

Abstract
Cancer immunotherapy is now a powerful clinical reality, with a steady progression of new drug approvals and a massive pipeline of additional treatments in clinical and preclinical development. However, modulation of the immune system can be a double-edged sword: Drugs that activate immune effectors are prone to serious non-specific systemic inflammation and autoimmune side effects. Drug delivery technologies have an important role to play in harnessing the power of immune therapeutics while avoiding on-target/off-tumor toxicities. Here we review mechanisms of toxicity for clinically-relevant immunotherapeutics, and discuss approaches based in drug delivery technology to enhance the safety and potency of these treatments. These include strategies to merge drug delivery with adoptive cellular therapies, targeting immunotherapies to tumors or select immune cells, and localizing therapeutics intratumorally. Rational design employing lessons learned from the drug delivery and nanomedicine fields has the potential to facilitate immunotherapy reaching its full potential.
Keywords: Cancer immunotherapy, checkpoint blockade, adoptive cell therapy, nanoparticles
1. Introduction
Immunotherapies, treatments that modulate the immune system, have long been proposed as a potentially powerful approach to “functional” or actual cures of disease, based on the natural function of the immune system in protecting the host and its cardinal features of potency, specificity, and memory [1]. Motivated by these features, immunotherapies are now in preclinical and clinical development for treatment of diverse infectious diseases, autoimmunity, allergies, transplant rejection, graft vs. host disease, and cancer. Among these therapeutic areas, cancer immunotherapy in particular has experienced dramatic recent progress in the clinic [2, 3]. For many years, cancer immunotherapies were plagued by high toxicity, low to negligible efficacy, or both. However, steady advances in fundamental cancer immunology and translational immunotherapy have now led to two classes of treatment with significant impact in advanced cancer patients – adoptive cell therapy (ACT), based on the injection of autologous tumor-directed T cells [4, 5]; and checkpoint blockade, treatment with antibodies that block the inhibitory receptors cytotoxic T lymphocyte antigen-4 (CTLA-4) or programmed death-1 (PD-1, or its counter-receptors PD-L1/PD-L2) [6, 7]. ACT therapy in patients with advanced metastatic melanoma and several hematologic cancers has shown a high proportion of complete responses (complete elimination of detectable tumor burden), some of which are durable responses lasting many years [8]. Treatment with ipilimumab, a fully human anti-CTLA-4 antibody, has led to complete responses in approximately 20% of advanced melanoma patients, with durations lasting more than 10 years [9]. Treatment with PD-1 blocking antibodies has elicited objective responses in a variety of solid tumors including melanoma, lung cancer, prostate cancer, breast cancer, ovarian cancer, head and neck cancer, and a subset of colorectal cancers [6]. Reflecting their complementary modes of action, combination therapy with anti-CTLA-4 and anti-PD1 has led to even greater response rates in melanoma patients, where a significant fraction of patients exhibit complete tumor regressions in a space of ~10 weeks [10, 11].
These findings have energized the field and motivated a massive effort to further explore combination immunotherapies that optimally arm the immune system against metastatic disease, but the power of the immune system creates the potential for not only a dramatic attack on tumors but also a significant danger to healthy tissues. For example, monotherapy with anti-CTLA-4, which both blocks a negative regulatory signal during T cell activation and inhibits the function of regulatory T cells, leads to a series of autoimmune side effects, including gastrointestinal toxicity, pruritis, and fatigue, side effects which become grade 3 or 4 serious adverse events in ~23% of patients [12]. When anti-CTLA-4 is combined with anti-PD-1, enhanced anti-tumor activity comes at the cost of synergistically exacerbated toxicity; ~55% of previously untreated melanoma patients given the combination experienced grade 3 or 4 adverse events [11, 12]. As discussed in detail in this review, serious toxicities are characteristic of a broad range of immunomodulatory drugs. Thus, a looming challenge in the field is the development of effective strategies to harness the potential of combination treatments while avoiding debilitating toxicities that prevent immunotherapies from reaching their full curative potential. Clinical studies are already underway seeking to optimize timing and dosing to limit the toxicity of these promising immunotherapy drugs, but in the setting of intravenous administration– believed to be key for systemically modulating the immune response against disseminated tumors– dosing schedules with high safety and high efficacy are often diametrically opposed.
In this review, we discuss the potential for drug delivery technologies spanning a range of approaches to enhance immunotherapies, with a particular emphasis on the potential for enhancing the safety of immunomodulatory drugs. We first review representative mechanisms of immune toxicity from immunotherapy agents of both clinical and preclinical interest, separating systemic and local (i.e. intratumoral) drug administration issues. We then discuss approaches to ameliorate these toxicities based in concepts from the field of drug delivery, employing technologies ranging from nanoparticles to synthetic biology. The immune system as a target for therapy presents several challenges and opportunities relative to somatic tissues: Immune cells circulate through the blood, creating the potential for efficient direct targeting of therapeutics to these cells (relative to, for example, targeting drugs to tumor cells); and immune cells proliferate, providing a source for self-amplification of small doses of appropriately-targeted drugs. However, there is a need to direct immunomodulatory drugs to tumor-specific cells rather than stimulating the entire leukocyte compartment non-specifically, and these cells may be preferentially enriched at tumor sites and tumor-draining lymph nodes. There are thus both challenges and opportunities for the field of drug delivery to impact cancer immunotherapy.
2. Mechanisms of toxicity elicited by immunotherapy drugs
To rationally approach strategies for increasing the safety of systemic immunotherapies, an understanding of mechanisms underlying the toxicity of systemically-administered immunoregulatory drugs is needed. In this section, we review the mechanisms of toxicity underlying several important classes of immunotherapy agents: interleukin-2, representative of several important γ-chain cytokines that promote lymphocyte proliferation and effector function; agonistic antibodies against the costimulatory receptors CD137 (also known as 41BB) and CD28, representative of agonistic antibodies against lymphocyte costimulatory molecules; and the checkpoint blockade agents anti-CTLA-4 and anti-PD-1. A discussion of all immunoregulatory agents in preclinical and clinical testing for cancer immunotherapy is beyond the scope of any single review, but these example biologics represent 3 important distinct mechanisms of immunomodulation relevant to much of the ongoing clinical development of immunotherapy.
2.1. Interleukin-2 as a paradigm for approved but toxic immunotherapy
Systemic high-dose interleukin-2 (IL-2) was one of the first immunotherapy agents to be licensed for cancer therapy, approved by the FDA for metastatic melanoma and renal cell carcinoma (RCC) treatment in 1992. IL-2 was first isolated as a factor promoting the growth of activated T cells, but also stimulates natural killer (NK) cells, both of which motivated its use as a cancer therapeutic. However, it is now also conversely known to also promote activation-induced cell death of stimulated T cells and maintains the survival and function of regulatory T-cells, which restrain the effector arms of the immune system to maintain tolerance and protect healthy tissues from autoimmune attack [13]. Interleukin-2 biology is further complicated by the nature of its tripartite receptor, which is comprised of the IL-2R α chain (CD25), β chain (CD122), and common γ chain (CD132) [13]. Differential expression of the three components of the IL-2R leads to different signaling and functional outcomes on different cell types at different stages of activation.
Based on dosing schedules established clinically in the 1980s, IL-2 is approved as a “high dose” (HD) IL-2 therapy for melanoma and RCC administered intravenously every 8 hours for up to 14 total doses [14]. Although much is made in the current renaissance of cancer immunotherapy around the “tail of the curve” effect, where a small proportion of patients treated with checkpoint blockade become long-term survivors [9], such durable increases in survival were already seen in the early 1990’s in patients treated with IL-2, where ~12% of patients treated with HDIL-2 at the National Cancer Institute had survival of at least 10 years [14]. Although HDIL-2 elicits objective responses in ~16% of patients, it is also extremely toxic. The very short half-life of IL-2 (~12 minutes [15]) leads to a requirement for high doses to be administered in order for functional levels to be maintained for a sufficient timespan. High level dosing in turn leads to dose-related toxicities including vascular leak syndrome (VLS) and cytokine release syndrome, a massive systemic cytokine release and inflammatory reaction caused by IL-2 immune stimulation [16]; lethal adverse events were found in 2% of patients [14]. These issues mean that IL-2 therapy requires careful selection of patients with analyses of baseline cardiac risk factors, performance status, and biomarker analysis. IL-2’s modes of action in expanding T-cells and NK cells make it a logical candidate for combination immunotherapies, but lethal toxicities were seen when HDIL-2 was administered after checkpoint blockade [17] or tyrosine kinase inhibitor (TKI) therapy [18].
The toxicity of IL-2 is multifactorial and involves a complex set of interactions, most notably between immune cells and the vascular endothelium. IL-2 therapy has an acute impact on circulating effector lymphocytes, with a rapid transient induction of lymphopenia. IL-2-activated cells strongly bind to endothelial cells (ECs), leading to endothelial cell cytotoxicity by NK cells and granulocytes [19–21]. In addition, IL-2-induced pulmonary edema is promoted by the interaction of IL-2 with functional IL-2 receptors (IL-2R) expressed on lung endothelial cells; blocking IL-2 interactions with IL-2Rα (CD25) abrogates IL-2-mediated pulmonary edema [22–24]. The endothelial cell damage caused by IL-2-activated host effector cells and/or cytokines and chemokines released in response to IL-2 (e.g., IFN-γ and TNF-α) contribute to VLS and systemic toxicity [25–30]. The role of cytokines in the development of VLS is related to both their direct effect on increasing the permeability of the vascular endothelium and their effects on leukocyte and EC activation [31]. Activation of both ECs and monocytes/neutrophils by cytokines results in the release of large amounts of nitric oxide (NO) [32, 33], which further damage ECs and enhance the adhesion of neutrophils through upregulation of adhesion receptors [34]. Administration of NO inhibitors has been reported to decrease IL-2-induced vascular toxicity in mice [30], and it has been shown that NK cell depletion protects mice from IL-2-induced VLS [35, 36]. Neutrophils also play a critical role in VLS by adhering to ECs and inducing lysis via reactive oxygen intermediates (ROI) and proteases [32, 37, 38]. IL-2 also leads to complement activation which induces mast cell degranulation, resulting in the release of vasoactive mediators and an increase in vascular permeability [39].
The efficacy of IL-2 has fueled much interest in strategies to mitigate its toxicity, as discussed in the subsequent sections below. Continuous intravenous infusion of low dose IL-2 is less toxic than bolus injection of high dose IL-2, but the therapeutic efficacy is also compromised [40, 41]. Strategies strictly localizing IL-2 to tumors or tumor-draining lymph nodes (discussed further below) lack the toxicity of i.v. administration [42–44], suggesting that IL-2’s toxicity is linked to systemic exposure. Thus, approaches to target IL-2 to specific cell types, engineer its interaction with different cell subsets, or localize it to tumors are all strategies of interest to improve its therapeutic index.
2.2. Checkpoint blockade
For many in the general clinical oncology world, the real breakthrough in cancer immunotherapy came with the results from recent clinical trials of so-called “checkpoint blockade” antibodies– antibodies that block negative regulatory signals restraining the host T-cell response against tumors. Two well studied checkpoint receptors, cytotoxic T lymphocyte antigen-4 (CTLA-4) and programmed death-1 (PD-1), are expressed on T-cells and act to diminish responses in early activated and mature peripherally disseminated T-cells, respectively. T-cells are activated when the T-cell receptor binds peptide presented in the major histocompatibility complex on antigen presenting cells (APCs, especially dendritic cells) and costimulatory receptors on the T-cell and APC are co-ligated (e.g., CD28 on the T-cell binding to CD80 and CD86 on APCs). CTLA-4 is upregulated on T-cells during early stages of activation, with expression increasing around 2 days after encounter with cognate peptide presented on APCs in lymphoid organs [12]. As a high affinity homologue of CD28, CTLA-4 serves to interrupt costimulation signals from CD28 to the newly activated T-cells by competition for CD28’s ligands, and thereby dampens the immune response (Figure 1A) [45]. Inhibition of CTLA-4 using monoclonal antibodies lifts the inhibitory effects of CTLA-4, but only on activated T-cells [46]. In addition, CTLA-4 is highly expressed on regulatory T cells (Tregs); therefore, in mice, administration of anti-CTLA-4 antibodies has been shown to deplete Tregs specifically from the tumor microenvironment where CTLA-4 is most highly expressed by these cells ([47–49]. In contrast to CTLA-4, PD-1 is primarily expressed on T cells in the periphery and in the tumor microenvironment (Figure 1B) [12]. Interaction with one of its two ligands (PD-L1 or PD-L2) downregulates antigen receptor signaling in mature T-cells and decreases the expression of pro-inflammatory cytokines. PD-1 expression is present during T-cell effector phases and on re-exposure to antigen [50]. Blockade of the PD-1 interaction with its ligand can transiently reverse exhaustion of T-cells in the periphery [51].
Figure 1. Mechanisms of action for immunomodulatory antibodies.
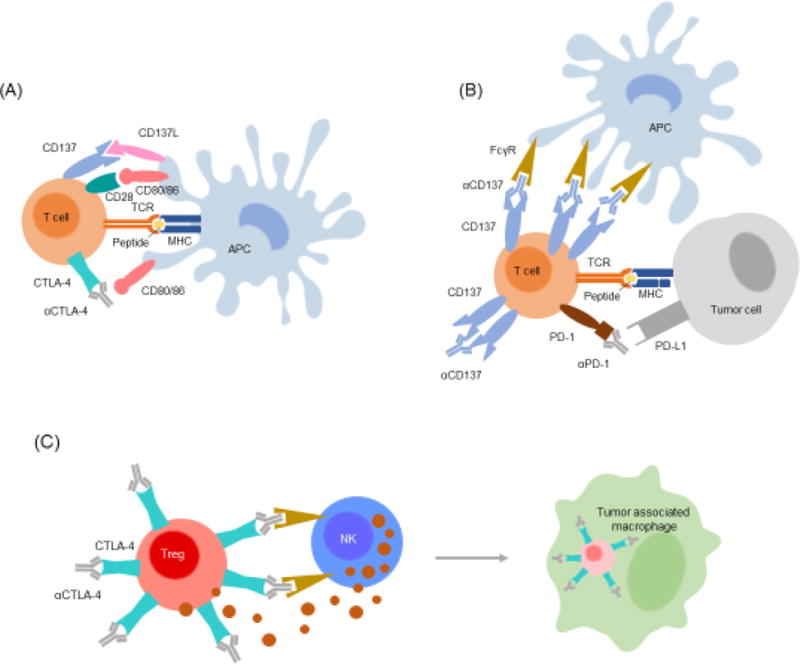
(A) T-cells are activated by APCs presenting specific peptide-MHC complexes in tandem with signals from both positive (e.g., CD28, CD137) and negative (e.g., CTLA-4) costimulatory receptors binding cognate partners on the APC surface. Anti-CTLA-4 blocks receipt of negative regulatory signals from CTLA-4 engagement to boost T-cell priming by APCs. (B) Anti-PD-1 augments T-cell function in the effector phase by blocking negative regulatory signals delivered by PD-L1 expressed on tumor cells or PD-L2 expressed by APCs and other cells. Anti-CD137 can also boost T-cell effector function through crosslinking of the CD137 costimulatory receptor and/or clustering receptors by antibody displayed on APCs through their Fcγ receptors. (C) In murine models antibodies have been utilized to bind cells and initiate their depletion/killing through complement mediated cytotoxicity and antibody dependent cellular cytotoxicity (ADCC). This mechanism can be applied directly to tumor cells. Anti-CTLA-4 antibodies also act to boost the immune response by triggering ADCC-mediated depletion of intratumoral Tregs that express high levels of CTLA-4 receptor.
These distinct mechanisms of action have led to therapeutic blocking antibodies targeting CTLA-4, PD-1, or PD-1’s ligands. Ipilimumab, an antibody that blocks CTLA-4 was approved in 2011 following a pivotal trial demonstrating its ability to improve long term overall survival in advanced metastatic melanoma- the first new drug for advanced melanoma approved in more than 30 years [52]. Ipilimumab therapy leads to complete tumor regressions lasting at least 10 years in ~20% of treated melanoma patients [9]. Following on the heels of ipilimumab’s approval, several antibodies blocking PD-1 (expressed by T-cells) binding to its ligands PD-L1 or PD-L2 (expressed by many cells, including tumor cells)– entered the clinic and have shown even more impressive initial responses, with 30–50% of patients in diverse diseases ranging from melanoma to lung cancer exhibiting objective tumor regressions [53–55]. The ability of PD-1 blockade to elicit responses in solid tumors not previously viewed as “immunogenic” (e.g., lung cancer) heralded a new era of promise for immunotherapies. However, “taking the brakes off” of the immune system with systemic checkpoint blockade, leads (not unexpectedly) to toxicities, which are amplified when these drugs are employed in combination.
Ipilimumab therapy elicits adverse events, with 60–65% of patients experiencing immune related adverse events (irAEs) at a moderate dose of 3 mg/kg every three weeks [52, 56]. In two large phase 1 trials, the most common adverse events included pruritus (25–35%), diarrhea (23–33%), rash (15–33%), and fatigue (15–28%) [11, 57]. Serious Grade 3 and 4 adverse events occur in 20–27% of patients, the most frequent being gastrointestinal toxicities resulting in enterocolitis and diarrhea (6–8% of grade 3–4 events) [11]. Of the 14 patients who died in the phase III clinical trial mentioned above, 7 died due to immune-related adverse events [11, 52, 56, 57]. Overall, ipilimumab elicits broad irAEs of the skin, gastrointestinal tract, and endocrine system.
Anti-CTLA-4 irAEs may manifest from depletion of regulatory T cells, as evidenced by the need for depleting antibody isotypes (such as IgG2a) capable of engaging Fcγ receptors to mediate antitumor activity in mouse models [48, 58, 59]. CTLA-4 blocking antibodies of IgG2a isotype yielded depletion of regulatory T-cells at tumor sites (Figure 1C) and an increase in CD8+ T effector cells in the periphery, while other isotypes expanded both regulatory and effector T-cells in the periphery [59]. Some evidence however demonstrates that the depletion of regulatory T-cells is context specific and limited to the tumor microenvironment, due to the high frequency of Fcγ receptor-expressing tumor associated macrophages, though depletion of regulatory T-cells in the periphery remains conceivable [48]. Regulatory T-cells are known to maintain tolerance and restrict lymphocytic infiltration to mucosal linings of organs including the lungs and gastrointestinal tract [60]. Depletion of such regulatory T-cells in the intestines may account for increased intraepithelial lymphocytes and leukocyte infiltration in the lamina propria of ipilimumab-treated patients as revealed by endoscopic analysis, thus causing irAEs such as diarrhea [61, 62].
PD-1 blockade antibodies have in general shown less serious side effects compared to anti-CTLA-4 in humans, with only ~15% of patients experiencing serious adverse events, primarily pneumonitis. For comparison, in phase I dose escalation studies, administration of nivolumab (one of the approved anti-PD-1 antibodies) resulted in grade 3–4 adverse events in only 14% of patients compared to the 20–27% in early ipilimumab trials. Lastly, irAEs of any grade affected only 41% of patients compared to ipilimumab’s 60–65% [63]. Consistent with their distinct mechanisms of action, the immune related adverse events differ between anti-CTLA-4 and anti-PD-1/PD-L1 therapeutics: anti-PD-1 irAEs include rash, hypothyroidism, hyperthyroidism, pneumonitis, diarrhea, and elevated aminotransferase levels [12]. A large phase III trial confirmed that both nivolumab and pembrolizumab (both now clinically approved anti-PD-1 antibodies) induce fewer adverse events than ipilimumab [64].
Mechanistic differences between PD-1 and CTLA-4 receptors are likely responsible for the differences in adverse outcomes and better tolerability of PD-1 checkpoint blockade. Given that CTLA-4 is expressed on all T cells around the time of activation in lymphoid tissue, broad spectrum autoimmunity can result from blockade of CTLA-4. Additionally, as noted above administration of anti-CTLA-4 antibodies in mice depletes regulatory T cells, which play an important role in restraining autoimmune attack of host tissue [12], and systemic depletion of Tregs leads to fatal autoimmune pathology. Such a mechanism would motivate strategies to localize the action of anti-CTLA-4 in vivo, but this phenomenon remains unconfirmed in humans so far. Unlike CTLA-4, the PD-1 receptor is largely expressed on antigen re-exposed cells in the periphery; therefore, a smaller pool of T-cells are likely affected by treatment with anti-PD-1 antibodies. Because expression of CTLA-4 is dependent on initial priming of T-cells, the onset of irAEs and therapeutic efficacy from anti-CTLA-4 antibodies is often delayed compared to PD-1 blockade, which reactivates T-cells already present in the tumor microenvironment [12]. The varying populations of T-cells targeted by checkpoint blockade antibodies may explain the severity and time to onset of immune-related adverse events.
