

Summary
Enteropathogenic Escherichia coli (EPEC) uses a type three secretion system to inject effector proteins into host intestinal epithelial cells causing diarrhea. EPEC infection redistributes basolateral proteins β1-integrin and Na+/K+ ATPase to the apical membrane of host cells. The Crumbs (Crb) polarity complex (Crb3/Pals1/Patj) is essential for epithelial cell polarization and tight junction (TJ) assembly. Here we demonstrate that EPEC displaces Crb3 and Pals1 from the apical membrane to the cytoplasm of cultured intestinal epithelial cells and colonocytes of infected mice. In vitro studies show that EspF, but not Map alters Crb3, while both effectors modulate Pals1. EspF perturbs polarity formation in cyst morphogenesis assays and induces endocytosis and apical redistribution of Na+/K+ ATPase. EspF binds to sorting nexin 9 (SNX9) causing membrane remodeling in host cells. Infection with ΔespF/pespFD3, a mutant strain that ablates EspF binding to SNX9, or inhibition of dynamin attenuates Crb3 endocytosis caused by EPEC. In addition, infection with ΔespF/pespFD3 has no impact on Na+/K+ ATPase endocytosis. These data support the hypothesis that EPEC perturbs apical-basal polarity in an EspF-dependent manner, which would contribute to EPEC-associated diarrhea by disruption of TJ and altering the crucial positioning of membrane transporters involved in the absorption of ions and solutes.
Introduction
Enteropathogenic Escherichia coli (EPEC) causes diarrhea in infants in developing countries. EPEC infection occurs in several steps: i) attachment to host cells, ii) delivery of bacterial effector proteins through a type III secretion system into host cells and iii) formation of actin pedestals and attaching/effacing lesions (A/E) with loss of microvilli (Lai, Rosenshine, Leong, & Frankel, 2013). The dramatic loss of the absorptive microvilli contributes to loss of water and altered intestinal transport of electrolytes and solutes leading to diarrhea, dehydration and sometimes death (Nataro & Kaper, 1998). Although the mechanisms by which EPEC induces diarrhea are not completely understood, it is known that EPEC effectors EspF, Map, Tir, and Intimin cooperate to induce a loss of the activity of the sodium-D-glucose co-transporter (SGLT-1), a major water pump in the small intestine (Dean et al., 2006). Map and NleH1 bind to Na+/H+ exchanger regulatory factors I and II (NHERF1/2), and Map regulates NHERF1 activity (Simpson et al., 2006; Martinez et al., 2010). EPEC also inhibits the activity of the Cl−/HCO3− exchanger, SLC26A3 (Down Regulated in Adenoma, DRA), and this process is mediated by EspG1/G2 effectors (Gill et al., 2007; Gujral et al., 2015).
In addition to altered ion transport, EPEC also disrupts the architecture and barrier function of intestinal epithelial tight junctions (TJ) (McNamara et al., 2001; Muza-Moons, Schneeberger, & Hecht, 2004) although the mechanisms have yet to be clearly elucidated. TJ form the intercellular barrier between epithelial cells and control paracellular permeability by regulating the flux of water and solutes across the epithelium. Select EPEC effectors EspF, Map, NleA, and EspG have been studied with respect to their specific effects on TJ, regulation of the size-selective paracellular permeability and TJ restoration in epithelial cells (McNamara et al., 2001; Dean & Kenny, 2004; Muza-Moons, Schneeberger, & Hecht, 2004; Singh & Aijaz, 2015; Matsuzawa, Kuwae, & Abe, 2005; Tomson et al., 2005; Thanabalasuriar et al., 2010; Glotfelty et al., 2014). EspF is a multifunctional molecule that interacts with several host proteins. The N-terminal of EspF contains the mitochondrial targeting signal (MTS) and nucleolar targeting domain (NTD) (Holmes, Muhlen, Roe, & Dean, 2010). EspF contains three proline-rich repeats (PRR), which bind to the SH3 domain of SNX9 (Marches et al., 2006; Alto et al., 2007). The interaction of EspF with SNX9 promotes the formation of elongated plasma membrane tubules (Weflen, Alto, Viswanathan, & Hecht, 2010; Alto et al., 2007) as well as the invasion of intestinal epithelial cells by EPEC (Weflen, Alto, Viswanathan, & Hecht, 2010). SNX9 localizes and binds to dynamin at the plasma membrane mediating endocytosis and cytoskeletal organization (Lundmark & Carlsson, 2003); this complex is recruited to clathrin-enriched areas before scission of the clathrin-coated vesicles (Soulet, Yarar, Leonard, & Schmid, 2005).
TJ contribute to the maintenance of apical-basal polarity by restricting the intermixing of apical and lateral plasma membrane components. Apical-basal polarity is crucial for cell morphology, directional vesicle transportation, ion and solute transport, and specific localization of proteins and lipids to specific membrane domains (Tepass 2012; Rodriguez-Boulan & Macara, 2014). Three polarity complexes are involved in the establishment and maintenance of epithelial cell apical-basal polarity. The Crumbs complex consists of Crumbs (Crb), protein associated with Lin-7 (Pals1), and Pals1-associated tight junction protein (Patj). Par3/Par6/aPKC/cdc42 form the Par complex and the third complex is comprised of Scribble (Scrib)/Lethal giant larvae (Lgl)/Disc large (Dlg) (Rodriguez-Boulan & Macara, 2014). Polarity proteins localize to the apical membrane and have been implicated in TJ formation and polarization. The Crb family of proteins possesses a single spanning transmembrane with a highly conserved cytoplasmic tail (Crb-CT) which contains a protein 4.1/Ezrin/Radixin/Moesin (FERM)-binding motif (FBM), a PDZ-binding motif (PBM), and a potential aPKC phosphorylation site (Makarova et al., 2003; Klebes & Knust, 2000; Klose, Flores-Benitez, Riedel, & Knust, 2013; Sotillos et al., 2004; Laprise et al., 2006). Overexpression of Crb3 disrupts polarity in MDCKII cysts and delays TJ assembly (Roh, Fan, Liu, & Margolis, 2003). MCF10A cells, which possess limited endogenous expression of Crb3, fail to assemble TJ; however, exogenous expression of Crb3 is sufficient for the development of functional TJ in these cells (Fogg, Liu, & Margolis, 2005; Tilston-Lunel et al., 2016). Mutation in either the PDZ or FERM sequence of Crb3 or expression of a dominant-negative chimeric protein impairs the development of apical polarity and TJ (Roh, Fan, Liu, & Margolis, 2003; Fogg, Liu, & Margolis, 2005).
Despite the large amount of data regarding the effects of EPEC on TJ, the impact of these effectors on epithelial apical-basal polarity has not been investigated. The basolateral proteins β1-integrin and Na+/K+ ATPase are also found on the apical membrane of EPEC-infected cells (Muza-Moons, Koutsouris, & Hecht, 2003; Muza-Moons, Schneeberger, & Hecht, 2004). In addition, aberrant TJ strands are present throughout the lateral membrane of EPEC-infected cells (Muza-Moons, Schneeberger, & Hecht, 2004). Similar to EPEC infection, Helicobacter pylori disrupts the organization and assembly of apical junctions in polarized epithelial cells. Cag-A, an effector protein of H. pylori, localizes to sites of TJ formation and associates with ZO-1 (Amieva et al., 2003; Bagnoli et al., 2005). It also perturbs apical-basal polarity and cell adhesion by interacting with and inhibiting Par1/MARK and PRK2 kinases activity, essential enzymes for apical-basal polarity and cell adhesion, respectively (Saadat et al., 2007; Zeaiter et al., 2008; Mishra et al., 2015) The hypothesis of this study is that EPEC modulates the integrity of the Crb complex thus disrupting epithelial polarity.
Results
EPEC infection promotes cytoplasmic accumulation of Crb polarity complex proteins
To determine the effect of EPEC on the polarity complexes of intestinal cells, SKCO-15 monolayers were plated on Transwell inserts and infected on the apical side for 2h. EPEC induced the movement of Crb3, and to a lesser extent Pals1, from the membrane to the cytoplasm (Figure 1A, arrows). In contrast, Patj and E-cadherin remain unchanged (Figure 1A). Fluorescence intensity indicates that EPEC significantly reduced the level of membrane-associated Crb3 and Pals1 (−60.0±5.0% and −43.0±8.0%, respectively) and increased their cytoplasmic accumulation (+100.0±16.0% and +131.0±17.0%, respectively) compared to uninfected monolayers (Figure 1B). Similar experiments were conducted using MDCKII cells, which were infected with EPEC for 1–4h and immunolocalization and western blot analysis were performed. Both Crb3 and Pals1 were redistributed following EPEC infection as was seen in SKCO-15 monolayers although the internalization of Pals1 was more pronounced in MDCKII cells (Supplemental Figure S1A). The level of protein expression was unchanged (Supplemental Figure S1B). These results indicate that EPEC promotes the redistribution of Crb3 and Pals1 to the cytoplasm without altering the level of protein expression.
Figure 1.
