

Abstract
Interstitial fibrosis serves a causal role in the development of heart failure following acute and chronic myocardial infarction, and anti-fibrotic therapy represents a promising strategy to mitigate this pathological process. Oxymatrine (OMT) exerts a number of pharmacological effects on the cardiovascular system, but its anti-cardiovascular disease mechanisms remain unclear. The purpose of the present study was to investigate the effect of OMT administration on transforming growth factor (TGF)-β1-induced cardiac fibroblast (CFB) proliferation and abnormal differentiation, and to elucidate the underlying mechanisms. Primary CFBs were isolated from neonatal rats and used for experimental treatments. TGF-β1 stimulation in CFBs resulted in increased proliferation, increased α-smooth muscle actin (SMA) and type I and type III collagen expression, and increased p38 mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK)1/2 phosphorylation. Treatment with OMT and SB431542 (a TGF-β1 receptor inhibitor) attenuated the proliferation and abnormal differentiation of CFBs induced by TGF-β1, and decreased p38MAPK and ERK1/2 phosphorylation. In addition, treatment with SB203580 (a p38MAPK inhibitor) or PD98059 (an ERK1/2 inhibitor), but not by SP600125 (a c-jun N-terminal kinase1/2/3 inhibitor), inhibited the TGF-β1 stimulated CFB proliferation, as well as the elevation of α-SMA and the deposition of type I and type III collagen, suggesting that ERK1/2 and p38MAPK signaling may be important in the in the process of myocardial fibrosis. In conclusion, the present study revealed that OMT treatment inhibited CFB proliferation and the CFB-myofibroblast transition induced by TGF-β1, at least in part through inhibition of ERK1/2 and p38MAPK signaling.
Keywords: oxymatrine, transforming growth factor-β1, myocardial fibrosis, cardiac fibroblasts, extracellular signal-regulated kinase 1/2, p38 mitogen-activated protein kinase, c-jun N-terminal kinase 1/2/3, myofibroblasts
Introduction
Myocardial fibrosis is the result of fibroblast proliferation and collagen deposition in various extracellular matrices, predominantly in myocardial interstitial cells, leading to myocardial tissue remodeling and causing cardiac diastolic and systolic dysfunction (1). There are a number of contributing factors to myocardial fibrosis, including increased pressure, immune injury, ischemia and hyperglycemia; it frequently involves the rennin-angiotensin-aldosterone system, the collagen production and degradation systems, apoptosis, inflammation, and a variety of cardiovascular active substances and cytokines (2). Myocardial fibrosis is a common pathological result of numerous diseases, eventually leading to increased myocardial stiffness (3), ventricular diastolic dysfunction, decreased coronary flow reserve, ventricular arrhythmia or sudden death (4). Therefore, investigation of the pathological characteristics and pathogenesis of myocardial fibrosis, and the identification of novel drugs with anti-myocardial fibrosis effects, is of importance for the prevention of severe clinical cardiovascular events.
Transforming growth factor (TGF)-β1 has been identified to be a pleiotropic multifunctional peptide (5). TGF-β1 exerts effects on a number of different types of cells, and is involved in various biological processes, including within the cardiovascular system; it primarily regulates the transformed phenotype of fibroblasts, and promotes the synthesis of collagen and fibrosis. TGF-β1 additionally induces the synthesis of endothelial nitric oxide and endothelin-1, and influences the barrier function of endothelial cells in vivo and in vitro. TGF-β1 inhibits matrix metalloproteinase activity, induces the synthesis of tissue inhibitor of metalloproteases (which inhibits extracellular matrix degradation), and regulates the extracellular matrix (6). TGF-β1 is involved in the regulation of various biological processes and upstream signal transduction pathways, and directly stimulates the proliferation of cardiac fibroblasts (CFBs) (7). Therefore, TGF-β1 is the common pathway of many factors inducing myocardial fibrosis. Numerous studies have confirmed that TGF-β1 may be the predominant stimulatory cytokine involved in exogenous and endogenous tissue fibrosis (8–10). A previous study demonstrated that TGF-β1 was able to induce the activation of reactive oxygen species (ROS), and activate the downstream signaling pathways p38 mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK)1/2 (11).
Oxymatrine (OMT) is one of the main bioactive ingredients of Kushen (Radix Sophorae), which is a traditional Chinese herbal medicine made from the dried root of Sophora flavescens Ait. Previous studies have demonstrated that OMT exhibits diuretic, immune-modulatory, antiviral and anticancer effects, and there is growing evidence that it may also regulate extracellular matrix remodeling (ECMR) in a variety of organs, tissues and cells, including the liver, kidney and lung (12–14). A previous study demonstrated that OMT may exhibit an inhibitory effect on experimental myocardial fibrosis induced by acute myocardial infarction (MI), and the associated TGF-β/SMAD signaling pathway (15). A previous study demonstrated that OMT attenuated fibrosis induced by acute lung injury in mice and inhibited the phosphorylation of p38MAPK (16). OMT decreased collagen production and deposition in rat hepatic tissue fibrosis induced by carbon tetrachloride, involving the p38MAPK signal pathway (17). The action of OMT against myocardial fibrosis and the underlying mechanism remain unclear. Therefore, the purpose of the present study was to investigate whether the inhibitory effect of OMT, a cardiovascular drug with numerous pharmacological effects, on CFB proliferation is associated with the inhibition of the p38MAPK/ERK1/2 signaling cascades induced by TGF-β1.
Materials and methods
Chemicals
OMT was purchased from Nanjing Zelang Medical Technology Co., Ltd. (Nanjing, China). TGF-β1 was purchased from PeproTech, Inc. (Rocky Hill, NJ, USA). p38MAPK inhibitor (SB203580), TGF-β1 receptor inhibitor (SB431542), ERK1/2 inhibitor (PD98059) and c-jun N-terminal kinase (JNK)1/2/3 inhibitor (SP600125) were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). All chemicals were obtained from Beijing Dingguo Changsheng Biotechnology Co., Ltd. (Beijing, China).