CTLA-4 and PD-1 are just the first successfully targeted inhibitory receptors on lymphocytes, two members of a large collection of negative regulatory receptors expressed by T-cells. A non-exhaustive list of T-cell-expressed negative regulatory receptors explored in preclinical studies and clinical trials includes Lymphocyte activation gene 3 (LAG-3), T cell membrane protein 3 (TIM-3), T-cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT), V-domain Ig suppressor of T-cell activation (VISTA), adenosine A2a receptor (A2aR), and B and T lymphocyte attenuator (BTLA) [65–68]. In addition, because of their distinct mechanisms of action (despite their classification together in the immune-oncology world as checkpoint blockade agents), therapies combining anti-CTLA-4 and anti-PD-1 were obvious to explore, and showed promising synergy in preclinical mouse models [69]. Recently, the first trials of ipilimumab combined with the PD-1 blocking antibody nivolumab in melanoma were completed, and showed that while the combination achieves a striking increase in efficacy– with 75% of patients experiencing objective responses– this enhanced efficacy was accompanied by a concomitant increase in serious toxicities, with 53% of patients experiencing grade 3 or 4 serious adverse events [10, 11, 70, 71]. Grade 3–4 toxicities were commonly characterized by elevated lipase, aspartate aminotransferase, and alanine aminotransferase levels, indicating pancreas and liver toxicities. Unexpected side effects including acute onset diabetes and diabetic ketoacidosis were noted in 17 patients co-treated with iplilimumab and nivolumab [72]. Often these autoimmune adverse effects do not present until weeks to months after treatment. Similar toxicities appear to be prevalent in a phase I study of a different anti-CTLA-4 antibody (tremelimumab) in combination with an anti-PD-L1 antibody (durvalumab) tested in NSCLC patients [73]. Recently reported interim results of an ongoing phase II trial of ipilimumab combined with nivolumab in recurrent small cell lung cancer reported a lower incidence rate of serious side effects but also lower levels of overall response [74]. Adjustment of dosing schedules in a phase I study in non-small cell lung cancer patients (nivolumab given every 2 weeks but ipilimumab given every 6 or 12 weeks) showed some reduction in toxicity compared to the initial melanoma trials, with only 33–37% grade 3–4 adverse events [75].
Combination treatment with checkpoint blockade antibodies epitomizes the promise and the challenge of immunotherapy– co-administration of these agents systemically leads to significant increases in anti-tumor efficacy, but also synergistic amplification of toxicity. Achieving the goal of long term durable remissions in a majority of patients is unlikely to be achieved by single drugs in difficult-to-treat solid tumors, and this has led much of the field to be convinced of the need for combination immunotherapy strategies combining multiple drugs acting via complementary mechanisms [76]. Solutions from the field of drug delivery may be critical to achieve this goal while avoiding life-threatening toxicities that plague many immunotherapy drugs administered using the systemic dosing strategies traditionally employed in oncology.
2.3. Agonist antibodies against immune costimulatory receptors
CTLA-4 and PD-1 represent important inhibitory receptors that restrain T-cell priming and effector functions. These negative regulatory receptors are counter-balanced by a suite of positive costimulatory receptors that support T-cell activation. Canonical examples include CD28, CD137 (also known as 4–1BB), and CD134 (also known as OX40 or Tumor necrosis factor receptor (TNFR) superfamily member 4), but like the negative regulatory receptors, a large collection of these receptors (the TNFR superfamily) has been discovered. These proteins are expressed by T-cells during activation by antigen presenting cells, and bind to counter receptors expressed on the APC surface, providing signaling synergistic to triggering of the T-cell receptor promoting T-cell expansion, survival, and effector functions [77]. These receptors are also expressed by other immune cells, such as natural killer cells. The natural mode of costimulation from these receptors occurs at cell-cell contacts, but this signaling can alternatively be induced by cross-linking of costimulatory receptors on T cells by agonistic antibodies. This represents an additional strategy for immunomodulation, and antibodies directed against costimulatory molecules such as CD137, OX40, and CD28, represent another class of promising cancer immunotherapies limited clinically by systemic toxicity. Though capable of amplifying tumor-specific cytotoxic T cells, agonistic antibodies to T-cell costimulatory molecules have on-target off-tumor effects due to the presence of their ligands on non-tumor-specific T-cells and other immune subtypes, as well as in some cases expression by other non-immune cell populations. Off-tumor effects and deregulated production of proinflammatory cytokines have led to dose limiting toxicities of both anti-CD137 and anti-CD28 antibodies. Their mode of action- actively triggering intracellular signaling rather than simply blocking a ligand-receptor interaction, means that such antibodies function distinctly from the checkpoint blockade antibodies described above. Antibodies are comprised of two antigen-binding Fab domains and a rear Fc domain that can be bound by Fc receptors (FcRs). Agonist antibodies against the TNFR superfamily member CD40 have formally been shown to require an ability for their Fc regions to bind Fcγ receptors for their activity in mice [78, 79], suggesting that the antibodies are “presented” from the surface of FcR-expressing cells and crosslink CD40 receptors on a neighboring cell in a manner mimicking the natural cell-cell engagement in T-cell/APC contacts (Figure 1B). Similarly, agonist antibodies against OX40, CD137, and CD27 require or show greatly enhanced activity if they are competent for FcR binding [80, 81].
CD137-targeting antibodies provide a useful case study of this class of agonist antibody therapeutics. CD137 is expressed on the surface of activated CD8+ T cells, and to a lesser extent on CD4+ T cells, natural killer (NK) cells, NKT cells, regulatory T cells, dendritic cells, macrophages, neutrophils, and eosinophils [82]. In addition, this receptor has been found to be expressed on vascular and lymphatic endothelial cells at sites of inflammation and tumor vasculature [83–85]. Its natural ligand, CD137L, is present on antigen presenting cells. As described above, engagement of CD137 on T-cells by CD137L results in enhanced T-cell proliferation, production of proinflammatory cytokines, and protection from activation-induced apoptosis [86]. Due to expansion of memory T-cells directed against tumor antigen, administration of anti-CD137 agonistic antibodies in preclinical mouse models has significant anti-tumor activity [87, 88]. For example, mice treated solely with anti-CD137 showed complete regression of tumors in a mastocytoma model [89]. As a monotherapy anti-CD137 activates endogenous tumor-reactive T-cells, but in combination with adoptive T-cell transfer yielded 80% survival in a thymoma model [87]. Similarly, combinations of anti-CD137 with chemotherapy, irradiation, and tumor lysate pulsed dendritic cells are also effective [90–92]. NK cell function is also enhanced by anti-CD137 therapy, though NK cells play an auxiliary role to T-cells in the anti-tumor activity of anti-CD137 agonists [86, 93]. Efficacy in most examined syngeneic mouse models is dependent on CD8+ T-cells though CD4+ T-cells, NK cells, cross-presenting dendritic cells, and IFN-γ production were proven to contribute in some tumor models [87, 88, 94].
Despite these promising therapeutic outcomes, anti-CD137 treatment was noted to induce dysregulated hematopoiesis and liver dysfunction in preclinical murine studies. Increased T-cell infiltrates in the liver, hepatitis, lymphopenia, thrombocytopenia, splenomegaly, hepatomegaly, and lymphadenopathy have been reported [94]. Additional side effects – namely alopecia, scaly skin, and increased AST and ALT levels – appear more characteristic of systemic inflammation [95]. Systemic cytokine release syndrome is also observed with elevated systemic levels of proinflammatory cytokines such as IFN-γ, TNF-α, IL-12, and type I interferons in mice treated with anti-CD137 monotherapy [93, 96]. TNF-α produced by CD8+ T-cells was shown to be critical in the development of splenomegaly, lymphadenopathy, hepatomegaly, and hepatitis. Conversely, IFN-γ and type I interferons contributed to the expansion of blood cells and mislocalization of T-cells, but were non-essential to the overall development of toxicity. These toxicities are T-cell- and CD137-dependent, as they are alleviated in Rag−/− and CD137−/− mice [94]. Of note, the increased mononuclear cell accumulation in the portal areas of the liver was dependent on polyclonal T-cell expansion: Due to the lack of oligoclonal T-cells in the liver, it is presumed that intra-liver T-cells are not directed against self-antigens [96]. Localization of these cytotoxic cells to the liver contributes to apoptosis of hepatocytes and subsequent hepatitis [94, 96]. The mechanisms governing anti-CD137 induced hepatotoxicity remain ill defined; however, preliminary evidence suggests IL-27 produced by anti-CD137 stimulated myeloid subsets mediates the recruitment and activation of liver damaging T cells. Additionally, depletion of FoxP3+ regulatory T cells aggravates liver toxicity, suggesting a role for Tregs in restraint of anti-CD137 initiated immune responses [97].
In spite of the adverse events in mice, studies by Bristol Myers Squibb in cynomolgus monkeys demonstrated tolerability of the anti-CD137 agonistic antibody urelumab, with no hepatic side effects at doses up to 100 mg/kg, warranting a transition to human clinical trials [98]. Urelumab was first tested in melanoma, non-small cell lung cancer, and other advanced solid tumors [99]. During dose escalation of urelumab, dose limiting grades 3 and 4 neutropenia were accompanied by frequent yet mild adverse outcomes including leukopenia, thrombocytopenia, and hyperbilirubinemia, mirroring toxicology results in mice. In this study, partial remissions and stabilized tumor growth justified further study of urelumab. Several clinical studies of urelumab in combination with checkpoint blockade and other cancer treatments such as chemotherapy and adoptive cell transfer have subsequently moved forward, despite urelumab toxicities resulting from broad expression of CD137 on many leukocyte populations (see clinicaltrials.gov). Most adverse events from urelumab have been managed with corticosteroids and anti-TNF-α antibodies; however, further clinical progress with anti-CD137 will require a deeper understanding of the mechanisms of toxicity [100].
A more dramatic cautionary tale for agonistic antibodies can be found in CD28 superagonists (CD28SAs). Preclinical studies in rats, non-human primates, and cultures of human cells failed to predict nearly lethal cytokine release syndrome in the six healthy volunteers first dosed with TeGenero’s CD28SA TGN1412 [101]. Anti-CD28 superagonist antibodies are capable of activating T-cells through the CD28 costimulatory receptor in the absence of the classical “signal one” stimulus from peptide-MHC binding the T cell receptor [102]. Crosslinking of CD28 using superagonist antibodies led to proliferation of all subsets T-cells in mouse and rat models [103]. In rodents CD28SAs also trigger the rapid expansion of Tregs, allowing for potential applications in autoimmune disease in addition to cancer [104]. However, when applied in healthy human volunteers, the release of proinflammatory cytokines was so significant that all six patients were hospitalized, indicating major shortcomings in the extensive preclinical research [105].
Six years of investigation were required to uncover the underlying causes of the toxicity and why the adverse outcomes weren’t predicted preclinically. Ultimately, discrepancies between the clinical and preclinical data were ascribed to 1) differences in the balance of Treg and T effector memory cells in humans and rodents, 2) loss of CD28 in T effector memory cells during CD4+ cell differentiation in primates but not humans, and 3) failure of human PBMC culture conditions to adequately recapitulate the tonic TCR signals found when T-cells are present at high densities promoting extensive cell-cell contacts as in lymphoid tissues [105]. In mice, two waves of T-cell expansion arise after administration of CD28SAs – the first a rise in conventional T cells and Tregs, the second a Treg-exclusive expansion [106]. The limited number of expanded conventional CD4+ T-cells and high percentage of Treg cells restrains an inflammatory immune response in rodents. The two wave model does not hold true in humans [107–109]. In humans, primarily CD4+ T effector memory cells are activated upon CD28SA administration [110]. These cells are tissue resident T-cells which accumulate over time to quickly respond to antigen rechallenge. The activation of T effector memory cells in humans without suppression by Tregs was one leading cause of cytokine release syndrome. Accumulation of T effector memory cells is common in humans which are constantly subjected to antigen exposure whereas they are less prominent in mice housed in clean caging, thus explaining one difference between the preclinical and clinical responses [110]. Eastwood et al showed that CD4+ T-cell differentiation into T effector memory cells causes loss of CD28 expression in macaque but not humans, explaining the inadequacy of the non-human primate studies [110]. Lastly, testing of CD28SAs on human PBMC in culture did not predict cytokine storm because PBMC grown in low density non-adherent culture fail to recapitulate the tonic TCR signal present in T-cell populations [111]. Treatment of high density human PBMC culture with CD28SAs revealed enhanced proliferation of T-cells and proinflammatory cytokine production lacking in earlier studies. Cell-cell contacts in these altered culture conditions allowed for minimal residual TCR activation in T-cells required for the function of CD28 superagonists [111]. The development of CD28SAs demonstrates the difficulty is translating safe immunotherapies from preclinical models to human trials.
Currently CD28SAs are not being clinically explored for cancer treatment, however, the agonist described above (TGN1412) is undergoing testing at lower doses for use in autoimmune diseases [112]. Newly developed human PBMC culture assays are utilized to test the compound, now named TAB08. At 1000-fold lower concentrations than used in the disastrous phase I clinical trial in 2006, TAB08 was well tolerated. At these low doses, TAB08 is expected to induce expansion of Treg populations and production of anti-inflammatory cytokines like IL-10 for use in autoimmune disease indications [112].
Altogether, similar to the checkpoint blockade antibodies, a general conclusion in the development of agonist immunostimulatory antibodies (and related recombinant agonist ligands) is that broad nonspecific stimulation of all leukocytes (or other cell types) expressing any given regulatory receptor is liable to be fraught with systemic toxicities arising from on-target, off-tumor stimulation of cells in the blood or healthy tissues. These observations motivate strategies to engineer control over multiple facets of immune stimulation: what cellular subsets are stimulated, where are they stimulated, and for what duration does stimulation last.
2.4. Tumor targeting antibodies
Antibodies recognizing tumor antigens can also be utilized as immunologic agents to promote tumor cell death. When directed against tumors, antibodies can facilitate a host of effects both immune system-dependent and -independent, including direct blockade of intracellular signaling, induction of signaling-based apoptosis, enhanced sensitivity to chemotherapy, complement mediated cytotoxicity (CMC), and antibody-dependent cellular cytotoxicity (ADCC) often mediated by NK cells [113]. In many cases, evidence suggests antibodies originally developed to block oncogenic receptor signaling also act through immune-dependent mechanisms. For example, trastuzumab is an approved anti-human epidermal growth factor receptor type 2 (HER2) monoclonal antibody; adjuvant administration of trastuzumab in breast cancer patients results in a 23–35% increase overall survival [114]. HER2 is a transmembrane tyrosine kinase receptor promoting a host of cellular functions including proliferation through activation of the MAPK pathway [115]. HER2 amplification is detectible in approximately 30% of human breast cancers; overexpression and mutations induce receptor dimerization and near-constitutive activation of proliferation and anti-apoptotic pathways [116, 117]. Trastuzumab binds to the extracellular portion of HER2, decreasing receptor dimerization and therefore intracellular signaling, increasing endocytosis of HER2, and inhibiting shedding of the extracellular domain of HER2 [118]. However, studies regarding Fc receptor polymorphisms in cancer patients also suggest that trastuzumab therapy may rely on immune effectors and ADCC by NK cells and monocytes for efficacy [119].
Many tumor targeting antibodies, trastuzumab included, have quite favorable safety profiles, though rare incidences of grade 3 and 4 toxicities have been noted. Trastuzumab, for example, is well-tolerated; however, cardiotoxicity remains a concern due to on-target off-tumor effects on HER2 expressing cardiomyocytes and cardiac stem cells [120]. Cardiac dysfunction is prevalent in 8% of patients treated with trastuzumab alone and increases to 30% in patients on concurrent anthracyclines [121, 122]. HER2 has been implicated in repair of cardiomyocytes following anthracycline-induced and reactive oxygen species induced damage, suggesting that combination of trastuzumab with anthracyclines leads to on-target cardiac damage [123, 124]. Cardiotoxicity ranges from decreased left ventricular ejection fraction (LVEF) to congestive heart failure. Comparison of a short 6 month trastuzumab regimen to a 12 month regimen reveals increased risk of LVEF decline with longer exposure to trastuzumab, though more study is needed to confirm these results [125]. Meta-analysis of 10,000 patients determined a risk ratio of 5.11 (p<0.0001) for congestive heart failure with trastuzumab compared to control populations [125]. While cardiotoxicity poses a serious threat to trastuzumab treated patients, identification of risk factors and patient surveillance are likely to limit treatment related deaths and hospitalizations [120].
Similarly, in the case of rituximab, an anti-CD20 human-mouse chimeric antibody developed for its ability to deplete CD20-expressing B cells via CMC and ADCC in patients with B-cell lymphoma, toxicities are typically mild, though serious adverse events are noted in a portion of the patient population. Common grade 1 and 2 events include pruritus, nausea, dizziness, and fevers. Serious infusion-related events such as anaphylaxis and myocardial infarction have occurred after initial rituximab administration, though these side effects are rarely fatal and can be managed with acetaminophen and antihistamines [126]. Additionally, increased rates of infection (8.1% of patients receiving rituximab, 3.9% in control arm) and neutropenia (13.4% of patients receiving rituximab, 6.3% in control arm) were noted [127], with the former likely related to loss of normal B cells during rituximab treatment. Patients on a rituximab maintenance regimen also experienced more infection-related adverse events compared to patients on observation alone (Risk ratio 1.99) [128]. Grade 3 and 4 infection-related adverse events all required hospitalization; patients on rituximab had more of these severe events than the control population (Risk ratio 2.90) [128]. Non-infection related respiratory adverse events including cough, dyspnea, and sinusitis afflict 38% of patients receiving rituximab. Meta-analysis of clinical studies up to June 2010 report 121 cases of rituximab-associated interstitial lung disease (ILD) characterized by diffuse bilateral lung infiltrates and hypoxaemia with ILD fatalities occurring in 18 patients [129]. Altogether, the side effects from tumor targeting antibodies are generally mild and grade 3/4 adverse events are rare, but the ability of antibodies to engage cellular components of the immune system remains an issue that requires careful consideration during antibody development, especially when overexpressed self-antigens present in healthy tissues are targeted.
2.5. Local administration of immunotherapy agents
One simple approach to mitigate immune toxicity has been the local injection of immunomodulatory drugs directly into accessible lesions, either primary tumors or metastases. Such an approach is predicated on the expectation that locally-administered drugs will be preferentially retained at the injected tumor site, and that such retention might be favored if concentrated local delivery allows the drug to be given at lower doses than used systemically. Local injections have thus been explored both preclinically and clinically for a variety of immunotherapy drugs. Local therapy can be considered in any cancer where primary or metastatic lesions are accessible either directly or through surgery, and thus a great variety of tumors have been treated clinically through local therapy administration, for example melanoma, breast cancer, ovarian cancer, bladder cancer, lymphoma, and lung metastases in multiple diseases.[130–135]
Local administration of immunotherapy is motivated by the hypothesis that the immune system, if stimulated locally, can disseminate from the treatment site to attack other tumors which did not directly receive any of the immunotherapy drug– if correct, this idea formalizes one of the cardinal distinctions between immunotherapy and traditional tumor-directed chemotherapy. Presently, several clinical and preclinical studies provide evidence in favor of this hypothesis. For many years, it has been known that some patients who receive radiation therapy at one selected tumor exhibit regressions of distal untreated lesions; this phenomenon was termed the abscopal effect in the 1950’s by Robert Mole [136]. Only recently, as the role of the immune system in the response to many traditional cancer therapies has become more clear, was it demonstrated in preclinical mouse models that the abscopal effect is dependent on the host immune system [137], and that in fact radiation treatment of tumors acts through the innate immune system to amplify the adaptive immune response [138–140]. Similar abscopal-like responses have now been reported in preclinical studies and clinical trials of several types of immunotherapy: For example, local intratumoral injection of an oncolytic virus combined with systemic anti-CTLA-4 led to rejection of both treated and untreated tumors [141]. Intratumoral injection of anti-CTLA-4 with the immune-agonist antibody anti-OX40 and the Toll like receptor agonist CpG led to depletion of regulatory T-cells in the injected tumors, followed by systemic tumor regression [142]. Phase I studies of intratumoral CpG combined with local radiation therapy elicited partial responses in uninjected lesions of both lymphoma and mycosis fungoides patients [134, 143]. Seung et al. found that a combination of localized radiation treatment with systemic IL-2 in melanoma patients led to a high proportion of complete or partial responses (74%), correlating with expanded CD4+ T-cell responses in the peripheral blood [144]. These are just a few examples of systemic responses to local immunotherapy, which has also been termed intratumoral vaccination or in situ vaccination based on the concept of the treated tumor itself serving as a source of antigens for priming new T-cell responses in draining lymph nodes [145–148].