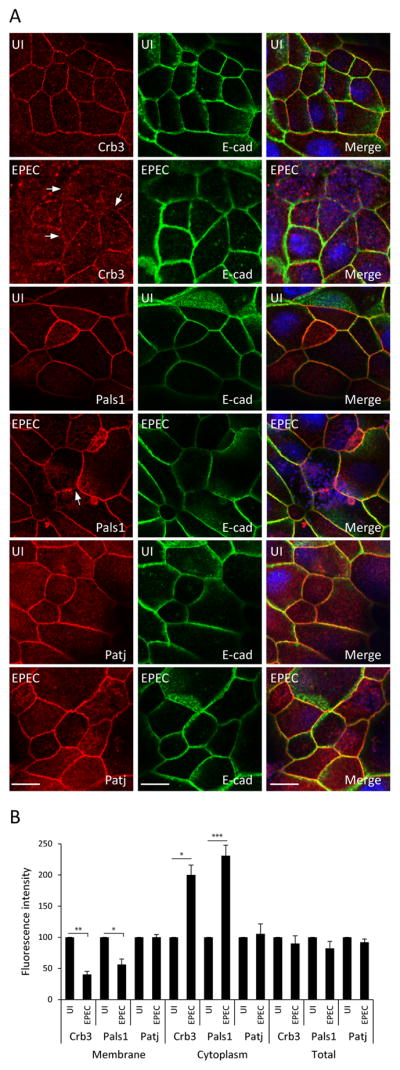
EPEC induces internalization of Crb polarity proteins Crb3 and Pals1, but not Patj in intestinal epithelial cells. SKCO-15 cells were plated on Transwells and infected on the apical side, or not (UI), with EPEC for 2h. Immunolocalization of endogenous Crb3/Pals1/Patj and E-cad was performed. (A and B) Representative confocal images and quantification of the fluorescence intensity are shown. Arrows indicate the loss of membrane-associated Crb3 and Pals1 induced by EPEC. Hoechst was used to mark the nuclei (blue) in all the images. Scale bar, 10 μm. Data represent the mean ± SEM (n=3); ***P < 0.001 values were calculated using ANOVA Tukey’s Multiple Comparison Test.
EPEC infection alters TJ structure, attenuates barrier function and increases paracellular permeability in vivo. The translocation of occludin and ZO-1 from cell-cell contacts to the cytoplasm of intestinal epithelial cells correlates with this functional phenotype (Shifflett et al., 2005; Zhang et al., 2010; Zhang et al., 2012). We questioned whether EPEC affects the localization of polarity proteins in vivo. Uninfected control mice show a well-delineated pattern of Crb3 at the apical membrane of intestinal epithelial cells (Figure 2A). In contrast, Crb3 in EPEC infected mice is more diffuse even when present in the membrane (Figure 2A). Similarly, Pals1 shifted from the apical membrane to the cytoplasm in EPEC-infected mice (Figure 2A). In concordance with our in vitro data from SKCO-15 and MDCKII cells (Figure 1A and Supplemental Figure S1A), Patj localization remained unchanged during EPEC infection in mice (Figure 2A). Fluorescence intensity of murine colonic tissues confirms that EPEC significantly reduced the level of membrane-associated Crb3 and Pals1 (−36.0±4.0% and −38.0±3.0%, respectively) and increased cytoplasmic accumulation (+82.0±5.0% and +106.0±6.0%, respectively) compared to uninfected mice (Figure 2B). In order to study the integrity of Crb complex using a murine pathogen similar to EPEC, mice infected with Citrobacter rodentium were also examined. Our data show that C. rodentium significantly reduced the amount of Crb3 in the apical membrane and increased its cytoplasmic accumulation (−36.0±4.0% and +186.0±3.0%, respectively) in a manner similar to that seen with EPEC infection (Figure 2C and D). Interestingly, the amount of Patj in the membrane slightly diminished during C. rodentium infection (−19.0±6.0%) (Figure 2C and D). These results indicate that EPEC and C. rodentium infection redistribute Crb polarity members from cell-cell contacts to the cytoplasm of murine colonic epithelial cells.
Figure 2.

EPEC and C. rodentium redistribute Crb complex proteins from the plasma membrane to the cytoplasm of murine colonocytes. Mice were infected with EPEC or C. rodentium strain by oral gavage, sacrificed on day 3 or day 10 post-infection respectively; intestinal tissues were processed for immunofluorescence. (A–D) Representative confocal images of Crb3/Pals1/Patj and quantification of the fluorescence intensity of the colonic tissues are shown. Scale bar, 40 μm. Data represent the mean ± SEM (n=3); ***P < 0.001 values were calculated using ANOVA Tukey’s Multiple Comparison Test.
Mislocalization of Crb3 from the membrane is EspF-dependent
To identify the EPEC effectors responsible for disruption of the Crb complex, in vitro models were used. SKCO-15 monolayers were infected with EPEC strains harboring mutations in effectors (Δmap and ΔespF) known to contribute to TJ disruption (McNamara et al., 2001; Dean & Kenny, 2004; Viswanathan et al., 2004a; Shifflett et al., 2005). Deletion of espF, but not map, prevented the mislocalization of Crb3 (Figure 3A). Complementation of espF (ΔespF/pespF) restored the wild-type EPEC phenotype of Crb3 internalization (Figure 3A). Deletion of either espF or map attenuated the redistribution of Pals1 (Figure 3B), while complemented strains (ΔespF/pespF or Δmap/pmap) reverted this phenotype to that of wild-type EPEC (Figure 3B). Patj and E-cadherin, as expected, were not affected by either mutant strain (Figure 3C). Total protein expression of Crb complex proteins remained unaltered following infection regardless of the infecting strain (Supplemental Figure 2A). However, fluorescence intensity demonstrated that EPEC, Δmap and the complemented strains (ΔespF/pespF or Δmap/pmap) significantly increased the cytoplasmic accumulation of Crb3 and reduced its membrane localization; both phenotypes are protected by ΔespF (Figure 3D). Although ΔespF and Δmap attenuated the membrane disruption of Pals1 caused by EPEC, these mutant strains do not significantly alter cytoplasmic accumulation (Figure 3E). The fluorescence intensity of Patj remained unchanged following infection by wild-type EPEC or mutant strains (Figure 3F).
Figure 3.

Deletion of espF protects against Crb3 mislocalization while deletion of either map or espF attenuates the internalization of Pals1. SKCO-15 cells were plated on Transwells and infected apically with wild-type EPEC, ΔespF, Δmap, or complemented strains (ΔespF/pespF and Δmap/pmap) for 2h. Cells were immunostained for Crb3/Pals1/Patj and E-cad was used to label the lateral membrane. (A–F) Representative confocal images and quantification of the fluorescence intensity of membrane, cytoplasm, and total protein are shown. Arrows show the absence of membrane-associated Crb3 and Pals1 in infected monolayers. Scale bar, 10 μm. Data represent the mean ± SEM (n=3); ***P < 0.001 values were calculated using ANOVA Tukey’s Multiple Comparison Test.
To determine if EspF has a direct role in Crb3 localization, pRetroX-Tight-Pur-EspF-HA or pRetroX-Tight-Pur-Map-HA was cloned into a Tet-inducible plasmid (Retro-X Tet-On Advanced) allowing for controlled expression of EspF-HA or Map-HA. Crb3 was localized as expected at the cell membrane of control cells (−dox), whereas induction of EspF (+dox), but not Map, disrupted Crb3 membrane localization and increased cytoplasmic accumulation (Figure 4A). In addition, transient expression of GFP-EspF displaced Crb3 from the membrane (Figure 4B). Consistent with our previous observations with EPEC infection, Patj remains at cell-cell contacts in monolayers expressing EspF (Figure 4B). Expression of HA or EspF was determined by immunodetection (Supplemental Figure S2B and C, respectively). These data suggest that EspF specifically promotes the membrane disruption of Crb3.
Figure 4.

Ectopic expression of EspF, but not Map, displaces Crb3 from the membrane. A) espF-HA or map-HA was cloned into the doxycycline-inducible pRetroX-Tight-Pur vector and transfected into SKCO-15 cells. Tet-On SKCO-15 cells were plated in absence of doxycycline (−dox) and transgene expression was induced with doxycycline (+dox) for 3 days. B) SKCO-15 cells were transiently transfected or not (control), with GFP-vector or GFP-EspF. Ectopic expression of EspF or Map was evaluated by immunodetection of HA (Green) or GFP (A and B, respectively). The impact of EspF or Map on localization of Crb3 and Patj was evaluated. Scale bar, 10 μm.
EPEC drives endocytosis of Crb3
The endocytosis of Crb3 is regulated by Rab5 (Lu & Bilder, 2005), an early endosome marker. To analyze whether EPEC promotes the endocytosis of Crb3 via the Rab5 pathway, SKCO-15 cells were infected with wild-type EPEC, ΔespF, or complemented espF (ΔespF/pespF) (Figure 5A). EPEC infection significantly increased the association of Crb3 with Rab5 in both intracellular and cell membrane-associated vesicles (Figure 5A and B). Deletion of espF reduced the association of Crb3 and Rab5 in both cytoplasmic and membrane-associated vesicles similar to uninfected cells, while complementation of espF (ΔespF/pespF) restored the wild-type EPEC phenotype (Figure 5A and B). To provide insight into the mechanism by which EspF disrupts Crb3 membrane localization, SKCO-15 monolayers were treated with inhibitors that block different endocytic pathways, then infected with EPEC for 2h. Inhibition of lipid rafts with MβCD or autophagic vacuoles with MDC failed to prevent Crb3 endocytosis induced by EPEC and showed a pattern similar to cells treated with DMSO alone (Supplemental Figure S3). Treatment of cells with dynasore, an inhibitor of dynamin GTPase activity, ablated the membrane disruption and attenuated the cytoplasmic accumulation of Crb3 caused by EPEC (Figure 5C), suggesting that EspF-induced clathrin-mediated endocytosis is involved. These data support the conclusion that EPEC EspF increases internalization of Crb3 via the clathrin pathway.
Figure 5.