Cell isolation and identification
The isolation of CFBs from neonatal rats was performed according to a modified protocol as described previously (4,18). The present study was approved by the Animal Care Welfare Committee of Guiyang Medical University (Guiyang, China). The hearts of 1–3 day-old Sprague Dawley rats (n=450; weight, 5–7 g; neonatal rats were used irrespective of gender; purchased from the Experimental Animal Center of Guizhou Medical University, Guiyang, Guizhou, China) were isolated and digested with 10 ml PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4; pH 7.2–7.4), containing 0.08% trypsin (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) for 10 min at 37°C, which was repeated 4–6 times. Following each digestion step, the medium containing suspended cells was removed and an equal volume of DMEM containing 10% fetal bovine serum (FBS; Zhejiang Tianhang Biotechnology Co., Ltd., Zhejiang, China) solution was added. Primary cultures of mouse cardiac stromal cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 20% FBS, penicillin (100 U/ml) and streptomycin (100 U/ml; all Sigma-Aldrich; Merck KGaA) at 37°C in a humidified atmosphere of 5% CO2. The differential adhesion method was used to separate out other cells (4,18), and primary neonatal rat CFBs were used for subculture. CFBs of the 2nd/3rd passage were used in the experiments. CFBs were cultured in 6-well plates at 37°C in a humidified atmosphere of 5% CO2 for 24 h and then fixed with ice-cold 4% paraformaldehyde for 30 min, followed by permeabilization with 0.1% Triton X-100 at room temperature for 20 min. Immunocytochemical staining was performed using the streptavidin peroxidase (SP) combined with Immunohistochemical 3′-3′-diaminobenzidine (DAB; ZSGB-BIO; OriGene Technologies, Inc., Beijing, China) staining method (19). The primary antibody, rabbit monoclonal anti-rat vimentin (Boster Biological Technology, Pleasanton, CA, USA; cat. no. BM0135) was added at a dilution of 1:500 and incubated with CFBs for 1 h at room temperature. Following incubation, CFBs were washed 3 times with PBS and then incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (ZSGB-BIO; OriGene Technologies, Inc.; cat. no. 111589; used at the working dilution recommended by the manufacturer) for 1 h at 37°C. This was followed by DAB and hematoxylin staining for 10 sec at room temperature, then observation under a light microscope (Leica DMi 1; Leica Microsystems GmbH, Wetzlar, Germany). Cells were visualized at ×200 magnification and 5 fields of view were analyzed.
Proliferation inhibition experiment
Following incubation in serum-free DMEM for 12 h, the cells (25,000 cells/well in a 96-well microtiter plate) were exposed to TGF-β1 (20 ng/ml) alone for 24 h, or pretreated with SB203580 (1×10−5 M), PD98059 (1×10−5 M), SP600125 (1×10−5 M) or OMT [1.89×10−4 M (low-dose; OMT-L), 3.78×10−4 M (medium-dose; OMT-M) or 7.56×10−4 M (high-dose; OMT-H)] for 1 h, prior to exposure to TGF-β1 for 24 h. An MTT assay was used to evaluate cell proliferation; 20 µl of 5 mg/ml MTT was added to each well and incubated at 37°C for 4 h. The media was removed and 150 µl dimethyl sulfoxide was added to each well to dissolve the formazan. Finally, the absorbance was measured at 490 nm using a standard plate reader.
Immunocytochemical experiments
Anti-α-smooth muscle actin (SMA) antibody (Boster Biological Technology; cat. no. BM0002) at a dilution of 1:500 was used for the immunocytochemical experiments. The immunohistochemical SP and DAB staining method (as described in the Cell isolation and identification section) was used to observe the expression of α-SMA in myofibroblasts, in order to evaluate the inhibitory effect of the differentiation of CFBs to myofibroblasts (19). Cells were visualized at ×200 magnification and 5 fields of view were analyzed.
Picrosirius red staining
CFBs from the 6-well plates were plated on cover slides according to the grouping requirements, following culturing, were washed 3 times with PBS, fixed with 4% paraformaldehyde at room temperature for 15 min, stained with celestine blue liquid dye for 10 min, and with picrosirius red liquid dye for 30 min. Hematoxylin was used as a nuclear counterstain for 15 min. The cells were subjected to ethanol dehydration and naturally dried, followed by observation under an inverted microscope (magnification, ×200). Type I collagen was stained bright red or yellow, and type III collagen, located around the type I collagen, was stained green.
Western blot analysis
Western blotting was used to measure the protein expression levels of p38MAPK, phosphorylated (p)-p38MAPK, ERK1/2, p-ERK1/2, p-JNK1/2/3, JNK1/2/3, α-SMA, type I collagen and type III collagen in CFBs. Following treatments as described above, CFBs were washed once in ice-cold PBS and lysed in radio immuno precipitation lysis buffer containing 1% protease inhibitor cocktail (Beijing Dingguo Changsheng Biotechnology Co., Ltd.) on ice. Protein concentrations were assessed using a bicinchoninic acid assay kit. Equal amounts of protein (50 µg/lane) were subjected to SDS-PAGE on a 10% gel, transferred onto polyvinylidene fluoride membranes using a Bio-Rad Western blot analysis apparatus (Bio-Rad Laboratories, Inc., Hercules, CA, USA), and the membranes were blocked in 5% non-fat dried milk in TBS with Tween-20 (TBST) at room temperature for 2 h. The membranes were subsequently incubated with polyclonal p38MAPK, p-p38MAPK, ERK1/2, p-ERK1/2, p-JNK1/2/3 and JNK1/2/3 antibodies (1:1,000 dilution; ImmunoWay Biotechnology Company, Plano, TX, USA; cat. nos. B1401, B1801, B2501, B0001, B4301 and B4101, respectively), α-SMA antibody (1:300; Boster Biological Technology; cat. no. BM0002), type I collagen and type III collagen antibodies (1:500; Abcam, Cambridge, UK; cat. nos. GR71249-7 and GR59128-1, respectively) and β-actin antibody (1:7,000 dilution; Abcam, Cambridge, UK; cat. no. P68032) overnight at 4°C. Following washing three times with TBST, the membranes were incubated with the corresponding HRP-conjugated Affinipure goat anti-rabbit secondary antibody (1:4,000; Bioworld Technology, Inc., St. Louis Park, MN, USA; cat. no. SA00001-2) for 2 h at room temperature, and the immunolabeled bands were visualized using Pierce ECL western blot substrate (Thermo Fisher Scientific, Inc.). Digital images of the blots were photographed using a ChemiDoc XRS+ system and analyzed with Image Lab™ Software, version 5.2 (Bio-Rad Laboratories, Inc.). The results were normalized to those of β-actin and the data from three independent experiments was analyzed.
Statistical analysis
SPSS v13.0 statistical software (SPSS, Inc., Chicago, IL, USA) was used for statistical analysis, which included one-way analysis of variance followed by a Dunnett's post hoc test (2-sided). P<0.05 was considered to indicate a statistically significant difference. Data are expressed as the mean ± standard error of the mean. Each experiment was performed in triplicate.
Results
Identification of primary CFBs
CFBs are flat and spindle-shaped in myocardial tissue; compared with other types of cells, CFBs are unique because they lack abasement membrane. Vimentin, an intermediate filament protein, is an important identifying biological marker of fibroblasts, which can be stained via immunocytochemistry to display its filamentous morphology. In the present study, primary CFBs were isolated from neonatal rats and used at passages 2–3 for experiments. In order to confirm that the isolated cells were indeed CFBs, the immunohistochemical DAB staining method combined with immunocytochemical SP staining was used for identification of vimentin expression in CFBs. Anti-vimentin antibody was used for the immunocytochemical experiments (Fig. 1). Control-stained cells exhibited a blue-purple nucleus and no yellow cytoplasmic staining (Fig. 1B). Isolated primary cells stained with the anti-vimentin antibody exhibited a blue-purple nucleus and a strong yellow cytoplasmic staining for vimentin (Fig. 1C). As the majority of cells were positive for vimentin, the present study successfully isolated primary CFBs.