Local immunotherapy thus has clinical relevancy for both primary and metastatic disease; however, intratumoral injection of free therapeutics does not necessarily limit systemic exposure to toxic immunotherapies. Compounds injected into the intratumoral/peritumoral space may reach systemic circulation via lymphatic drainage or by direct access through leaky tumor vasculature. By definition, such systemic dissemination raises the potential for systemic toxicity mirroring direct intravenous administration. For example, intratumoral injections of agonist antibodies or cytokines in mouse models of solid tumors has resulted in the rapid appearance of high serum concentrations of these agents [43, 149, 150]. The dissemination of these compounds into the systemic circulation can result in significant weight loss, systemic cytokine storms, and even lethality from systemic immunotoxicity [43]. Intratumoral administration also does not provide persistent stimulation at the tumor site; for example 48 hours after intratumoral injection of an agonistic anti-CD40, the antibody was nearly undetectable in tumors by immunohistochemistry [149]. Similarly, intratumoral or peritumoral injections of other cytokines, antibodies, and TLR agonists have all been shown to lead to systemic dissemination of these agents and often, systemic toxicity in mouse models [149–152]. These preclinical results echo findings in the clinic: In phase I studies of recombinant IL-12 and TNF-α, patients receiving intratumoral injections showed the recombinant cytokines at high levels in plasma within 30 minutes after injection, indicating a lack of local retention [153, 154]; systemic levels of IFN-γ and IL-10 and fever-like systems were elevated within 4–8 hours post injection and did not return to background levels for 48 hours [154]. Other studies of intratumorally-injected cytokines where dissemination of the drug was not characterized reported toxicities equivalent to systemic injections, suggesting systemic exposure [155]. Trials of low doses of IFN-γ injected intratumorally have shown good safety profiles, but also lacked efficacy, which may reflect the low doses and/or poor retention of the therapeutic in the injected lesions [156]. Thus, local injection is a well characterized strategy to alter the pharmacokinetics of drug treatments, but this simple approach does not fully isolate immunotherapies from the systemic circulation. Taking full advantage of abscopal-like effects of immunotherapies while mitigating systemic toxicities requires strategies to locally target and retain drugs in the tumor microenvironment.
3. Engineering safer local therapies
The previous two sections highlight a variety of challenges associated with the yin and yang of efficacy vs. toxicity in both systemic and local immunotherapy. Though it is clear that dosing parameters have a significant impact on safety and therapeutic outcome [157], these challenges often cannot be solved by optimizing dosing and timing alone (e.g., lowering dose increases safety but lowers efficacy). Drug delivery technologies provide many potential solutions to these issues. While enhancing the safety of systemic immunotherapies is important, we first discuss the conceptually simpler problem of enhancing the safety and efficacy of local immunotherapy. A key objective is promoting better local retention of immunotherapeutics and blocking their dissemination into the circulation. Approaches include the use of local drug depots that match release rates of drugs to their uptake by target immune/tumor cells, blocking therapeutic diffusion through locally-injected biomaterial anchors, and confining therapeutics to tumors through localized intratumoral gene delivery (e.g., using oncolytic viral vectors). We discuss in turn examples of each of these approaches applied to immunotherapy. The use of drug delivery technologies to enhance the safety of cancer vaccine formulations, e.g., through enhanced delivery to lymph nodes or targeting of specific APC populations, is a subject of much research effort, but as this topic could fill a review on its own, we have chosen to focus on treatments focused on the tumor or tumor-draining lymph nodes, and refer interested readers to other recent reviews on the subject of vaccine technologies [158–162].
3.1. Intratumoral drug depots
Because many immunotherapy toxicities (e.g., vascular leak syndrome) are linked to systemic stimulation of circulating leukocytes and/or direct action of immunomodulators on endothelial cells, an obvious strategy to enhance local therapy is to better confine therapeutics to a chosen target lesion. One way to achieve this is through local controlled release of drug from intra- or peritumorally injected drug delivery matrices (Figure 2A). One of the first demonstrations of this approach was work by Egilmez et al. seeking to improve on the safety and efficacy of IL-12 as an immunotherapeutic [163, 164]. IL-12’s potent anticancer effects are limited by dose- and temporal regimen-dependent toxicity when administered systemically [165, 166]. Egilmez showed that a single intratumoral (i.t.) injection of biodegradable polylactic acid microspheres exhibiting controlled release of IL-12 was safe and led to complete regression of transplanted lung tumors, prevented metastatic spread to the lung, and enabled animals to reject a subsequent re-challenge with live tumor cells, indicating the development of systemic antitumor immunity [163]. More recently, chitosan matrices have been similarly used to provide sustained localized dosing of IL-12 in several tumor models, including as a neoadjuvant treatment in a breast cancer model prior to surgical resection of the primary tumor [167–169]. Hanes et al. used crosslinked gelatin/chondroitin sulfate microspheres as delivery vehicles for intratumoral delivery of IL-2 in models of brain or hepatic melanoma metastases [151]. IL-2 delivered by these microspheres persisted in tumors for 3 weeks, compared to only 24–48 hrs for bolus-injected drug. At the same time, bolus-injected IL-2 was found in the blood, spleen, and other organs minutes after bolus i.t. injection of IL-2, while microsphere-delivered cytokine led to very low or undetectable cytokine outside of the tumor microenvironment. Hori et al. designed self-crosslinking alginate gels [170] and used these to provide sustained local release of IL-15 following peritumoral injection [150]. This strategy lowered the systemic exposure to the cytokine by ~2-fold and increased the dosing within the tumor 40-fold relative to a bolus injection of IL-15.
Figure 2. Strategies for enhancing localized immunotherapy.
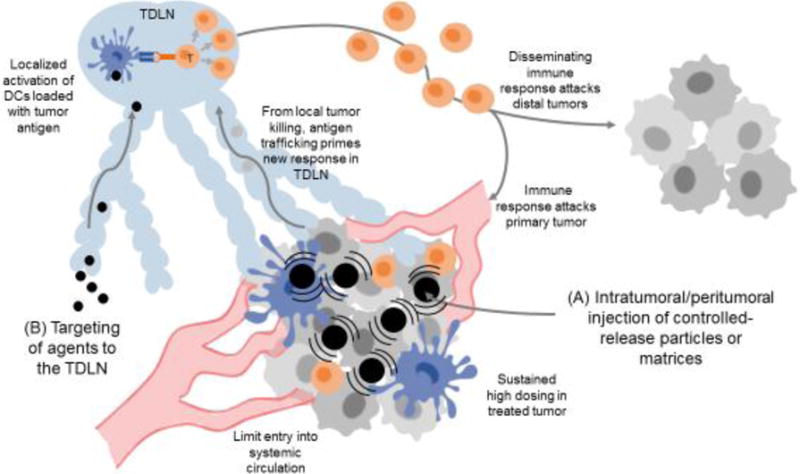
(A) Synthetic particles/matrices administered directly to accessible lesions can provide sustained local dosing of immunomodulatory drugs with greatly lowered systemic exposure. Local immune activation leads to tumor cell death in the treated tumor and tumor antigen delivery to the TDLN, priming new T-cell responses that return to the treated tumor as well as disseminate to attack other metastases that were not directly treated. (B) Nanoparticles can be used to safely target immunomodulators to the TDLN, activating tumor antigen-loaded dendritic cells to prime T-cell responses that disseminate to attack tumors systemically.
Checkpoint blockade antibodies have also been shown to benefit from localized slow-release delivery at tumors. Peritumoral injection of water-in-oil emulsions (Montanide) containing anti-CTLA-4 allowed a low dose of the checkpoint blockade antibody to effectively drive anti-tumor immunity while providing greatly decreased systemic exposure and reduced liver toxicity compared to traditional systemic anti-CTLA-4 dosing [152]. Recently, Wang et al. developed dissolving microneedles that deposited slow-release dextran nanoparticles into melanoma lesions in the skin [171]. These particles dissolved in response to local tissue glucose mediated by incorporated glucose oxidase, releasing anti-PD-1 in a sustained manner that enhanced survival in treatment of B16F10 tumors. A similar microneedle-based transdermal patch was applied to synergistically co-deliver anti-PD-1 and an inhibitor of the immunosuppressive enzyme indoleamine 2,3-dioxygenase (IDO) to mouse melanoma, achieving effective T cell immunity and reduced immunosuppression in the local site (Figure 3A i–iii) [172].
Figure 3. Strategies for enhancing local delivery of immunotherapy.
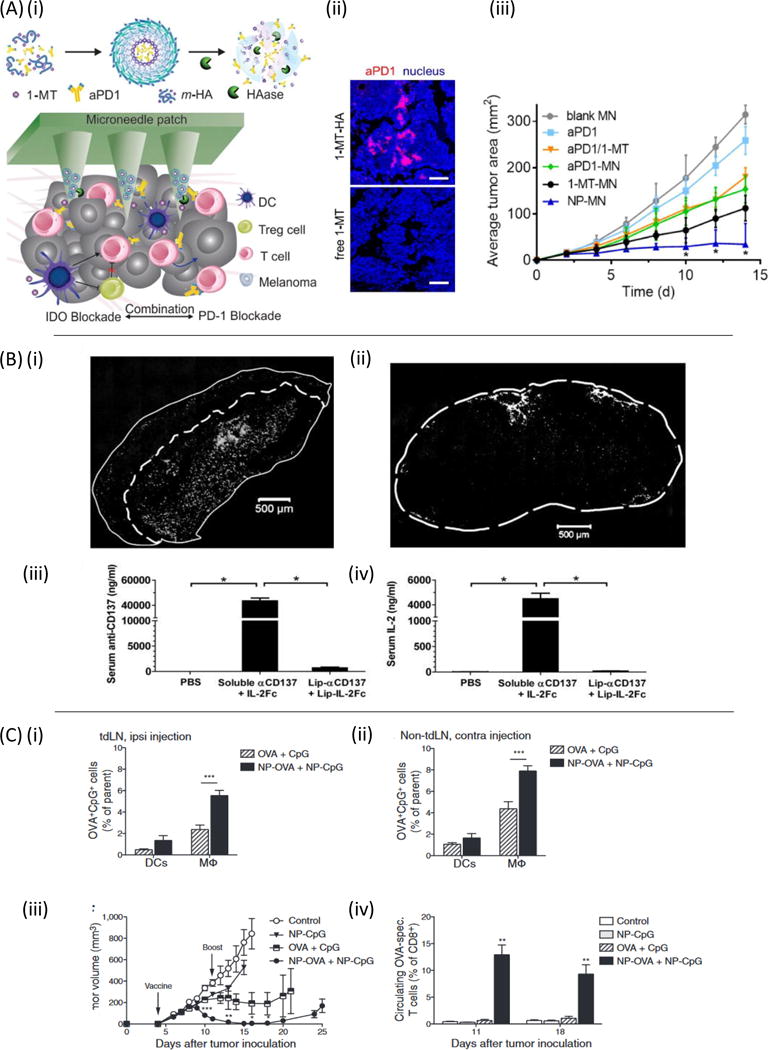
(A)(i) Microneedle-based delivery platform containing self-assembled nanocarriers (m-HA or NP) loaded with IDO inhibitor 1-MT and anti-PD-1. (ii) Immunofluorescence comparison of anti-PD-1/1-MT nanocarrier loaded microneedles and free anti-PD-1/1-MT loaded microneedles in tumors days post drug administration. (iii) B16F10 tumor growth versus time for 1-MT and anti-PD-1 delivered from microneedles (MN) as free drugs or encapsulated within nanocarriers (NP). Reproduced with permission from [172]. (B)(i) Retention of anti-CD137 and IL-2Fc-conjugated liposomes (white) within subcutaneous B16F10 tumors 24 hours after intratumoral injection. (ii) Cryosection of fluorescently labeled immuno-liposomes in the TDLN. (iii-iv) Serum levels of anti-CD137 and IL-2Fc 18 hours following intratumoral injection in soluble or liposome-bound form. Reproduced from [43] with permission. (C) Conjugation of CpG and antigen to PPS nanoparticles improves the efficacy of a TDLN-targeted cancer vaccine. (i) 7 days after E.G7-OVA tumor inoculation, fluorescently labeled nanoparticle-conjugated OVA and CpG were delivered intradermally ipsilateral or contralateral (ii) to the tumor and presence of signal was analyzed 24 hours later in the brachial lymph node. (iii) Tumor volume versus time with nanoparticle delivery of antigen and adjuvant to the tumor draining lymph node. (iv) Resulting antigen specific T-cell frequency following lymph node draining vaccine. Reproduced from [198] with permission.
Intratumoral depots of cytokines or checkpoint blockade antibodies primarily act to enhance the action of a pre-existing immune response against tumors. Another approach is to create intratumoral depots of agents aiming to promote de novo tumor killing, antigen capture and priming of new T-cell responses by tumor-localized immune cells using innate immune stimulating “danger signals”. Imidazoquinolines are a class of small molecule drugs that bind to Toll like receptors (TLRs) 7 and 8, promoting dendritic cell and macrophage activation. Injection of unformulated forms of these compounds leads to rapid dissemination into the blood and systemic cytokine storm signatures [173, 174]. However, acylated forms of these TLR agonists can be formulated into liposomes or oil/water emulsions for localized retention in tissue [173, 175]. Singh et al. demonstrated that an acylated TLR7 agonist delivered in an oil emulsion into solid tumors could both arrest the growth of the treated tumor and prime a disseminated immune response that attacked distal, untreated tumors [175].
Related to these intratumoral treatments, another strategy is to introduce controlled-release biomaterials into a tumor resection site, with the goal of stimulating local immunity to residual tumor cells in the absence of the bulk immunosuppressive factors derived from a large primary tumor: Stephan et al. used alginate gels as a matrix for co-delivery of tumor-specific T-cells and mesoporous silicon microparticles to resection sites of primary breast tumors. The microparticles carried anti-CD3/CD28 stimulators for T-cells and slowly released IL-15, providing localized TCR and cytokine support for the T-cells and leading to elimination of tumor recurrence that was not achieved if T-cells were administered systemically or lacking the matrix of supporting factors [176]. Thus, a number of approaches can be used to augment local immunotherapy while improving safety profiles of immunoregulatory drugs through local controlled release materials.
3.2. Intratumoral gene delivery
An alternative approach to slow-release depots of immunotherapy drugs is to locally produce the agent of interest through gene transfection in the tumor microenvironment. This is perhaps best exemplified by oncolytic viral vectors, viruses which specifically replicate within tumor cells and promote tumor cell death, often in tandem with expression of immunomodulatory proteins delivered in the viral genome. Talimogene Laherparepvec (T-Vec), a granulocyte-macrophage colony-stimulating factor (GM-CSF)-encoding oncolytic herpes simplex virus, is the first example of this strategy to receive FDA approval, in the setting of metastatic melanoma. T-vec is administered by direct intratumoral injection, and was shown to trigger complete regression of both injected and uninjected lesions in 16% of treated patients, suggesting intratumoral administration of an oncolytic virus can effectively cross-prime and amplify antitumor immunity [177, 178]. In this setting, GM-CSF expression from infected tumor cells is thought to promote the chemoattraction and differentiation of dendritic cell precursors to the tumor site, combined with immunogenic tumor cell death leading to enhanced presentation of antigen to prime new T-cell responses against the tumor. Similarly, intratumoral injections of escalating doses of a GM-CSF-expressing vaccinia virus to patients with cutaneous melanoma or non-hepatocellular carcinoma, result in favorable immune responses and tumor regression [179, 180]. These intratumorally-injected viruses only spread locally within the tumor microenvironment due to their large size, and do not spread systemically to distant sites of tumor growth. Thus, the systemic toxicity observed has been infrequent and rapidly resolving [179].
Therapeutic efficacy can be achieved through intratumoral injection of viruses, DNA, or RNA expressing immunoregulatory factors even without direct oncolytic activity of the nucleic acid vector. For example, i.t. injection of an adenovirus or alphavirus expressing IL-12 induced highly localized cytokine expression in tumors, leading to tumor regression and long-term immunity [181, 182]. Intralesional injection of adenovirus encoding human CC chemokine ligand (CCL) 16 inhibited mammary tumor growth and prevented metastatic spread in mice bearing 4T1 mammary adenocarcinoma [183]. Polyplexes of DNA plasmids encoding IL-2 and folate-targeted polyethyleneimine-cyclodextrin were shown to be an effective and safe therapy for melanoma in mice, with an efficacy comparable to that of recombinant adenoviruses expressing IL-2 (rAdv-IL2) [184]. In clinical studies, intratumoral injection of naked DNA encoding cytokines (IL-2, IL-12) has shown some clinical benefits for melanoma patients [185, 186]. A particularly promising approach is to combine localized tumor microenvironment modulation through intralesional gene or oncolytic virus delivery with systemic administration of checkpoint blockade antibodies or other immune modulators with known/acceptable systemic toxicities, enabling an immune response primed by local therapy to be protected as it disseminates to attack untreated lesions [141]. Thus, a variety of vectors and approaches are being explored for local expression of immunostimulatory cytokines and chemokines in tumors.
3.3. Anchored drugs
The examples above are based on releasing immunomodulators at controlled rates within tumor sites, which can only avoid systemic exposure if release rates are in careful balance with drug consumption/degradation rates in the tumor. An alternative is to deliver immunotherapy agents into the tumor microenvironment bound to particles, synthetic matrices, or extracellular components of the tumor microenvironment itself that present these molecules to surrounding immune cells but physically prevent their free diffusion out of the tumor site. An early example of this strategy utilized the injection of lipidated recombinant costimulatory receptors to “paint” tumors with a costimulatory ligand that would promote T-cell recognition of tumor cells [187, 188]. Insertion of the lipid tail of these recombinant proteins into the membranes of tumor cells following intratumoral administration led to retention of these proteins at the tumor site, enabling engineering of tumor cell recognition by immune cells or induction of chemotactic signals to recruit more immune effectors to the tumor microenvironment. Lipid conjugation to DNA oligonucleotides has similarly been used to anchor immunostimulatory CpG DNA (a TLR9 agonist) in tumors, promoting retention in the microenvironment that improved the safety profile and efficacy of intratumoral CpG therapy [189].
A second approach to “anchoring” therapeutics in the tumor is to utilize nanoparticles not for their capacity to home to tumors spontaneously but rather for their tendency to become entrapped in the tumor ECM following intratumoral injection. This approach has been demonstrated with combinations of potent cytokines and innate immune stimulators. As noted in section 2, both IL-2 and anti-CD137 elicit potent antitumor immune responses, but their clinical use is limited by inflammatory toxicities upon systemic administration. These toxicities are further amplified in combination treatment with these drugs, which elicits lethal systemic toxicities even following intratumoral administration at therapeutic doses [43]. To block the systemic dissemination that drives this toxicity, Kwong et al. conjugated anti-CD137 and an engineered IL-2-Fc fusion protein to the surface of PEGylated liposomes. Intratumoral injection of these immunoliposomes restricted the immunotherapeutics to the tumor and tumor-draining lymph nodes, but completely blocked their entry into the systemic circulation by virtue of physical trapping of the liposomes in the tumor extracellular matrix (Figure 3B i–iv). Treatment with these particles eliminated injected primary tumors, elicited systemic antitumor immunity, and eliminated systemic inflammatory toxicity compared to equivalent intratumoral doses of soluble immunotherapy. A similar approach was used to deliver liposome-anchored anti-CD40 and CpG intratumorally, leading to significant tumor growth inhibition and enhanced survival similar to their soluble counterparts after intratumoral injection, while avoiding systemic exposure [149]. Immunotherapy agents may also be intrinsically nanoparticulate in nature, promoting their local retention following intratumoral injection. For example, Lizotte et al. used cowpea mosaic viruses for in situ vaccination of tumors [190]. Although the precise mechanism remains to be defined, these plant virus-derived nanoparticles stimulated intratumoral inflammation that led to a systemic anti-tumor immune response. Thus, particulate immunostimulatory therapies administered intratumorally can significantly decrease or eliminate systemic inflammatory side effects, while retaining the anti-tumor efficacy of free soluble drugs.
3.4. Tumor draining lymph node-targeted drugs
Besides the tumor itself, tumor-draining lymph nodes (TDLNs, or sentinel lymph nodes) are of interest as a target for localized immunomodulatory drugs, because despite evidence for lymphatic dysfunction in some tumor models, TDLNs are known to accumulate antigens from dying tumor cells that could be used to prime de novo anti-tumor T-cell responses. However, tumor-induced dendritic cell dysfunction within TDLNs is a known mechanism of immune evasion [191, 192]. Concentration of innate immune-stimulatory adjuvants within sentinel lymph nodes allows for the activation and maturation of dendritic cells exposed to tumor-associated antigen while preventing cytokine storm-like symptoms which occurs from systemic administration of these agents [193, 194]. Local injections near a tumor can be used to target TDLNs through the local lymphatic tree (Figure 2B). Lymphatic drainage from the interstitial space is highly dependent on molecule or particle size. Small particles less than 30 nm diam. injected intradermally can be found within 50% of lymph node-resident dendritic cells, while larger 100 nm particles only reach 6% of the same population, suggesting that larger particles may be engulfed by phagocytic cells prior to lymph localization and/or impeded in convection through the ECM [195, 196]. Conversely, while small particles can traverse through dense interstitial matrix to directly reach the lymph nodes, they may not be retained within lymphoid organs – as evidenced by significant blood concentrations of 30 nm sized particles 12 hours post-intradermal injection [197]. Therefore, favorable lymphatic localization is reliant upon a balance of drainage from the injection site and capture within local lymph nodes.