Depletion of espF or treatment with dynasore inhibits EPEC-induced cytoplasmic internalization of Crb3. (A–B) SKCO-15 cells were infected with wild-type EPEC, ΔespF or complemented strain (ΔespF/pespF) for 4h and processed for immunodetection of Crb3 and Rab5. (A and B) Representative confocal images and quantification of the area of the co-localization vesicles Crb3/Rab5 are shown. Data represent the mean ± SEM (n=3); ***P < 0.001 values were calculated using ANOVA Tukey’s Multiple Comparison Test. C) SKCO-15 cells were plated in Transwells and treated with DMSO or Dyn, then cells were infected or not (UI) with EPEC for 2h. Samples were processed for immunodetection of Crb3. Scale bar, 10 μm.
The interaction of EspF with SNX9 is critical for Crb3 endocytosis
EspF has several functions in host cells including formation of a complex with 14-3-3ζ and cytokeratin 18 (CK-18) and interaction with the SH3 domain of SNX9 through its RxAPxxP motif (Viswanathan et al., 2004b; Marches et al., 2006; Alto et al., 2007). To determine the contribution of these interactions to Crb3 endocytosis, SKCO-15 cells were incubated for 2h with wild-type EPEC, ΔespF, or ΔespF complemented to express specific site-directed EspF mutations (ΔespF/pSer47A, ΔespF/pSer47/50A and ΔespF/pespFD3). Infection with ΔespF/pSer47A and ΔespF/pSer47/50A, which prevent 14-3-3ζ binding, induced the redistribution of Crb3 from the membrane to the cytoplasm in a similar manner to that seen in cells infected with wild-type EPEC (Figure 6A), indicating that EspF/14-3-3ζ interactions are not involved in Crb3 endocytosis. In contrast, cells infected with ΔespF/pespFD3, a mutant that prevents the interaction of EspF with SNX9, blocked the mislocalization of Crb3 (Figure 6A). Fluorescence intensity shows that EPEC, ΔespF/pSer47A and ΔespF/pSer47/50A strains, diminished the level of membrane-associated Crb3 and increased its cytoplasmic accumulation to the same degree as wild-type EPEC (Figure 6B). In contrast, infection with ΔespF/pespFD3 afforded the same level of protection against Crb3 redistribution as the ΔespF mutant strain (Figure 6B). These data suggest that binding of EspF to SNX9 is crucial for promoting the endocytosis of Crb3.
Figure 6.

The interaction of EspF with SNX9 is crucial for Crb3 endocytosis. SKCO-15 cells were infected with wild-type EPEC, ΔespF, or ΔespF complemented to express specific site-directed EspF mutations (ΔespF/pSer47A, ΔespF/pSer47/50A and ΔespF/pespFD3) for 2h. (A and B) Representative images of Crb3 localization and quantification of the fluorescence intensity are shown. Only infection with ΔespF and ΔespF/pespF-D3, which cannot bind SNX9, preserved Crb3 localization. Scale bar, 10 μm. Data represent the mean ± SEM (n=3); ***P < 0.001 values were calculated using ANOVA Tukey’s Multiple Comparison Test.
EspF alters apical-basal polarity
Polarity proteins are the main regulators of cyst formation. Since the data presented above demonstrate that EspF perturbs the membrane localization of Crb3, the impact of EspF on cyst formation in 3D cultures of MDCKII cells was investigated. Cells were transfected to express various GFP-EspF constructs and cyst formation was assessed. As expected, cells transfected with GFP-vector alone developed cysts containing a single lumen exhibiting cortical actin, the TJ markers ZO-1 and occludin at apical cell-cell contacts, as well as E-cadherin basolaterally (Figure 7A and Supplemental Figure S4, A and B), indicating no defect in cell polarity. In contrast, cells expressing GFP-EspF showed evidence of perturbed polarity (Figure 7A and Supplemental Figure S4, A and B) with the presence of ZO-1 and occludin along the lateral membrane (Figure 7A and Supplemental Figure S4A, arrows). In addition, cells transfected with GFP-EspF formed a significantly higher number of multi-lumen cysts and fewer single lumen cysts compared to cells expressing GFP-vector alone (Figure 7B). Expression of EspF in 2D MDCKII cultures induced a redistribution of Crb3 without changing its expression levels (Supplemental Figure S4, C and D).
Figure 7.

EspF prevents cyst formation. MDCKII cells were transfected with GFP-vector, GFP-EspF, GFP-EspF-L16E or GFP-EspF-D3, and after 24h placed as a single-cell suspension in Matrigel; cyst formation occurred after 2–3 days. Cysts were fixed and phalloidin (red) and ZO-1 (magenta) were used to label the lumen and the TJ region, respectively. Visualization of GFP was used to determine cysts containing transfected cells. A) Representative confocal images of cysts. EspF and EspF-L16E displaced ZO-1 from tight junctions to the lateral membrane (A, arrows). In contrast, cyst morphogenesis and ZO-1 localization in the presence of GFP-EspF-D3 expression was not different from that seen with GFP-vector. Scale bar, 10 μm. B) Twenty cysts from each group harboring transfected cells were analyzed for the number of lumens formed: single-lumen, 2–3 lumens and multi-lumen. Expression of GFP-EspF and EspF-L16E increased the number of multi-lumen (>3) cysts compared to GFP-vector. Data represent the mean ± SEM (n=3); *P < 0.05 values were calculated using Student t-test analysis.
The N-terminal sequence of EspF targets mitochondria and promotes apoptosis (Nougayrede & Donnenberg, 2004). Substitution of leucine at position 16 to glutamic acid (L16E) within the mitochondria targeting sequence prevents EspF localization to the mitochondria and cell death (Nougayrede & Donnenberg, 2004; Nagai, Abe, & Sasakawa, 2005). To investigate whether the effect of EspF on polarity disruption is due to increased cell death, GFP-EspF-L16E was expressed in MDCKII cells and cyst formation was analyzed. Similar to cells expressing wild-type EspF, cells expressing GFP-EspF-L16E formed significantly more multi-lumen cysts compared to those transfected with GFP-vector and displayed a redistribution of ZO-1 and occludin (Figure 7, A and B and Supplemental Figure S4A, arrows). In addition, the formation of single lumen cysts was reduced by GFP-EspF-L16E expression compared to GFP-vector (Figure 7B). Expression of GFP-EspF-L16E also mislocalized Crb3 from the cell-cell contacts without altering its protein expression in a similar manner to that of wild-type EspF (Supplemental Figure S4, C and D). To verify that EspF-L16E expression reduced apoptosis caused by wild-type EspF, cleaved caspase-3 was examined. GFP-EspF-L16E diminished the number of cells positive for cleaved caspase-3 compared to GFP-EspF expression (Supplemental Figure S5). These results indicate that the interference of EspF with the formation of apical-basal polarity is independent of its apoptotic function.
Since infection with ΔespF/pespFD3 mutant protects the mislocalization of Crb3 (Figure 6A), we questioned the impact of EspF/SNX9 interaction on cyst formation and polarity. Interestingly, cells expressing GFP-EspF-D3 formed normal single lumen cysts with the apical marker actin outlining a single central lumen, ZO-1 and occludin restricted to the TJ, and E-cadherin distributed to the basolateral membrane in a similar manner to cells transfected with GFP-vector (Figure 7A and Supplemental Figure S4, A and B). No significant differences were observed in cyst formation between cells transfected with GFP-vector and those expressing GFP-EspF-D3 (Figure 7B). Expression of GFP-EspF-D3 did not alter the redistribution of Crb3 from the plasma membrane or the level of protein expression (Supplemental Figure S4, C and D). These results indicate that EspF via its interaction with SNX9 impairs the development of apical-basal polarity in epithelial cells.
EPEC infection induces the redistribution of the basolateral proteins β1-integrin and Na+/K+ ATPase to the apical membrane of T-84 intestinal cells (Muza-Moons, Koutsouris, & Hecht, 2003; Muza-Moons, Schneeberger, & Hecht, 2004). To determine the impact of EPEC on the apical-basal polarity of intestinal epithelial cells, SKCO-15 were plated on Transwells, infected with wild-type EPEC or EspF mutant strains (ΔespF, ΔespF/pespFD3, ΔespF/pespF), and the localization of the basolateral protein Na+/K+ ATPase was determined. EPEC induced the endocytosis of Na+/K+ ATPase after 2h infection and re-distributed it from the basolateral membrane to the apical domain by 3h post-infection (Figure 8). Infection of monolayers with ΔespF or ΔespF/pespFD3 ablated the apical redistribution of Na+/K+ ATPase (Figure 8). In contrast, complementation of ΔespF with wild-type espF (ΔespF/pespF) restored the wild-type phenotype of Na+/K+ ATPase mislocalization to the apical domain (Figure 8). Together these results indicate that EPEC EspF plays a major role in the perturbation of intestinal epithelial apical-basal polarity.
Figure 8.

EPEC induces the internalization of the basolateral protein Na+/K+ ATPase and its redistribution to the apical membrane in an EspF-dependent manner. SKCO-15 cells were infected wild-type EPEC, ΔespF, ΔespF/pespFD3 or complemented strain (ΔespF/pespF) for 2–3h, and monolayers immunostained for Na+/K+ ATPase. Figure shows representative confocal images of three independent experiments. Scale bar, 10 μm.