Figure 1.
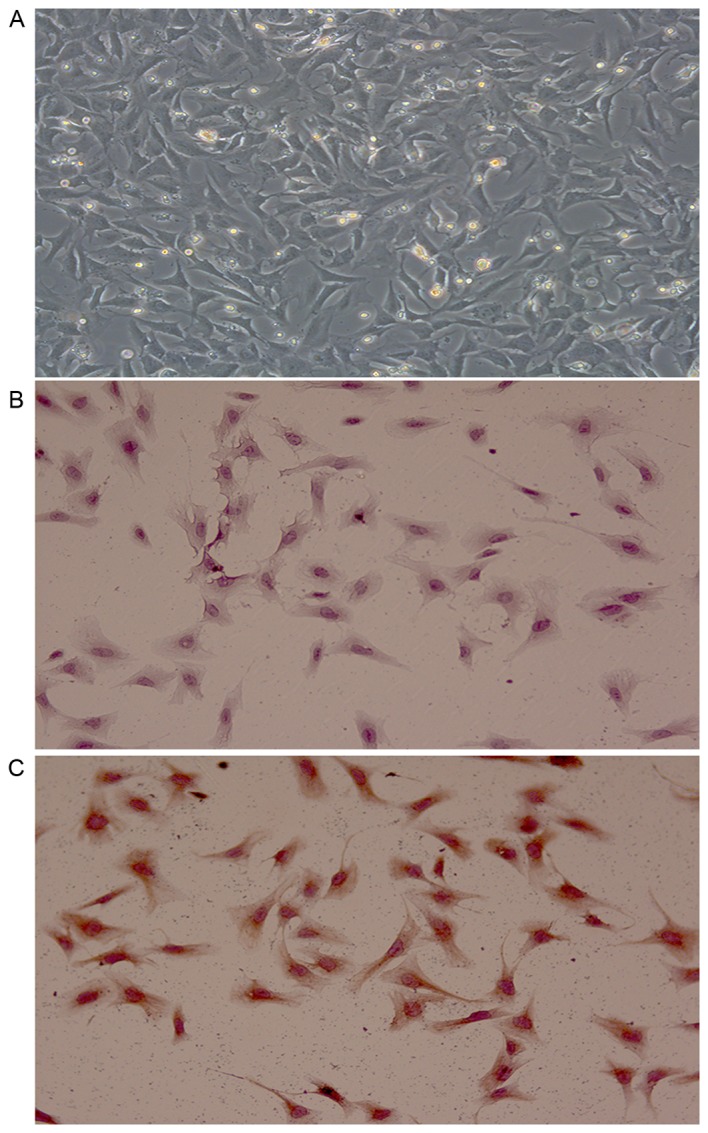
Identification of CFBs (magnification, ×200). (A) Bright-field image of the 2nd passage CFBs. (B) Representative image of negative control stained cells (PBS was used instead of primary antibody). (C) Representative image of cells stained with the anti-vimentin antibody. CFBs, cardiac fibroblasts.
Inhibitory effect of OMT on TGF-β1-induced CFB proliferation
CFBs were stimulated with TGF-β1 alone or following pretreatment with OMT or the specific inhibitors. Then, the effect of each treatment group on CFB proliferation was examined after 24 h. TGF-β1 stimulation induced a significant increase in CFB proliferation compared with control untreated cells (Table I). This TGF-β1-induced increase was significantly inhibited by pretreatment with the p38MAPK inhibitor SB203580, the ERK1/2 inhibitor PD98059 and OMT (Table I); however, the JNK1/2/3 inhibitor SP600125 did not exert any apparent effect on TGF-β1 induced proliferation (Table I).
Table I.
Inhibitory effect of OMT, SB203580, PD98059 and SP600125 on TGF-β1-induced cardiac fibroblast proliferation.
| Experimental group | Concentration | OD490 | Rate of inhibition of cell proliferation (%) |
|---|---|---|---|
| Control | – | 0.343±0.007 | – |
| TGF-β1 | 20 ng/ml | 0.369±0.013a | – |
| TGF-β1+SB203580 | 1×10−5 M | 0.318±0.015b | 13.82 |
| TGF-β1+PD98059 | 1×10−5 M | 0.279±0.023b | 24.39 |
| TGF-β1+SP600125 | 1×10−5 M | 0.351±0.039 | 4.88 |
| TGF-β1+OMT-L | 1.89×10−4 M | 0.347±0.025 | 5.96 |
| TGF-β1+OMT-M | 3.78×10−4 M | 0.288±0.057b | 21.95 |
| TGF-β1+OMT-H | 7.56×10−4 M | 0.236±0.057b | 36.04 |
Data are expressed as mean ± standard error of the mean (n=3).
P<0.05 vs. control
P<0.01 vs. TGF-β1. OMT, oxymatrine; TGF-β1, transforming growth factor-β1; OD, optical density; L, low-dose; M, medium-dose; H, high-dose.
Inhibitory effect of OMT on TGF-β1-induced α-SMA expression in CFB differentiation
α-SMA is a protein marker of myofibroblasts. The differentiation from CFBs to myofibroblasts is promoted by TGF-β1 (20). The effect of OMT on the expression of α-SMA induced by TGF-β1 was measured by immunocytochemistry and western blot analysis. The results of the immunocytochemistry experiment demonstrated that the expression of α-SMA increased in the TGF-β1 treatment group compared with the control group (Fig. 2). The expression of α-SMA in the OMT-H, SB203580, and PD98059 pretreatment groups was downregulated compared with the TGF-β1 alone group (Fig. 2). The results of the western blot analysis demonstrated that α-SMA protein expression levels were significantly increased in the TGF-β1 group compared with the control group (P<0.01; Fig. 3). Compared with the TGF-β1 alone group, SB203580, PD98059 and OMT pretreatment significantly inhibited the TGF-β1-induced α-SMA upregulation in myofibroblasts, while SP600125 exerted no significant effect on the expression of α-SMA (Fig. 3).
Figure 2.

Effects of TGF-β1, SB203580, PD98059, SP600125 and OMT treatments on α-smooth muscle actin protein expression in myofibroblasts examined by immunocytochemistry (magnification, ×200). Cells were treated with TGF-β1 (20 ng/ml) for 24 h, with or without pretreatment with SB203580 (1×10−5 M), PD98059 (1×10−5 M), SP600125 (1×10−5 M), or different doses of OMT (L, 1.89×10−4 M; M, 3.78×10−4 M; or H, 7.56×10−4 M) for 1 h. TGF-β1, transforming growth factor-β1; OMT, oxymatrine; L, low-dose; M, medium-dose; H, high-dose.
Figure 3.
