

Abstract
During a search for inhibitors of oncogenic K-Ras, we detected two known and two new examples of the rare neoantimycin structure class from a liquid cultivation of Streptomyces orinoci, and reassigned/assigned structures to all based on detailed spectroscopic analysis and microscale C3 Marfey’s and C3 Mosher chemical degradation/derivatization/analysis. SAR investigations inclusive of the biosynthetically related antimycins and respirantin, and synthetic benzoxazolone, documented a unique N-formyl amino-salicylamide pharmacophore as a potent inhibitor of oncogenic K-Ras.
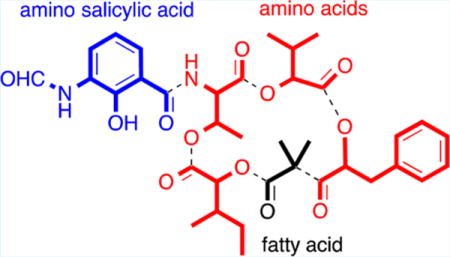
Graphical abstract
Ras GTPases, which are ubiquitously expressed as three isoforms (H-Ras, N-Ras, and K-Ras), act as key molecular switches that regulate cell growth, proliferation, and differentiation.1 Constitutively activated K-Ras is involved in 90% of pancreatic, 45% of colorectal, and 35% of lung carcinomas.2 Ras signaling mechanisms have been researched extensively, and studies have shown that preventing Ras plasma membrane (PM) localization blocks all oncogenic activity.3 Although an attractive anticancer target, the development of potent and selective inhibitors of Ras protein PM localization with clinical application has proven both challenging and elusive.
To address this challenge, we employed a high-throughput, high-content assay to screen a library of microbial extracts, establishing Streptomyces orinoci (MST-AS4461) as a source of secondary metabolites that inhibit Ras protein PM localization.4 Bioassay-guided fractionation of an 8 L liquid fermentation yielded the known secondary metabolites neoantimycin (1) and neoantimycin F (2), along with two new exemplars, neoantimycin G (3) and neoantimycin H (4) (Figure 1), as exceptionally potent (IC50 3–10 nM) inhibitors of K-Ras PM localization. Planar structures were assigned to 1–4 on the basis of detailed spectroscopic analysis, with microscale C3 Marfey’s and C3 Mosher analyses permitting assignment of absolute configurations, including revisions to 15 and 2.6 To support structure–activity relationship (SAR) investigations into the K-Ras modulatory properties of neoantimycins, we obtained the biosynthetically related microbial metabolites, antimycin A2a (5), antimycin A4a (6), and respirantin (7), and prepared the novel benzoxazolone analog 8 (Figure 1). SAR studies established that the natural products 1–7 were all potent inhibitors of K-Ras PM localization and correlated this activity with the presence of an N-formyl amino-salicylamide moiety. Of note, this latter functionality is restricted in the natural products literature to the neoantimycins, antimycins, and respirantin. What follows is an account of our chemical and biological studies into 1–8.
Figure 1.
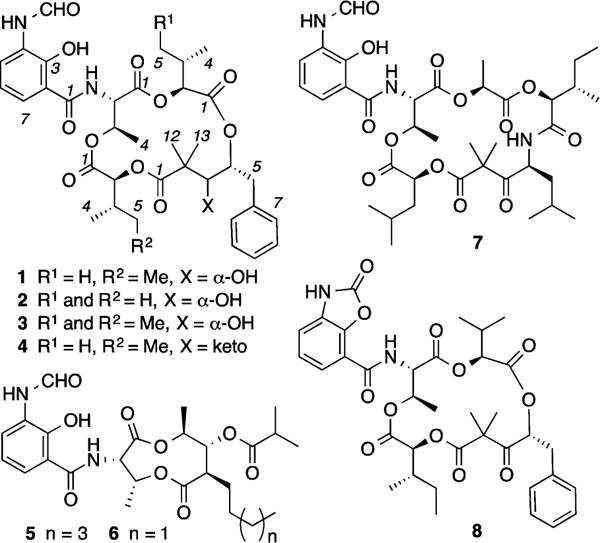
Neoantimycins and analogs (available to this study).