Discussion
EPEC infection disrupts intestinal epithelial cell architecture and TJ complexes leading to altered barrier and fence function (Simonovic, Rosenberg, Koutsouris, & Hecht, 2000; McNamara et al., 2001; Dean & Kenny, 2004; Shifflett et al., 2005; Guttman et al., 2006; Tomson et al., 2005; Matsuzawa, Kuwae, & Abe, 2005; Thanabalasuriar et al., 2010; Singh & Aijaz, 2015). EPEC also modulates intestinal epithelial cell electrolyte transport, reducing the expression of sodium hydrogen exchanger 3 (NHE3), the major intestinal transporter for Na+ absorption (Gawenis et al., 2002; Hecht et al., 2004; Hodges et al., 2008). Furthermore, EPEC alters the activity and surface expression of the intestinal Cl−/HCO3− exchanger, SLC26A3/DRA, resulting in a reduced Cl− uptake and its accumulation in the lumen, driving water loss (Gill et al., 2007; Gujral et al., 2015). EPEC effectors EspF, Map, Tir, and Intimin, act together to inactivate SGLT-1, a cotransporter responsible for 70% of the total fluid uptake by the small intestine (Dean et al., 2006; Meinild et al., 1998). Although the effect of EPEC on TJ barrier function has been widely investigated, few studies have focused on the impact on TJ fence function and maintenance of apical-basal polarity. EPEC infection redistributes the basolateral proteins β1-integrin and Na+/K+ ATPase to the apical surface (Muza-Moons, Koutsouris, & Hecht, 2003) and induces aberrant TJ strands throughout the lateral membrane (Muza-Moons, Schneeberger, & Hecht, 2004), indicating a loss in apical-basal polarity. Other pathogens have been reported to induce the loss of polarity. For example, the H. pylori effector, CagA, interacts with and inhibits Par1 kinase activity causing junctional and polarity defects (Saadat et al., 2007). Neisseria meningitidis recruits the Par3/Par6/aPKC complex beneath colonies, thus disorganizing cell-cell junctions and perturbing the endothelial barrier (Coureuil et al., 2009). This is the first study to demonstrate that EPEC perturbs apical-basal polarity by targeting Crb polarity complex.
In polarized epithelial cells, the Crb complex located at the apical membrane is important for the development and maintenance of apical-basal cell polarity (Roman-Fernandez & Bryant, 2016). Crb3 is regulated by multiple mechanisms, including endocytosis, recycling, degradation, and interaction with cytoplasmic partners. For example, expression of Snail, down-regulation of either Patj/Cdc42/Lgl/Adaptor Protein 2 (AP-2), or mutant aPKC with reduced kinase activity, all destabilize Crb inducing its internalization from the membrane into sub-apical vesicles causing polarity defects (Harder et al., 2012; Michel et al., 2005; Harris & Tepass, 2008; Kim et al., 2009; Fletcher et al., 2012; de Vreede et al., 2014; Whiteman, Liu, Fearon, & Margolis, 2008; Lin et al., 2015). Our findings show that internalized Crb3 induced by EPEC infection is recruited to the endosomal pathway and that this phenotype is dependent on the interaction of EspF and SNX9. EspF regulates actin cytoskeletal dynamics through its binding and activation of neuronal Wiskott-Aldrich syndrome protein (N-WASP) and dynamically colocalizes to clathrin-coated pits (CCPs) (Alto et al., 2007). SNX9 associates with N-WASP and dynamin, and together with Arp2/3 and associated proteins, these complexes are recruited to CCPs promoting the clathrin-mediated endocytosis (CME) of certain plasma membrane receptors (Badour et al., 2007; Merrifield, Qualmann, Kessels, & Almers, 2004; Benesch et al., 2005; Yarar, Waterman-Storer, & Schmid, 2007; Lundmark & Carlsson, 2003; Soulet, Yarar, Leonard, & Schmid, 2005; Shin et al., 2007). AP-2, also involved in CME, binds to and controls Crb3 levels at the plasma membrane by regulating its internalization (Lin et al., 2015). Our data demonstrate that inhibition of either dynamin GTPase activity or EspF/SNX9 interaction (EspF-D3 mutant) blocks EPEC-induced Crb3 endocytosis suggesting that the EspF/SNX9 complex and possibly other binding partners, as NWASP/Arp2/3/dynamin/clathrin, promote Crb3 endocytosis.
Little is known about the endocytosis of the other Crb polarity complex proteins, Pals1 and Patj. Amot is an adaptor protein that binds both Patj and Pals1 (Sugihara-Mizuno et al., 2007; Wells et al., 2006) and promotes their internalization to endosomes and induces the loss of TJ integrity (Wells et al., 2006; Heller et al., 2010; Campbell et al., 2016). Amot is found in Rab11 positive endosomes but not with Rab5 or Rab7 in MDCK cells, indicating that Amot has an important role in protein recycling (Heller et al., 2010). Our data show that Pals1, but not Patj, moves to the cytoplasm following EPEC infection and that this phenotype is diminished by deletion of espF or map. The finding that EspF, but not Map, alters Crb3 while both effectors modulate Pals1 suggests that the internalization of Crb3 and Pals1 is differentially regulated. The maintenance and regulation of apical-basal polarity depends largely on the interaction of Crb3 with Pals1 and ezrin (Makarova et al., 2003; Roh, Fan, Liu, & Margolis, 2003; Li et al., 2014; Whiteman et al., 2014). Pals1 binds to and regulates ezrin localization at the apical membrane of gastric parietal cells (Cao et al., 2005). EPEC activates ezrin, thus contributing to disruption of intestinal barrier function (Simonovic et al., 2001). In addition, reduced expression of ezrin decreases transepithelial electrical resistance (TER) and augments permeability of MCF10A cells overexpressing Crb3b (Tilston-Lunel et al., 2016). EPEC induces microvillus effacement of the intestinal epithelium of infected human and mice (Savkovic et al., 2005). Our in vivo data correlate with our in vitro findings, demonstrating that EPEC redistributes Crb3 and Pals1 from cell-cell contacts to the cytoplasm of colonocytes. Similar to EPEC, infection with C. rodentium also induces the redistribution of Crb3. Interestingly, membrane-associated Patj is slightly diminished in murine colonocytes infected with C. rodentium indicating that despite the similarities between EPEC and C. rodentium strains, there are some pathophysiological differences between these two organisms. The difference in this phenotype could simply be the increase in attached C. rodentium as compared to EPEC. Interestingly, the intestines of Crb3- or ezrin-null mice show similar defects to those caused by EPEC, intestinal villus fusion and disrupted microvilli (Whiteman et al., 2014; Charrier, Loie, & Laprise, 2015; Saotome, Curto, & McClatchey, 2004; Casaletto, Saotome, Curto, & McClatchey, 2011). Although the mechanism by which EPEC displaces Crb3 and Pals1 from plasma membrane is unknown, it is possible that EPEC destabilizes the cytoplasmic interaction of Crb3 with Pals1/ezrin leading to a loss of cell polarity and barrier function.
The establishment of apical-basal polarity allows the asymmetric segregation of proteins and lipids into apical and basolateral domains and contributes to the development of lumens allowing vectorial transport, an essential process for the balance of ions, solutes and water along epithelia (Shivas, Morrison, Bilder, & Skop, 2010; Rodriguez-Boulan & Macara, 2014). EPEC infection is characterized by an increased loss of ions and water, and profound diarrhea. In the intestine, the assimilation of electrolytes, nutrients, and fluid occurs via Na-dependent cotransport processes (Rose & Valdes, 1994). The basolateral protein Na+/K+ ATPase provides for the export of Na+ across the basolateral membrane, thus controlling the osmotic balance and volume regulation of cells (Robinson & Flashner, 1979; Pavlov & Sokolov, 2000). The proper function of Na+/K+ ATPase is essential for efficient nutrient and ion absorption. Decreased Na+/K+ ATPase activity during acute and transient enteritis diminishes the reabsorption of sodium and water (Allgayer et al., 1988; Ejderhamn, Finkel, & Strandvik, 1989; Rachmilewitz, Karmeli, & Sharon, 1984). In an animal model of chronic ileal inflammation, decreased Cl−/HCO−3 and stimulated Na+/K+ ATPase exchange in villus and crypts cells, respectively, has been shown, suggesting that chronic inflammation alters electrolyte transport (Sundaram & West, 1997). Na-glucose uptake, Na-alanine, Na-glutamine and Na-taurocholate cotransport are also down regulated during inflammation due to diminished expression and activity of Na+/K+ ATPase (Sundaram, Wisel, Rajendren, & West, 1997; Sundaram, Wisel, & Fromkes, 1998; Saha, Arthur, Kekuda, & Sundaram, 2012; Coon, Kekuda, Saha, & Sundaram, 2010). Campylobacter jejuni, an intestinal pathogen, decreases Na+/K+ ATPase activity in the rat ileum, causing impaired electrolyte absorption (Kanwar et al., 1994). This underscores the importance of apical-basal polarity maintenance and supports the conclusion that the perturbation of Na+/K+ ATPase activity contributes to EPEC pathophysiology.
Although the mechanisms are not well defined, several studies have focused on the importance of Na+/K+ ATPase endocytosis. Na+/K+ ATPase interacts with Src leading to the subsequent activation of PI3K, Ras/Raf/ERKs, PLC/PKC and ERK1/2 pathways (Liang et al., 2006; Tian et al., 2006). PI3K activation, as well as treatment with ouabain, a cardiac glycoside that inhibits Na+/K+ ATPase, facilitates the binding of Na+/K+ ATPase to AP-2, recruiting Na+/K+ ATPase into CCPs and triggering its localization to both early and late endosomes (Yudowski et al., 2000; Liu et al., 2004). Our findings show that EPEC induces Na+/K+ ATPase endocytosis in an EspF-dependent manner. Interestingly, the ablation of EspF/SNX9 interaction inhibits Na+/K+ ATPase redistribution to the apical membrane indicating that EspF/SNX9 interaction plays an important role in maintaining apical-basal polarity.
