreover, in vivo E7107 treatment of isogenic murine myeloid leukemias with or without mutant Srsf2 expression, revealed preferential cell death of leukemias bearing mutant Srsf263. Similar synthetic lethal interactions between expression of mutant U2AF1 and exposure to sudemycins has also been reported in in vitro and in vivo studies65. Further genomic analysis in isogenic cancer cells with and without splicing factor mutations will be important to identify if there are specific pre-mRNA sequence features which predict responsiveness to these compounds and/or if specific mis-spliced targets can be identified which are responsible for the preferential lethality in the context of splicing factor-mutant cells.
In addition to the interest in general splicing inhibition for splicing factor-mutant cancers, there has also been an interest to understand if general splicing inhibition might have utility in the setting of cancers driven by specific pathologic splicing events. As one example, nearly 30% of human melanomas with a BRAFV600E mutation develop BRAF inhibitor resistance through expression of a splice variant of BRAFV600E which lacks the RAS binding domain (RBD)66. Recent work has identified that at least one human melanoma cell line developed acquired BRAF inhibitor resistance through an intronic mutation in BRAF that results in in-frame skipping of exons 3-5 encoding the RBD. Interestingly, use of SSA or a related analogue meamycin49 (Table 1), promotes inclusion of exons encoding the RBD and can overcome splicing mediated BRAF inhibitor resistance67. Although this study elegantly demonstrates the utility of reversing pathologic splicing events in cancer, this work also highlights the potential for more focused modulation of specific pathologic splicing events in cancer (described below).
In parallel to biochemical studies, preclinical studies, and analogue efforts, clinical trials to understand the therapeutic potential of SF3B1 inhibitors have been initiated. E7107 has now been studied in two separate phase I, open-label, single-arm, dose-escalation studies in patients with locally advanced or metastatic solid tumors. A total of 40 and 26 patients were treated in Europe68 and the U.S.69 (Study E7107-A001-101; Trial registration ID: NCT00499499), respectively. The results of these studies have been both encouraging as well as concerning. First, pharmacodynamic analysis of the effects of E7107 on splicing of target mRNAs in peripheral blood mononuclear cells revealed that splicing inhibition was achieved in vivo and was commensurate to E7107 dose68. At the same time, while the drug was generally well tolerated and a maximal tolerated dose was established, an unexpected toxicity of bilateral optic neuritis was identified and resulted in suspension of both trials68,69. Currently it is unclear if this toxicity, which was not encountered in preclinical animal studies63,70, is an on-target effect of SF3B1 inhibition or a specific toxicity associated with E7107. Future clinical trial efforts will clearly be needed to understand the safety and potential therapeutic efficacy of any of the other structurally distinct forms of pharmacologic SF3B1 inhibition that have been described.
Potential for spliceosome inhibition in MYC-dependent cancers
While mutations in splicing factors have highlighted the potential for cancers bearing these alterations to be targeted by general spliceosome inhibition, numerous reports have suggested that cancer cells dependent on MYC activation may also be preferentially vulnerable to spliceosome inhibition. First, a number of splicing regulatory proteins that promote transformation are direct transcriptional targets of MYC. This includes MYC-induced upregulation of hnRNP A1 and hnRNP A2, which in turn, regulate alternative splicing of pyruvate kinase to promote expression of the cancer-associated pyruvate kinase M2 (PKM2) isoform71. In addition, SRSF1 is also directly upregulated by MYC and can drive transformation of mammary epithelial cells in collaboration with MYC72. More recently, an analysis of the direct transcriptional targets of MYC in human and murine lymphomas have identified that MYC directly upregulates the transcription of several core snRNP genes as well as snRNP assembly genes, including Prmt5, an arginine methyltransferase that methylates the Sm proteins of U2 snRNP73 (Figure 3). Prmt5 knockdown in MYC-driven lymphomas resulted in exon skipping and IR as well as abrogation of lymphomagenesis73. These data suggest that MYC overexpression may require cells to rely on high levels of PRMT5 and mature snRNPs to sustain splicing fidelity. This is a therapeutically exciting possibility, given the recent development of pharmacologic inhibitors of PRMT574. At the same time, it is important to note that PRMT5 has many other substrates beyond splicing-related proteins, which are likely critical for cellular function (reviewed recently75,76). Moreover, several other mechanisms have been recently proposed whereby cancer cells are sensitized to PRMT5 inhibition which are unrelated to RNA processing or splicing77,78. Further efforts to understand the effects of PRMT5 pharmacologic inhibition on splicing, its therapeutic index, and roles in MYC-dependent cancers will be critical.