HRESI(+)MS measurements supported by analysis of NMR (DMSO-d6 and CDCl3) data (Supporting Information (SI), Tables S1a-b and S2a-b) permitted assignment of planar structures to 1 (C36H46N2O12) and 2 (C35H44N2O12), consistent with those previously ascribed to neoantimycin5,7 and neoantimycin F.6 Comparison of [α]D measurements for the cometabolites 1 (+58.7) and 2 (+55.2), with that published for neoantimycin (+58.3),7b supported the proposition that these compounds belong to a common antipodal series. Comparable HRESI(+)MS and NMR analyses (SI, Tables S3 and S4) permitted assignment of planar structures to 3 (C37H48N2O12) and 4 (C36H44N2O12), consistent with two new neoantimycin homologues allocated the trivial names neoantimycin G and H respectively. All the cometabolites 1–4 incorporate an aromatic and two aliphatic hydroxycarboxylic acid residues, embedded in a macrolactone ring featuring a threonine bearing a pendant N-formyl aminosalicylic acid residue. Identification and assembly of these residues were greatly assisted by diagnostic COSY and HMBC correlations (Figure 2). In advance of chemical degradation, derivatization, and analytical studies designed to assign absolute configurations to 1–4, it is useful to briefly review the history of the neoantimycin scaffold.
Figure 2.
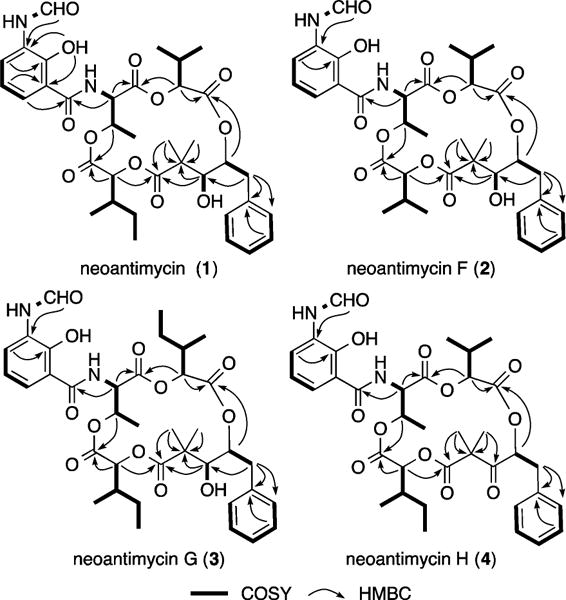
Diagnostic 2D NMR (DMSO-d6) correlations for 1–4.
The neoantimycins are a rare class of microbial natural product, biosynthetically related to the ring-contracted antimycins (e.g., 5–6) and ring-expanded respirantin (e.g., 7).8 Isolated in 1967 from a South American soil isolate of Streptomyces (formerly Streptoverticillium) orinoci,7c the partial configuration of neoantimycin was assigned in 1969 by preparative-scale (1 g) degradation that yielded methyl (S)-2-hydroxyisovalerate and methyl (2S,3S)-2-hydroxy-3-methyl-valerate.7b At that time an L-Thr configuration was also asserted, but not proven.7b A subsequent 1998 study used NMR to (incorrectly, see below) attribute a 3S,4S configuration to the 3,4-dihydroxy-2,2-dimethyl-5-phenylvaleric acid (DDPVA) residue in neoantimycin.5 Only a handful of neoantimycin-like metabolites have since been reported in the scientific literature (Figure 3). Planar structures for SW-163A (9) and SW-163B (10) were reported in 20019 from a Japanese soil Streptomyces sp. (SNA15986), while the planar structures for prunustatin A (11), and JBIR-04 (12) and JBIR-05 (13), were reported in 200510 and 200711 respectively from the Okinawan soil Streptomyces violaceoniger (4521-SVS3), as down-regulators of the molecular chaperone GRP-78. The absolute configurations of 9 and 11 were subsequently determined in 200712 by chemical degradation and derivatization. Neoantimycins D (14), E (15), and F (2) were reported in 20136 using a genome mining approach on S. orinoci, although configurational assignments made at that time were inferred rather than proven. In summary, despite a history extending back several decades, inclusive of chemical, spectroscopic, biosynthetic, and pharmacological investigations across nine metabolites (1–2 and 9–15), knowledge of the neoantimycin structures (in particular relative and absolute configuration) remains incomplete. In furthering our investigations into the chemistry and pharmacology of 1–4, we applied a rapid microscale analytical methodology capable of unambiguously assigning absolute configurations across the full array of chiral centers within the neoantimycins (and related classes).