In summary, this study shows that EPEC induces the redistribution of Crb3 and Pals1 in an EspF-dependent manner (Figure 9). The data presented herein support the conclusion that binding of EspF to SNX9 is crucial for both preventing the formation of polarity (cyst multi-lumen structures) and disrupting established epithelial polarity (movement of basolateral Na+/K+ ATPase to the apical domain). We therefore speculate that EspF targeting and disruption of the Crb polarity complex may initiate crucial signaling processes that ultimately impact host epithelial physiology and integrity contributing to EPEC pathogenesis.
Figure 9.

Model depicting the effect of EPEC on apical-basal polarity. Polarized epithelial cells consist of the apical membrane facing the lumen and the basolateral domain contacting the underlying basement membrane. The apical polarity complex Crb (Crb3/Pals1/Patj) localizes at the TJ (Control). i) During EPEC infection, the injected EPEC effectors, including EspF and Map, induce the endocytosis of polarity proteins Crb3 and Pals1 and the basolateral protein Na+/K+ ATPase. ii) Increased endocytosis of Crb3, Pals1 and likely other polarity proteins leads to a loss in the apical-basal polarity, as demonstrated by the redistribution of Na+/K+ ATPase to the apical membrane.
Experimental procedures
Tissue culture
SKCO-15 and MDCKII epithelial cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/I penicillin and 100 U/I streptomycin at 5% CO2.
Antibodies and Reagents
Antibodies used include (species-antigen): rat-Crb3 (ab180835, Abcam), rabbit-Patj (ab102113, Abcam), rabbit-Pals1 (07-708, Millipore), rabbit-ZO-1 (61-7300, Zymed), rabbit-EspF (Hecht Lab), rabbit-cleaved caspase-3 (9661S, Cell Signaling), rabbit-actin (A2066, Sigma), rabbit-HA (C29F4, Cell Signaling), rabbit-GFP (A11122, Life Technologies), mouse-Rab5 (sc-46692, Santa Cruz), mouse-E-cad (610181, BD), mouse-occludin (33-1500, ThermoFisher Scientific), mouse-Na+/K+ ATPase (610992, BD), Phalloidin Alexa Fluor 568 (Life Technologies). Secondary antibodies used for immunofluorescence were Alexa fluor (Life Technologies). Drugs used were from Sigma, 10μM methyl-beta-cyclodextrin (MβCD), 400 μM monodansyl cadaverin (MDC) and 80μM dynasore (Dyn).
Bacteria culture
The following EPEC strains were used: wild-type EPEC 0127:H6 E2348/69, ΔespF (McNamara & Donnenberg, 1998), ΔespF/pespF, Δmap (Kenny & Jepson, 2000), Δmap/pmap, ΔespF/pespFD3 (Alto et al., 2007), ΔespF/pSer47A and ΔespF/pSer47/50A (Hecht unpublished). Bacterial strains were grown at 37°C in Luria broth overnight and with addition of appropriate selective antibiotics. For infections, overnight bacterial cultures were diluted in serum- and antibiotic-free T84 1:1 (vol/vol) mixture of low glucose Dulbecco’s modified Eagle’s medium (DMEM) and Ham’s F-12 (Gibco) medium containing 0.5% mannose and grown to mid-log growth phase. Monolayers were infected with EPEC at a MOI of 50 for the indicated times.
Murine infection
For murine infection, six to eight-week-old male C57BL/6J mice were used (Jackson Laboratory Bar Harbor, ME, USA) and housed in a specific pathogen-free facility at Loyola University Chicago, Maywood, IL for 7–14 days with free access to food and water. Mice were infected with EPEC by oral gavage as previously described (Rhee et al., 2011) and sacrificed on day 3 post-infection. All animal protocols were approved by LU Animal Care and Use Committee. Mouse intestines were removed, cleaned, processed as “Swiss rolls”, and fixed in 10% phosphate-buffered formalin for 24h. Fixed tissues were embedded in paraffin, cut into 5 μm sections, and processed for immunofluorescence. Paraffin embedded sections of intestine from C. rodentium infected mice were obtained from the laboratory of Dr. Katherine Knight (Department of Microbiology; Loyola University School of Medicine). Infection and tissue preparation were performed as previously described (Jones & Knight, 2012).
DNA plasmids
For transient transfection, the following plasmids were used: pGFP (vector alone), pEGFP-EspF and pEGFP-EspF-D3 (Alto et al., 2007). The pEGFP-C1-EspF-L16E plasmid was constructed using QuikChange site-directed mutagenesis kit according to manufacturer’s protocol (Agilent Technologies) using pEGFP-EspF as template and the primer pair (5′-cgacttgcgatacctacttcctgccgccctagtgtag-3′ and 5′-ctacactagggcggcaggaagtaggtatcgcaagtcg-3′). For inducible cell lines, EspF-HA and Map-HA were generated by amplifying EPEC genomic DNA using the following primers: EspF-FOR (5′-cttgaagcggccgcatgcttaatggaattagtaacgc-3′ and EspF-REV-HA (5′-aatcaggaattcttaagcgtaatctggaacatcgtatgggtaccctttcttcgattgctc-3′); Map-FOR (5′-cttgaagcggccgcatgtttagtccaacggcaatgg-3′) and Map-REV-HA (5′-aatcaggaattcctaagcgtaatctggaacatcgtatgggtacagccgagtatcctgcacatt-3′) and separately cloned into the NotI/EcoRI sites of the retroviral plasmid pRetroX-Tight-Pur (Clontech), then co-transfected with pRetroX-Tet- On-Advance Inducible Expression System (632104, Clontech) in SKCO-15 cells.
Western blotting
For protein isolation, cells were washed twice with ice-cold phosphate buffered saline (PBS), and lysed in 0.25 ml of radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 40 mM Tris-HCl, pH 7.5, 2 mM EDTA, 10% glycerol, 1% Triton X-100, 0.5% sodium deoxycholate and 0.2% SDS) and protease inhibitor cocktail (P8340, Sigma). Proteins were boiled in SDS sample buffer, and analyzed by SDS-PAGE. For immunoblotting, nitrocellulose or ImmobilonR-FL (IPFL00010, Millipore) membranes were incubated in blocking solution (Invitrogen) for 1h, and probed with primary antibodies overnight at 4°C. Membranes were washed three times for 10 min with TBS + 0.05% Tween and incubated for 1h with secondary antibodies horseradish peroxidase-conjugated (Sigma) or IRDyeR 800CW (915-32213, LI-COR). Chemiluminescent exposure was performed with ECL (GE Healthcare) or using LI-COR Odyssey Classic (Image Studio software).
Immunofluorescence
Cells were plated on Transwell inserts or glass coverslips and fixed with cold methanol at −20°C. Fixation of cysts in Matrigel (Corning) was performed using 3% PFA and permeabilized with 0.1% Triton X-100 in PBS. For mouse tissue samples, paraffin embedded tissues were deparaffinized in xylene, rehydrated with ethanol and H2O washes, and antigen retrieval performed in Tris-EDTA plus Tween20 buffer (pH 9) (Crb3 and Pals1) and in sodium citrate plus Tween20 buffer (Patj). Samples were blocked and incubated with primary antibodies overnight at 4°C or 30°C. Tissue samples were blocked and incubated with primary and secondary antibodies in 5% normal goat serum in 1x PBS plus 0.1% Tween20 and 0.1% saponin. Samples were washed with PBS, and then processed for immunofluorescence using Alexa fluor conjugated goat secondary antibodies and counterstained with Hoechst to visualize nuclei. Slides were analyzed using a Leica DM 4000B microscope (MetaMorph software) or confocal Leica TCS SPE DMI 4000B (LAS X software) microscope. Images were processed using Adobe Photoshop and ImageJ software.
Tet-On system and transient transfections
To generate SKCO-15 Tet-On system, cells were transfected with p-RetroX-Tet-On Advanced and pRetroX-Tight-Pur-EspF-HA or pRetroX-Tight-Pur-Map-HA using the Xfect nanoparticles polymer kit (ST0153, Clontech) following manufacturer’s instructions. After transfection, cells were selected with G418 and puromycin for 2–3 weeks. Expression of pRetroX-Tight-Pur-EspF-HA or pRetroX-Tight-Pur-Map-HA was induced in presence of Doxycycline (500 ng/ml) for three days. For transient transfection, MDCKII and SKCO-15 cells were transfected with Lipofectamine 2000 (Invitrogen) or nanoparticles according to manufacturer instructions. Cells were processed for immunofluorescence 72–96h post-transfection.
Cyst formation assay
MDCKII cells were plated and transfected with GFP-vector, GFP-EspF, GFP-EspF-L16E or GFP-EspF-D3 DNA constructs using the Xfect nanoparticles polymer kit. The following day, cells were trypsinized and plated in Matrigel to allow cyst formation according to (Debnath, Muthuswamy, & Brugge, 2003). Immunostaining was performed 3–4 days post-transfection.
Supplementary Material
Acknowledgments
This research was supported by National Institutes of Health grant (DK097043 to GH) and Edward Hines JR VA Hospital grant (BX002687 to GH). We thank Dr. Ben Margolis, University of Michigan, for training our laboratory members to perform cyst morphogenesis assays. We thank Dr. Katherine Knight, Loyola University Chicago-Stritch School of Medicine for providing the C. rodentium tissue samples.