Exploiting these principles, Jeanbart et al. synthesized 25nm diam. pluronic-stabilized poly(propylene sulfide) (PPS) nanoparticles capable of concentrating within TDLNs following intradermal injection [198]. These nanoparticles are carried via interstitial flow from the injection site into lymphatic capillary beds and from there to TDLN-resident dendritic cells. Conjugation of the TLR9 agonist CpG DNA to these particles elicited activation of dendritic cells in tumor draining lymph nodes, and primed anti-tumor adaptive immune responses in murine thymoma and melanoma models, while limiting systemic pro-inflammatory responses (Figure 3C i–iv) [198]. CpG has also been targeted to tumor draining lymph nodes through association with cationic gelatin nanoparticles and cationic polyethylenimine (PEI) coated poly(lactic-co-glycolic acid) (PLGA) nanoparticles, alone and in combination with IL-10 siRNA [199–201]. TLR7/8 agonists have also been successfully delivered to tumor draining lymph nodes in nanoparticle format to focus the production of pro-inflammatory cytokines within the site of T cell priming and prevent systemic inflammation [173, 202].
In addition to size-dependent trafficking of drug through lymphatics, molecular targeting to dendritic cells which migrate to and reside within lymph nodes has improved the safety and efficacy of immunotherapies. Cruz et al. describe in detail dendritic cell targeting via DEC205, CD11c, and CD40 dendritic cell receptors [162]. Lymph node targeting by physical, chemical, and molecular properties has been extensively studied for applications in both prophylactic and therapeutic vaccines as vaccination depends on the delivery of antigen and adjuvant to these sites of immune cell education [161].
4. Engineering safer systemic immunotherapies
The administration of immunotherapy agents systemically is desired for treatment of metastatic disease, but faces limitations due to non-tumor-targeted stimulation of leukocytes and other cell types expressing immunoregulatory receptors. An ongoing challenge is the design of strategies to deliver immune-modulating drugs to appropriate immune cells in target tissue sites (e.g., tumors and tumor-draining lymph nodes) while minimizing non-specific systemic stimulation.
4.1. Molecularly-targeted immunotherapy
A common strategy to target therapeutics to tumors employs conjugation of drug to a tumor-antigen specific ligand, antibody, or other engineered binding molecule to achieve local accumulation of the drug following systemic delivery. The fusion of pro-inflammatory cytokines to tumor-associated antigen specific antibodies, known as immunocytokines, represents a common approach to direct the delivery of cytokines to the tumor microenvironment. Cytokines can be fused to either the N or C termini of heavy or light chains of IgG molecules (Figures 4A and B) [203]. In these formats, functions of the antibody such as antigen binding, interaction with Fc receptors, and participation in the complement cascade can be maintained. Alternatively, cytokines can be fused to diabodies or single chain variable fragment (scFv) antibodies to solely maintain the antigen binding property of the antibody (Figure 4C–E). One proposed mechanism of action of immunocytokines is the bridging of tumor cells to leukocytes [204–207]. In the case of IL-2 immunocytokines, the antibody interacts with tumor surface antigens while the IL-2 binds to the IL-2 receptor (IL-2R) on T-cells and NK cells, thereby promoting their proliferation and effector function in the tumor microenvironment. Antibody-dependent cellular cytotoxicity (ADCC) afforded by interaction with the Fc domain of the antibody component has also been shown to be important for the efficacy of immunocytokines [208]. Finally, immunocytokines have longer blood half-lives compared to the parent cytokine molecules due to their increased size and endocytic recycling of the fusion protein through the Fc neonatal receptor [209]. This enhanced half-life enables immunocytokines to be administered at decreased doses, which in some cases enhances their safety profile [210].
Figure 4. Tumor targeting with immunocytokines.
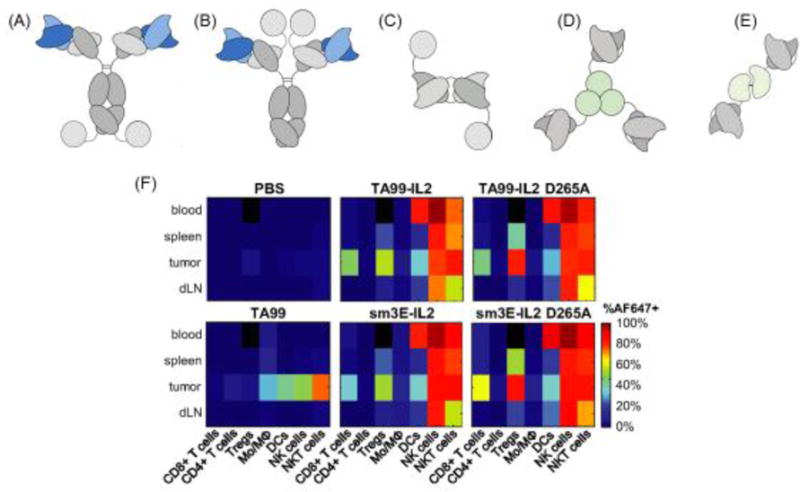
(A–E) Immunocytokine formats based on IL-2, IL-12, and TNF-α. (A) IgG format with IL-2 cytokine covalently linked at the c-terminus of the heavy or (B) light chains. (C) Diabody fusion protein featuring IL-2. (D) Homotrimeric scFv-TNF fusion protein. (E) Heterodimer featuring scFv fused to p40 and p35 subunits of IL-12. Reproduced from [203] with permission. (F) Biodistribution of TA99-IL-2 immunocytokine of format (A) targeting Trp-1 melanoma antigen in mice bearing subcutaneous B16F10 tumors. 24 hours post injection of Alexa Fluor 647-labeled proteins, organs were dissociated into single cell suspensions and stained for immune lineage markers. D265A indicates a mutation in the Fc portion of TA99 abrogating interaction with Fc receptors. The sm3E antibody targeting carcinoembryonic antigen is an irrelevant antibody in model. This irrelevant immunocytokine features similar biodistribution to the melanoma-targeted TA99 immunocytokine. Used with permission from [210].
Antibody-targeted cytokines have shown promising results in preclinical mouse models of cancer. The immunocytokine hu14.18-IL2, a fusion of two molecules of IL-2 with an antibody (14.18) recognizing the GD2 disialoganglioside expressed on the surface of melanomas and neuroblastomas [211], has shown enhanced anti-tumor activity in preclinical melanoma models than equivalent amounts of un-fused 14.18 antibody and IL2 [212]. In a study of 33 melanoma patients, hu14.18-IL2 given as 4-hour intravenous infusion daily for three days resulted in an increase in lymphocyte counts, NK lysis, and ADCC when peripheral blood samples were monitored. Immunocytokines have also been targeted to components of the extracellular matrix overexpressed in the tumor microenvironment. For example, F8-IL2, an immunocytokine based on the F8 antibody in diabody format allows for targeting of IL-2 to the alternatively spliced extra-domain A of fibronectin in the lung tumor microenvironment [213]. F8-IL2 was shown to selectively localize at the tumor site in vivo following intravenous administration, and to mediate tumor growth retardation of non-small cell lung cancer [213]. Other common tumor environment targets include fibronectin extra-domain B, which is highly expressed in tumor vasculature, tenascin C A1 domain, an alternatively spliced form of the tenascin glycoprotein in angiogenic vasculature, and extracellular DNA, found frequently as a result of cell death in the necrotic cores of tumor [211, 214–219].
Clinically, immunocytokines have shown improved efficacy and reduced toxicity compared to soluble pro-inflammatory cytokines; however, the fusion protein format does not abrogate systemic toxicity. Following IL-2 as a model case, hu14.18-IL2 elicits dose limiting toxicities of hypoxia, hypotension, hyperglycemia, and elevated ALT and AST levels [220]. While toxicity was reduced relative to parental IL-2 therapy, patients treated with the hu14.18-IL2 immunocytokine still experienced dose limiting toxicities [221]. Recently, Tzeng et al. demonstrated that a major factor limiting the efficacy and safety of immunocytokines is the dominant role played by binding of the fusion protein to circulating cytokine receptor-expressing leukocytes in the blood, prior to arrival in tumors [210]: A single injection of a fusion of the melanoma-targeting monoclonal antibody (mAb) TA99 with IL-2 labeled 2% of tumor cells, while an equimolar dose of the parental antibody labeled 40% of tumor cells. Further, replacing TA99 with another antibody (sm3E) targeting carcinoembryonic antigen (CEA), an onco-fetal antigen not present in the tumor line used, revealed that antigen specificity was dispensable for the limited tumor targeting observed for the immunocytokine. Loss of tumor targeting by the mAb-IL-2 fusion was due to dominant uptake of the fusion protein by circulating IL-2 receptor-expressing immune cells including DCs, NK cells, NKT cells, CD8+ T-cells, and Tregs, indicating that the cytokine rather than the tumor antigen-specific antibody component of the immunocytokines dictated its in vivo cellular biodistribution (Figure 4F) [210]. From this case study, it is clear that the immunocytokine format can be important for reducing the toxicity of proinflammatory cytokines, but the “targeting” behavior of both components of the fusion must be considered to understand the ultimate biodistribution. An alternative is to administer immunocytokines directly into the tumor microenvironment through intralesional injection, using the tumor-binding antibody component to enhance retention of the cytokine in the local site. This approach has shown promise in melanoma, and allowed high doses of a tumor matrix-binding IL-2 immunocytokine to be administered intratumorally with minimal systemic toxicity [133].
In addition to directing cytokine to the tumor microenvironment, tumor-targeted antibodies have also been used to home innate immune stimulatory danger signals to tumors. In a mouse model of pancreatic cancer, CpG DNA (TLR9 agonist) conjugated to an antibody directed against the tumor antigen mucin-1 reduced tumor burden via activation of Natural Killer (NK) cells and promotion of ADCC [222]. CpG-antibody conjugates have also been formulated to target CD20 on B cells for applications in non-Hodgkin lymphoma and the Her2/neu receptor found in Her2 positive breast cancers [223]. This approach has also been used with other danger signals including polyinosine/polycytosine (pIC, a TLR3 agonist), which was successfully targeted to EGFR- and HER2-overexpressing tumors. Schrand et al. generated bispecific aptamers that bound vascular endothelial growth factor, a product of tumor stroma, and agonized CD137 [224]. Systemic administration of these aptamers elicited tumor regression in multiple tumor models with lower toxicity than untargeted CD137-binding aptamers or anti-CD137 antibodies. Natural ligands for receptors overexpressed by tumor cells can also be used to guide immunomodulators to tumors. For example, a ternary conjugate of epidermal growth factor, melittin (a peptide promoting cytosolic delivery), and PEG coupled to a polyethyleneimine backbone was complexed with pIC to deliver the TLR agonist to EGFR-overexpressing cells, resulting in apoptosis and inflammation in the tumor site [225, 226].
4.2. Nanoparticle delivery of immunotherapy agents to tumors
Targeting toxic compounds to tumors has historically been pursued as a strategy to increase the efficacy and limit systemic toxicity of cancer therapeutics. To this end, much effort has been invested in the development of nanoparticles that passively promote the accumulation of small molecule chemotherapies and targeted drugs in tumors [227, 228]. This nanomedicine approach is based on the concept of the enhanced permeability and retention (EPR) effect, which predicts that particles of a suitable size (large enough to avoid clearance from the blood through the kidneys but small enough to avoid rapid filtration by the reticuloendothelial system) can enter tumors through their leaky vasculature, and accumulate due to defective lymphatic drainage [229, 230]. The efficiency of the EPR effect in heterogeneous human tumors has been debated [231], but nanomedicine-based immunotherapy approaches remain of significant interest because potent immunoregulatory drugs can be active at doses much lower than chemotherapy or anti-tumor targeted drugs (e.g., kinase inhibitors), and do not need to accumulate in every tumor cell for their mode of action.
A number of studies have explored the use of nanoparticles to concentrate immunotherapy drugs in tumors. PLGA-PEG block copolymer nanoparticles ~80 nm in diameter were used to promote accumulation of the kinase inhibitor sunitinib in tumors, which among other activities inhibits the key immunosuppressive transcription factor STAT3 [232]. Nanoparticle delivery of the inhibitor led to a pro-immune remodeling of the tumor microenvironment that synergized with therapeutic vaccination in a murine melanoma model [233]. With similar goals in mind, it has also been shown that delivery of siRNA targeting the immunosuppressive cytokine TGF-β to tumors using lipid/protamine/hyaluronic acid nanoparticles led to knockdown of TGF-β in tumors but not lymph nodes, and reversed accumulation of Tregs and MDSCs in tumors following therapeutic vaccination [234]. To mitigate the systemic side effects of intravenously administered checkpoint blockade antibodies, cationic polymeric nanoparticles have been used to deliver siRNA targeting CTLA-4 to tumors, thus targeting the checkpoint blockade inhibition to tumor infiltrating lymphocytes [235]. Here, siRNA was complexed with a cationic surfactant and encapsulated in poly(ethylene glycol)-block-poly(D,L-lactide) nanoparticles, providing the size requirements for tumor accumulation, and leading to reduced CTLA-4 expression by tumor-infiltrating lymphocytes.
Another advantage of nanoparticle drug delivery is the capacity to co-deliver multiple factors to the same cell/tissue site, and this feature has been demonstrated to be particularly useful for immunotherapy. To exploit the synergy of providing immune-stimulatory signals together with blockade of immunosuppressive pathways, Fahmy and colleagues generated liposome-encapsulated nanogels capable of co-delivering both a small molecule hydrophobic TGF-β inhibitor and water-soluble IL-2 (Figure 5A i) [236]. These nanolipogels enhanced the efficacy of IL-2/TGF-β inhibitor co-therapy both as intratumoral agents and administered i.v. for direct targeting of melanoma metastases, increasing NK cell activity and intratumoral-activated CD8+ T-cell infiltration [236]. Enhanced anti-tumor activity and survival was achieved by these particles without eliciting evidence for systemic toxicity or lung tissue damage (in the case of treating lung metastases) (Figure 5A i–iv). Micellar nanocarriers comprised of PEG and stabilizing Fmoc chemical moieties were used to co-deliver the chemotherapeutic paclitaxel and indoleamine 2,3-dioxygenase (IDO) inhibitor to the tumor microenvironment in both breast cancer and melamona models. Co-delivery resulted in cytotoxic effects in the tumor by paclitaxel and a reversal of tumor immune suppression by the IDO inhibitor yielding a significant survival benefit [237].
Figure 5. Nanoparticles for systemic immunotherapy.
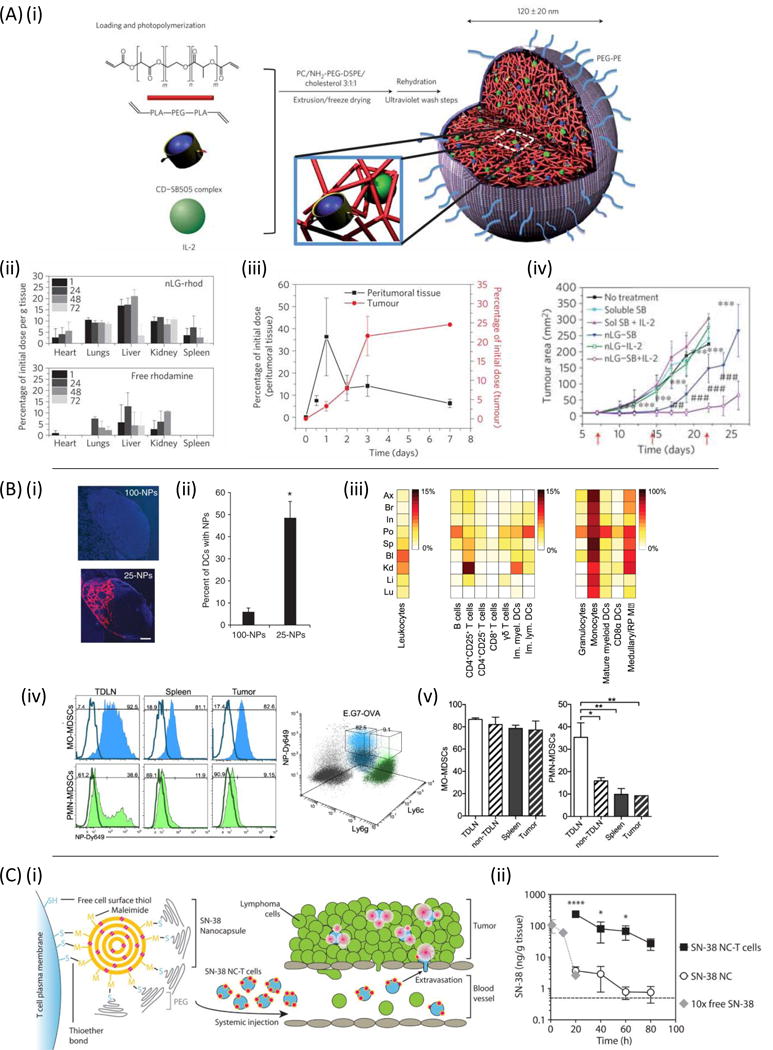
(A)(i) Photoinduced polymerization of nanoscale liposomal polymeric gels forms a core-shell structure of biodegradable polymer (red) with a PEGylated liposomal coating (grey) capable of co-encapsulating IL-2 cytokine (green) and coated TGF-β inhibitor, SB505 (blue). (ii) Biodistribution of rhodamine loaded nanoscale liposomal gels and soluble rhodamine following a single injection into mice with euthanasia time points of 1, 24, 48, and 72 hours post-injection. (iii) Accumulation of rhodamine following peritumoral injection of rhodamine loaded nanoscale liposomal gels in B6 mice. (iv) Subcutaneous tumor area versus time. Used with permission of [236]. (B)(i) Pluronic-stabilized poly(propylene sulfide) (PPS) nanoparticles of 25nm but not 100nm are detectable in lymph nodes 24 hours after injection. (ii) Quantified flow cytometric analysis of CD11c+ dendritic cells that have internalized fluorescently labeled PPS nanoparticles. (iii) Nanoparticle-positive percentages of cells found in organs and lymph nodes 12 hours after intradermal injection. Ax: axillary, Br: brachial, In: inguinal, Po: popliteal; Sp: spleen, Bl: blood, Kd: kidneys, Li: liver, Lu: lungs. (iv) Flow cytometry of monocytic (MO) and polymorphonuclear (PMN) MDSCs from E.G7-OVA tumor bearing mice injected with Dy649-labeled PPS nanoparticles. (v) Quantification of nanoparticle accumulation in MO-MDSCs and PMN-MDSCs of the tumor draining lymph node (TDLN), non-TDLN, spleen, and tumor. Used with permission from [195, 197]. (C)(i) Schematic of T cell functionalization with SN-38 chemotherapy loaded nanocapsules and homing to sites of lymphoma. (ii) Retention of SN-38 chemotherapeutic in tumor draining lymph nodes. Free SN-38 was given at a 10-fold lower dose than in the nanocapsule conditions. Used from [250] with permission.
It has recently become appreciated that nanocarrier accumulation in tumors is often mediated by uptake of particles in phagocytic myeloid cells [197, 238, 239]. These myeloid populations include tumor-associated macrophages and myeloid-derived suppressor cells, which support tumor growth and have numerous mechanisms of promoting immunosuppression [240]. Particle accumulation in these tumor-associated populations provides another strategy for nanomedicine-mediated immunotherapy. For example, sub-100 nm sized pluronic-stabilized poly(propylene sulfide) (PPS) micelles administered intradermally drain through the lymphatic system to tumors, tumor draining lymph nodes, and the spleen (Figure 5B i and ii). These polymeric nanoparticles are efficiently taken up by myeloid-derived suppressor cells in lymphoid organs and tumors (Figure 5B iii–v) [197]. Loading of PPS particles with 6-thioguanine, a purine analog capable of depleting MDSCs, enhanced the efficacy of adoptive T-cell immunotherapy in two mouse models through reprogramming of the immunosuppressive cellular tumor microenvironment [239]. Some nanomaterials may also have intrinsic immunomodulatory effects on myeloid cells: Dextran-coated iron oxide nanoparticles are FDA-approved therapeutics for treatment of iron deficiency [241]. Recently, it has been shown that such nanoparticles, which accumulate in tumors, are capable of repolarizing macrophages from an immunosuppressive M2 phenotype to M1 subtypes, leading to TNF-α production and inducing formation of reactive oxygen species through Fenton reactions with the iron oxide [242–244]. Huang et al. exploited elevated expression of macrophage galactose-type lectin receptor to target galactosylated polyplexes of cationic dextran complexed with alginate, CpG DNA, and anti-sense oligonucleotides against IL-10 and IL-10 receptor to tumor-associated macrophages [245]. To overcome the problem of promiscuous expression of target receptors by myeloid cells in other tissue compartments, Zhe et al. designed PLGA nanoparticles carrying both mannose (to target the mannose receptor expressed broadly by macrophages) and acid-cleavable PEG chains [246]. Shedding of the acid-labile PEG chains in the acidic tumor microenvironment was proposed to promote enhanced accumulation in tumors followed by mannose-mediated targeting to TAMs; enhanced colocalization of particles functionalized with both sheddable PEG and mannose was demonstrated in B16F10 melanomas.