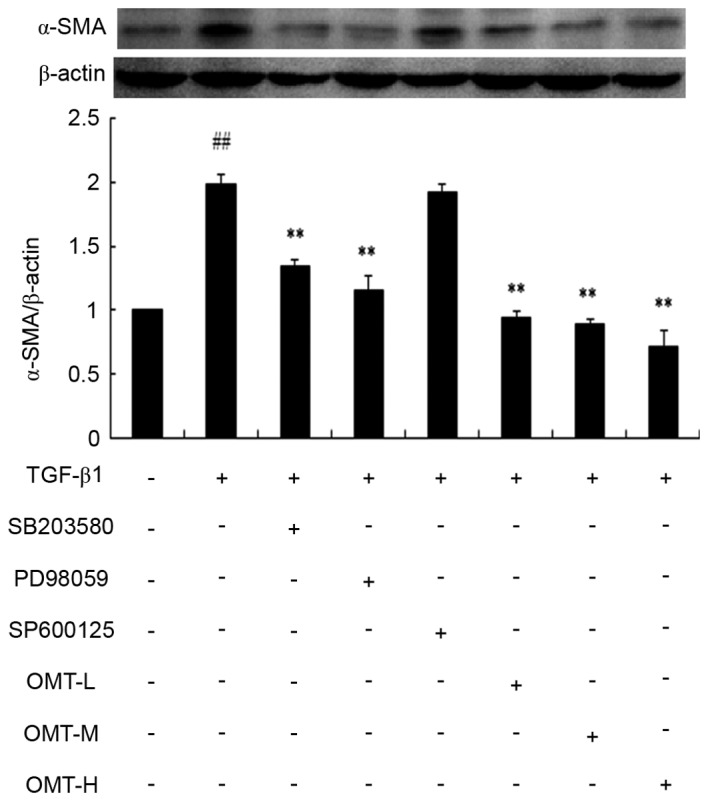
Effects of TGF-β1, SB203580, PD98059, SP600125 and OMT on α-SMA protein expression in myofibroblasts examined by western blotting. Cells were treated with TGF-β1 (20 ng/ml) for 24 h, with or without pretreatment with SB203580 (1×10−5 M), PD98059 (1×10−5 M), SP600125 (1×10−5 M), or different doses of OMT (L, 1.89×10−4 M; M, 3.78×10−4 M; or H, 7.56×10−4 M) for 1 h. Data are expressed as the mean ± standard error of the mean (n=3). ##P<0.01 vs. control; **P<0.01 vs. TGF-β1 group. TGF-β1, transforming growth factor-β1; OMT, oxymatrine; α-SMA, α-smooth muscle actin; L, low-dose; M, medium-dose; H, high-dose.
Effect of OMT on type I and type III collagen synthesis following TGF-β1 stimulation
Synthesis and deposition of collagen is an indicator of fibrosis, particularly type I collagen. TGF-β1 binding with the corresponding TGF-β1 response element has been demonstrated to activate procollagen gene expression, including type I and type III collagen and fibronectin, leading to myocardial fibrosis (1). Picrosirius red staining and western blotting were used in the present study to determine the deposition and expression, respectively, of type I and type III collagen. The results of the picrosirius red staining demonstrated that the bright red/yellow staining (type I collagen) inside the cells, as well as in the extracellular space between the cells, increased in the TGF-β1 group compared with the control, while the green staining (type III collagen; pericellular membrane) was increased compared with control, however, it was less overall than the red staining (Fig. 4). Compared with the TGF-β1 alone, the number of cells in the OMT-L, OMT-M and OMT-H groups was reduced, the intercellular space increased in size, and the amount of bright red/yellow staining was decreased, indicating that OMT pretreatment inhibited proliferation and collagen deposition in CFBs (Fig. 4). Collagen deposition was also decreased following pretreatment with p38MAPK and ERK1/2 blocking agents, while JNK1/2/3 inhibition had no effect (Fig. 4). The results from the western blotting experiments confirmed the immunocytochemistry results; OMT, SB203580 and PD98059 pretreatments significantly inhibited type I and type III collagen protein expression levels compared with TGF-β1 alone (Fig. 5).
Figure 4.

Effects of TGF-β1, SB203580, PD98059, SP600125 and OMT on type I and type III collagen deposition in CFBs (magnification, ×200). Cells were treated with TGF-β1 (20 ng/ml) for 24 h, with or without pretreatment with SB203580 (1×10−5 M), PD98059 (1×10−5 M), SP600125 (1×10−5 M), or different doses of OMT (L, 1.89×10−4 M; M, 3.78×10−4 M; or H, 7.56×10−4 M) for 1 h. TGF-β1, transforming growth factor-β1; OMT, oxymatrine; CFBs, cardiac fibroblasts.
Figure 5.

Effects of TGF-β1, SB203580, PD98059, SP600125 and OMT on (A) type I and (B) type III collagen protein expression in CFBs as examined by western blotting. Data are expressed as the mean ± standard error of the mean (n=3). ##P<0.01 vs. control; **P<0.01 vs. TGF-β1 group. TGF-β1, transforming growth factor-β1; OMT, oxymatrine; CFBs, cardiac fibroblasts; Coll I, type I collagen; Coll III, type III collagen; L, low-dose; M, medium-dose; H, high-dose.
Effect of OMT pretreatment on p38MAPK, ERK1/2 and JNK1/2/3 phosphorylation
Western blot analysis revealed that the protein expression levels of p-p38MAPK and p-ERK1/2 over total p38MAPK and total ERK1/2 levels, respectively, were significantly increased in the TGF-β1 group compared with control (P<0.01; Figs. 6 and 7, respectively). Pretreatment with SB431542, SB203580, PD98059 or different concentrations of OMT were able to decrease p38MAPK and ERK1/2 phosphorylation levels compared with the TGF-β1 alone group (P<0.01; Figs. 6 and 7, respectively). Compared with the TGF-β1+SB431542 group, p-p38MAPK protein expression exhibited no significant difference in the TGF-β1+SB203580 group (Fig. 6). Similarly, compared with the TGF-β1+SB431542 group, p-ERK1/2 protein expression exhibited no significant difference in the TGF-β1+PD98059 group (Fig. 7). Overall, these results indicated that OMT pretreatment significantly inhibited the TGF-β1-induced upregulation of the active forms of p38MAPK and ERK1/2 in CFBs (p-p38MAPK and p-ERK1/2, respectively), while exhibiting no effect on the total p38MAPK and ERK1/2 expression.
Figure 6.