Related to the above, several RNA interference (RNAi) screens have identified a unique preferential vulnerability of MYC-expressing glioblastoma as well as mammary epithelial cells to downregulation of splicing factors compared with cells transformed by other oncogenes such as RAS, EGFR, or HER2. First, an effort to identify genes required for growth and viability of patient-derived glioblastoma stem cells (GSCs) revealed a preferential requirement for PHF5A (PHD Finger Protein 5A)79 in GSCs over untransformed neural stem cells (NSCs). PHF5A encodes a protein required to facilitate interaction between U2 snRNP and ATP-dependent helicases80. Accordingly, pharmacologic inhibition of U2 snRNP with SSA or sudemycin C revealed potent therapeutic efficacy in GSCs. Efforts to understand the molecular alterations which might underlie responsiveness to PHF5A knockdown or U2 snRNP inhibition led the authors to identify that expression of MYC in NSCs induced sensitivity to U2 snRNP perturbation79.
A more recent effort to identify genes whose downregulation are synthetic lethal with MYC in the context of human mammary epithelial cells by Hsu et al. similarly identified several splicing factors as required in cells in order to tolerate MYC. This includes expression of the protein BUD31 whose function in mammalian cells had not previously been elucidated81. This study revealed that BUD31 associates with multiple subcomplexes of the spliceosome and BUD31 depletion is associated with global IR, suggesting a role for BUD31 in normal splicing function. This then led the authors to a series of experiments which suggested that MYC-dependency (as indicated by sensitivity to MYC downregulation by RNAi) correlates with dependency on spliceosomal proteins (as indicated by shRNA's targeting spliceosomal components)81. Moreover, pharmacologic inhibition of SF3B1 with sudemycin D in vivo increased survival and limited metastatic potential in breast xenograft models, suggesting a clear potential for therapeutic translation of these findings.
Overall, the data above suggest an enhanced dependency for spliceosomal protein expression in MYC-driven cancers. Nonetheless, the therapeutic index of this approach and specificity for MYC-driven cancers versus other genetic subsets of cancer needs to be clarified further. Each of the above studies report distinct mechanisms by which MYC-expressing cells become preferentially dependent on spliceosome function which appears to differ based on tissue context73,79,81. While this may be related to the known context-dependent regulation of splicing23, further understanding of the mechanisms by which MYC affects splicing may be important for future therapeutic efforts to target MYC-dependencies.
Targeting splicing regulatory proteins in cancer
In addition to modulating splicing through inhibition of the core spliceosome, the identification that splicing regulatory proteins promote oncogenesis through overexpression14,15,82,83 as well as alteration-of-function36,84 mutations (Figure 1), have highlighted the potential for therapeutic targeting of these proteins as novel cancer therapies. In particular, inhibition of phosphorylation of SR proteins has emerged as a potential means to modulate splicing through altering the function of splicing regulatory proteins. SR proteins are a family of proteins required for constitutive splicing as well as modulating AS. They are highly conserved in eukaryotes and have 1-2 RNA recognition motifs (RRMs) at the amino terminus and an RS (Arginine/Serine rich) domain at the C-terminus (reviewed recenty27,28). The RS domains consist of multiple consecutive RS/SR dipeptide repeats which undergoes extensive phosphorylation by multiple kinases including the SRPK family kinases (SR protein kinases 1 and 2 (SRPK1 and 2)), hPRP4, topoisomerase I, and the CLK (CDC2-like) kinase family (CLK members 1-4) (reviewed recently85,86). While the precise physiologic function of phosphorylation of SR proteins is not well defined, experimental induction of either hyper- or hypophosphorylation of SR proteins inhibits splicing87. For example, expression of kinase-dead mutant forms of SRPKs or CLKs results in global inhibition of splicing and enlargement of nuclear speckles88,89.
A variety of kinase screens have been performed to identify chemical compounds that inhibit SR protein phosphorylation. A screen of 100,000 chemical compounds for effects on in vitro phosphorylation of Clk kinases, identified the benzothiazole compound, TG-003, as an inhibitor of CLK kinases 1, 2, and 490 (Table). While the genome-wide effects of TG-003 are not well characterized, the drug appears to affect expression of functional isoforms of SRSF2 and CLK1, alterations that may themselves be therapeutically important targets90.
A recent large-scale screen for compounds that inhibit SRPK kinase activity uncovered a series of three compounds (currently named “Cpd-1” to “Cpd-3”; Table) that appear to inhibit SPRK1-2 and/or CLK1-291. Comparison of the inhibitory effects of Cpd compounds against SPRK, CLK, and 28 other kinases suggests some specificity of these compounds for SRPKs and/or CLKs but larger efforts to understand their kinase specificity will be important91. In addition, analysis of the transcriptome-wide effects of Cpd compounds revealed a global effect on cassette exon skipping which is potentially different from the IR consistently observed with SF3B1 inhibition91. Direct comparison of the effects on splicing of SF3B1 inhibitors versus compounds affecting SR protein phosphorylation may help elucidate distinct effects of these 2 therapeutic approaches. In addition, further preclinical studies are needed to understand the potential therapeutic relevance of SR protein inhibitors as cancer therapy and if any genetic or histologic subtypes of cancer are most susceptible to cell killing by these compounds.