The safety of immune-stimulating nanomedicine can be augmented by following nanoparticle delivery with exogenous localized activation of a therapeutic at the tumor site. Examples of “remote” activation of therapeutics include photodynamic and photothermal therapies, where photosensitive compounds are activated by tissue-penetrating light to produce local radical species or heating that cause damage to tumor tissues. For example, PLGA nanoparticles carrying the phototheramlly-active dye indocyanine green were delivered systemically, followed by localized near-infrared light of a tumor to stimulate localized energy absorption by the dye, subsequent heating, and tumor ablation [247]. This photothermal therapy generated a local depot of tumor antigen, which functioned as an in situ vaccine when combined with co-delivery of a TLR agonist adjuvant and checkpoint blockade inhibitors.
The EPR effect is mediated by particle size, and thus macromolecules can also exhibit EPR-mediated accumulation in tumors. Nektar is developing an immunotherapy prodrug, NKTR-214, which uses sheddable PEG chains linked to IL-2 to promote tumor accumulation and limit early “bolus” activation of systemic IL-2 receptors following infusion of the PEGylated cytokine [248]. NKTR-214 is IL-2 linked to releasable PEG chains conjugated near residues in the IL-2:IL-2Rα binding pocket, which initially prevent interaction of the IL-2 molecule with the high-affinity IL-2 receptor-α chain (CD25). NKTR-214 accumulates in tumor sites and serves an IL-2 reservoir as the PEG chains hydrolyze with half-lives of 20 hours per chain. In the B16F10 mouse melanoma model, NKTR-214 provided a 500-fold greater tumor exposure to IL-2 as well as dose sparing compared to soluble IL-2. This cytokine prodrug was well tolerated in mice and non-human primates, with no vascular leak syndrome detected at maximum tolerated doses, unlike soluble IL-2 [248]. In a related approach, Puskas et al. generated masked IL-2 molecules formed by fusing IL-2 to a soluble form of the IL-2Rα chain through a peptide linker containing a motif recognized by tumor-associated proteases [249]. Similar to PEGylated IL-2, this fusion protein is designed to limit systemic stimulation and localized active IL-2 to the tumor site. This fusion had some anti-tumor activity but was not compared to systemic free IL-2.
4.3. Systemic gene delivery of immunomodulators to tumors
An alternative to physical targeting of immunomodulators to the tumor microenvironment is to deliver genes encoding these factors. As noted in section 3.2, oncolytic viruses that preferentially replicate in tumor cells and co-express immunoregulatory cytokines are under intense study, and the first example of this class of immunotherapies recently received FDA approval [251]. However, expression of immunocytokines by viruses administered systemically has been found to lead to significant toxicity in animal models, due to early transfection events occurring outside of tumors (e.g., in the spleen), which lead to transient but highly toxic systemic cytokine expression outside of tumors [42, 44]. Banaszynski et al. developed a promising strategy to overcome this issue: these investigators fused a destabilizing domain, a mutant of the FK506- and rapamycin-binding protein, to a cytokine of interest [252]. When transfected into cells this intrinsically unstable domain leads to constitutive degradation of the fusion protein by the proteasome. However, in the presence of a small molecule drug (termed Shld-1) that binds to the peptide, the fusion protein is stabilized, secreted, and has bioactivity [252]. Using this approach, it was shown that an oncolytic virus encoding either tumor necrosis factor-α or IL-2 could be safely delivered intravenously, with immunoctyokine expression activated by administration of Shld-1 a few days later, when viral expression is confined to transfected tumors [42, 44]. This approach is an elegant and general strategy that should be compatible with many types of oncolytic viruses and synthetic nucleic acid delivery vectors under study for cancer immunotherapy.
4.4. Immune cells as drug carriers
A variety of cellular therapies based on the injection of autologous immune cells primed for anti-tumor activity are in preclinical and clinical development. Immunotherapies based on the adoptive transfer of autologous tumor-specific T cells (adoptive cell therapy, or ACT) have been shown to be effective in hematologic cancers and a subset of solid tumors [4, 253]. Clinical trials are also underway using adoptively transferred natural killer cells [254]. In preclinical studies, adoptively transferred macrophages have been shown to migrate to and accumulate in tumors [255]. These findings have motivated studies of a second strategy for focusing immunotherapies on specific sites or immune cells, using transferred immune cells themselves as living drug carriers [256]. These approaches, while clinically much more complex to implement than the simple injection of a drug or nanoparticle construct, have a significant advantage over drug delivery mediated by convective diffusion or the EPR effect: while traditional drug targeting is hampered by variation in tumor vasculature, lymphatics, ECM content and interstitial pressures both within a single tumor and between different tumor types [229, 257], cell-mediated targeting of therapy has the potential to overcome these heterogeneities and disseminate drugs throughout tumor tissue through the active processes of cellular extravasation from blood vessels and migration.
A first approach is based on the concept of employing immune cells as active chaperones to deliver immunotherapeutics to tumors based on their intrinsic tissue homing patterns. For example, MDSCs are actively recruited from the circulation by tumors to promote active suppression of anti-tumor immunity [240, 258, 259]. Eisenstein et al. demonstrated that MDSCs loaded with a oncolytic vesicular stomatitis virus (VSV) (using a non-neutralizing anti-VSV antibody to link the viral particles to the cells through MDSC Fc receptors) efficiently home to hepatic lung tumor metastases following adoptive transfer into tumor-bearing mice [258]. MDSC-mediated delivery enabled the VSV vector, which had no therapeutic efficacy as a systemically-administered agent alone, to cure 80% of treated animals. This approach has also been employed with adaptive immune cells: T-cells expressing CD62L and CCR7 naturally home to lymph nodes, a primary site of lymphoma/leukemia accumulation and a common site for metastasis of many solid tumors. Exploiting this natural trafficking pattern, Qiao et al. used naive T-cells with surface-adsorbed VSV to chaperone the oncolytic virus into lymphoid organs harboring metastatic tumor cells. Adoptive transfer of virus-loaded T-cells effectively reduced metastatic tumor burden and vaccinated the affected individual against micrometastatic disease via the release of immunostimulatory tumor antigens in situ [260]. Using synthetic nanoparticles as the therapeutic payload, Huang et al. conjugated T-cells with drug-loaded lipid nanocapsules as vehicles to ferry the chemotherapy agent SN-38 to tumor-ridden lymph nodes in a murine model of disseminated lymphoma. These “backpacked” T-cells significantly re-directed the biodistribution and pharmacokinetic profiles of SN-38 in vivo, enabling anti-tumor efficacy at doses of drug that showed no systemic toxicity. T-cell-mediated delivery conferred a 90-fold greater SN-38 accumulation in lymph nodes comparing to free drug systemically administered at 10-fold higher doses, leading to significantly reduced tumor burden and enhanced survival; while free SN-38 and SN-38-loaded nanocapsules alone were ineffective (Figure 5C i and ii) [250].
A variation on the use of cell-intrinsic homing programs is to load immune cells with particles that permit external guidance, e.g., using magnetic fields: Muthana et al. recently described a strategy to direct cells carrying an oncolytic virus to tumors using magnetic resonance imaging (MRI)-based magnetic guidance: Exploiting their phagocytic capacity, macrophages were loaded ex vivo with magnetic iron oxide nanoparticles, followed by infection with an oncolytic HSV vector. Following i.v. injection, a pulsed magnetic field gradient was applied by MRI to provide a magnetic force promoting accumulation of the iron-loaded macrophages at primary or metastatic tumor sites, leading to a ~6-fold increase in donor cell accumulation throughout tumors. While injection of free oncolytic virus led to only minor inhibition of tumor growth, substantial sustained tumor growth blockade was achieved through magnetic field-mediated targeting of iron oxide-loaded infected macrophages to tumors [261].
A third strategy is to payload immunotherapy agents on tumor-specific T-cells, which instead of relying on an intrinsic tissue-homing program accumulate in tumors following recognition of tumor antigen with their T-cell receptors. In adoptive cell therapy (ACT), the effector function of infused activated T-cells often undergoes rapid decay due to the highly immunosuppressive environment in tumors. Systemic administration of supporting adjuvant immunomodulators together with ACT T-cells has been used to boost the function of infused ACT T-cells, but these simultaneous adjuvant treatments are often accompanied by serious toxicities, especially cytokine storms that have caused patient deaths in multiple clinical trials [262, 263]. As an alternative to systemic adjuvant drug therapy, Stephan et al. conjugated nanoparticles loaded with interleukin cytokines or immunosuppression-blocking drugs to the surface of ACT T-cells ex vivo prior to transfer into tumor-bearing recipients (Figure 6). These T-cell-bound particles provided autocrine drug delivery to the transferred T-cells while minimizing systemic exposure to these inflammatory signals, leading to enhanced efficacy of drug-modified ACT T-cells and reduced systemic toxicity in multiple cancer models [264, 265]. This strategy is currently in commercial/clinical translation by Torque Pharmaceuticals. In a similar vein, Prussian blue nanoparticles (PBNPs) that exhibit photothermal responsiveness were conjugated to Epstein-Barr virus (EBV)-specific CD8+ T-cells, to enable homing of these photothermal agents to EBV-associated tumor malignancies [266]. Wayteck et al. designed a reversible coupling strategy linking siRNA-carrying liposomes to the surface of CD8+ T cells via disulfide bonds [267]. Levels of reductants such as glutathione are low in the circulation, but tumors are thought to be a thiolytic milieu [268], providing the potential for specific release of these cell-bound particles in the tumor microenvironment. To enhance the delivery of viral vectors for immunotherapy, Cole et al. adsorbed viruses encoding IL-12 or Herpes Simplex Virus thymidine kinase (HSVtk) to the cell surface glycosaminoglycans of tumor antigen-specific T-cells. These retroviral vectors could release from T-cell surfaces at the tumor site after in vivo cell transfer, leading to productive infection of tumor cells. This strategy protected the therapeutic viral particles from neutralization in circulation, and allowed virus to specifically accumulate at the sites of antigen expression, providing high levels of selectivity for viral transfer to metastatic tumors [269]. Altogether, these various approaches to surface engineering of T-cells show much promise for enhancing adoptive cell therapy of cancer.
Figure 6. Targeting immunomodulators to tumors or immune cells.
Overview of strategies for targeted delivery of immunotherapy: Molecular or nanoparticle targeting can be directed to circulating immune cells or directly to the tumor; alternatively, nanoparticles can be payloaded ex vivo on immune cells for adoptive transfer.
4.5. Targeting immunotherapy to immune cells
Payloading immunotherapy agents on leukocytes during adoptive cell therapy provides exquisite control over the initial state of the cells and their cargo, but does not allow for repeated or serial stimulation of the transferred cells. An alternative is to target immunotherapy agents directly to immune cells in vivo. Immune cell targeting is motivated not only by the requirement for many immunomodulatory drugs to act directly on leukocytes, but also by the fact that many immune cell populations recirculate through the blood or are present at high densities in the spleen, sites efficiently accessed by intravenous administration that avoid the multiple and heterogeneous barriers to accessing cells deep within tumors. Careful selection of targeting strategies can enable such systemic immune cell targeting to both enhance efficacy and improve the safety profile of immunotherapy drugs.
As already discussed, immunoregulatory cytokines naturally target their cognate receptors on lymphocytes but often stimulate a broad range of immune cell subsets and may stimulate non-immune cells, complicating their safe therapeutic use. One approach to this challenge is to engineer cytokines to confer narrowed target cell specificities, altering the intensity and/or duration of signaling to mitigate toxicity [270]. This strategy has been extensively studied with IL-2: T-cells, NK cells, endothelial cells, and Tregs each have distinct expression patterns for the 3 chains (α, β, and γ) of the IL-2 receptor. Complexation of IL-2 with an anti-IL-2 antibody that blocks the IL-2Rα binding site but leaves the β and γ chain epitopes accessible allowed IL-2 to be redirected away from stimulating endothelial cells and Tregs, focusing its action on effector T-cells and NK cells and greatly reducing vascular leak syndrome in mice [271]. Protein engineering has also been used to redesign IL-2 for enhanced binding to the IL-2R β chain, creating an IL-2 “superkine” exhibiting greatly enhanced stimulation of cytotoxic T-cells, while unexpectedly also achieving lower induction of pulmonary edema in vivo [272]. This latter beneficial side effect might reflect preferential uptake of the superkine by circulating CD8+ T-cells. Other lymphocyte targeting modalities are in development, including aptamers that target surface molecules to directly modulate T-cell function. Berezhnoy et al. generated aptamer-siRNA conjugates where the aptamer binds to CD137 on activated T-cells and delivers siRNA to knock down expression of the key metabolic regulator mTOR, leading to enhanced memory generation during therapeutic cancer vaccination against tumors [273]. Another approach to controllably target lymphocyte subpopulations is to combine molecular targeting with extrinsic therapeutic activation. This approach was impressively demonstrated by Sato et al., who targeted a near infrared-photoactivated radical-generating photodynamic drug to regulatory T-cells via an anti-CD25 targeting antibody [274]. Although this targeting moiety systemically labels Tregs and activated T-cells, within immunosuppressive tumors the only cells expressing high levels of CD25 are Tregs. By applying NIR light to an accessible tumor, depletion of intratumoral Tregs and induction of systemic immunity was demonstrated, with only brief systemic inflammation– in stark contrast to the lethal autoimmunity that ensues when Tregs are systemically depleted [275].
Bispecific antibodies bearing variable domains that bind to a tumor antigen and CD3, a key signaling component of the T-cell receptor complex, have been developed to redirect endogenous T-cells against tumors– enabling in theory T-cells of any antigen specificity to be stimulated on contact with a bispecific-decorated tumor cell. The first such Bispecific T-cell Engager (BiTE), an anti-CD19/anti-CD3 bispecific called Blinatumomab, was recently approved for treatment of acute lymphoblastic leukemia [276]. Similar BiTE molecules are in preclinical and clinical development targeting antigens expressed by a variety of solid tumors, leukemias, and lymphomas [277]. Shen et al. reported on a strategy to create a modular and drug-regulatable form of multispecific T-cell engager based on self-assembled nanorings [278]. In this system, eight copies of a dihydrofolate reductase (DHFR) dimer fused to a single-chain antibody are assembled into a nanoscale ring structure through DFHR binding to bismethotrexate. By self-assembling DHFR dimers fused to an anti-CD3 and an anti-tumor antibody, bi- or multi-specific constructs are obtained that can bind to T-cells and prime them for rapid and high level effector responses on recognition of tumor cells [278].
Nanoparticle drug carriers can also be targeted to adoptively transferred or endogenous lymphocytes. Using PLGA particles as an analog to BiTEs, Schütz et al. showed that nanoparticles bi-functionalized with an antibody targeting a tumor cell surface antigen together with a peptide-MHC complex to target pre-existing memory T-cells could be used to redirect influenza-specific memory T-cells against lymphomas in a humanized mouse model [279]. This approach may have advantages relative to CD3-targeting BiTEs, by allowing specific memory T-cell populations to be targeted that could be selected for known favorable effector profiles (e.g., flu or CMV-specific T-cell populations). Zheng et al. functionalized PEGylated liposomes with IL-2 or antibody fragments targeting an isoform of Thy1 uniquely expressed by adoptively-transferred T-cells, to assess the ability of immunoliposomes to be targeted to ACT T-cells [280]. A single injection of immunoliposomes labeled nearly 100% of circulating target T-cells, with low off-target binding. IL-2-functionalized liposomes served not only to target the vesicles to transferred T-cells but also could be repeatedly injected to directly stimulate the transferred cells following transfer. Frick et al. coupled recombinant IL-2 to the surface of hydroxyethyl starch nanocapsules via copper-free click coupling, and demonstrated specific T-cell targeting in vitro and in vivo by IL-2 receptor-mediated internalization [281]. Use of Thy1-targeting to direct liposomes carrying a small molecule TGF-β inhibitor to ACT T-cells allowed doses of the drug that had no effect when administered systemically as a free drug to enhance the T-cells’ anti-tumor activity [282]. Recently, single-walled carbon nanotubes functionalized with antibodies targeting the GITR receptor (expressed at particularly high levels on intratumoral Tregs) were used to target intratumoral Tregs preferentially over peripheral Tregs in the spleen, relying on a combination of the EPR effect and ligand-mediated targeting [283]. These materials might serve as the basis for targeting specific depletion of intratumoral Tregs, with the goal of achieving therapeutic effects similar to anti-CTLA-4 therapy without systemic autoimmunity.
Immunotherapies have also been targeted to innate immune cells to enhance anti-tumor activity. The strategy of piggybacking oncolytic viruses on immune cells described in the previous section has recently been extended to direct in vivo loading of antibody-opsonized oncolytic viruses onto myeloid cells that home to tumors. Monocytes and macrophages mobilized from the bone marrow by a systemic injection of GM-CSF (an approved cytokine therapy [284, 285]) were found to bind opsonized virus injected after the cytokine, and subsequently homed into tumors and transfer virus to the tumor cells [286]. Neutrophils can also home to tumors in some settings. Chu et al. targeted photodynamic drugs and other therapeutics to circulating neutrophils using nanoparticles formed from denatured albumin. By administering a monoclonal antibody against gp75 antigen expressed in melanoma tumors, neutrophils were recruited to the tumors, bringing the albumin nanoparticle-associated drugs to the tumor site [287]. Smith et al. recently reported an unexpected strong tropism of i.v.-injected single-walled carbon nanotubes for circulating Ly-6Chi cells, which subsequently transported the internalized SWNTs to tumors [288]. This tumor homing by monocytic cells was further enhanced when SWNTs were functionalized with the integrin-binding peptide RGD, which perhaps may reflect selective targeting of the SWNTs to cells primed for tumor homing. Such an innate cell-selective nanomaterial could be well suited to delivery of immunomodulatory drugs to the tumor microenvironment. Exploiting the fact that tumors attract myeloid-derived suppressor cells to promote immunosuppression, Kullberg et al. developed a method to target MDSCs using endogenous activated complement C3 of tumor bearing animals [289]. PEGylated liposomes bearing an orthopyridyl disulfide were injected intravenously, which will react with the exposed sulfhydryl of activated C3, leading to binding and uptake by C3 receptor-expressing granulocytic MDSCs present at high levels in the blood of tumor-bearing animals and patients [289]. Use of an endogenous molecule to mediate targeting avoids issues of immunogenicity and stability that can occur using antibodies and other engineered targeting molecules. These examples illustrate the potential for fine-tuned targeting of circulating immune cell populations as a potentially very powerful approach to obtain potent anti-tumor activity of immunotherapeutics while mitigating immunotoxiciy.
5. Conclusions and future perspectives
As summarized here, many approaches founded in rational controlled drug delivery technologies offer promise for simultaneously enhancing the efficacy and safety of immunotherapies for cancer. This includes strategies based on protein and genetic engineering, controlled drug delivery materials, and nanomedicine. As with any cancer therapy, a key to achieving optimal results will be finding the right marriage of these approaches to achieve robust activity of immunoregulatory drugs while avoiding on-target/off-tumor toxicities. There remain a number of challenges/opportunities in the field- two examples being limitations of animal models and further improving on strategies to control the targeting of immunotherapy drugs. For example, a limitation of current animal models is their limited capacity to reliably predict the toxicity profile that will be observed in human patients. Mice are the predominant preclinical species used to evaluate candidate cancer immunotherapies, and while mouse models have been successfully used to guide the development of all of the currently approved immunotherapy drugs, the immune toxicity profiles of many agents in mice are clearly distinct from humans. For example, anti-CTLA-4 in mice does not elicit the same level of gastrointestinal toxicity observed in humans, and mice tolerate a number of immune agonist compounds better than humans. One solution to this issue could be increased use of humanized mouse models to study immunotherapy, but humanized mouse models do not yet fully recapitulate the complete range of functions of an intact healthy immune system [290] and have largely been used only for testing adoptive cell therapy treatments where engineered T-cells are infused into animals lacking any other human immune system components. Other groups are seeking to improve the predictive power of murine immunotherapy models by using partial regulatory T-cell depletion to make animals more susceptible to autoimmune side effects observed in patients [291]. Improvements in animal models will be a key part for future success in the development of treatments that maximize the potential of immunotherapy.