Abbreviations used
- EPEC
Enteropathogenic Escherichia coli
- TJ
tight junctions
- Crb3
Crumbs3
References
- Allgayer H, Kruis W, Paumgartner G, Wiebecke B, Brown L, Erdmann E. Inverse relationship between colonic (Na+ + K+)-ATPase activity and degree of mucosal inflammation in inflammatory bowel disease. Digestive diseases and sciences. 1988;33:417–422. doi: 10.1007/BF01536025. [DOI] [PubMed] [Google Scholar]
- Alto NM, Weflen AW, Rardin MJ, Yarar D, Lazar CS, Tonikian R, … Dixon JE. The type III effector EspF coordinates membrane trafficking by the spatiotemporal activation of two eukaryotic signaling pathways. The Journal of cell biology. 2007;178:1265–1278. doi: 10.1083/jcb.200705021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science (New York, NY ) 2003;300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badour K, McGavin MK, Zhang J, Freeman S, Vieira C, Filipp D, … Siminovitch KA. Interaction of the Wiskott-Aldrich syndrome protein with sorting nexin 9 is required for CD28 endocytosis and cosignaling in T cells. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:1593–1598. doi: 10.1073/pnas.0610543104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagnoli F, Buti L, Tompkins L, Covacci A, Amieva MR. Helicobacter pylori CagA induces a transition from polarized to invasive phenotypes in MDCK cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:16339–16344. doi: 10.1073/pnas.0502598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesch S, Polo S, Lai FP, Anderson KI, Stradal TE, Wehland J, Rottner K. N-WASP deficiency impairs EGF internalization and actin assembly at clathrin-coated pits. Journal of cell science. 2005;118:3103–3115. doi: 10.1242/jcs.02444. [DOI] [PubMed] [Google Scholar]
- Campbell CI, Samavarchi-Tehrani P, Barrios-Rodiles M, Datti A, Gingras AC, Wrana JL. The RNF146/Tankyrase pathway maintains the junctional Crumbs complex through regulation of Angiomotin. Journal of cell science. 2016 doi: 10.1242/jcs.188417. [DOI] [PubMed] [Google Scholar]
- Cao X, Ding X, Guo Z, Zhou R, Wang F, Long F, … Yao X. PALS1 specifies the localization of ezrin to the apical membrane of gastric parietal cells. The Journal of biological chemistry. 2005;280:13584–13592. doi: 10.1074/jbc.M411941200. [DOI] [PubMed] [Google Scholar]
- Casaletto JB, Saotome I, Curto M, McClatchey AI. Ezrin-mediated apical integrity is required for intestinal homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:11924–11929. doi: 10.1073/pnas.1103418108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrier LE, Loie E, Laprise P. Mouse Crumbs3 sustains epithelial tissue morphogenesis in vivo. Scientific reports. 2015;5:17699. doi: 10.1038/srep17699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon S, Kekuda R, Saha P, Sundaram U. Glucocorticoids differentially regulate Na-bile acid cotransport in normal and chronically inflamed rabbit ileal villus cells. American journal of physiology Gastrointestinal and liver physiology. 2010;298:G675–82. doi: 10.1152/ajpgi.00176.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coureuil M, Mikaty G, Miller F, Lecuyer H, Bernard C, Bourdoulous S, … Nassif X. Meningococcal type IV pili recruit the polarity complex to cross the brain endothelium. Science (New York, NY ) 2009;325:83–87. doi: 10.1126/science.1173196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vreede G, Schoenfeld JD, Windler SL, Morrison H, Lu H, Bilder D. The Scribble module regulates retromer-dependent endocytic trafficking during epithelial polarization. Development (Cambridge, England) 2014;141:2796–2802. doi: 10.1242/dev.105403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean P, Kenny B. Intestinal barrier dysfunction by enteropathogenic Escherichia coli is mediated by two effector molecules and a bacterial surface protein. Molecular microbiology. 2004;54:665–675. doi: 10.1111/j.1365-2958.2004.04308.x. [DOI] [PubMed] [Google Scholar]
- Dean P, Maresca M, Schuller S, Phillips AD, Kenny B. Potent diarrheagenic mechanism mediated by the cooperative action of three enteropathogenic Escherichia coli-injected effector proteins. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:1876–1881. doi: 10.1073/pnas.0509451103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods (San Diego, Calif ) 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- Ejderhamn J, Finkel Y, Strandvik B. Na, K-ATPase activity in rectal mucosa of children with ulcerative colitis and Crohn’s disease. Scandinavian Journal of Gastroenterology. 1989;24:1121–1125. doi: 10.3109/00365528909089265. [DOI] [PubMed] [Google Scholar]
- Fletcher GC, Lucas EP, Brain R, Tournier A, Thompson BJ. Positive feedback and mutual antagonism combine to polarize Crumbs in the Drosophila follicle cell epithelium. Current biology: CB. 2012;22:1116–1122. doi: 10.1016/j.cub.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Fogg VC, Liu CJ, Margolis B. Multiple regions of Crumbs3 are required for tight junction formation in MCF10A cells. Journal of cell science. 2005;118:2859–2869. doi: 10.1242/jcs.02412. [DOI] [PubMed] [Google Scholar]
- Gawenis LR, Stien X, Shull GE, Schultheis PJ, Woo AL, Walker NM, Clarke LL. Intestinal NaCl transport in NHE2 and NHE3 knockout mice. American journal of physiology Gastrointestinal and liver physiology. 2002;282:G776–84. doi: 10.1152/ajpgi.00297.2001. [DOI] [PubMed] [Google Scholar]
- Gill RK, Borthakur A, Hodges K, Turner JR, Clayburgh DR, Saksena S, … Dudeja PK. Mechanism underlying inhibition of intestinal apical Cl/OH exchange following infection with enteropathogenic E. coli. The Journal of clinical investigation. 2007;117:428–437. doi: 10.1172/JCI29625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glotfelty LG, Zahs A, Hodges K, Shan K, Alto NM, Hecht GA. Enteropathogenic E. coli effectors EspG1/G2 disrupt microtubules, contribute to tight junction perturbation and inhibit restoration. Cellular microbiology. 2014;16:1767–1783. doi: 10.1111/cmi.12323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gujral T, Kumar A, Priyamvada S, Saksena S, Gill RK, Hodges K, … Dudeja PK. Mechanisms of DRA recycling in intestinal epithelial cells: effect of enteropathogenic E. coli. American journal of physiology Cell physiology. 2015;309:C835–46. doi: 10.1152/ajpcell.00107.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman JA, Li Y, Wickham ME, Deng W, Vogl AW, Finlay BB. Attaching and effacing pathogen-induced tight junction disruption in vivo. Cellular microbiology. 2006;8:634–645. doi: 10.1111/j.1462-5822.2005.00656.x. [DOI] [PubMed] [Google Scholar]
- Harder JL, Whiteman EL, Pieczynski JN, Liu CJ, Margolis B. Snail destabilizes cell surface Crumbs3a. Traffic (Copenhagen, Denmark) 2012;13:1170–1185. doi: 10.1111/j.1600-0854.2012.01376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KP, Tepass U. Cdc42 and Par proteins stabilize dynamic adherens junctions in the Drosophila neuroectoderm through regulation of apical endocytosis. The Journal of cell biology. 2008;183:1129–1143. doi: 10.1083/jcb.200807020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht G, Hodges K, Gill RK, Kear F, Tyagi S, Malakooti J, … Dudeja PK. Differential regulation of Na+/H+ exchange isoform activities by enteropathogenic E. coli in human intestinal epithelial cells. American journal of physiology Gastrointestinal and liver physiology. 2004;287:G370–8. doi: 10.1152/ajpgi.00432.2003. [DOI] [PubMed] [Google Scholar]
- Heller B, Adu-Gyamfi E, Smith-Kinnaman W, Babbey C, Vora M, Xue Y, … Wells CD. Amot recognizes a juxtanuclear endocytic recycling compartment via a novel lipid binding domain. The Journal of biological chemistry. 2010;285:12308–12320. doi: 10.1074/jbc.M109.096230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges K, Alto NM, Ramaswamy K, Dudeja PK, Hecht G. The enteropathogenic Escherichia coli effector protein EspF decreases sodium hydrogen exchanger 3 activity. Cellular microbiology. 2008;10:1735–1745. doi: 10.1111/j.1462-5822.2008.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A, Muhlen S, Roe AJ, Dean P. The EspF effector, a bacterial pathogen’s Swiss army knife. Infection and immunity. 2010;78:4445–4453. doi: 10.1128/IAI.00635-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SE, Knight KL. Bacillus subtilis-mediated protection from Citrobacter rodentium-associated enteric disease requires espH and functional flagella. Infection and immunity. 2012;80:710–719. doi: 10.1128/IAI.05843-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanwar RK, Ganguly NK, Kanwar JR, Kumar L, Walia BN. Impairment of Na+, K(+)-ATPase activity following enterotoxigenic Campylobacter jejuni infection: changes in Na+, Cl- and 3-O-methyl-D-glucose transport in vitro, in rat ileum. FEMS microbiology letters. 1994;124:381–385. doi: 10.1111/j.1574-6968.1994.tb07312.x. [DOI] [PubMed] [Google Scholar]
- Kenny B, Jepson M. Targeting of an enteropathogenic Escherichia coli (EPEC) effector protein to host mitochondria. Cellular microbiology. 2000;2:579–590. doi: 10.1046/j.1462-5822.2000.00082.x. [DOI] [PubMed] [Google Scholar]