Figure 3.
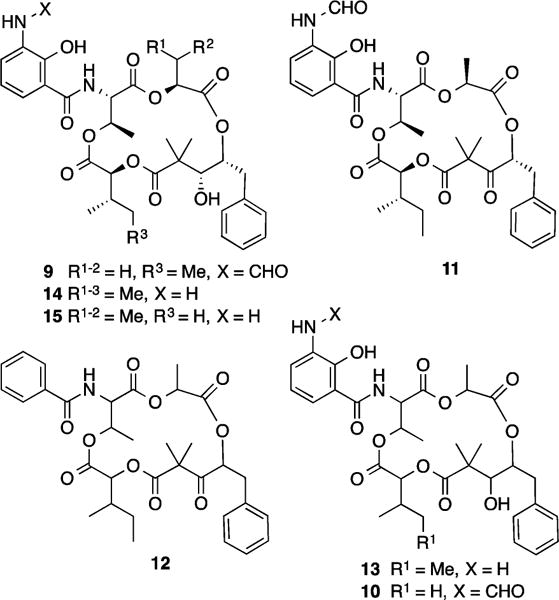
Reported neoantimycin-like metabolites (9–15).
To assign the threonine configuration in 1–4 we employed a C3 Marfey’s analysis. Microscale (50 μg) acid hydrolysates from 1–4 were derivatized with the L enantiomer of Marfey’s reagent (L-FDAA), as were authentic standards of L-Thr and L-allo-Thr. To prepare chromatographically equivalent surrogates for L-FDAA derivatives of D-Thr and D-allo-Thr, authentic standards of L-Thr and L-allo-Thr were derivatized with the D enantiomer of Marfey’s reagent (D-FDAA). Subsequent C3 Marfey’s analyses (HPLC-DAD-ESIMS) confirmed that 1–4 incorporate an L-Thr residue (SI Figure S8).
In addition to L-Thr, the microscale acid hydrolysis of 1 yielded (S)-2-hydroxyisovaleric acid (16) and (2S,3S)-2-hydroxy-3-methylvaleric acid (17) (Figure 4).7b By contrast, acid hydrolysis of 2–4 yielded one or both of these carboxylic acids, or their enantiomers. To assign absolute configurations to these residues in 2–4, a sample of the acid hydrolysate from 1 was derivatized with the R enantiomer of Mosher’s reagent to yield authentic standards of the (R)-MTPA esters 16a and 17a, while a duplicate sample was derivatized with (S)-MTPA to yield authentic standards of the (S)-MTPA esters 16b and 17b (the latter being chromatographic surrogates for the (R)-MTPA esters of the enantiomers of 16 and 17 respectively) (Figure 4). Both sets of standards were subjected to C3 Mosher analysis, confirming resolution of all four diastereomeric MTPA esters (Figure 5A). Subsequent derivatization of acid hydrolysates from 2–4 with (R)-MTPA followed by C3 Mosher analysis confirmed that 2 incorporates two residues of 16 (Figure 5B), 3 incorporates two residues of 17 (Figure 5C), and 4 incorporate one residue each of 16 and 17 (Figure 5D).