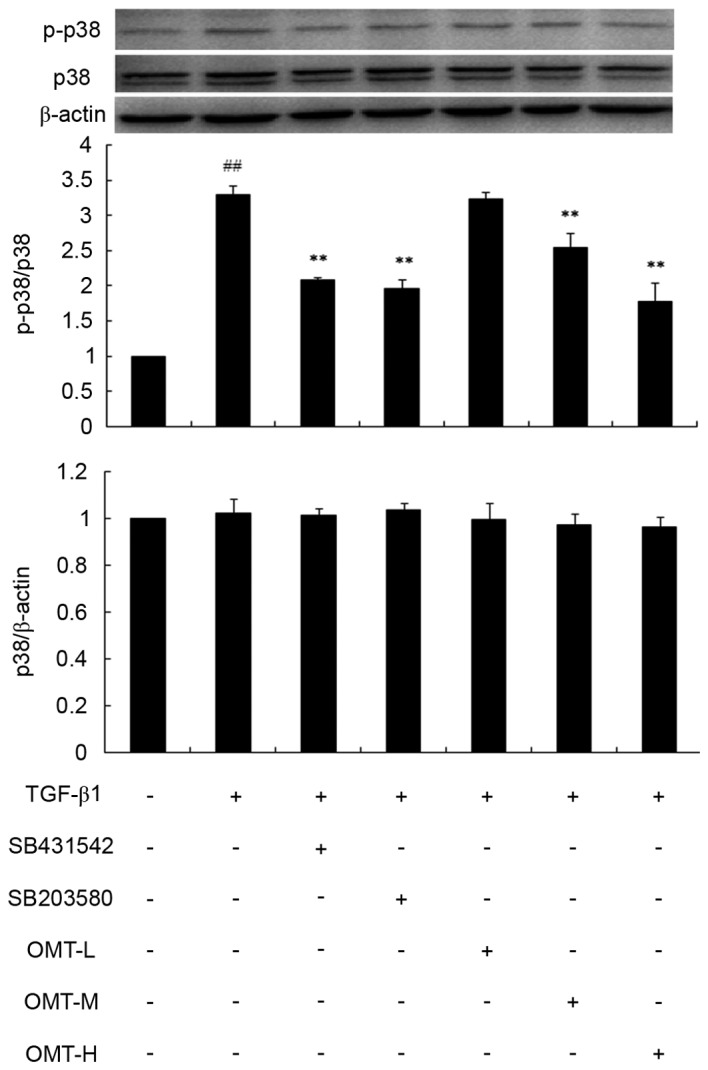
Effects of TGF-β1, SB431542, SB203580 and OMT on p38 mitogen-activated protein kinase phosphorylation in CFBs. Representative western blot images and quantification are shown. Data are expressed as the mean expression relative to control ± standard error of the mean (n=3). ##P<0.01 vs. control; **P<0.01 vs. TGF-β1 group. TGF-β1, transforming growth factor-β1; OMT, oxymatrine; CFBs, cardiac fibroblasts; p-, phosphorylated; p38, p38 mitogen-activated protein kinase; L, low-dose; M, medium-dose; H, high-dose.
Figure 7.
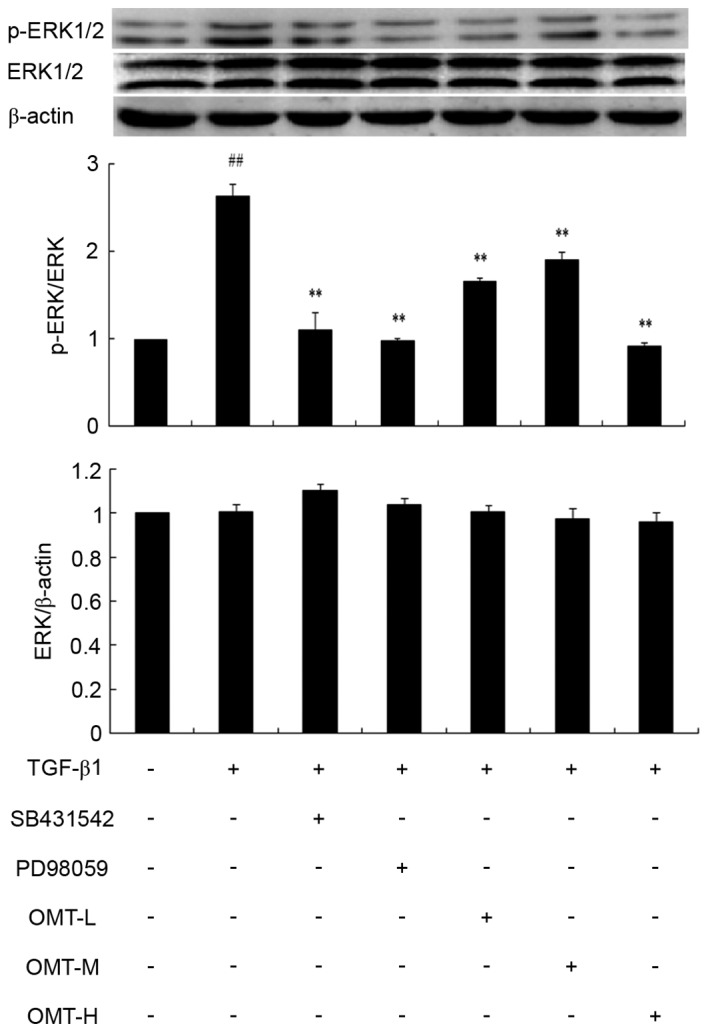
Effects of TGF-β1, SB431542, PD98059 and OMT on ERK1/2 phosphorylation in CFBs. Representative western blot images and quantification are shown. Data are expressed as the mean expression relative to control ± standard error of the mean (n=3). ##P<0.01 vs. control; **P<0.01 vs. TGF-β1 group. TGF-β1, transforming growth factor-β1; OMT, oxymatrine; ERK1/2, extracellular signal-regulated kinase 1/2; CFBs, cardiac fibroblasts; p-, phosphorylated; L, low-dose; M, medium-dose; H, high-dose.
As far as the JNK pathway is concerned, JNK1/2/3 phosphorylation in the TGF-β1 group was not significantly altered compared with control (Fig. 8), indicating that TGF-β1 did not activate JNK1/2/3 signaling. In addition, the TGF-β1+SP600125 and TGF-β1+OMT groups exhibited no difference in p-JNK levels compared with control, suggesting that JNK signaling is not involved in the effects of TGF-β1 and OMT in CFBs (Fig. 8).
Figure 8.