Oligonucleotide therapy to modulate splicing in cancer
Given the widespread alterations of splicing expected to result from global spliceosome inhibition and the increased identification of specific pathologic splicing events important in cancer, there has been continued interest in targeting individual splicing events with oligonucleotide-based therapeutics. Such oligonucleotides can be designed to hybridize to RNA in a sequence-specific manner through Watson-Crick base-pairing and alter splicing. The advantages of oligonucleotide-based therapies to modulate splicing include the vast myriad of ways they can potentially modify splicing as described below. At the same time, it is important to note that despite dozens of years of effort, no form of oligonucleotide-based therapy has yet to meet FDA-approval in the treatment of cancer (the use of oligonucleotides to modulate gene expression through induction of RNA cleavage or suppression of translation will not be discussed here but have been reviewed recently92-94). Nonetheless, oligonucleotide-based therapies appear extremely promising in the correction of specific pathologic splicing events underlying non-cancer monogenic disorders. Indeed, such therapies are currently in late-stage clinical trials for patients affected by Duchenne muscular dystrophy (DMD)95 and spinal muscular atrophy (SMA)96, providing continued motivation to explore modulation of splicing as a potential cancer therapy.
Thus far, oligonucleotides have been identified to modulate splicing to repair defective mRNA, restore the production of essential proteins, generate novel proteins, and regulate the presence of disease-related splice variants. So-called splice-switching oligonucleotides (SSOs) have been used most commonly to manipulate splicing by preventing the interaction of splicing molecules with pre-mRNA through steric hindrance, without inducing cleavage of the RNAs. For example, some exons are poorly included in mRNA because they contain ESS sequences or are adjacent to ISS sequences, which recruit splicing inhibitory factors to prevent inclusion of the exons in pre-mRNA. Binding of SSOs to such sites to block recruitment of splicing inhibitory factors would be expected to promote exon inclusion. In fact, the oligonucleotide therapy that is most advanced in clinical trials of SMA blocks an ISS element to promote exon inclusion in a manner which restores effective expression of the protein which is pathologically reduced and causative of SMA97. Conversely, efforts to manipulate splicing through binding of SSOs to ESEs or ISEs have also been demonstrated to promote exon exclusion. In fact, the use of SSO's to promote skipping of exons containing disease-causative frameshift or nonsense mutations constitutes the therapeutic strategy that has been utilized clinically in DMD95. Exon skipping mediated by SSO's in this manner is associated with slower disease progression as it promotes expression of an internally deleted, partially functional version of the protein which is pathologically reduced in DMD98,99.
While the success and safety of SSO's to treat neuromuscular disorders is promising, the therapeutic impact of modulating splicing of a single event within cancer cells is unclear, especially given the widespread alterations in splicing which have been described in most common forms of cancer. However, the discovery of alteration-of-function mutations and overexpression of splicing factors in cancer highlights the potential use of SSO's to target the function of wildtype and/or mutant splicing factors themselves. For instance, recent studies have highlighted that mutations in SRSF2 alter its RNA binding and splicing preference in a sequence-specific manner36,84 and that expression of mutant SRSF2 is sufficient to drive a clonal hematopoietic disorder36. This opens the possibility that SSO's targeting the ESE preferred by the mutant SRSF2 protein may have therapeutic impact in SRSF2-mutant malignancies. At the same time, recent work has also identified that the wildtype SRSF2 protein is essential for the survival of cells expressing mutant SRSF263. This suggests that SSO's targeting the ESE recognized primarily by wildtype SRSF2 might be an alternative therapeutic approach for targeting SRSF2 mutant cells. Similarly, the identification of the ESE motifs recognized by SRSF1100,101 provide a potential ability to target SRSF1 binding in cancer types thought to be driven by SRSF1 overexpression. It is important to note, however, that the sequences recognized by SR, hnRNP, and other splicing regulatory proteins may not provide great specificity and could result in similar widespread alterations in splicing as expected with SF3B1 or SR kinase inhibitors. Thus, further efforts to understand and prioritize the pathologic splicing events driven by altered splicing factors in cancer are greatly needed. For example, several recent studies have identified that SF3B1 mutations result in the widespread use of cryptic 3′ splice sites and suggest that this is basis for its pathologic effects in cancer102,103. In fact, altered splicing of mRNA's encoding specific iron-transport proteins (such as ABCB7102 and Mitoferrin-1104) have been suggested to mediate the hematopoietic disorder most closely linked to SF3B1 mutations, termed RARS (refractory anemia with ring sideroblasts, a subtype of the myelodysplastic syndromes). Thus, use of SSOs to correct splicing of these specific events within SF3B1-mutant leukemias may be a novel and targeted therapeutic approach to correct mutation-associated splicing events driving the disease phenotype.