Figure 4.
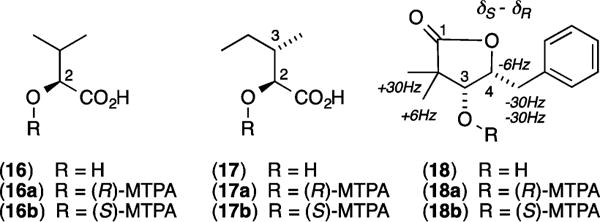
Hydrolysis products and Mosher esters from 1–4.
Figure 5.
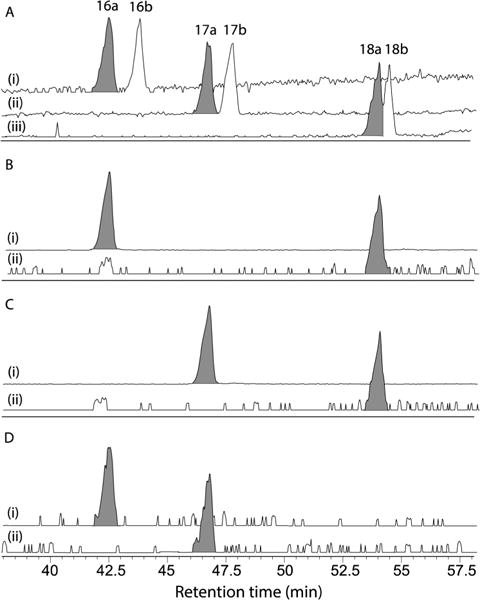
Single ion extracts (SIE) from C3 Mosher HPLC-ESIMS chromatograms of analytes prepared from (A) 1 [(i) m/z 333 [M–H]− (16a–b), (ii) m/z 347 [M–H]− (17a–b), (iii) m/z 437 [M + H]+ (18a–b)]; (B) 2 [(i) 333 [M–H]− (16a) and (ii) m/z 437 [M + H]+ (18a)]; (C) 3, [(i) m/z 347 [M–H]− (17a) and (ii) m/z 437 [M + H]+ (18a)]; and (D) 4 [(i) m/z 333 [M–H]− (16a) and (ii) m/z 347 [M–H]− (17a)].
Alkaline hydrolysis of 1 had been reported in 1975 to yield the DDPVA lactone 18,7a for which a 1998 study applied NOE measurements to assign a 3S,4S configuration,5 enantiomeric with the 3R,4R lactone produced during a 2007 alkaline hydrolysis of SW-163A (9).12 Given this unexpected stereo-complexity, we elected to use alkaline hydrolysis to independently investigate the absolute configuration of the DDPVA residues in 1–4. Alkaline hydrolysis of 1 (5 mg) yielded the lactone 18, which was derivatized to yield authentic samples of the (R)-MTPA ester 18a and the (S)-MTPA ester 18b. The 1H NMR (CDCl3) data for 18a–b recovered from 1 proved identical with esters reported from 9 (SI Table S5),12 with a Mosher NMR (δS – δR) analysis (Figure 4) establishing a 3R,4R configuration (Figure 1), revising the 1998 3S,4S assignment.5 Application of a microscale (50 μg) alkaline hydrolysis and C3 Mosher analysis revealed a common 3R,4R configuration about the DDPVA residue in 2 and 3 (Figure 5B and 5C).