- Kim S, Gailite I, Moussian B, Luschnig S, Goette M, Fricke K, … Wodarz A. Kinase-activity-independent functions of atypical protein kinase C in Drosophila. Journal of cell science. 2009;122:3759–3771. doi: 10.1242/jcs.052514. [DOI] [PubMed] [Google Scholar]
- Klebes A, Knust E. A conserved motif in Crumbs is required for E-cadherin localisation and zonula adherens formation in Drosophila. Current biology: CB. 2000;10:76–85. doi: 10.1016/s0960-9822(99)00277-8. [DOI] [PubMed] [Google Scholar]
- Klose S, Flores-Benitez D, Riedel F, Knust E. Fosmid-based structure-function analysis reveals functionally distinct domains in the cytoplasmic domain of Drosophila crumbs. G3 (Bethesda, Md ) 2013;3:153–165. doi: 10.1534/g3.112.005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Rosenshine I, Leong JM, Frankel G. Intimate host attachment: enteropathogenic and enterohaemorrhagic Escherichia coli. Cellular microbiology. 2013;15:1796–1808. doi: 10.1111/cmi.12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprise P, Beronja S, Silva-Gagliardi NF, Pellikka M, Jensen AM, McGlade CJ, Tepass U. The FERM protein Yurt is a negative regulatory component of the Crumbs complex that controls epithelial polarity and apical membrane size. Developmental cell. 2006;11:363–374. doi: 10.1016/j.devcel.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wei Z, Yan Y, Wan Q, Du Q, Zhang M. Structure of Crumbs tail in complex with the PALS1 PDZ-SH3-GK tandem reveals a highly specific assembly mechanism for the apical Crumbs complex. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:17444–17449. doi: 10.1073/pnas.1416515111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang M, Cai T, Tian J, Qu W, Xie ZJ. Functional characterization of Src-interacting Na/K-ATPase using RNA interference assay. The Journal of biological chemistry. 2006;281:19709–19719. doi: 10.1074/jbc.M512240200. [DOI] [PubMed] [Google Scholar]
- Lin YH, Currinn H, Pocha SM, Rothnie A, Wassmer T, Knust E. AP-2-complex-mediated endocytosis of Drosophila Crumbs regulates polarity by antagonizing Stardust. Journal of cell science. 2015;128:4538–4549. doi: 10.1242/jcs.174573. [DOI] [PubMed] [Google Scholar]
- Liu J, Kesiry R, Periyasamy SM, Malhotra D, Xie Z, Shapiro JI. Ouabain induces endocytosis of plasmalemmal Na/K-ATPase in LLC-PK1 cells by a clathrin-dependent mechanism. Kidney international. 2004;66:227–241. doi: 10.1111/j.1523-1755.2004.00723.x. [DOI] [PubMed] [Google Scholar]
- Lu H, Bilder D. Endocytic control of epithelial polarity and proliferation in Drosophila. Nature cell biology. 2005;7:1232–1239. doi: 10.1038/ncb1324. [DOI] [PubMed] [Google Scholar]
- Lundmark R, Carlsson SR. Sorting nexin 9 participates in clathrin-mediated endocytosis through interactions with the core components. The Journal of biological chemistry. 2003;278:46772–46781. doi: 10.1074/jbc.M307334200. [DOI] [PubMed] [Google Scholar]
- Makarova O, Roh MH, Liu CJ, Laurinec S, Margolis B. Mammalian Crumbs3 is a small transmembrane protein linked to protein associated with Lin-7 (Pals1) Gene. 2003;302:21–29. doi: 10.1016/s0378111902010843. [DOI] [PubMed] [Google Scholar]
- Marches O, Batchelor M, Shaw RK, Patel A, Cummings N, Nagai T, … Frankel G. EspF of enteropathogenic Escherichia coli binds sorting nexin 9. Journal of Bacteriology. 2006;188:3110–3115. doi: 10.1128/JB.188.8.3110-3115.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez E, Schroeder GN, Berger CN, Lee SF, Robinson KS, Badea L, … Frankel G. Binding to Na(+)/H(+) exchanger regulatory factor 2 (NHERF2) affects trafficking and function of the enteropathogenic Escherichia coli type III secretion system effectors Map, EspI and NleH. Cellular microbiology. 2010;12:1718–1731. doi: 10.1111/j.1462-5822.2010.01503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa T, Kuwae A, Abe A. Enteropathogenic Escherichia coli type III effectors EspG and EspG2 alter epithelial paracellular permeability. Infection and immunity. 2005;73:6283–6289. doi: 10.1128/IAI.73.10.6283-6289.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara BP, Donnenberg MS. A novel proline-rich protein, EspF, is secreted from enteropathogenic Escherichia coli via the type III export pathway. FEMS microbiology letters. 1998;166:71–78. doi: 10.1111/j.1574-6968.1998.tb13185.x. [DOI] [PubMed] [Google Scholar]
- McNamara BP, Koutsouris A, O’Connell CB, Nougayrede JP, Donnenberg MS, Hecht G. Translocated EspF protein from enteropathogenic Escherichia coli disrupts host intestinal barrier function. The Journal of clinical investigation. 2001;107:621–629. doi: 10.1172/JCI11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinild A, Klaerke DA, Loo DD, Wright EM, Zeuthen T. The human Na+-glucose cotransporter is a molecular water pump. The Journal of physiology. 1998;508(Pt 1):15–21. doi: 10.1111/j.1469-7793.1998.015br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrifield CJ, Qualmann B, Kessels MM, Almers W. Neural Wiskott Aldrich Syndrome Protein (N-WASP) and the Arp2/3 complex are recruited to sites of clathrin-mediated endocytosis in cultured fibroblasts. European journal of cell biology. 2004;83:13–18. doi: 10.1078/0171-9335-00356. [DOI] [PubMed] [Google Scholar]
- Michel D, Arsanto JP, Massey-Harroche D, Beclin C, Wijnholds J, Le Bivic A. PATJ connects and stabilizes apical and lateral components of tight junctions in human intestinal cells. Journal of cell science. 2005;118:4049–4057. doi: 10.1242/jcs.02528. [DOI] [PubMed] [Google Scholar]
- Mishra JP, Cohen D, Zamperone A, Nesic D, Muesch A, Stein M. CagA of Helicobacter pylori interacts with and inhibits the serine-threonine kinase PRK2. Cellular microbiology. 2015;17:1670–1682. doi: 10.1111/cmi.12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muza-Moons MM, Koutsouris A, Hecht G. Disruption of cell polarity by enteropathogenic Escherichia coli enables basolateral membrane proteins to migrate apically and to potentiate physiological consequences. Infection and immunity. 2003;71:7069–7078. doi: 10.1128/IAI.71.12.7069-7078.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muza-Moons MM, Schneeberger EE, Hecht GA. Enteropathogenic Escherichia coli infection leads to appearance of aberrant tight junctions strands in the lateral membrane of intestinal epithelial cells. Cellular microbiology. 2004;6:783–793. doi: 10.1111/j.1462-5822.2004.00404.x. [DOI] [PubMed] [Google Scholar]
- Nagai T, Abe A, Sasakawa C. Targeting of enteropathogenic Escherichia coli EspF to host mitochondria is essential for bacterial pathogenesis: critical role of the 16th leucine residue in EspF. The Journal of biological chemistry. 2005;280:2998–3011. doi: 10.1074/jbc.M411550200. [DOI] [PubMed] [Google Scholar]
- Nataro JP, Kaper JB. Diarrheagenic Escherichia coli. Clinical microbiology reviews. 1998;11:142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nougayrede JP, Donnenberg MS. Enteropathogenic Escherichia coli EspF is targeted to mitochondria and is required to initiate the mitochondrial death pathway. Cellular microbiology. 2004;6:1097–1111. doi: 10.1111/j.1462-5822.2004.00421.x. [DOI] [PubMed] [Google Scholar]
- Pavlov KV, Sokolov VS. Electrogenic ion transport by Na+, K+-ATPase. Membrane & cell biology. 2000;13:745–788. [PubMed] [Google Scholar]
- Rachmilewitz D, Karmeli F, Sharon P. Decreased colonic Na-K-ATPase activity in active ulcerative colitis. Israel journal of medical sciences. 1984;20:681–684. [PubMed] [Google Scholar]
- Rhee KJ, Cheng H, Harris A, Morin C, Kaper JB, Hecht G. Determination of spatial and temporal colonization of enteropathogenic E. coli and enterohemorrhagic E. coli in mice using bioluminescent in vivo imaging. Gut microbes. 2011;2:34–41. doi: 10.4161/gmic.2.1.14882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JD, Flashner MS. The (Na+ + K+)-activated ATPase. Enzymatic and transport properties. Biochimica et biophysica acta. 1979;549:145–176. doi: 10.1016/0304-4173(79)90013-2. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Boulan E, Macara IG. Organization and execution of the epithelial polarity programme. Nature reviews Molecular cell biology. 2014;15:225–242. doi: 10.1038/nrm3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh MH, Fan S, Liu CJ, Margolis B. The Crumbs3-Pals1 complex participates in the establishment of polarity in mammalian epithelial cells. Journal of cell science. 2003;116:2895–2906. doi: 10.1242/jcs.00500. [DOI] [PubMed] [Google Scholar]
- Roman-Fernandez A, Bryant DM. Traffic (Copenhagen, Denmark) 2016. Complex polarity: building multicellular tissues through apical membrane traffic. [DOI] [PubMed] [Google Scholar]
- Rose AM, Valdes R., Jr Understanding the sodium pump and its relevance to disease. Clinical chemistry. 1994;40:1674–1685. [PubMed] [Google Scholar]
- Saadat I, Higashi H, Obuse C, Umeda M, Murata-Kamiya N, Saito Y, … Hatakeyama M. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature. 2007;447:330–333. doi: 10.1038/nature05765. [DOI] [PubMed] [Google Scholar]