Conclusion
A number of diverse mechanisms by which splicing is dyregulated in cancer have been described and have highlighted the need to understand and identify means to therapeutically manipulate splicing as cancer therapy. While a number of chemical compounds inhibiting splicing catalysis have been described to date, nearly all of these drugs inhibit early spliceosome assembly or SR protein phosphorylation. However, it is quite likely that additional chemical screens will identify compounds that inhibit the spliceosome at later stages of splicing catalysis than the currently described inhibitors of SF3B1. Indeed, several such compounds with novel mechanistic effects on splicing have already been described105-107 and will be important to evaluate in the context of cancer in vitro and in vivo. It is also hopeful that increased structural understanding of the spliceosome, as has been achieved recently20-22, will help to elucidate the mechanistic effects of these compounds on splicing and potentially suggest novel means of splicing perturbation.
In parallel to identifying new therapeutic modalities to target splicing, further clarification of the therapeutic index of existing methods of general spliceosome inhibition and the genetic subsets of cancer most susceptible to the effects of these compounds is needed. Numerous preclinical models have suggested the somewhat surprising safety of general spliceosome inhibition in vivo as well as a potential rationale for the use of these compounds in cancers driven by MYC. At this point, clinical evaluation of compounds beyond E7107 will be necessary to definitively address the safety, potential therapeutic effects, and toxicities of splicing inhibition in patients. In addition to MYC-driven cancers, several studies have now suggested that cancer cells bearing splicing factor mutations are actually dependent on the presence of the wildtype splicing allele63,64, providing a rationale for therapeutically targeting wildtype splicing in these cells. The use of oligonucleotides to manipulate splicing may be very attractive in cells bearing spliceosomal gene mutations, given the altered splicing preferences generated by these mutations. Further biological studies to prioritize and functionally characterize the altered splicing events which link mutated splicing factors to the cancer phenotype may be very important for future targeted therapeutic approaches. For instance, it is possible that novel, aberrant proteins generated in the context of splicing factor mutations may render these cells susceptible to pharmacologic and immunologic agents targeting these proteins or their associated pathways. Moreover, the widespread alterations of splicing generated by inhibition of SF3B1 and SR protein phosphorylation may themselves generate aberrant proteins that could be utilized for immunologic targeting of cancer. Further efforts to understand the effects of these compounds on the proteome could identify aberrant proteins recurrently generated in the presence of these drugs and may also be important in this regard. Thus, an increased understanding of splicing in cancer is highly likely to advance our understanding of cancer pathogenesis in addition to nominating a broad array of novel therapeutic approaches.
Acknowledgments
SC-WL is supported by a Leukemia and Lymphoma Society (LLS) Special Fellow Award. OAW is supported by grants from the Edward P. Evans Foundation, the Dept. of Defense Bone Marrow Failure Research Program (BM150092 and W81XWH-12-1-0041), NIH/NHLBI (R01 HL128239), an NIH K08 Clinical Investigator Award (1K08CA160647-01),), the Josie Robertson Investigator Program, a Damon Runyon Clinical Investigator Award, an award from the Starr Foundation (I8-A8-075), the Mr. William H. Goodwin and Mrs. Alice Goodwin Commonwealth Foundation for Cancer Research, and The Experimental Therapeutics Center of MSKCC.