To complete our configurational investigations, a sample of 1 was subjected to Dess-Martin oxidation to yield a product identical with 4 (SI Figure S9), confirming both the structure and absolute configuration of 4 (Figure 1). An unexpected byproduct of Dess-Martin oxidation was identified as the benzoxazolone 8 (C36H42N2O12). Key differences in the 1H NMR (DMSO-d6) data for 8 versus 4 included the absence of formamide (NHCHO) and phenolic (ArOH) resonances and changes in the chemical shifts for the aminosalicylic acid protons H-5 (Δδ −0.76), H-6 (Δδ +0.32), and H-7 (Δδ −0.68) (SI Table S6). As neoantimycins bearing benzoxazolones have not been described previously, the formation of 8 represented a valuable SAR opportunity.
To advance SAR investigations further, we sourced and characterized authentic samples of the biosynthetically related antimycins A2a (5) and A4a (6), and respirantin (7) (Figure 1, SI Figures S10–S11). The antimycin structure class was first reported in 1949 (as an inseparable complex),13 with the planar structure for antimycin A1 assigned in 196114 and the absolute configuration in 1972.15 With several dozen antimycins reported to date, selected antimycins have been attributed antifungal,13 insecticidal,16 nematocidal,17 and piscicidal18 activity, as well as exhibiting binding to cytochrome c oxidoreductase to inhibit the electron-transfer activity of ubiquinol in the mitochondrial respiratory chain19 and to Bcl-2 to induce apoptotic cell death.20 By comparison, literature accounts of respirantin (7) are limited and include its initial 1993 discovery as an insecticidal antibiotic from a Japanese soil Streptomyces sp.21 and its 2007 rediscovery (along with kitastatin) as a cytotoxic anticancer agent from an Alaskan soil Kitasatospora sp.22
Historically, the assignment of absolute configurations to antimycins and respirantin has proven problematic as in the case for neoantimycins (see above) and would clearly benefit from a more efficient and reliable methodology. Significantly, the C3 Marfey’s and C3 Mosher methodology described above meet all these criteria and are equally applicable to neoantimycins, antimycins, and respirantin.
We employed quantitative confocal imaging to measure the ability of 1–8 to mislocalize oncogenic mutant K-Ras (mGFP-K-RasG12 V) from the PM of intact Madin–Darby canine kidney (MDCK) cells.4 These experiments established 1–7 as exceptionally potent inhibitors of K-Ras PM localization (Table 1), as compared to the reported inhibitory properties of spirooxanthromicin (IC50 27 μM)23 and staurosporine (IC50 0.42 nM).4 These results were particularly informative as they indicate that the N-formyl amino-salicylic acid residue, common to 1–7 (but not 8), and restricted in the natural products literature to the neoantimycins, antimycins and respirantin, correlates with K-Ras mislocalization. Furthermore, we determined that selected neoantimycins, antimycins, and respirantin exhibited increased cytotoxicity toward human lung cancer cells that feature a mutated oncogenic K-Ras (e.g., A549) versus those that are not K-Ras mutated (e.g., H522) (1, 50-fold; 5, >26-fold; and 7, >33-fold).
Table 1.
K-Ras PM Mislocalization and MTT Cytotoxicity Assay Results for 1–8
| K-Ras PM mislocalization
|
MTT cytotoxicity
|
||||
|---|---|---|---|---|---|
| Emaxa | IC50 (nM) | SW620 IC50 (μM) |
SW620 Ad300 IC50 (μM) |
FRb | |
| 1 | 0.60 ± 0.03 | 2.9 ± 0.7 | 0.06 | 0.23 | 3.8 |
| 2 | 0.56 ± 0.01 | 9.9 ± 0.2 | 0.16 | 0.61 | 3.8 |
| 3 | 0.62 ± 0.01 | 7.9 ± 0.5 | 0.04 | 0.26 | 6.5 |
| 4 | 0.60 ± 0.02 | 6.4 ± 0.5 | 0.07 | 0.19 | 2.7 |
| 5 | 0.78 ± 0.02 | 7.9 ± 0.3 | 0.15 | 0.32 | 2.1 |
| 6 | 0.79 ± 0.02 | 3.7 ± 0.3 | 0.12 | 0.44 | 3.7 |
| 7 | 0.78 ± 0.02 | 0.7 ± 0.1 | 0.01 | 0.03 | 3.0 |
| 8 | inactive | inactive | >30 | c | |
Emax quantifies the maximum extent of mislocalization of K-Ras from the PM to the endomembrane.