A second challenge/opportunity is the potential for immunotherapies to be enhanced by leveraging promising strategies developed in other areas of drug delivery to the problems of immune-modulatory drugs. As one example, the systemic exposure and/or limited efficacy often observed following local bolus injection of immunotherapies is a characteristic of many biologic therapeutics that have minimal interactions with the extracellular matrix, which exhibit rapid tissue diffusion and/or proteolysis. However, other signaling proteins such as cytokines and chemokines have the ability to interact with binding sites presented by ECM proteins and cell surface proteoglycans, providing a means to sequester or retain these factors following secretion in tissue [292]. Biomaterial matrices are being developed to mimic these key characteristics of the ECM, including those that are derived from naturally occurring molecules and those that recapitulate key motifs of biomolecules. For instance, the 12th–14th fibronectin type III region (FN III12-14) can bind a variety of cytokines with high affinity, without blocking their activity [293]. Matrix-binding variants of FN III12–14 have been designed as universal binding anchors for cytokine retention in matrices for the application of tissue engineering and regenerative medicine, improving efficacy, safety, and cost-effectiveness for growth factor delivery [292] – such approaches should be of interest for modulating local immunotherapies. Conversely, immunotherapy agents are being designed to bind efficiently to tumor microenvironment ECM components to retain therapeutics locally, as demonstrated recently for engineered growth factors designed to be retained locally in tissue regeneration sites [294]. The advent of controlled delivery properties in a single molecular entity is attractive for clinical translation.
Altogether, there is reason to hope that within the foreseeable future, immunotherapy-based treatments will lead to the transition of cancer from a fatal diagnosis to a chronic, manageable disease, if not curable condition for a large number of patients. Strategies as discussed in this review will likely play a major role in helping the field attain this goal of enhanced survival with a high quality of life for cancer patients worldwide.
Acknowledgments
DJI is an investigator of the Howard Hughes Medical Institute. This work was supported in part by the Ragon Institute of MIT, MGH, and Harvard, the NIH (awards CA206218, CA172164, and CA174795), and Stand Up 2 Cancer.
References
- 1.Waldmann TA. Immunotherapy: past, present and future. Nat Med. 2003;9:269–277. doi: 10.1038/nm0303-269. [DOI] [PubMed] [Google Scholar]
- 2.Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. 2016;13:394. doi: 10.1038/nrclinonc.2016.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 4.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62–68. doi: 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16:566–581. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv324. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 8.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, Morton KE, Laurencot CM, Steinberg SM, White DE, Dudley ME. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lebbe C, Weber JS, Maio M, Neyns B, Harmankaya K, Hamid O, O’Day SJ, Konto C, Cykowski L, McHenry MB, Wolchok JD. Survival follow-up and ipilimumab retreatment of patients with advanced melanoma who received ipilimumab in prior phase II studies. Ann Oncol. 2014;25:2277–2284. doi: 10.1093/annonc/mdu441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, Izzeddine H, Marabelle A, Champiat S, Berdelou A, Lanoy E, Texier M, Libenciuc C, Eggermont AM, Soria JC, Mateus C, Robert C. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol. 2016;13:473–486. doi: 10.1038/nrclinonc.2016.58. [DOI] [PubMed] [Google Scholar]
- 13.Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. 2013;38:13–25. doi: 10.1016/j.immuni.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, Paradise C, Kunkel L, Rosenberg SA. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 15.Konrad MW, Hemstreet G, Hersh EM, Mansell PW, Mertelsmann R, Kolitz JE, Bradley EC. Pharmacokinetics of recombinant interleukin 2 in humans. Cancer Res. 1990;50:2009–2017. [PubMed] [Google Scholar]
- 16.Alwan LM, Grossmann K, Sageser D, Van Atta J, Agarwal N, Gilreath JA. Comparison of acute toxicity and mortality after two different dosing regimens of high-dose interleukin-2 for patients with metastatic melanoma. Target Oncol. 2014;9:63–71. doi: 10.1007/s11523-013-0276-7. [DOI] [PubMed] [Google Scholar]
- 17.Buchbinder EI, Gunturi A, Perritt J, Dutcher J, Aung S, Kaufman HL, Ernstoff MS, Miletello GP, Curti BD, Daniels GA, Patel SP, Kirkwood JM, Hallmeyer S, Clark JI, Gonzalez R, Richart JM, Lutzky J, Morse MA, Sullivan RJ, McDermott DF. A retrospective analysis of High-Dose Interleukin-2 (HD IL-2) following Ipilimumab in metastatic melanoma. J Immunother Cancer. 2016;4:52. doi: 10.1186/s40425-016-0155-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lam ET, Wong MK, Agarwal N, Redman BG, Logan T, Gao D, Flaig TW, Lewis K, Poust J, Monk P, Jarkowski A, Sendilnathan A, Bolden M, Kuzel TM, Olencki T. Retrospective analysis of the safety and efficacy of high-dose interleukin-2 after prior tyrosine kinase inhibitor therapy in patients with advanced renal cell carcinoma. J Immunother. 2014;37:360–365. doi: 10.1097/CJI.0000000000000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Damle NK, Doyle LV, Bender JR, Bradley EC. Interleukin 2-activated human lymphocytes exhibit enhanced adhesion to normal vascular endothelial cells and cause their lysis. J Immunol. 1987;138:1779–1785. [PubMed] [Google Scholar]
- 20.Amador JF, Vazquez AM, Cabrera L, Barral AM, Gendelman R, Jondal M. Toxic effects of interleukin-2-activated lymphocytes on vascular endothelial cells. Nat Immun Cell Growth Regul. 1991;10:207–215. [PubMed] [Google Scholar]
- 21.Aronson FR, Libby P, Brandon EP, Janicka MW, Mier JW. IL-2 rapidly induces natural killer cell adhesion to human endothelial cells. A potential mechanism for endothelial injury. J Immunol. 1988;141:158–163. [PubMed] [Google Scholar]
- 22.Krieg C, Letourneau S, Pantaleo G, Boyman O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc Natl Acad Sci U S A. 2010;107:11906–11911. doi: 10.1073/pnas.1002569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cotran RS, Pober JS, Gimbrone MA, Jr, Springer TA, Wiebke EA, Gaspari AA, Rosenberg SA, Lotze MT. Endothelial activation during interleukin 2 immunotherapy. A possible mechanism for the vascular leak syndrome. J Immunol. 1988;140:1883–1888. [PubMed] [Google Scholar]
- 24.Pober JS. Warner-Lambert/Parke-Davis award lecture. Cytokine-mediated activation of vascular endothelium. Physiology and pathology. Am J Pathol. 1988;133:426–433. [PMC free article] [PubMed] [Google Scholar]
- 25.Grimm EA, Mazumder A, Zhang HZ, Rosenberg SA. Lymphokine-activated killer cell phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes. J Exp Med. 1982;155:1823–1841. doi: 10.1084/jem.155.6.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grimm EA, Robb RJ, Roth JA, Neckers LM, Lachman LB, Wilson DJ, Rosenberg SA. Lymphokine-activated killer cell phenomenon. III. Evidence that IL-2 is sufficient for direct activation of peripheral blood lymphocytes into lymphokine-activated killer cells. J Exp Med. 1983;158:1356–1361. doi: 10.1084/jem.158.4.1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grimm EA, Wilson DJ. The human lymphokine-activated killer cell system. V. Purified recombinant interleukin 2 activates cytotoxic lymphocytes which lyse both natural killer-resistant autologous and allogeneic tumors and trinitrophenyl-modified autologous peripheral blood lymphocytes. Cell Immunol. 1985;94:568–578. doi: 10.1016/0008-8749(85)90280-1. [DOI] [PubMed] [Google Scholar]
- 28.Miltenburg AM, Meijer-Paape ME, Daha MR, Paul LC. Endothelial cell lysis induced by lymphokine-activated human peripheral blood mononuclear cells. Eur J Immunol. 1987;17:1383–1386. doi: 10.1002/eji.1830170926. [DOI] [PubMed] [Google Scholar]
- 29.Montesano R, Orci L, Vassalli P. Human endothelial cell cultures: phenotypic modulation by leukocyte interleukins. J Cell Physiol. 1985;122:424–434. doi: 10.1002/jcp.1041220313. [DOI] [PubMed] [Google Scholar]
- 30.Orucevic A, Lala PK. NG-nitro-L-arginine methyl ester, an inhibitor of nitric oxide synthesis, ameliorates interleukin 2-induced capillary leakage and reduces tumour growth in adenocarcinoma-bearing mice. Br J Cancer. 1996;73:189–196. doi: 10.1038/bjc.1996.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baluna R, Vitetta ES. Vascular leak syndrome: a side effect of immunotherapy. Immunopharmacology. 1997;37:117–132. doi: 10.1016/s0162-3109(97)00041-6. [DOI] [PubMed] [Google Scholar]
- 32.Stevens P, Piazza DE. Interleukin-2 increases the oxidative activity and induces migration of murine polymorphonuclear leukocytes in vivo. Int J Immunopharmacol. 1990;12:605–611. doi: 10.1016/0192-0561(90)90097-7. [DOI] [PubMed] [Google Scholar]
- 33.Suschek C, Rothe H, Fehsel K, Enczmann J, Kolb-Bachofen V. Induction of a macrophage-like nitric oxide synthase in cultured rat aortic endothelial cells. IL-1 beta-mediated induction regulated by tumor necrosis factor-alpha and IFN-gamma. J Immunol. 1993;151:3283–3291. [PubMed] [Google Scholar]
- 34.Mulvin DW, Grosso MA, Kruse CA, Howard RB, Repine JE, Johnston MR. The role of toxic oxygen metabolites in interleukin-2-induced vascular leak syndrome. Curr Surg. 1989;46:396–398. [PubMed] [Google Scholar]
- 35.Anderson TD, Hayes TJ, Gately MK, Bontempo JM, Stern LL, Truitt GA. Toxicity of human recombinant interleukin-2 in the mouse is mediated by interleukin-activated lymphocytes. Separation of efficacy and toxicity by selective lymphocyte subset depletion. Lab Invest. 1988;59:598–612. [PubMed] [Google Scholar]
- 36.Gately MK, Anderson TD, Hayes TJ. Role of asialo-GM1-positive lymphoid cells in mediating the toxic effects of recombinant IL-2 in mice. J Immunol. 1988;141:189–200. [PubMed] [Google Scholar]
- 37.Elliott MJ, Finn AH. Interaction between neutrophils and endothelium. Ann Thorac Surg. 1993;56:1503–1508. doi: 10.1016/0003-4975(93)90741-y. [DOI] [PubMed] [Google Scholar]
- 38.Edwards MJ, Miller FN, Sims DE, Abney DL, Schuschke DA, Corey TS. Interleukin 2 acutely induces platelet and neutrophil-endothelial adherence and macromolecular leakage. Cancer Res. 1992;52:3425–3431. [PubMed] [Google Scholar]
- 39.Edwards MJ, Heniford BT, Miller FN. Mast cell degranulation inhibits IL-2-induced microvascular protein leakage. J Surg Res. 1992;52:429–435. doi: 10.1016/0022-4804(92)90307-l. [DOI] [PubMed] [Google Scholar]
- 40.Schwartz RN, Stover L, Dutcher J. Managing toxicities of high-dose interleukin-2. Oncology (Williston Park) 2002;16:11–20. [PubMed] [Google Scholar]
- 41.McDermott DF. Update on the application of interleukin-2 in the treatment of renal cell carcinoma. Clin Cancer Res. 2007;13:716s–720s. doi: 10.1158/1078-0432.CCR-06-1872. [DOI] [PubMed] [Google Scholar]
- 42.Chen H, Sampath P, Hou W, Thorne SH. Regulating cytokine function enhances safety and activity of genetic cancer therapies. Mol Ther. 2013;21:167–174. doi: 10.1038/mt.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kwong B, Gai SA, Elkhader J, Wittrup KD, Irvine DJ. Localized immunotherapy via liposome-anchored Anti-CD137 + IL-2 prevents lethal toxicity and elicits local and systemic antitumor immunity. Cancer Res. 2013;73:1547–1558. doi: 10.1158/0008-5472.CAN-12-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banaszynski LA, Sellmyer MA, Contag CH, Wandless TJ, Thorne SH. Chemical control of protein stability and function in living mice. Nat Med. 2008;14:1123–1127. doi: 10.1038/nm.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 46.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 47.Sutmuller RP, Van Duivenvoorde LM, Van Elsas A, Schumacher TN, Wildenberg ME, Allison JP, Toes RE, Offringa R, Melief CJ. Synergism of cytotoxic T lymphocyte–associated antigen 4 blockade and depletion of CD25+ regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. The Journal of Experimental Medicine. 2001;194:823–832. doi: 10.1084/jem.194.6.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti–CTLA-4 therapy against melanoma. The Journal of Experimental Medicine. 2013;210:1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 50.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJM, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. New England Journal of Medicine. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lipson EJ, Sharfman WH, Drake CG, Wollner I, Taube JM, Anders RA, Xu H, Yao S, Pons A, Chen L, Pardoll DM, Brahmer JR, Topalian SL. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res. 2013;19:462–468. doi: 10.1158/1078-0432.CCR-12-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen LP, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu HY, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. New England Journal of Medicine. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and Activity of Anti-PD-L1 Antibody in Patients with Advanced Cancer. The New England journal of medicine. 2012 doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, Waterfield W, Schadendorf D, Smylie M, Guthrie TJ, Grob JJ, Chesney J, Chin K, Chen K, Hoos A, O’Day SJ, Lebbe C. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. The Lancet Oncology. 2010;11:155–164. doi: 10.1016/S1470-2045(09)70334-1. [DOI] [PubMed] [Google Scholar]
- 57.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, Blank CU, Hamid O, Mateus C, Shapira-Frommer R, Kosh M, Zhou H, Ibrahim N, Ebbinghaus S, Ribas A. Pembrolizumab versus Ipilimumab in Advanced Melanoma. New England Journal of Medicine. 2015;372:2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 58.Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, Wilson NS, Dranoff G, Brogdon JL. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med. 2013;210:1685–1693. doi: 10.1084/jem.20130573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ. Anti-CTLA-4 Antibodies of IgG2a Isotype Enhance Antitumor Activity through Reduction of Intratumoral Regulatory T Cells. Cancer immunology research. 2013;1:32–42. doi: 10.1158/2326-6066.CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- 60.Lan RY, Mackay IR, Gershwin ME. Regulatory T cells in the prevention of mucosal inflammatory diseases: Patrolling the border. Journal of Autoimmunity. 2009;29:272–280. doi: 10.1016/j.jaut.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Romain PES, Salama AKS, Ferguson NL, Burbridge RA. Ipilimumab-associated lymphocytic colitis: a case report. Translational gastrointestinal cancer. 2015;5:44–46. [Google Scholar]
- 62.Weber J, Thompson JA, Hamid O, Minor D, Amin A, Ron I, Ridolfi R, Assi H, Maraveyas A, Berman D, Siegel J, O’Day SJ, Randomized A. Double-Blind, Placebo-Controlled, Phase II Study Comparing the Tolerability and Efficacy of Ipilimumab Administered with or without Prophylactic Budesonide in Patients with Unresectable Stage III or IV Melanoma. Clinical Cancer Research. 2009;15:5591–5598. doi: 10.1158/1078-0432.CCR-09-1024. [DOI] [PubMed] [Google Scholar]
- 63.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. The New England journal of medicine. 2012 doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH, Lao CD, Linette GP, Thomas L, Lorigan P, Grossmann KF, Hassel JC, Maio M, Sznol M, Ascierto PA, Mohr P, Chmielowski B, Bryce A, Svane IM, Grob J, Krackhardt AM, Horak C, Lambert A, Yang AS, Larkin J. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. The Lancet Oncology. 2015;16:375–384. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 65.Deng J, Le Mercier I, Kuta A, Noelle RJ. A New VISTA on combination therapy for negative checkpoint regulator blockade. Journal for ImmunoTherapy of Cancer. 2016;4 doi: 10.1186/s40425-016-0190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sakuishi K, Jayaraman P, Behar SM, Anderson AC, Kuchroo VK. Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol. 2011;32:345–349. doi: 10.1016/j.it.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, Kirkwood JM, Olive D, Kuchroo V, Zarour HM. CD8+ T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Research. 2012;72:887–896. doi: 10.1158/0008-5472.CAN-11-2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano D, Vogel P, Liu CL, Tangsombatvisit S, Grosso JF, Netto G, Smeltzer MP, Chaux A, Utz PJ, Workman CJ, Pardoll DM, Korman AJ, Drake CG, Vignali DA. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T cell function to promote tumoral immune escape. Cancer Research. 2012;72:917–927. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proceedings of the National Academy of Sciences. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, Shaheen M, Ernstoff MS, Minor D, Salama AK, Taylor M, Ott PA, Rollin LM, Horak C, Gagnier P, Wolchok JD, Hodi FS. Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma. New England Journal of Medicine. 2015;372:2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sznol M, Kluger HM, Callahan MK, Postow MA, Gordon RA, Segal NH, Rizvi NA, Lesokhin AM, Atkins MB, Kirkwood JM, Burke MM, Ralabate AL, Rivera AL, Kronenberg A, Agunwamba B, Feely W, Hong Q, Krishnan S, Gupta AK, Wolchok JD. Survival, response duration, and activity by BRAF mutation (MT) status of nivolumab (NIVO, anti-PD-1, BMS-936558, ONO-4538) and ipilimumab (IPI) concurrent therapy in advanced melanoma (MEL) Journal of Clinical Oncology. 2014;32 [Google Scholar]
- 72.Hofmann L, Forschner A, Loquai C, Goldinger SM, Zimmer L, Ugurel S, Schmidgen MI, Gutzmer R, Utikal JS, Goppner D, Hassel JC, Meier F, Tietze JK, Thomas I, Weishaupt C, Leverkus M, Wahl R, Dietrich U, Garbe C, Kirchberger MC, Eingentler T, Berking C, Gesierich A, Krackhardt AM, Schadendorf D, Schuler G, Dummer R, Heinzerling LM. Cutaneous, gastrointestinal, hepatic, endocrine, and renal side-effects of anti-PD-1 therapy. European Journal of Cancer. 2016;60:190–209. doi: 10.1016/j.ejca.2016.02.025. [DOI] [PubMed] [Google Scholar]
- 73.Antonia S, Goldberg SB, Balmanoukian A, Chaft JE, Sanborn RE, Gupta A, Narwal R, Steele K, Gu Y, Karakunnel JJ, Rizvi NA. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: a multicentre, phase 1b study. Lancet Oncol. 2016;17:299–308. doi: 10.1016/S1470-2045(15)00544-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Antonia SJ, Lopez-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, Jager D, Pietanza MC, Le DT, de Braud F, Morse MA, Ascierto PA, Horn L, Amin A, Pillai RN, Evans J, Chau I, Bono P, Atmaca A, Sharma P, Harbison CT, Lin CS, Christensen O, Calvo E. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016;17:883–895. doi: 10.1016/S1470-2045(16)30098-5. [DOI] [PubMed] [Google Scholar]