References
- 1.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature genetics. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 2.Wang ET, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Supek F, Miñana B, Valcárcel J, Gabaldón T, Lehner B. Synonymous mutations frequently act as driver mutations in human cancers. Cell. 2014;156:1324–1335. doi: 10.1016/j.cell.2014.01.051. [DOI] [PubMed] [Google Scholar]
- 4.Jung H, et al. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nature genetics. 2015;47:1242–1248. doi: 10.1038/ng.3414. [DOI] [PubMed] [Google Scholar]
- 5.Yoshida K, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 6.Papaemmanuil E, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. The New England journal of medicine. 2011;365:1384–1395. doi: 10.1056/NEJMoa1103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. The New England journal of medicine. 2011;365:2497–2506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graubert TA, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nature genetics. 2012;44:53–57. doi: 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harbour JW, et al. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nature genetics. 2013;45:133–135. doi: 10.1038/ng.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quesada V, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nature genetics. 2012;44:47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 11.Imielinski M, et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin M, et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nature genetics. 2013;45:933–936. doi: 10.1038/ng.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karni R, et al. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nature structural & molecular biology. 2007;14:185–193. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jensen MA, Wilkinson JE, Krainer AR. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nature structural & molecular biology. 2014;21:189–197. doi: 10.1038/nsmb.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anczuków O, et al. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nature structural & molecular biology. 2012;19:220–228. doi: 10.1038/nsmb.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Danan-Gotthold M, et al. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res. 2015;43:5130–5144. doi: 10.1093/nar/gkv210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L, Tovar-Corona JM, Urrutia AO. Increased levels of noisy splicing in cancers, but not for oncogene-derived transcripts. Hum Mol Genet. 2011;20:4422–4429. doi: 10.1093/hmg/ddr370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dvinge H, Bradley RK. Widespread intron retention diversifies most cancer transcriptomes. Genome medicine. 2015;7:45. doi: 10.1186/s13073-015-0168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen TH, et al. The architecture of the spliceosomal U4/U6.U5 tri-snRNP. Nature. 2015;523:47–52. doi: 10.1038/nature14548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan C, et al. Structure of a yeast spliceosome at 3.6-angstrom resolution. Science. 2015;349:1182–1191. doi: 10.1126/science.aac7629. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen TH, et al. Cryo-EM structure of the yeast U4/U6.U5 tri-snRNP at 3.7 A resolution. Nature. 2016;530:298–302. doi: 10.1038/nature16940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wan R, et al. The 3.8 A structure of the U4/U6.U5 tri-snRNP: Insights into spliceosome assembly and catalysis. Science. 2016;351:466–475. doi: 10.1126/science.aad6466. [DOI] [PubMed] [Google Scholar]
- 23.Fu XD, Ares M., Jr Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet. 2014;15:689–701. doi: 10.1038/nrg3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 25.Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matera AG, Wang Z. A day in the life of the spliceosome. Nature reviews Molecular cell biology. 2014;15:108–121. doi: 10.1038/nrm3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shepard PJ, Hertel KJ. The SR protein family. Genome Biol. 2009;10:242. doi: 10.1186/gb-2009-10-10-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Busch A, Hertel KJ. Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip Rev RNA. 2012;3:1–12. doi: 10.1002/wrna.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simon JM, et al. Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Res. 2014;24:241–250. doi: 10.1101/gr.158253.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma PC, et al. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;63:6272–6281. [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas Research, N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Puente XS, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526:519–524. doi: 10.1038/nature14666. [DOI] [PubMed] [Google Scholar]