FR: fold resistance is calculated as the IC50 ratio for SW620 Ad300 versus SW620.
Not tested due to limited amount of compound.
Finally, as up-regulation of ABC transporters such as P-glycoprotein (P-gp) in cancers can lead to accelerated drug efflux and multidrug resistance (MDR), we assayed 1–7 against the colon cancer cell line SW620 and its P-gp overexpressing daughter cell line SW620 Ad300. In control studies we confirmed that SW620 Ad300 cells were 22-fold (FR = 22) less sensitive to doxorubicin than SW620 cells and that coadministration with the P-gp inhibitor verapamil quenched this efflux effect (FR = 2.4). By comparison, 1–7 exhibited comparable cytotoxicity toward both SW620 and SW620 Ad300 cell lines (FR = 2.1–6.5), confirming that they were largely unaffected by P-gp mediated-MDR (Table 1).
In conclusion, this study explored the rare neoantimycin, antimycin, and respirantin structure classes, developing new microanalytical methodology for assigning absolute configurations, revising structures for known, and discovering and assigning structures to new neoantimycins. Significantly, this study discovered a new class of low nM K-Ras inhibitor that is not subject to P-gp mediated drug efflux.
Supplementary Material
Acknowledgments
We thank S. E. Bates and R. W. Robey (NIH, Bethesda, MD) for providing SW620 and SW620 Ad300, A. M. Piggott (UQ) for HRMS and NMR spectroscopic support, and D. Vuong and A. E. Lacey (MST) for assistance with the fermentation and metabolite purification. This research was funded in part by The University of Queensland, the Institute for Molecular Bioscience, the Cancer Prevention and Research Institute of Texas (RP130059), and the Australian Research Council (DP120100183 and LP120100088).
Footnotes
Supporting Information
These data include general experimental procedures, NMR spectroscopic data, C3 Marfey’s and C3 Mosher methods, and K-Ras and MTT cytotoxicity assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Hancock JF. Nat Rev Mol Cell Biol. 2003;4:373. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 2.Bodemann BO, White MA. Curr Biol. 2013;23:R17. doi: 10.1016/j.cub.2012.11.054. [DOI] [PubMed] [Google Scholar]
- 3.Baines AT, Xu D, Der CJ. Future Med Chem. 2011;3:1787. doi: 10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho KJ, Park JH, Piggott AM, Salim AA, Gorfe AA, Parton RG, Capon RJ, Lacey E, Hancock JF. J Biol Chem. 2012;287:43573. doi: 10.1074/jbc.M112.424457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takeda Y, Masuda T, Matsumoto T, Takechi Y, Shingu T, Floss HG. J Nat Prod. 1998;61:978. doi: 10.1021/np9800194. [DOI] [PubMed] [Google Scholar]
- 6.Li X, Zvanych R, Vanner SA, Wang W, Magarvey NA. Bioorg Med Chem Lett. 2013;23:5123. doi: 10.1016/j.bmcl.2013.07.031. [DOI] [PubMed] [Google Scholar]