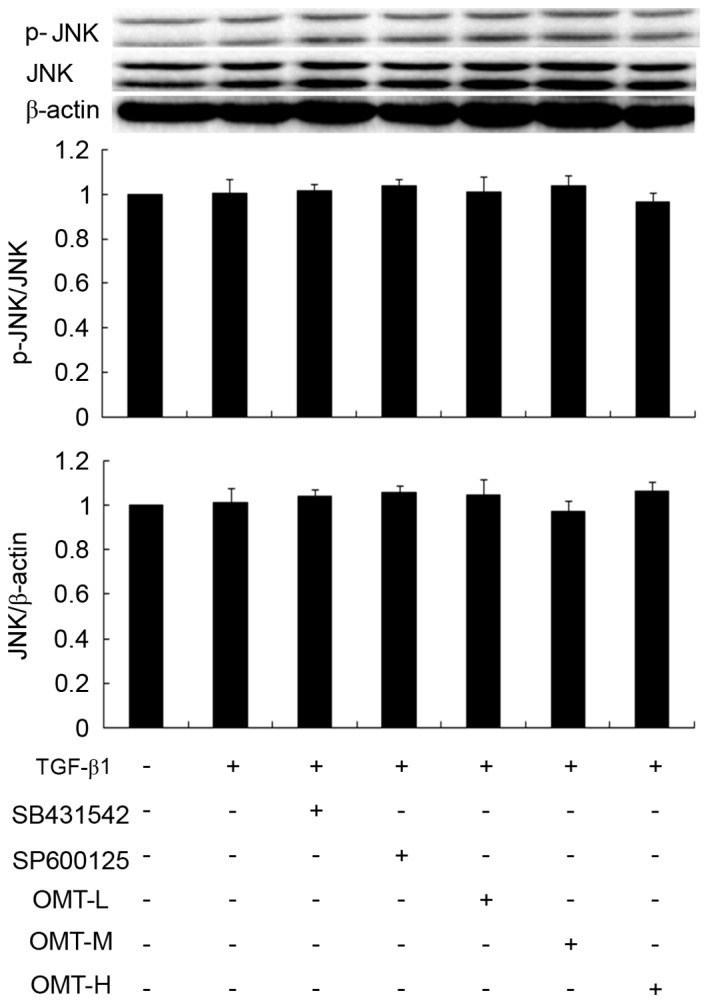
Effects of TGF-β1, SB431542, SP600125 and OMT on JNK protein phosphorylation in CFBs. Representative western blot images and quantification are shown. Data are expressed as the mean expression relative to control ± standard error of the mean (n=3). No significant differences were observed. TGF-β1, transforming growth factor-β1; OMT, oxymatrine; JNK, c-jun N-terminal kinase; CFBs, cardiac fibroblasts; p-, phosphorylated; L, low-dose; M, medium-dose; H, high-dose.
Discussion
In the early stages, fibrosis following MI is an integral component of the reparative process, maintaining the structural integrity of the necrotic area through the formation of a connective tissue scar. However, in the long term, the continuous remodeling of the infarct left ventricle may lead to progressive ventricular dilatation, decreased cardiac performance and chronic heart failure (21,22). During fibrosis, the accumulation of collagenous material remote from the site of the infarction contributes the decrease in myocardial elasticity and impaired contractility.
TGF-β1 is an important signaling molecule that induces cardiac fibrosis by activating the proliferation and collagen production of CFBs (23). Previous studies have investigated TGF-β-induced ECMR and fibrosis in various diseases, including CFB-myofibroblast transition (24–26). A number of lines of evidence suggest a role for excessive TGF-β1 signaling in the pathogenesis of maladaptive remodeling and fibrosis. TGF-β1 binds to receptors and induces a number of responses, including the upregulation of mesenchymal-associated proteins, such as collagen and α-SMA (27). The TGF-β signaling pathways include the Smad signaling pathway and the atypical Smad signaling pathway (20,28). In a rat model of experimental myocardial fibrosis, the mRNA expression of the TGF-β1 receptor, and Smad-2, −3 and −4 increased; however, the mRNA expression of Smad-7 was markedly decreased (29). TGF-β1 signal transduction additionally activates the intracellular MAPK signaling pathways (ERK1/2, p38MAPK and JNK1/2/3 being the three main branches of the stress-induced activation of the MAPK system), which has been demonstrated to serve an important role in the pathogenesis of cardiac fibrosis (30). The administration of a p38MAPK inhibitor to long-term MI rats resulted in suppressed interstitial fibrosis and improved cardiac function (31). The MAPK signaling pathway is involved in the CFB to myofibroblast transition process, and activated myofibroblasts secrete collagen, which is associated with the process of myocardial fibrosis (32). The balance between collagen synthesis and degradation following tissue injury, as occurs during MI, is regulated by myofibroblasts (33). Myofibroblasts predominantly originate from CFBs, and respond to mechanical stress, ischemia, and autocrine and paracrine factors, including TGF-β1, angiotensin II and pro-inflammatory cytokines, by increasing the synthesis and deposition of extracellular matrix proteins to replace the necrotic myocardium with a scar (1,34). The most typical feature of myofibroblasts is the novel formation and deployment of stress fibers, the expression of smooth muscle genes, such as α-SMA, and collagen synthesis and deposition. Although myofibroblasts are required to stabilize the infarcted area and promote scar tissue contraction, their persistence maybe associated with abnormal myocardial stiffness and impairment of ventricular function (35). Accordingly, identification of the factors and the underlying mechanisms may lead to prevention of myofibroblast generation, and defining their molecular targets may be important for the design of therapeutic strategies aimed at inhibiting excessive left ventricular stiffness following MI. α-SMA is a protein marker of myofibroblasts; the differentiation of CFBs to myofibroblasts is promoted by TGF-β1, cytokines, the extracellular matrix and other growth factors (36). TGF-β 1inducesthe differentiation of CFBs into myofibroblasts, altering the homeostasis of the extracellular matrix, which increases the synthesis and deposition of collagen protein and the expression of α-SMA (19). It has been reported that TGF-β1 may activate the MAPK signaling cascades, increase collagen protein deposition and increase α-SMA expression via a positive feedback regulatory mechanism, which may facilitate myocardial fibrosis during myocardial remodeling (20,37). The results of the present study demonstrated that SB203580 (a p38MAPK inhibitor), PD98059 (an ERK1/2 inhibitor) and OMT were able to decrease α-SMA generation and the deposition of type I and type III in TGF-β1-induced CFBs. MAPK signaling may be affected by cytokines, inflammation, growth factors and oxidative stress. A previous study reported that TGF-β1 may induce ROS generation via oxidative stress reactions which, in turn, activate the p38MAPK and ERK1/2 signaling pathways; ROS, ERK1/2 and p38MAPK molecules are involved in myocardial fibrosis formation (38). The present study further demonstrated that TGF-β1-induced myocardial fibrosis may be predominantly mediated by the phosphorylation of p38MAPK and ERK1/2.
OMT has been used for the clinical treatment of viral hepatitis and liver fibrosis in China, and a number of studies have confirmed that it exerts beneficial effects on the cardiovascular system, particularly anti-arrhythmic effects (39,40). Previous studies demonstrated that OMT exerts a positive effect on ventricular remodeling following acute MI, which is associated with the expression of TGF-β1 (15), and that OMT was able to decrease the mRNA expression of TGF-β1 and the TGF-β receptor, which may activate the MAPK signaling pathway. Xiao et al (41) reported that OMT exerted a protective effect against aldosterone-mediated cardiomyocyte injury. Fu et al (4) reported that OMT exerted an inhibitive effect against aldosterone-induced rat cardiac fibroblast proliferation and differentiation by attenuating Smad-2, −3 and −4 expression. As demonstrated by a study of OMT on improving ventricular remodeling, OMT may decrease interstitial and perivascular collagen deposition and inhibit the expression of TGF-β1, and the phosphorylation of p38MAPK, JNK and ERK1/2, in spontaneous hypertensive rats (42). The present study demonstrated that OMT pretreatment decreased the TGF-β1-induced expression of p-p38MAPK and p-ERK1/2 in CFBs, although it exerted no apparent effect on the phosphorylation of JNK1/2/3. In addition, the results from the western blot analysis verified that SB203580 and PD98059 pretreatments also decreased p-p38MAPK and ERK1/2 expression in TGF-β1-induced CFBs. It may be hypothesized that TGF-β1 binds to the TGF-β1 receptor, which in turn activates ROS, stimulating increased phosphorylation of p38MAPK and ERK1/2, and leading to CFB proliferation and abnormal differentiation; however, the enhanced phosphorylation of p38MAPK and ERK1/2 in cultured CFBs challenged by TGF-β1 was inhibited by treatment with OMT.