- 75.Hellmann MD, Rizvi NA, Goldman JW, Gettinger SN, Borghaei H, Brahmer JR, Ready NE, Gerber DE, Chow LQ, Juergens RA, Shepherd FA, Laurie SA, Geese WJ, Agrawal S, Young TC, Li X, Antonia SJ. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 2017;18:31–41. doi: 10.1016/S1470-2045(16)30624-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nirschl CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clinical Cancer Research. 2013;19:4917–4924. doi: 10.1158/1078-0432.CCR-12-1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li F, Ravetch JV. Inhibitory Fcγ receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science. 2011;333:1030–1034. doi: 10.1126/science.1206954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.White AL, Chan HT Claude, Roghanian A, French RR, Mockridge CI, Tutt AL, Dixon SV, Ajona D, Verbeek JS, Al-Shamkhani A, Cragg MS, Beers SA, Glennie MJ. Interaction with FcγRIIB Is Critical for the Agonistic Activity of Anti-CD40 Monoclonal Antibody. The Journal of Immunology. 2011;187:1754–1763. doi: 10.4049/jimmunol.1101135. [DOI] [PubMed] [Google Scholar]
- 80.Vitale LA, He L, Thomas LJ, Widger J, Weidlick J, Crocker A, O’Neill T, Storey J, Glennie MJ, Grote DM, Ansell SM, Marsh H, Keler T. Development of a Human Monoclonal Antibody for Potential Therapy of CD27-Expressing Lymphoma and Leukemia. Clinical Cancer Research. 2012;18:3812–3821. doi: 10.1158/1078-0432.CCR-11-3308. [DOI] [PubMed] [Google Scholar]
- 81.Morris NP, Peters C, Montler R, Hu HM, Curti BD, Urba WJ, Weinberg AD. Development and characterization of recombinant human Fc:OX40L fusion protein linked via a coiled-coil trimerization domain. Molecular Immunology. 2007;44:3112–3121. doi: 10.1016/j.molimm.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bartkowiak T, Curran MA. 4-1BB Agonists: Multi-Potent Potentiators of Tumor Immunity. Front Oncol. 2015;5:117. doi: 10.3389/fonc.2015.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Teijeira A, Palazon A, Garasa S, Marre D, Auba C, Rogel A, Murillo O, Martinez-Forero I, Lang F, Melero I, Rouzaut A. CD137 on inflamed lymphatic endothelial cells enhances CCL21-guided migration of dendritic cells. FASEB J. 2012;26:3380–3392. doi: 10.1096/fj.11-201061. [DOI] [PubMed] [Google Scholar]
- 84.Palazón A, Teijeira A, Martínez-Forero I, Hervás-Stubbs S, Roncal C, Peñuelas I, Dubrot J, Morales-Kastresana A, Pérez-Gracia JL, Ochoa MC, Ochoa-Callejero L, Martínez A, Luque A, Dinchuk J, Rouzaut A, Jure-Kunkel M, Melero I. Agonist anti-CD137 mAb act on tumor endothelial cells to enhance recruitment of activated T lymphocytes. Cancer Research. 2011;71:801–811. doi: 10.1158/0008-5472.CAN-10-1733. [DOI] [PubMed] [Google Scholar]
- 85.Drenkard D, Becke FM, Langstein J, Spruss T, Kunz-Schughart LA, Tan TE, Lim YC, Schwarz H. CD137 is expressed on blood vessel walls at sites of inflammation and enhances monocyte migratory activity. FASEB J. 2007;21:456–463. doi: 10.1096/fj.05-4739com. [DOI] [PubMed] [Google Scholar]
- 86.Hernandez-Chacon JA, Li Y, Wu RC, Bernatchez C, Wang Y, Weber JS, Hwu P, Radvanyi LG. Costimulation through the CD137/4-1BB pathway protects human melanoma tumor-infiltrating lymphocytes from activation-induced cell death and enhances antitumor effector function. J Immunother. 2011;34:236–250. doi: 10.1097/CJI.0b013e318209e7ec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lin GH, Liu Y, Ambagala T, Kwon BS, Ohashi PS, Watts TH. Evaluating the cellular targets of anti-4-1BB agonist antibody during immunotherapy of a pre-established tumor in mice. PLoS One. 2010;5:e11003. doi: 10.1371/journal.pone.0011003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Narazaki H, Zhu Y, Luo L, Zhu G, Chen L. CD137 agonist antibody prevents cancer recurrence: contribution of CD137 on both hematopoietic and nonhematopoietic cells. Blood. 2010;115:1941–1948. doi: 10.1182/blood-2008-12-192591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tsuchihashi Z, Hu B, Girit E. Assessment of tissue biomarkers following anti-CD137 treatment in the EMT-6 mouse tumor model. Proceedings of the American Association for Cancer Research. 2006;47:5556. [Google Scholar]
- 90.Ascierto PA, Simeone E, Sznol M, Fu YX, Melero I. Clinical Experiences With Anti-CD137 and Anti-PD1 Therapeutic Antibodies. Seminars in Oncology. 2010;37:508–516. doi: 10.1053/j.seminoncol.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 91.Ito F, Li Q, Shreiner AB, Okuyama R, Jure-Kunkel MN, Teitz-Tennenbaum S, Chang AE. Anti-CD137 Monoclonal Antibody Administration Augments the Antitumor Efficacy of Dendritic Cell-Based Vaccines. Cancer Research. 2004;64:8411–8419. doi: 10.1158/0008-5472.CAN-04-0590. [DOI] [PubMed] [Google Scholar]
- 92.Kim YH, Choi BK, Kim KH, Kang SW, Kwon BS. Combination Therapy with Cisplatin and Anti–4-1BB: Synergistic Anticancer Effects and Amelioration of Cisplatin-Induced Nephrotoxicity. Cancer Research. 2008;68:7264–7269. doi: 10.1158/0008-5472.CAN-08-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wilcox RA. Ligation of CD137 receptor prevents and reverses established anergy of CD8+ cytolytic T lymphocytes in vivo. Blood. 2004;103:177–184. doi: 10.1182/blood-2003-06-2184. [DOI] [PubMed] [Google Scholar]
- 94.Dubrot J, Milheiro F, Alfaro C, Palazón A, Martínez-Forero I, Perez-Gracia JL, Morales-Kastresana A, Romero-Trevejo JL, Ochoa MC, Hervás-Stubbs S, Prieto J, Jure-Kunkel M, Chen L, Melero I. Treatment with anti-CD137 mAbs causes intense accumulations of liver T cells without selective antitumor immunotherapeutic effects in this organ. Cancer Immunology, Immunotherapy. 2010;59:1223–1233. doi: 10.1007/s00262-010-0846-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sznol M, Hodi FS, Margolin K, McDermott DF, Ernstoff MS, Kirkwood JM, Wojtaszek C, Feltquate D, Logan T. Phase I study of BMS-663513, a fully human anti-CD137 agonist monoclonal antibody, in patients (pts) with advanced cancer (CA) Journal of Clinical Oncology. 2008;26:3007. [Google Scholar]
- 96.Niu L, Strahotin S, Hewes B, Zhang B, Zhang Y, Archer D, Spencer T, Dillehay D, Kwon B, Chen L, Vella AT, Mittler RS. Cytokine-mediated disruption of lymphocyte trafficking, hemopoiesis, and induction of lymphopenia, anemia, and thrombocytopenia in anti-CD137-treated mice. Journal of immunology (Baltimore, Md : 1950) 2007;178:4194–4213. doi: 10.4049/jimmunol.178.7.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bartkowiak T, Jaiswal AR, Ai M, Budhani P, Ager C, Curran MA. Mechanisms underlying 4-1BB agonist antibody mediated hepatotoxicity. The Journal of Immunology. 2016;196:188.185. [Google Scholar]
- 98.Melero I, Hervas-STubbs S, Glennie MJ, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nature Reviews Cancer. 2007;7:95–106. doi: 10.1038/nrc2051. [DOI] [PubMed] [Google Scholar]
- 99.Chester C, Ambulkar S, Kohrt HE. 4-1BB agonism: adding the accelerator to cancer immunotherapy. Cancer Immunology, Immunotherapy. 2016;65:1243–1248. doi: 10.1007/s00262-016-1829-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Makkouk A, Chester C, Kohrt HE. Rationale for anti-CD137 cancer immunotherapy. European Journal of Cancer. 2016;54:112–119. doi: 10.1016/j.ejca.2015.09.026. [DOI] [PubMed] [Google Scholar]
- 101.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine Storm in a Phase 1 Trial of the Anti-CD28 Monoclonal Antibody TGN1412. New England Journal of Medicine. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 102.Schraven B, Kalinke U. CD28 Superagonists: What Makes the Difference in Humans? Immunity. 2008;28:591–595. doi: 10.1016/j.immuni.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 103.Tacke M, Hanke G, Hanke T, Hünig T. CD28-mediated induction of proliferation in resting T cells in vitro and in vivo without engagement of the T cell receptor: Evidence for functionally distinct forms of CD28. European Journal Of Immunology. 1997;27:239–247. doi: 10.1002/eji.1830270136. [DOI] [PubMed] [Google Scholar]
- 104.Beyersdorf N, Balbach K, Hünig T, Kerkau T. Large-scale expansion of rat CD4(+) CD25(+) T(reg) cells in the absence of T-cell receptor stimulation. Immunology. 2006;119:441–450. doi: 10.1111/j.1365-2567.2006.02455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hunig T. The storm has cleared: lessons from the CD28 superagonist TGN1412 trial. Nature Reviews Immunology. 2012;12:317–318. doi: 10.1038/nri3192. [DOI] [PubMed] [Google Scholar]
- 106.Muller N, van den Brandt J, Odoardi F, Tischner D, Herath J, Flugel A, Reichardt HM. A CD28 superagonistic antibody elicits 2 functionally distinct waves of T cell activation in rats. The Journal of Clinical Investigation. 2008;118:1405–1416. doi: 10.1172/JCI32698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sester U, Wabnitz GH, Kirchgessner H, Samstag Y. Ras/PI3Kinase/cofilin-independent activation of human CD45RA+ and CD45RO+ T cells by superagonistic CD28 stimulation. European Journal of Immunology. 2007;37:2881–2891. doi: 10.1002/eji.200737206. [DOI] [PubMed] [Google Scholar]
- 108.Stebbings R, Findlay L, Edwards C, Eastwood D, Bird C, North D, Mistry Y, Dilger P, Liefooghe E, Cludts I, Fox B, Tarrant G, Robinson J, Meager T, Dolman C, Thorpe SJ, Bristow A, Wadhwa M, Thorpe R, Poole S. Cytokine storm” in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. The Journal of Immunology. 2007;179:3325–3331. doi: 10.4049/jimmunol.179.5.3325. [DOI] [PubMed] [Google Scholar]
- 109.Waibler Z, Sender LY, Merten C, Hartig R, Kliche S, Gunzer M, Reichardt P, Kalinke U, Schraven B. Signaling signatures and functional properties of anti-human CD28 superagonistic antibodies. PLoS One. 2008;3:e1708. doi: 10.1371/journal.pone.0001708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Eastwood D, Findlay L, Poole S, Bird C, Wadhwa M, Moore M, Burns C, Thorpe R, Stebbings R. Monoclonal antibody TGN1412 trial failure explained by species differences in CD28 expression on CD4+ effector memory T-cells. British Journal of Pharmacology. 2010;161:512–526. doi: 10.1111/j.1476-5381.2010.00922.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Romer PS, Berr S, Avota E, Battaglia M, ten Berge I, Einsele H, Hunig T. Preculture of PBMCs at high cell density increases sensitivity of T-cell responses, revealing cytokine release by CD28 superagonist TGN1412. Blood. 2011;118 doi: 10.1182/blood-2010-12-319780. [DOI] [PubMed] [Google Scholar]
- 112.Tyrsin D, Chuvpilo S, Matskevich A, Nemenov D, Romer PS, Tabares P, Hunig T. From TGN1412 to TAB08: the return of CD28 superagonist therapy to clinical development for the treatment of rheumatoid arthritis. Clinical and Experimental Rheumatology. 2016;34:45–48. [PubMed] [Google Scholar]
- 113.Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat Rev Cancer. 2015;15:361–370. doi: 10.1038/nrc3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gianni L, Dafni U, Gelber RD, Azambuja E, Muehlbauer S, Goldhirsch A, Untch M, Smith I, Baselga J, Jackisch C, Cameron D, Mano M, Pedrini JL, Veronesi A, Mendiola C, Pluzanska A, Semiglazov V, Vrdoljak E, Eckart MJ, Shen Z, Skiadopoulos G, Procter M, Pritchard KI, Piccart-Gebhart MJ, Bell R. Treatment with trastuzumab for 1 year after adjuvant chemotherapy in patients with HER2-positive early breast cancer: a 4-year follow-up of a randomised controlled trial. The lancet oncology. 2011;12:236–244. doi: 10.1016/S1470-2045(11)70033-X. [DOI] [PubMed] [Google Scholar]
- 115.Hudis CA. Trastuzumab — Mechanism of Action and Use in Clinical Practice. New England Journal of Medicine. 2007;357:39–51. doi: 10.1056/NEJMra043186. [DOI] [PubMed] [Google Scholar]
- 116.Slamon D, Godolphin W, Jones L, Holt J, Wong S, Keith D, Levin W, Stuart S, Udove J, Ullrich A, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–712. doi: 10.1126/science.2470152. [DOI] [PubMed] [Google Scholar]
- 117.Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, Shak S, Stewart SJ, Press M. Efficacy and Safety of Trastuzumab as a Single Agent in First-Line Treatment of HER2-Overexpressing Metastatic Breast Cancer. Journal of Clinical Oncology. 2002;20:719–726. doi: 10.1200/JCO.2002.20.3.719. [DOI] [PubMed] [Google Scholar]
- 118.Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Annals of Oncology. 2007;18:977–984. doi: 10.1093/annonc/mdl475. [DOI] [PubMed] [Google Scholar]
- 119.Musolino A, Naldi N, Bortesi B, Pezzuolo D, Capelletti M, Missale G, Laccabue D, Zerbini A, Camisa R, Bisagni G, Neri TM, Ardizzoni A. Immunoglobulin G Fragment C Receptor Polymorphisms and Clinical Efficacy of Trastuzumab-Based Therapy in Patients With HER-2/neu–Positive Metastatic Breast Cancer. Journal of Clinical Oncology. 2008;26:1789–1796. doi: 10.1200/JCO.2007.14.8957. [DOI] [PubMed] [Google Scholar]
- 120.Onitilo AA, Engel JM, Stankowski RV. Cardiovascular toxicity associated with adjuvant trastuzumab therapy: prevalence, patient characteristics, and risk factors. Therapeutic Advances in Drug Safety. 2014;5:154–166. doi: 10.1177/2042098614529603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Seidman A, Hudis C, Pierri MK, Shak S, Paton V, Ashby M, Murphy M, Stewart SJ, Keefe D. Cardiac Dysfunction in the Trastuzumab Clinical Trials Experience. Journal of Clinical Oncology. 2002;20:1215–1221. doi: 10.1200/JCO.2002.20.5.1215. [DOI] [PubMed] [Google Scholar]
- 122.Slamon D, Eiermann W, Robert N, Pienkowski T, Martin M, Press M, Mackey J, Glaspy J, Chan A, Pawlicki M, Pinter T, Valero V, Liu M-C, Sauter G, von Minckwitz G, Visco F, Bee V, Buyse M, Bendahmane B, Tabah-Fisch I, Lindsay M-A, Riva A, Crown J, G. The Breast Cancer International Research Adjuvant Trastuzumab in HER2-Positive Breast Cancer. The New England journal of medicine. 2011;365:1273–1283. doi: 10.1056/NEJMoa0910383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Crone SA, Zhao YY, Fan L, Gu Y, Minamisawa S, Liu Y, Peterson KL, Chen J, Kahn R, Condorelli G, R J, Jr, Chien KR, Lee KF. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat Med. 2002;8:459–465. doi: 10.1038/nm0502-459. [DOI] [PubMed] [Google Scholar]
- 124.Farolfi A, Melegari E, Aquilina M, Scarpi E, Ibrahim T, Maltoni R, Sarti S, Cecconetto L, Pietri E, Ferrario C, Fedeli A, Faedi M, Nanni O, Frassineti GL, Amadori D, Rocca A. Trastuzumab-induced cardiotoxicity in early breast cancer patients: a retrospective study of possible risk and protective factors. Heart. 2013;99:634–639. doi: 10.1136/heartjnl-2012-303151. [DOI] [PubMed] [Google Scholar]
- 125.Moja L, Tagliabue L, Balduzzi S, Parmelli E, Pistotti V, Guarneri V, D’Amico R. Trastuzumab containing regimens for early breast cancer. Cochrane Database of Systematic Reviews. 2012 doi: 10.1002/14651858.CD006243.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dotan E, Aggarwal C, Smith MR. Impact of Rituximab (Rituxan) on the Treatment of B-Cell Non-Hodgkin’s Lymphoma. Pharmacy and Therapeutics. 2010;35:148–157. [PMC free article] [PubMed] [Google Scholar]
- 127.Aksoy S, Dizdar Ö, Hayran M, Harputluoğlu H. Infectious complications of rituximab in patients with lymphoma during maintenance therapy: a systematic review and meta-analysis. Leukemia & Lymphoma. 2009;50:357–365. doi: 10.1080/10428190902730219. [DOI] [PubMed] [Google Scholar]
- 128.Vidal L, Gafter-Gvili A, Leibovici L, Shpilberg O. Rituximab as maintenance therapy for patients with follicular lymphoma. Cochrane Database of Systematic Reviews. 2009 doi: 10.1002/14651858.CD006552.pub2. [DOI] [PubMed] [Google Scholar]
- 129.Hadjinicolaou AV, Nisar MK, Parfrey H, Chilvers ER, Östör AJK. Non-infectious pulmonary toxicity of rituximab: a systematic review. Rheumatology. 2012;51:653–662. doi: 10.1093/rheumatology/ker290. [DOI] [PubMed] [Google Scholar]
- 130.Stearns V, Mori T, Jacobs LK, Khouri NF, Gabrielson E, Yoshida T, Kominsky SL, Huso DL, Jeter S, Powers P, Tarpinian K, Brown RJ, Lange JR, Rudek MA, Zhang Z, Tsangaris TN, Sukumar S. Preclinical and clinical evaluation of intraductally administered agents in early breast cancer. Sci Transl Med. 2011;3:106ra108. doi: 10.1126/scitranslmed.3002368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Galanis E, Hartmann LC, Cliby WA, Long HJ, Peethambaram PP, Barrette BA, Kaur JS, Haluska PJ, Jr, Aderca I, Zollman PJ, Sloan JA, Keeney G, Atherton PJ, Podratz KC, Dowdy SC, Stanhope CR, Wilson TO, Federspiel MJ, Peng KW, Russell SJ. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010;70:875–882. doi: 10.1158/0008-5472.CAN-09-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Posch C, Weihsengruber F, Bartsch K, Feichtenschlager V, Sanlorenzo M, Vujic I, Monshi B, Ortiz-Urda S, Rappersberger K. Low-dose inhalation of interleukin-2 bio-chemotherapy for the treatment of pulmonary metastases in melanoma patients. Br J Cancer. 2014;110:1427–1432. doi: 10.1038/bjc.2014.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Weide B, Eigentler TK, Pflugfelder A, Zelba H, Martens A, Pawelec G, Giovannoni L, Ruffini PA, Elia G, Neri D, Gutzmer R, Becker JC, Garbe C. Intralesional treatment of stage III metastatic melanoma patients with L19-IL2 results in sustained clinical and systemic immunologic responses. Cancer Immunol Res. 2014;2:668–678. doi: 10.1158/2326-6066.CIR-13-0206. [DOI] [PubMed] [Google Scholar]
- 134.Brody JD, Ai WZ, Czerwinski DK, Torchia JA, Levy M, Advani RH, Kim YH, Hoppe RT, Knox SJ, Shin LK, Wapnir I, Tibshirani RJ, Levy R. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol. 2010;28:4324–4332. doi: 10.1200/JCO.2010.28.9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Biot C, Rentsch CA, Gsponer JR, Birkhauser FD, Jusforgues-Saklani H, Lemaitre F, Auriau C, Bachmann A, Bousso P, Demangel C, Peduto L, Thalmann GN, Albert ML. Preexisting BCG-specific T cells improve intravesical immunotherapy for bladder cancer. Sci Transl Med. 2012;4:137ra172. doi: 10.1126/scitranslmed.3003586. [DOI] [PubMed] [Google Scholar]
- 136.Mole RH. Whole body irradiation; radiobiology or medicine? Br J Radiol. 1953;26:234–241. doi: 10.1259/0007-1285-26-305-234. [DOI] [PubMed] [Google Scholar]
- 137.Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, Formenti SC. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. 2004;58:862–870. doi: 10.1016/j.ijrobp.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 138.Surace L, Lysenko V, Fontana AO, Cecconi V, Janssen H, Bicvic A, Okoniewski M, Pruschy M, Dummer R, Neefjes J, Knuth A, Gupta A, van den Broek M. Complement is a central mediator of radiotherapy-induced tumor-specific immunity and clinical response. Immunity. 2015;42:767–777. doi: 10.1016/j.immuni.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 139.Gupta A, Probst HC, Vuong V, Landshammer A, Muth S, Yagita H, Schwendener R, Pruschy M, Knuth A, van den Broek M. Radiotherapy promotes tumor-specific effector CD8+ T cells via dendritic cell activation. J Immunol. 2012;189:558–566. doi: 10.4049/jimmunol.1200563. [DOI] [PubMed] [Google Scholar]
- 140.Lugade A, Moran J, Gerber S, Rose R, Frelinger J, Lord E. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. Journal of immunology (Baltimore, Md : 1950) 2005;174:7516–7523. doi: 10.4049/jimmunol.174.12.7516. [DOI] [PubMed] [Google Scholar]