- 7.(a) Benedetti E, Ganis P, Bombieri G, Caglioti L, Germain G. Acta Crystallogr, Sect B. 1975;B31:1097. [Google Scholar]; (b) Caglioti L, Misiti D, Mondelli R, Selva A, Arcamone F, Cassinelli G. Tetrahedron. 1969;25:2193. doi: 10.1016/s0040-4020(01)82768-x. [DOI] [PubMed] [Google Scholar]; (c) Cassinelli G, Grein A, Orezzi P, Pennella P, Sanfilippo A. Arch Mikrobiol. 1967;55:358. doi: 10.1007/BF00406442. [DOI] [PubMed] [Google Scholar]
- 8.Vanner SA, Li X, Zvanych R, Torchia J, Sang J, Andrews DW, Magarvey NA. Mol BioSyst. 2013;9:2712. doi: 10.1039/c3mb70219g. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi K, Tsuda E, Kurosawa K. J Antibiot. 2001;54:867. doi: 10.7164/antibiotics.54.867. [DOI] [PubMed] [Google Scholar]
- 10.Umeda Y, Chijiwa S, Furihata K, Furihata K, Sakuda S, Nagasawa H, Watanabe H, Shin-ya K. J Antibiot. 2005;58:206. doi: 10.1038/ja.2005.25. [DOI] [PubMed] [Google Scholar]
- 11.Izumikawa M, Ueda J-Y, Chijiwa S, Takagi M, Shin-ya K. J Antibiot. 2007;60:640. doi: 10.1038/ja.2007.82. [DOI] [PubMed] [Google Scholar]
- 12.Umeda Y, Furihata K, Sakuda S, Nagasawa H, Ishigami K, Watanabe H, Izumikawa M, Takagi M, Doi T, Nakao Y, Shin-ya K. Org Lett. 2007;9:4239. doi: 10.1021/ol7017856. [DOI] [PubMed] [Google Scholar]
- 13.Dunshee BR, Leben C, Keitt GW, Strong FM. J Am Chem Soc. 1949;71:2436. [Google Scholar]
- 14.(a) Birch AJ, Cameron DW, Harada Y, Rickards RW. J Chem Soc. 1961:889. [Google Scholar]; (b) Van Tamelen EE, Dickie JP, Loomans ME, Dewey RS, Strong FM. J Am Chem Soc. 1961;83:1639. [Google Scholar]
- 15.Kinoshita M, Aburaki S, Umezawa S. J Antibiot. 1972;25:373–376. doi: 10.7164/antibiotics.25.373. [DOI] [PubMed] [Google Scholar]
- 16.Kido GS, Spyhalski E. Science. 1950;112:172. doi: 10.1126/science.112.2902.172. [DOI] [PubMed] [Google Scholar]
- 17.Shiomi K, Hatae K, Hatano H, Matsumoto A, Takahashi Y, Jiang CL, Tomoda H, Kobayashi S, Tanaka H, Omura S. J Antibiot. 2005;58:74. doi: 10.1038/ja.2005.10. [DOI] [PubMed] [Google Scholar]
- 18.Cumming KB, Burress RM, Gilderhus PA. Prog Fish Cult. 1975;37:81. [Google Scholar]
- 19.Wikstrom MKF, Berden JA. Biochim Biophys Acta, Bioenerg. 1972;283:403. doi: 10.1016/0005-2728(72)90258-7. [DOI] [PubMed] [Google Scholar]
- 20.Tzung SP, Kim KM, Basanez G, Giedt CD, Simon J, Zimmerberg J, Zhang KYJ, Hockenbery DM. Nat Cell Biol. 2001;3:183. doi: 10.1038/35055095. [DOI] [PubMed] [Google Scholar]
- 21.Urushibata I, Isogai A, Matsumoto S, Suzuki A. J Antibiot. 1993;46:701. doi: 10.7164/antibiotics.46.701. [DOI] [PubMed] [Google Scholar]
- 22.Pettit GR, Tan R, Pettit RK, Smith TH, Feng S, Doubek DL, Richert L, Hamblin J, Weber C, Chapuis JC. J Nat Prod. 2007;70:1069. doi: 10.1021/np068072c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salim AA, Xiao X, Cho KJ, Piggott AM, Lacey E, Hancock JF, Capon RJ. Org Biomol Chem. 2014;12:4872. doi: 10.1039/c4ob00745j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.