- 34.Graubert TA, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2012;44:53–57. doi: 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ilagan JO, et al. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome research. 2015;25:14–26. doi: 10.1101/gr.181016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim E, et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer cell. 2015;27:617–630. doi: 10.1016/j.ccell.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alsafadi S, et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun. 2016;7:10615. doi: 10.1038/ncomms10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J, Manley JL. Misregulation of pre-mRNA alternative splicing in cancer. Cancer discovery. 2013;3:1228–1237. doi: 10.1158/2159-8290.CD-13-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sotillo E, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015;5:1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakajima H, et al. New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J Antibiot (Tokyo) 1996;49:1204–1211. doi: 10.7164/antibiotics.49.1204. [DOI] [PubMed] [Google Scholar]
- 41.Nakajima H, et al. New antitumor substances, FR901463, FR901464 and FR901465. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. J Antibiot (Tokyo) 1996;49:1196–1203. doi: 10.7164/antibiotics.49.1196. [DOI] [PubMed] [Google Scholar]
- 42.Sakai Y, et al. GEX1 compounds, novel antitumor antibiotics related to herboxidiene, produced by Streptomyces sp. I. Taxonomy, production, isolation, physicochemical properties and biological activities. J Antibiot (Tokyo) 2002;55:855–862. doi: 10.7164/antibiotics.55.855. [DOI] [PubMed] [Google Scholar]
- 43.Mizui Y, et al. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J Antibiot (Tokyo) 2004;57:188–196. doi: 10.7164/antibiotics.57.188. [DOI] [PubMed] [Google Scholar]
- 44.Sakai T, Asai N, Okuda A, Kawamura N, Mizui Y. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. II. Physico-chemical properties and structure elucidation. J Antibiot (Tokyo) 2004;57:180–187. doi: 10.7164/antibiotics.57.180. [DOI] [PubMed] [Google Scholar]
- 45.Bonnal S, Vigevani L, Valcarcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov. 2012;11:847–859. doi: 10.1038/nrd3823. [DOI] [PubMed] [Google Scholar]
- 46.Webb TR, Joyner AS, Potter PM. The development and application of small molecule modulators of SF3b as therapeutic agents for cancer. Drug Discov Today. 2013;18:43–49. doi: 10.1016/j.drudis.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lagisetti C, et al. Pre-mRNA splicing-modulatory pharmacophores: the total synthesis of herboxidiene, a pladienolide-herboxidiene hybrid analog and related derivatives. ACS Chem Biol. 2014;9:643–648. doi: 10.1021/cb400695j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mandel AL, Jones BD, La Clair JJ, Burkart MD. A synthetic entry to pladienolide B and FD-895. Bioorg Med Chem Lett. 2007;17:5159–5164. doi: 10.1016/j.bmcl.2007.06.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Albert BJ, Sivaramakrishnan A, Naka T, Czaicki NL, Koide K. Total syntheses, fragmentation studies, and antitumor/antiproliferative activities of FR901464 and its low picomolar analogue. J Am Chem Soc. 2007;129:2648–2659. doi: 10.1021/ja067870m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thompson CF, Jamison TF, Jacobsen EN. FR901464: total synthesis, proof of structure, and evaluation of synthetic analogues. J Am Chem Soc. 2001;123:9974–9983. doi: 10.1021/ja016615t. [DOI] [PubMed] [Google Scholar]
- 51.Arai K, et al. Total synthesis of 6-deoxypladienolide D and Assessment of Splicing Inhibitory Activity in a Mutant SF3B1 cancer cell line. Org Lett. 2014;16:5560–5563. doi: 10.1021/ol502556c. [DOI] [PubMed] [Google Scholar]
- 52.Kotake Y, Sagane K, Owa T, Mimori-Kiyosue Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nature chemical biology. 2007;3:570–575. doi: 10.1038/nchembio.2007.16. [DOI] [PubMed] [Google Scholar]
- 53.Kaida D, et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol. 2007;3:576–583. doi: 10.1038/nchembio.2007.18. [DOI] [PubMed] [Google Scholar]
- 54.Fan L, Lagisetti C, Edwards CC, Webb TR, Potter PM. Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem Biol. 2011;6:582–589. doi: 10.1021/cb100356k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kotake Y, et al. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat Chem Biol. 2007;3:570–575. doi: 10.1038/nchembio.2007.16. [DOI] [PubMed] [Google Scholar]
- 56.Corrionero A, Minana B, Valcarcel J. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev. 2011;25:445–459. doi: 10.1101/gad.2014311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hasegawa M, et al. Identification of SAP155 as the target of GEX1A (Herboxidiene), an antitumor natural product. ACS Chem Biol. 2011;6:229–233. doi: 10.1021/cb100248e. [DOI] [PubMed] [Google Scholar]
- 58.Yokoi A, et al. Biological validation that SF3b is a target of the antitumor macrolide pladienolide. The FEBS journal. 2011;278:4870–4880. doi: 10.1111/j.1742-4658.2011.08387.x. [DOI] [PubMed] [Google Scholar]
- 59.Folco EG, Coil KE, Reed R. The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev. 2011;25:440–444. doi: 10.1101/gad.2009411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roybal GA, Jurica MS. Spliceostatin A inhibits spliceosome assembly subsequent to prespliceosome formation. Nucleic Acids Res. 2010;38:6664–6672. doi: 10.1093/nar/gkq494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Effenberger KA, Urabe VK, Prichard BE, Ghosh AK, Jurica MS. Interchangeable SF3B1 inhibitors interfere with pre-mRNA splicing at multiple stages. RNA. 2016;22:350–359. doi: 10.1261/rna.053108.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kashyap MK, et al. Targeting the spliceosome in chronic lymphocytic leukemia with the macrolides FD-895 and pladienolide-B. Haematologica. 2015;100:945–954. doi: 10.3324/haematol.2014.122069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee SCW, et al. Therapeutic targeting of spliceosomal mutant myeloid leukemias through modulation of splicing catalysis (Abstract 4) Blood. 2015;126 [Google Scholar]