The current results demonstrated that OMT inhibited proliferation, α-SMA expression and type I and type III collagen deposition induced by TGF-β1 stimulation in CFBs. On a molecular level, OMT pretreatment inhibited the phosphorylation of p38MAPK and ERK1/2 induced by TGF-β1 in CFBs. Therefore, the present study indicated that OMT may prevent CFB proliferation and abnormal differentiation by decreasing TGF-β1-induced p38MAPK and ERK1/2 activation, and thereby reversing the process of myocardial fibrosis.
Acknowledgements
The present study was supported by grants from the National Natural Science Foundation of China (grant nos. 81173586 and 81560588), the Key Project for Science and Technology Foundation of Guizhou Province (grant no. JZ-2015-2039), the Innovated Team of the Education Department of Guizhou Province (grant no. 2014-31), the Program for New Century Excellent Talents in University (grant no. NCET-13-0747), the High level Innovation Talents (grant no. 2015-4029), the 2011 Modern Drug of Cooperation Innovation (grant no. [2013] 04), and the Science and Technology Project of Guizhou Province (grant no. LH [2014] 7098).
Glossary
Abbreviations
- OMT
oxymatrine
- α-SMA
α-smooth muscle actin
- CFBs
cardiac fibroblasts
- MI
myocardial infarction
- OMT-L
low-dose oxymatrine
- OMT-M
medium-dose oxymatrine
- OMT-H
high-dose oxymatrine
- p38MAPK
p38 mitogen-activated protein kinases
- ERK1/2
extracellular signal-regulated protein kinases l/2
- JNK
c-jun N-terminal kinase
- TGF-β1
transforming growth factor-β1
References
- 1.Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair. 2012;5:15. doi: 10.1186/1755-1536-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zamilpa R, Navarro MM, Flores I, Griffey S. Stem cell mechanisms during left ventricular remodeling post-myocardial infarction: Repair and regeneration. World J Cardiol. 2014;6:610–620. doi: 10.4330/wjc.v6.i7.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moor-Morris T, Guimarães-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest. 2014;124:2921–2934. doi: 10.1172/JCI74783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fu L, Xu Y, Tu L, Huang H, Zhang Y, Chen Y, Tao L, Shen X. Oxymatrine inhibits aldosterone-induced rat cardiac fibroblast proliferation and differentiation by attenuating smad-2,-3 and-4 expression: An in vitro study. BMC Complement Altern Med. 2016;16:241. doi: 10.1186/s12906-016-1231-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu CP, Dandapat A, Liu Y, Hermonat PL, Mehta JL. Blockade of hypoxia-reoxygenation-mediated collagen type I expression and MMP activity by overexpression of TGF-beta1 delivered by AAV in mouse cardiomyocytes. Am J Physiol Heart Circ Physiol. 2007;293:H1833–H1838. doi: 10.1152/ajpheart.00488.2007. [DOI] [PubMed] [Google Scholar]
- 6.Purnomo Y, Piccart Y, Coenen T, Prihadi JS, Lijnen PJ. Oxidative stress and transforming growth factor-β1-induced cardiac fibrosis. Cardiovasc Hematol Disord Drug Targets. 2013;13:165–172. doi: 10.2174/1871529X11313020010. [DOI] [PubMed] [Google Scholar]
- 7.Li B, Chen H, Yang X, Wang Y, Qin L, Chu Y. Knockdown of elF3a ameliorates cardiac fibrosis by inhibiting the TGF-β1/Smad3 signaling pathway. Cell Mol Biol (Noisy-le-grand) 2016;62:97–101. [PubMed] [Google Scholar]
- 8.Deng YL, Xiong XZ, Cheng NS. Organ fibrosis inhibited by blocking transforming growth factor-β signaling via peroxisome proliferator-activated receptor γ agonists. Hepatobiliary Pancreat Dis Int. 2012;11:467–478. doi: 10.1016/S1499-3872(12)60210-0. [DOI] [PubMed] [Google Scholar]
- 9.Yong KW, Li Y, Liu F, Bin Gao, Lu TJ, Abas Wan WA, Safwani Wan WK, Pingguan-Murphy B, Ma Y, Xu F, Huang G. Paracrine effects of adipose-derived stem cells on matrix stiffness-induced cardiac myofibroblast differentiation via angiotensin II type 1 receptor and smad7. Sci Rep. 2016;6:33067. doi: 10.1038/srep33067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, Lan HY. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-β/Smad3 signaling. Mol Ther. 2014;22:974–985. doi: 10.1038/mt.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das D, Holmes A, Murphy GA, Mishra K, Rosenkranz AC, Horowitz JD, Kennedy JA. TGF-beta1-Induced MAPK activation promotes collagen synthesis, nodule formation, redox stress and cellular senescence in porcine aortic valve interstitial cells. J Heart Valve Dis. 2013;22:621–630. [PubMed] [Google Scholar]
- 12.Liang JX, Qu XF, Zeng WT, Zhu KL, Zhang H, Wei JJ. Mechanism of oxymatrine in preventing hepatic fibrosis formation in patients with chronic hepatitis B. Nan Fang Yi Ke Da Xue Xue Bao. 2010;30:1871–1873. (In Chinese) [PubMed] [Google Scholar]
- 13.Chen C, Jin Y. Effects of oxymatrine on expression of nuclear factor kappa B in kidneys of rats with adriamycin-induced chronic renal fibrosis. Nan Fang Yi Ke Da Xue Xue Bao. 2007;27:345–348. (In Chinese) [PubMed] [Google Scholar]
- 14.Xu GL, Yao L, Rao SY, Gong ZN, Zhang SQ, Yu SQ. Attenuation of acute lung injury in mice by oxymatrine in associated with inhibition of phosphorylated p38 mitogen-activated protein kinase. J Ethnopharmacol. 2005;98:177–183. doi: 10.1016/j.jep.2005.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen XC, Yang YP, Xiao TT, Peng J, Liu XD. Protective effect of oxymatrine on myocardial fibrosis induced by acute myocardial infarction in rats involved in TGF-β1-Smads signal pathway. J Asian Nat Prod Res. 2011;13:215–224. doi: 10.1080/10286020.2010.550883. [DOI] [PubMed] [Google Scholar]
- 16.Guo CY, Wang Y, Fang YN, Li Q, Chen Y. Antifibrotic effects of oxymatrine in mice with chronic viral myocarditis. Zhonghua Er Ke Za Zhi. 2010;48:273–278. (In Chinese) [PubMed] [Google Scholar]