- Saha P, Arthur S, Kekuda R, Sundaram U. Na-glutamine co-transporters B(0)AT1 in villus and SN2 in crypts are differentially altered in chronically inflamed rabbit intestine. Biochimica et biophysica acta. 2012;1818:434–442. doi: 10.1016/j.bbamem.2011.11.005. [DOI] [PubMed] [Google Scholar]
- Saotome I, Curto M, McClatchey AI. Ezrin is essential for epithelial organization and villus morphogenesis in the developing intestine. Developmental cell. 2004;6:855–864. doi: 10.1016/j.devcel.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Savkovic SD, Villanueva J, Turner JR, Matkowskyj KA, Hecht G. Mouse model of enteropathogenic Escherichia coli infection. Infection and immunity. 2005;73:1161–1170. doi: 10.1128/IAI.73.2.1161-1170.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifflett DE, Clayburgh DR, Koutsouris A, Turner JR, Hecht GA. Enteropathogenic E. coli disrupts tight junction barrier function and structure in vivo. Laboratory investigation; a journal of technical methods and pathology. 2005;85:1308–1324. doi: 10.1038/labinvest.3700330. [DOI] [PubMed] [Google Scholar]
- Shin N, Lee S, Ahn N, Kim SA, Ahn SG, YongPark Z, Chang S. Sorting nexin 9 interacts with dynamin 1 and N-WASP and coordinates synaptic vesicle endocytosis. The Journal of biological chemistry. 2007;282:28939–28950. doi: 10.1074/jbc.M700283200. [DOI] [PubMed] [Google Scholar]
- Shivas JM, Morrison HA, Bilder D, Skop AR. Polarity and endocytosis: reciprocal regulation. Trends in cell biology. 2010;20:445–452. doi: 10.1016/j.tcb.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonovic I, Arpin M, Koutsouris A, Falk-Krzesinski HJ, Hecht G. Enteropathogenic Escherichia coli activates ezrin, which participates in disruption of tight junction barrier function. Infection and immunity. 2001;69:5679–5688. doi: 10.1128/IAI.69.9.5679-5688.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonovic I, Rosenberg J, Koutsouris A, Hecht G. Enteropathogenic Escherichia coli dephosphorylates and dissociates occludin from intestinal epithelial tight junctions. Cellular microbiology. 2000;2:305–315. doi: 10.1046/j.1462-5822.2000.00055.x. [DOI] [PubMed] [Google Scholar]
- Simpson N, Shaw R, Crepin VF, Mundy R, FitzGerald AJ, Cummings N, … Frankel G. The enteropathogenic Escherichia coli type III secretion system effector Map binds EBP50/NHERF1: implication for cell signalling and diarrhoea. Molecular microbiology. 2006;60:349–363. doi: 10.1111/j.1365-2958.2006.05109.x. [DOI] [PubMed] [Google Scholar]
- Singh AP, Aijaz S. Generation of a MDCK cell line with constitutive expression of the Enteropathogenic E. coli effector protein Map as an in vitro model of pathogenesis. Bioengineered. 2015;6:335–341. doi: 10.1080/21655979.2015.1096456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillos S, Diaz-Meco MT, Caminero E, Moscat J, Campuzano S. DaPKC-dependent phosphorylation of Crumbs is required for epithelial cell polarity in Drosophila. The Journal of cell biology. 2004;166:549–557. doi: 10.1083/jcb.200311031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulet F, Yarar D, Leonard M, Schmid SL. SNX9 regulates dynamin assembly and is required for efficient clathrin-mediated endocytosis. Molecular biology of the cell. 2005;16:2058–2067. doi: 10.1091/mbc.E04-11-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugihara-Mizuno Y, Adachi M, Kobayashi Y, Hamazaki Y, Nishimura M, Imai T, … Tsukita S. Molecular characterization of angiomotin/JEAP family proteins: interaction with MUPP1/Patj and their endogenous properties. Genes to cells: devoted to molecular & cellular mechanisms. 2007;12:473–486. doi: 10.1111/j.1365-2443.2007.01066.x. [DOI] [PubMed] [Google Scholar]
- Sundaram U, West AB. Effect of chronic inflammation on electrolyte transport in rabbit ileal villus and crypt cells. The American Journal of Physiology. 1997;272:G732–41. doi: 10.1152/ajpgi.1997.272.4.G732. [DOI] [PubMed] [Google Scholar]
- Sundaram U, Wisel S, Fromkes JJ. Unique mechanism of inhibition of Na+-amino acid cotransport during chronic ileal inflammation. The American Journal of Physiology. 1998;275:G483–9. doi: 10.1152/ajpgi.1998.275.3.G483. [DOI] [PubMed] [Google Scholar]
- Sundaram U, Wisel S, Rajendren VM, West AB. Mechanism of inhibition of Na+-glucose cotransport in the chronically inflamed rabbit ileum. The American Journal of Physiology. 1997;273:G913–9. doi: 10.1152/ajpgi.1997.273.4.G913. [DOI] [PubMed] [Google Scholar]
- Tepass U. The apical polarity protein network in Drosophila epithelial cells: regulation of polarity, junctions, morphogenesis, cell growth, and survival. Annual Review of Cell and Developmental Biology. 2012;28:655–685. doi: 10.1146/annurev-cellbio-092910-154033. [DOI] [PubMed] [Google Scholar]
- Thanabalasuriar A, Koutsouris A, Weflen A, Mimee M, Hecht G, Gruenheid S. The bacterial virulence factor NleA is required for the disruption of intestinal tight junctions by enteropathogenic Escherichia coli. Cellular microbiology. 2010;12:31–41. doi: 10.1111/j.1462-5822.2009.01376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Cai T, Yuan Z, Wang H, Liu L, Haas M, … Xie ZJ. Binding of Src to Na+/K+-ATPase forms a functional signaling complex. Molecular biology of the cell. 2006;17:317–326. doi: 10.1091/mbc.E05-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilston-Lunel AM, Haley KE, Schlecht NF, Wang Y, Chatterton AL, Moleirinho S, … Reynolds PA. Crumbs 3b promotes tight junctions in an ezrin-dependent manner in mammalian cells. Journal of molecular cell biology. 2016 doi: 10.1093/jmcb/mjw020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomson FL, Viswanathan VK, Kanack KJ, Kanteti RP, Straub KV, Menet M, … Hecht G. Enteropathogenic Escherichia coli EspG disrupts microtubules and in conjunction with Orf3 enhances perturbation of the tight junction barrier. Molecular microbiology. 2005;56:447–464. doi: 10.1111/j.1365-2958.2005.04571.x. [DOI] [PubMed] [Google Scholar]
- Viswanathan VK, Koutsouris A, Lukic S, Pilkinton M, Simonovic I, Simonovic M, Hecht G. Comparative analysis of EspF from enteropathogenic and enterohemorrhagic Escherichia coli in alteration of epithelial barrier function. Infection and immunity. 2004a;72:3218–3227. doi: 10.1128/IAI.72.6.3218-3227.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VK, Lukic S, Koutsouris A, Miao R, Muza MM, Hecht G. Cytokeratin 18 interacts with the enteropathogenic Escherichia coli secreted protein F (EspF) and is redistributed after infection. Cellular microbiology. 2004b;6:987–997. doi: 10.1111/j.1462-5822.2004.00416.x. [DOI] [PubMed] [Google Scholar]
- Weflen AW, Alto NM, Viswanathan VK, Hecht G. E. coli secreted protein F promotes EPEC invasion of intestinal epithelial cells via an SNX9-dependent mechanism. Cellular microbiology. 2010;12:919–929. doi: 10.1111/j.1462-5822.2010.01440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells CD, Fawcett JP, Traweger A, Yamanaka Y, Goudreault M, Elder K, … Pawson T. A Rich1/Amot complex regulates the Cdc42 GTPase and apical-polarity proteins in epithelial cells. Cell. 2006;125:535–548. doi: 10.1016/j.cell.2006.02.045. [DOI] [PubMed] [Google Scholar]
- Whiteman EL, Fan S, Harder JL, Walton KD, Liu CJ, Soofi A, … Margolis B. Crumbs3 is essential for proper epithelial development and viability. Molecular and cellular biology. 2014;34:43–56. doi: 10.1128/MCB.00999-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman EL, Liu CJ, Fearon ER, Margolis B. The transcription factor snail represses Crumbs3 expression and disrupts apico-basal polarity complexes. Oncogene. 2008;27:3875–3879. doi: 10.1038/onc.2008.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarar D, Waterman-Storer CM, Schmid SL. SNX9 couples actin assembly to phosphoinositide signals and is required for membrane remodeling during endocytosis. Developmental cell. 2007;13:43–56. doi: 10.1016/j.devcel.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Yudowski GA, Efendiev R, Pedemonte CH, Katz AI, Berggren PO, Bertorello AM. Phosphoinositide-3 kinase binds to a proline-rich motif in the Na+, K+-ATPase alpha subunit and regulates its trafficking. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:6556–6561. doi: 10.1073/pnas.100128297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeaiter Z, Cohen D, Musch A, Bagnoli F, Covacci A, Stein M. Analysis of detergent-resistant membranes of Helicobacter pylori infected gastric adenocarcinoma cells reveals a role for MARK2/Par1b in CagA-mediated disruption of cellular polarity. Cellular microbiology. 2008;10:781–794. doi: 10.1111/j.1462-5822.2007.01084.x. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Li Q, Wang C, Li N, Li J. Redistribution of tight junction proteins during EPEC infection in vivo. Inflammation. 2012;35:23–32. doi: 10.1007/s10753-010-9285-1. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Li Q, Wang C, Liu X, Li N, Li J. Enteropathogenic Escherichia coli changes distribution of occludin and ZO-1 in tight junction membrane microdomains in vivo. Microbial pathogenesis. 2010;48:28–34. doi: 10.1016/j.micpath.2009.10.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.