- 64.Zhou Q, et al. A chemical genetics approach for the functional assessment of novel cancer genes. Cancer Res. 2015;75:1949–1958. doi: 10.1158/0008-5472.CAN-14-2930. [DOI] [PubMed] [Google Scholar]
- 65.Shirai CL, et al. Preclinical Activity of Splicing Modulators in U2AF1 Mutant MDS/AML (Abstract 1653) Blood. 2015;126 [Google Scholar]
- 66.Poulikakos PI, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salton M, et al. Inhibition of vemurafenib-resistant melanoma by interference with premRNA splicing. Nat Commun. 2015;6:7103. doi: 10.1038/ncomms8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eskens FA, et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin Cancer Res. 2013;19:6296–6304. doi: 10.1158/1078-0432.CCR-13-0485. [DOI] [PubMed] [Google Scholar]
- 69.Hong DS, et al. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest New Drugs. 2014;32:436–444. doi: 10.1007/s10637-013-0046-5. [DOI] [PubMed] [Google Scholar]
- 70.Masao Iwata YO, et al. E7107, a new 7-urethane derivative of pladienolide D, displays curativeeffect against several human tumor xenografts. Proc Am Assoc Cancer Res. 2004;45 [Google Scholar]
- 71.David CJ, Chen M, Assanah M, Canoll P, Manley JL. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature. 2010;463:364–368. doi: 10.1038/nature08697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Das S, Anczukow O, Akerman M, Krainer AR. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep. 2012;1:110–117. doi: 10.1016/j.celrep.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koh CM, et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature. 2015;523:96–100. doi: 10.1038/nature14351. [DOI] [PubMed] [Google Scholar]
- 74.Chan-Penebre E, et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol. 2015;11:432–437. doi: 10.1038/nchembio.1810. [DOI] [PubMed] [Google Scholar]
- 75.Stopa N, Krebs JE, Shechter D. The PRMT5 arginine methyltransferase: many roles in development, cancer and beyond. Cell Mol Life Sci. 2015;72:2041–2059. doi: 10.1007/s00018-015-1847-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37–50. doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- 77.Kryukov GV, et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science. 2016;351:1214–1218. doi: 10.1126/science.aad5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mavrakis KJ, et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science. 2016;351:1208–1213. doi: 10.1126/science.aad5944. [DOI] [PubMed] [Google Scholar]
- 79.Hubert CG, et al. Genome-wide RNAi screens in human brain tumor isolates reveal a novel viability requirement for PHF5A. Genes & development. 2013;27:1032–1045. doi: 10.1101/gad.212548.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rzymski T, Grzmil P, Meinhardt A, Wolf S, Burfeind P. PHF5A represents a bridge protein between splicing proteins and ATP-dependent helicases and is differentially expressed during mouse spermatogenesis. Cytogenet Genome Res. 2008;121:232–244. doi: 10.1159/000138890. [DOI] [PubMed] [Google Scholar]
- 81.Hsu TY, et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015;525:384–388. doi: 10.1038/nature14985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Karni R, Hippo Y, Lowe SW, Krainer AR. The splicing-factor oncoprotein SF2/ASF activates mTORC1. Proc Natl Acad Sci U S A. 2008;105:15323–15327. doi: 10.1073/pnas.0801376105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Karni R, et al. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007;14:185–193. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang J, et al. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E4726–4734. doi: 10.1073/pnas.1514105112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Giannakouros T, Nikolakaki E, Mylonis I, Georgatsou E. Serine-arginine protein kinases: a small protein kinase family with a large cellular presence. FEBS J. 2011;278:570–586. doi: 10.1111/j.1742-4658.2010.07987.x. [DOI] [PubMed] [Google Scholar]
- 86.Zhou Z, Fu XD. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma. 2013;122:191–207. doi: 10.1007/s00412-013-0407-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Prasad J, Colwill K, Pawson T, Manley JL. The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing. Mol Cell Biol. 1999;19:6991–7000. doi: 10.1128/mcb.19.10.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Colwill K, et al. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J. 1996;15:265–275. [PMC free article] [PubMed] [Google Scholar]
- 89.Lai MC, Lin RI, Tarn WY. Differential effects of hyperphosphorylation on splicing factor SRp55. Biochem J. 2003;371:937–945. doi: 10.1042/BJ20021827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Muraki M, et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J Biol Chem. 2004;279:24246–24254. doi: 10.1074/jbc.M314298200. [DOI] [PubMed] [Google Scholar]