- 17.Wu XL, Zeng WZ, Jiang MD, Qin JP, Xu H. Effect of oxymatrine on the TGFbeta-Smad signaling pathway in rats with CCl4-induced hepatic fibrosis. World J Gastroenterol. 2008;14:2100–2105. doi: 10.3748/wjg.14.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiao TT, Wang YY, Zhang Y, Bai CH, Shen XC. Similar to spironolactone, oxymatrine is protective in aldosterone-induced cardiomyocyte injury via inhibition of calpain and apoptosis-inducing factor signaling. PLoS One. 2014;9:e88856. doi: 10.1371/journal.pone.0088856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sassoli C, Chellini F, Pini A, Tani A, Nistri S, Nosi D, Zecchi-Orlandini S, Bani D, Formigli L. Relaxin prevents cardiac fibroblast-myofibroblast transition via notch-1-mediated inhibition of TGF-β/Smad3 signaling. PLoS One. 2013;8:e63896. doi: 10.1371/journal.pone.0063896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrov VV, Fagard RH, Lijnen PJ. Stimulation of collagen production by transforming growth factor-beta1 during differentiation of cardiac fibroblasts to myofibroblasts. Hypertension. 2002;39:258–263. doi: 10.1161/hy0202.103268. [DOI] [PubMed] [Google Scholar]
- 21.Weber KT. Extracellular matrix remodeling in heart failure: A role for de novo angiotensin II generation. Circulation. 1997;96:4065–4082. doi: 10.1161/01.CIR.96.11.4065. [DOI] [PubMed] [Google Scholar]
- 22.White HD, Braunwald E. Applying the open artery theory: Use of predictive survival markers. Eur Heart J. 1998;19:1132–1139. doi: 10.1053/euhj.1998.1017. [DOI] [PubMed] [Google Scholar]
- 23.Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–195. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tao Z, Ge Y, Zhou N, Wang Y, Cheng W, Yang Z. Puerarin inhibits cardiac fibrosis via monocyte chemoattractant protein (MCP)-1 and the transforming growth factor-β1 (TGF-β1) pathway in myocardial infarction mice. Am J Transl Res. 2016;8:4425–4433. [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez D, Contreras O, Rebolledo DL, Espinoza JP, Van Zundert B, Brandan E. ALS skeletal muscle shows enhanced TGF-β signaling, fibrosis and induction of fibro/adipogenic progenitor markers. PLoS One. 2017;12:e0177649. doi: 10.1371/journal.pone.0177649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu W, Wang X, Mei Z, Gong J, Huang L, Gao X, Zhao Y, Ma J, Qian L. BNIP3L promotes cardiac fibrosis in cardiac fibroblasts through [Ca+]i-TGF-β-Smad2/3 pathway. Sci Rep. 2017;7:1906. doi: 10.1038/s41598-017-01936-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X, Xu J, Jiang B, Liu D. Bone morphogenetic protein-7 antagonizes myocardial fibrosis induced by atrial fibrillation by restraining transforming growth factor-β (TGF-β)/Smads signaling. Med Sci Monit. 2016;22:3457–3468. doi: 10.12659/MSM.897560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang YE. Non-smad pathway in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen X, Yang Y, Xu Y, Xu L, Fang T. Protective effect of TGF-beta-Smads signal-based oxymatrine on myocardial fibrosis induced by acute myocardial infarction in rats. Zhongguo Zhong Yao Za Zhi. 2012;37:632–636. (In Chinese) [PubMed] [Google Scholar]
- 30.Muslin AJ. MAPK signaling in cardiovascular health and disease: Molecular mechanisms and therapeutic targets. Clin Sci (Lond) 2008;115:203–218. doi: 10.1042/CS20070430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.See F, Thomas W, Way K, Tzanidis A, Kompa A, Lewis D, Itescu S, Krum H. p38 mitogen-activated protein kinase inhibition improves cardiac function and attenuates left ventricular remodeling following myocardial infarction in the rat. J Am Coll Cardiol. 2004;44:1679–1689. doi: 10.1016/j.jacc.2004.07.038. [DOI] [PubMed] [Google Scholar]
- 32.Biernacka A, Dobaczewski M, Frangogiannis NG. TGF-β signaling in fibrosis. Growth Factors. 2011;29:196–202. doi: 10.3109/08977194.2011.595714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van den Borne SW, Diez J, Blankesteijin WM, Verjans J, Hofstra L, Narula J. Myocardial remodeling after infarction: The role of myofibroblasts. Nat Rev Cardiol. 2010;7:30–37. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 34.Kolonin MG, Evans KW, Mani SA, Gomer RH. Alternative origins of stroma in normal organs and disease. Stem Cell Res. 2012;8:312–323. doi: 10.1016/j.scr.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Driesen RB, Nagaruju CK, Abi-Char J, Coenen T, Lijnen PJ, Fagard RH, Sipido KR, Petrov VV. Reversible and irreversible differentiation of cardiac fibroblasts. Cardiovasc Res. 2014;101:411–422. doi: 10.1093/cvr/cvt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tamaoki M, Imannaka-Yoshida K, Yokoyama K, Nishioka T, Inada H, Hiroe M, Sakakura T, Yoshida T. Tenascin-C regulates recruitment of myofibroblasts during tissue repair after myocardial injury. Am J Pathol. 2005;167:71–80. doi: 10.1016/S0002-9440(10)62954-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodeling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 38.Fujii T, Onohara N, Maruyama Y, Tanabe S, Kobayashi H, Fukutomi M, Nagamatsu Y, Nishihara N, Inoue R, Sumimoto H, et al. Galpha12/13-mediated production of reactive oxygen species is critical for angiotensin receptor-induced NFAT activation in cardiac fibroblasts. J Biol Chem. 2005;280:23041–23047. doi: 10.1074/jbc.M409397200. [DOI] [PubMed] [Google Scholar]
- 39.Runtao G, Guo D, Jiangbo Y, Xu W, Shusen Y. Oxymatrine, the main alkaloid component of Sophora roots, protects heart against arrhythmias in rats. Planta Med. 2011;77:226–230. doi: 10.1055/s-0030-1250256. [DOI] [PubMed] [Google Scholar]
- 40.Cao YG, Jing S, Li L, Gao JQ, Shen ZY, Liu Y, Xing Y, Wu ML, Wang Y, Xu CQ, Sun HL. Antiarrhythmic effects and ionic mechanisms of oxymatrine from Sophora flavescens. Phytother Res. 2010;24:1844–1849. doi: 10.1002/ptr.3206. [DOI] [PubMed] [Google Scholar]
- 41.Xiao TT, Wang YY, Zhang Y, Bai CH, Shen XC. Similar to spironolactone, oxymatrine is protective in aldosterone-induced cardiomyocyte injury via inhibition of calpain and apoptosis-inducing factor signaling. PLoS One. 2014;9:e88856. doi: 10.1371/journal.pone.0088856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang XY, Chen CX. Effect of oxymatrine, the active component from Radix Sophorae flavescentis (Kushen), on ventricular remodeling in spontaneously hypertensive rats. Phytomedicine. 2013;20:202–212. doi: 10.1016/j.phymed.2012.10.012. [DOI] [PubMed] [Google Scholar]