- 91.Araki S, et al. Inhibitors of CLK protein kinases suppress cell growth and induce apoptosis by modulating pre-mRNA splicing. PLoS One. 2015;10:e0116929. doi: 10.1371/journal.pone.0116929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Castanotto D, Stein CA. Antisense oligonucleotides in cancer. Curr Opin Oncol. 2014;26:584–589. doi: 10.1097/CCO.0000000000000127. [DOI] [PubMed] [Google Scholar]
- 93.McClorey G, Wood MJ. An overview of the clinical application of antisense oligonucleotides for RNA-targeting therapies. Curr Opin Pharmacol. 2015;24:52–58. doi: 10.1016/j.coph.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 94.Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012;11:125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cirak S, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zanetta C, et al. Molecular therapeutic strategies for spinal muscular atrophies: current and future clinical trials. Clin Ther. 2014;36:128–140. doi: 10.1016/j.clinthera.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 97.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lu QL, et al. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat Med. 2003;9:1009–1014. doi: 10.1038/nm897. [DOI] [PubMed] [Google Scholar]
- 99.Goyenvalle A, et al. Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science. 2004;306:1796–1799. doi: 10.1126/science.1104297. [DOI] [PubMed] [Google Scholar]
- 100.Anczukow O, et al. SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol Cell. 2015;60:105–117. doi: 10.1016/j.molcel.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pandit S, et al. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell. 2013;50:223–235. doi: 10.1016/j.molcel.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Darman RB, et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Reports. 2015;13:1033–1045. doi: 10.1016/j.celrep.2015.09.053. [DOI] [PubMed] [Google Scholar]
- 103.DeBoever C, et al. Transcriptome sequencing reveals potential mechanism of cryptic 3′ splice site selection in SF3B1-mutated cancers. PLoS computational biology. 2015;11:e1004105. doi: 10.1371/journal.pcbi.1004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Visconte V, et al. Distinct iron architecture in SF3B1-mutant myelodysplastic syndrome patients is linked to an SLC25A37 splice variant with a retained intron. Leukemia. 2015;29:188–195. doi: 10.1038/leu.2014.170. [DOI] [PubMed] [Google Scholar]
- 105.Effenberger KA, et al. The Natural Product N-Palmitoyl-l-leucine Selectively Inhibits Late Assembly of Human Spliceosomes. J Biol Chem. 2015;290:27524–27531. doi: 10.1074/jbc.M115.673210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Effenberger KA, et al. A high-throughput splicing assay identifies new classes of inhibitors of human and yeast spliceosomes. J Biomol Screen. 2013;18:1110–1120. doi: 10.1177/1087057113493117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Berg MG, et al. A quantitative high-throughput in vitro splicing assay identifies inhibitors of spliceosome catalysis. Mol Cell Biol. 2012;32:1271–1283. doi: 10.1128/MCB.05788-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Aukema KG, Chohan KK, Plourde GL, Reimer KB, Rader SD. Small molecule inhibitors of yeast pre-mRNA splicing. ACS Chem Biol. 2009;4:759–768. doi: 10.1021/cb900090z. [DOI] [PubMed] [Google Scholar]
- 109.Pilch B, et al. Specific inhibition of serine- and arginine-rich splicing factors phosphorylation, spliceosome assembly, and splicing by the antitumor drug NB-506. Cancer Res. 2001;61:6876–6884. [PubMed] [Google Scholar]
- 110.Kim H, et al. Identification of a novel function of CX-4945 as a splicing regulator. PLoS One. 2014;9:e94978. doi: 10.1371/journal.pone.0094978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sehgal PB, Darnell JE, Jr, Tamm I. The inhibition by DRB (5,6-dichloro-1-beta-Dribofuranosylbenzimidazole) of hnRNA and mRNA production in HeLa cells. Cell. 1976;9:473–480. doi: 10.1016/0092-8674(76)90092-1. [DOI] [PubMed] [Google Scholar]
- 112.Liu X, et al. Genomics-guided discovery of thailanstatins A, B, and C As pre-mRNA splicing inhibitors and antiproliferative agents from Burkholderia thailandensis MSMB43. J Nat Prod. 2013;76:685–693. doi: 10.1021/np300913h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Seki-Asano M, et al. Isolation and characterization of a new 12-membered macrolide FD-895. J Antibiot (Tokyo) 1994;47:1395–1401. doi: 10.7164/antibiotics.47.1395. [DOI] [PubMed] [Google Scholar]
- 114.Sakai T, et al. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. I. Taxonomy, fermentation, isolation and screening. J Antibiot (Tokyo) 2004;57:173–179. doi: 10.7164/antibiotics.57.173. [DOI] [PubMed] [Google Scholar]
- 115.Lagisetti C, et al. Optimization of antitumor modulators of pre-mRNA splicing. J Med Chem. 2013;56:10033–10044. doi: 10.1021/jm401370h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fukuhara T, et al. Utilization of host SR protein kinases and RNA-splicing machinery during viral replication. Proc Natl Acad Sci U S A. 2006;103:11329–11333. doi: 10.1073/pnas.0604616103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Siqueira RP, et al. Potential Antileukemia Effect and Structural Analyses of SRPK Inhibition by N-(2-(Piperidin-1-yl)-5-(Trifluoromethyl)Phenyl)Isonicotinamide (SRPIN340) PLoS One. 2015;10:e0134882. doi: 10.1371/journal.pone.0134882. [DOI] [PMC free article] [PubMed] [Google Scholar]