- 141.Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6:226ra232. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, Rajapaksa R, Green MR, Torchia J, Brody J, Luong R, Rosenblum MD, Steinman L, Levitsky HI, Tse V, Levy R. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest. 2013;123:2447–2463. doi: 10.1172/JCI64859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kim YH, Gratzinger D, Harrison C, Brody JD, Czerwinski DK, Ai WZ, Morales A, Abdulla F, Xing L, Navi D, Tibshirani RJ, Advani RH, Lingala B, Shah S, Hoppe RT, Levy R. In situ vaccination against mycosis fungoides by intratumoral injection of a TLR9 agonist combined with radiation: a phase 1/2 study. Blood. 2012;119:355–363. doi: 10.1182/blood-2011-05-355222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Seung SK, Curti BD, Crittenden M, Walker E, Coffey T, Siebert JC, Miller W, Payne R, Glenn L, Bageac A, Urba WJ. Phase 1 study of stereotactic body radiotherapy and interleukin-2--tumor and immunological responses. Sci Transl Med. 2012;4:137ra174. doi: 10.1126/scitranslmed.3003649. [DOI] [PubMed] [Google Scholar]
- 145.Aznar MA, Tinari N, Rullan AJ, Sanchez-Paulete AR, Rodriguez-Ruiz ME, Melero I. Intratumoral Delivery of Immunotherapy-Act Locally, Think Globally. J Immunol. 2017;198:31–39. doi: 10.4049/jimmunol.1601145. [DOI] [PubMed] [Google Scholar]
- 146.Singh M, Overwijk WW. Intratumoral immunotherapy for melanoma. Cancer Immunol Immunother. 2015;64:911–921. doi: 10.1007/s00262-015-1727-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.van den Boorn JG, Hartmann G. Turning tumors into vaccines: co-opting the innate immune system. Immunity. 2013;39:27–37. doi: 10.1016/j.immuni.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 148.Marabelle A, Kohrt H, Caux C, Levy R. Intratumoral immunization: a new paradigm for cancer therapy. Clin Cancer Res. 2014;20:1747–1756. doi: 10.1158/1078-0432.CCR-13-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kwong B, Liu H, Irvine DJ. Induction of potent anti-tumor responses while eliminating systemic side effects via liposome-anchored combinatorial immunotherapy. Biomaterials. 2011;32:5134–5147. doi: 10.1016/j.biomaterials.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hori Y, Stern PJ, Hynes RO, Irvine DJ. Engulfing tumors with synthetic extracellular matrices for cancer immunotherapy. Biomaterials. 2009;30:6757–6767. doi: 10.1016/j.biomaterials.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hanes J, Sills A, Zhao Z, Suh KW, Tyler B, DiMeco F, Brat DJ, Choti MA, Leong KW, Pardoll DM, Brem H. Controlled local delivery of interleukin-2 by biodegradable polymers protects animals from experimental brain tumors and liver tumors. Pharm Res. 2001;18:899–906. doi: 10.1023/a:1010963307097. [DOI] [PubMed] [Google Scholar]
- 152.Fransen MF, van der Sluis TC, Ossendorp F, Arens R, Melief CJ. Controlled local delivery of CTLA-4 blocking antibody induces CD8+ T-cell-dependent tumor eradication and decreases risk of toxic side effects. Clin Cancer Res. 2013;19:5381–5389. doi: 10.1158/1078-0432.CCR-12-0781. [DOI] [PubMed] [Google Scholar]
- 153.Pfreundschuh MG, Steinmetz HT, Tuschen R, Schenk V, Diehl V, Schaadt M. Phase I study of intratumoral application of recombinant human tumor necrosis factor. Eur J Cancer Clin Oncol. 1989;25:379–388. doi: 10.1016/0277-5379(89)90034-5. [DOI] [PubMed] [Google Scholar]
- 154.Mulder CMLvH, van der Voort Robbert, van der Laak Jeroen AWM, Klasen Ina S, de Graaf Aniek O, van Kempen Léon CL, de Vries I Jolanda M, Boer Tjitske Duiveman-de, Dolstra Harry, Torensma Ruurd, van Krieken Johan H, Adema Gosse J, De Pieter HM. Intratumoral rhIL-12 administration in head and neck squamous cell carcinoma patients induces B cell activation. International Journal of Cancer. 2008;123:2354–2361. doi: 10.1002/ijc.23756. [DOI] [PubMed] [Google Scholar]
- 155.Bartsch HH, Pfizenmaier K, Schroeder M, Nagel GA. Intralesional application of recombinant human tumor necrosis factor alpha induces local tumor regression in patients with advanced malignancies. Eur J Cancer Clin Oncol. 1989;25:287–291. doi: 10.1016/0277-5379(89)90021-7. [DOI] [PubMed] [Google Scholar]
- 156.Mauldin IS, Wages NA, Stowman AM, Wang E, Smolkin ME, Olson WC, Deacon DH, Smith KT, Galeassi NV, Chianese-Bullock KA, Dengel LT, Marincola FM, Petroni GR, Mullins DW, Slingluff CL., Jr Intratumoral interferon-gamma increases chemokine production but fails to increase T cell infiltration of human melanoma metastases. Cancer Immunol Immunother. 2016;65:1189–1199. doi: 10.1007/s00262-016-1881-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tzeng A, Kauke MJ, Zhu EF, Moynihan KD, Opel CF, Yang NJ, Mehta N, Kelly RL, Szeto GL, Overwijk WW, Irvine DJ, Wittrup KD. Temporally Programmed CD8alpha+ DC Activation Enhances Combination Cancer Immunotherapy. Cell Rep. 2016;17:2503–2511. doi: 10.1016/j.celrep.2016.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Moyer TJ, Zmolek AC, Irvine DJ. Beyond antigens and adjuvants: formulating future vaccines. J Clin Invest. 2016;126:799–808. doi: 10.1172/JCI81083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gause KT, Wheatley AK, Cui J, Yan Y, Kent SJ, Caruso F. Immunological Principles Guiding the Rational Design of Particles for Vaccine Delivery. ACS Nano. 2017;11:54–68. doi: 10.1021/acsnano.6b07343. [DOI] [PubMed] [Google Scholar]
- 160.Benne N, van Duijn J, Kuiper J, Jiskoot W, Slutter B. Orchestrating immune responses: How size, shape and rigidity affect the immunogenicity of particulate vaccines. J Control Release. 2016;234:124–134. doi: 10.1016/j.jconrel.2016.05.033. [DOI] [PubMed] [Google Scholar]
- 161.Irvine DJ, Swartz MA, Szeto GL. Engineering synthetic vaccines using cues from natural immunity. Nat Mater. 2013;12:978–990. doi: 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cruz LJ, Rueda F, Tacken P, Albericio F, Torensma R, Figdor CG. Enhancing immunogenicity and cross-reactivity of HIV-1 antigens by in vivo targeting to dendritic cells. Nanomedicine (Lond) 2012;7:1591–1610. doi: 10.2217/nnm.12.131. [DOI] [PubMed] [Google Scholar]
- 163.Egilmez NK, Jong YS, Sabel MS, Jacob JS, Mathiowitz E, Bankert RB. In situ tumor vaccination with interleukin-12-encapsulated biodegradable microspheres: induction of tumor regression and potent antitumor immunity. Cancer Res. 2000;60:3832–3837. [PubMed] [Google Scholar]
- 164.Egilmez NK, Jong YS, Iwanuma Y, Jacob JS, Santos CA, Chen FA, Mathiowitz E, Bankert RB. Cytokine immunotherapy of cancer with controlled release biodegradable microspheres in a human tumor xenograft/SCID mouse model. Cancer Immunol Immunother. 1998;46:21–24. doi: 10.1007/s002620050455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, Ryan JL. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90:2541–2548. [PubMed] [Google Scholar]
- 166.Atkins MB, Robertson MJ, Gordon M, Lotze MT, DeCoste M, DuBois JS, Ritz J, Sandler AB, Edington HD, Garzone PD, Mier JW, Canning CM, Battiato L, Tahara H, Sherman ML. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res. 1997;3:409–417. [PubMed] [Google Scholar]
- 167.Smith SG, Koppolu BP, Ravindranathan S, Kurtz SL, Yang L, Katz MD, Zaharoff DA. Intravesical chitosan/interleukin-12 immunotherapy induces tumor-specific systemic immunity against murine bladder cancer. Cancer Immunol Immunother. 2015;64:689–696. doi: 10.1007/s00262-015-1672-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Vo JL, Yang L, Kurtz SL, Smith SG, Koppolu BP, Ravindranathan S, Zaharoff DA. Neoadjuvant immunotherapy with chitosan and interleukin-12 to control breast cancer metastasis. Oncoimmunology. 2014;3:e968001. doi: 10.4161/21624011.2014.968001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yang L, Zaharoff DA. Role of chitosan co-formulation in enhancing interleukin-12 delivery and antitumor activity. Biomaterials. 2013;34:3828–3836. doi: 10.1016/j.biomaterials.2013.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hori Y, Winans AM, Irvine DJ. Modular injectable matrices based on alginate solution/microsphere mixtures that gel in situ and co-deliver immunomodulatory factors. Acta Biomater. 2009;5:969–982. doi: 10.1016/j.actbio.2008.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wang C, Ye Y, Hochu GM, Sadeghifar H, Gu Z. Enhanced Cancer Immunotherapy by Microneedle Patch-Assisted Delivery of Anti-PD1 Antibody. Nano Lett. 2016;16:2334–2340. doi: 10.1021/acs.nanolett.5b05030. [DOI] [PubMed] [Google Scholar]
- 172.Ye Y, Wang J, Hu Q, Hochu GM, Xin H, Wang C, Gu Z. Synergistic Transcutaneous Immunotherapy Enhances Antitumor Immune Responses through Delivery of Checkpoint Inhibitors. ACS Nano. 2016;10:8956–8963. doi: 10.1021/acsnano.6b04989. [DOI] [PubMed] [Google Scholar]
- 173.Smirnov D, Schmidt JJ, Capecchi JT, Wightman PD. Vaccine adjuvant activity of 3M-052: an imidazoquinoline designed for local activity without systemic cytokine induction. Vaccine. 2011;29:5434–5442. doi: 10.1016/j.vaccine.2011.05.061. [DOI] [PubMed] [Google Scholar]
- 174.Hanson MC, Crespo MP, Abraham W, Moynihan KD, Szeto GL, Chen SH, Melo MB, Mueller S, Irvine DJ. Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants. J Clin Invest. 2015;125:2532–2546. doi: 10.1172/JCI79915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Singh M, Khong H, Dai Z, Huang XF, Wargo JA, Cooper ZA, Vasilakos JP, Hwu P, Overwijk WW. Effective innate and adaptive antimelanoma immunity through localized TLR7/8 activation. J Immunol. 2014;193:4722–4731. doi: 10.4049/jimmunol.1401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Stephan SB, Taber AM, Jileaeva I, Pegues EP, Sentman CL, Stephan MT. Biopolymer implants enhance the efficacy of adoptive T-cell therapy. Nat Biotechnol. 2015;33:97–101. doi: 10.1038/nbt.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G, Gonzalez R, Glaspy J, Whitman E, Harrington K, Goldsweig H, Marshall T, Love C, Coffin R, Nemunaitis JJ. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol. 2009;27:5763–5771. doi: 10.1200/JCO.2009.24.3675. [DOI] [PubMed] [Google Scholar]
- 178.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mastrangelo MJ, Maguire HC, Jr, Eisenlohr LC, Laughlin CE, Monken CE, McCue PA, Kovatich AJ, Lattime EC. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999;6:409–422. doi: 10.1038/sj.cgt.7700066. [DOI] [PubMed] [Google Scholar]
- 180.Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC, Oh SY, Han SY, Yoon JH, Hong SH, Moon A, Speth K, Park C, Ahn YJ, Daneshmand M, Rhee BG, Pinedo HM, Bell JC, Kirn DH. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9:533–542. doi: 10.1016/S1470-2045(08)70107-4. [DOI] [PubMed] [Google Scholar]
- 181.Bramson JL, Hitt M, Addison CL, Muller WJ, Gauldie J, Graham FL. Direct intratumoral injection of an adenovirus expressing interleukin-12 induces regression and long-lasting immunity that is associated with highly localized expression of interleukin-12. Hum Gene Ther. 1996;7:1995–2002. doi: 10.1089/hum.1996.7.16-1995. [DOI] [PubMed] [Google Scholar]
- 182.Kramer MG, Masner M, Casales E, Moreno M, Smerdou C, Chabalgoity JA. Neoadjuvant administration of Semliki Forest virus expressing interleukin-12 combined with attenuated Salmonella eradicates breast cancer metastasis and achieves long-term survival in immunocompetent mice. BMC Cancer. 2015;15:620. doi: 10.1186/s12885-015-1618-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Guiducci C, Di Carlo E, Parenza M, Hitt M, Giovarelli M, Musiani P, Colombo MP. Intralesional injection of adenovirus encoding CC chemokine ligand 16 inhibits mammary tumor growth and prevents metastatic-induced death after surgical removal of the treated primary tumor. J Immunol. 2004;172:4026–4036. doi: 10.4049/jimmunol.172.7.4026. [DOI] [PubMed] [Google Scholar]
- 184.Yao H, Ng SS, Huo LF, Chow BK, Shen Z, Yang M, Sze J, Ko O, Li M, Yue A, Lu LW, Bian XW, Kung HF, Lin MC. Effective melanoma immunotherapy with interleukin-2 delivered by a novel polymeric nanoparticle. Mol Cancer Ther. 2011;10:1082–1092. doi: 10.1158/1535-7163.MCT-10-0717. [DOI] [PubMed] [Google Scholar]
- 185.Heinzerling L, Burg G, Dummer R, Maier T, Oberholzer PA, Schultz J, Elzaouk L, Pavlovic J, Moelling K. Intratumoral injection of DNA encoding human interleukin 12 into patients with metastatic melanoma: clinical efficacy. Hum Gene Ther. 2005;16:35–48. doi: 10.1089/hum.2005.16.35. [DOI] [PubMed] [Google Scholar]
- 186.Heinzerling L, Dummer R, Pavlovic J, Schultz J, Burg G, Moelling K. Tumor regression of human and murine melanoma after intratumoral injection of IL-12-encoding plasmid DNA in mice. Exp Dermatol. 2002;11:232–240. doi: 10.1034/j.1600-0625.2001.110306.x. [DOI] [PubMed] [Google Scholar]
- 187.Liu S, Breiter DR, Zheng G, Chen A. Enhanced antitumor responses elicited by combinatorial protein transfer of chemotactic and costimulatory molecules. J Immunol. 2007;178:3301–3306. doi: 10.4049/jimmunol.178.5.3301. [DOI] [PubMed] [Google Scholar]
- 188.Zheng G, Chen A, Sterner R, Zhang P, Pan T, Kiyatkin N, Tykocinski M. Induction of antitumor immunity via intratumoral tetra-costimulator protein transfer. Cancer research. 2001;61:8127. [PubMed] [Google Scholar]
- 189.Liu H, Kwong B, Irvine DJ. Membrane anchored immunostimulatory oligonucleotides for in vivo cell modification and localized immunotherapy. Angew Chem Int Ed Engl. 2011;50:7052–7055. doi: 10.1002/anie.201101266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lizotte PH, Wen AM, Sheen MR, Fields J, Rojanasopondist P, Steinmetz NF, Fiering S. In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nat Nanotechnol. 2016;11:295–303. doi: 10.1038/nnano.2015.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Cochran AJ, Morton DL, Stern S, Lana AMA, Essner R, Wen DR. Sentinel Lymph Nodes Show Profound Downregulation of Antigen-Presenting Cells of the Paracortex: Implications for Tumor Biology and Treatment. Mod Pathol. 2001;14:604–608. doi: 10.1038/modpathol.3880358. [DOI] [PubMed] [Google Scholar]
- 192.Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4:941–952. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- 193.Vicari AP, Chiodoni C, Vaure C, Ait-Yahia S, Dercamp C, Matsos F, Reynard O, Taverne C, Merle P, Colombo MP, O’Garra A, Trinchieri G, Caux C. Reversal of tumor-induced dendritic cell paralysis by CpG immunostimulatory oligonucleotide and anti-interleukin 10 receptor antibody. J Exp Med. 2002;196:541–549. doi: 10.1084/jem.20020732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.van Mierlo GJD, Boonman ZFHM, Dumortier HMH, den Boer AT, Fransen MF, Nouta J, van der Voort EIH, Offringa R, Toes REM, Melief CJM. Activation of Dendritic Cells That Cross-Present Tumor-Derived Antigen Licenses CD8<sup>+</sup> CTL to Cause Tumor Eradication. The Journal of Immunology. 2004;173:6753–6759. doi: 10.4049/jimmunol.173.11.6753. [DOI] [PubMed] [Google Scholar]
- 195.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neil CP, Lee LK, Swartz MA, Hubbell JA. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007;25:1159–1164. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 196.Reddy ST, Rehor A, Schmoekel HG, Hubbell JA, Swartz MA. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. Journal of Controlled Release. 2006;112:26–34. doi: 10.1016/j.jconrel.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 197.Kourtis IC, Hirosue S, de Titta A, Kontos S, Stegmann T, Hubbell JA, Swartz MA. Peripherally administered nanoparticles target monocytic myeloid cells, secondary lymphoid organs and tumors in mice. PLoS One. 2013;8:e61646. doi: 10.1371/journal.pone.0061646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Jeanbart L, Ballester M, de Titta A, Corthesy P, Romero P, Hubbell JA, Swartz MA. Enhancing efficacy of anticancer vaccines by targeted delivery to tumor-draining lymph nodes. Cancer immunology research. 2014;2:436–447. doi: 10.1158/2326-6066.CIR-14-0019-T. [DOI] [PubMed] [Google Scholar]
- 199.Pradhan P, Qin H, Leleux J, Gwak D, Sakamaki I, Kwak LW, Roy K. The effect of combined IL10 siRNA and CpG ODN as pathogen-mimicking microparticles on Th1/Th2 cytokine balance in dendritic cells and protective immunity against B cell lymphoma. Biomaterials. 2014;35:5491–5504. doi: 10.1016/j.biomaterials.2014.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bourquin C, Anz D, Zwiorek K, Lanz AL, Fuchs S, Weigel S, Wurzenberger C, von der Borch P, Golic M, Moder S, Winter G, Coester C, Endres S. Targeting CpG Oligonucleotides to the Lymph Node by Nanoparticles Elicits Efficient Antitumoral Immunity. The Journal of Immunology. 2008;181:2990–2998. doi: 10.4049/jimmunol.181.5.2990. [DOI] [PubMed] [Google Scholar]
- 201.Zwiorek K, Bourquin C, Battiany J, Winter G, Endres S, Hartmann G, Coester C. Delivery by Cationic Gelatin Nanoparticles Strongly Increases the Immunostimulatory Effects of CpG Oligonucleotides. Pharmaceutical Research. 2008;25:551–562. doi: 10.1007/s11095-007-9410-5. [DOI] [PubMed] [Google Scholar]
- 202.Ilyinskii PO, Roy CJ, O’Neil CP, Browning EA, Pittet LA, Altreuter DH, Alexis F, Tonti E, Shi J, Basto PA. Adjuvant-carrying synthetic vaccine particles augment the immune response to encapsulated antigen and exhibit strong local immune activation without inducing systemic cytokine release. Vaccine. 2014;32:2882–2895. doi: 10.1016/j.vaccine.2014.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Neri D, Sondel PM. Immunocytokines for cancer treatment: past, present and future. Current Opinion in Immunology. 2016;40:96–102. doi: 10.1016/j.coi.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Naramura M, Gillies SD, Mendelsohn J, Reisfeld RA, Mueller BM. Mechanisms of cellular cytotoxicity mediated by a recombinant antibody-IL2 fusion protein against human melanoma cells. Immunol Lett. 1993;39:91–99. doi: 10.1016/0165-2478(93)90169-3. [DOI] [PubMed] [Google Scholar]
- 205.Dorai H, Mueller BM, Reisfeld RA, Gillies SD. Aglycosylated chimeric mouse/human IgG1 antibody retains some effector function. Hybridoma. 1991;10:211–217. doi: 10.1089/hyb.1991.10.211. [DOI] [PubMed] [Google Scholar]
- 206.Gubbels JA, Gadbaw B, Buhtoiarov IN, Horibata S, Kapur AK, Patel D, Hank JA, Gillies SD, Sondel PM, Patankar MS, Connor J. Ab-IL2 fusion proteins mediate NK cell immune synapse formation by polarizing CD25 to the target cell-effector cell interface. Cancer Immunol Immunother. 2011;60:1789–1800. doi: 10.1007/s00262-011-1072-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Buhtoiarov IN, Neal ZC, Gan J, Buhtoiarova TN, Patankar MS, Gubbels JA, Hank JA, Yamane B, Rakhmilevich AL, Reisfeld RA, Gillies SD, Sondel PM. Differential internalization of hu14.18-IL2 immunocytokine by NK and tumor cell: impact on conjugation, cytotoxicity, and targeting. J Leukoc Biol. 2011;89:625–638. doi: 10.1189/jlb.0710422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sondel PM, Gillies SD. Current and Potential Uses of Immunocytokines as Cancer Immunotherapy. Antibodies. 2012;1:149–171. doi: 10.3390/antib1020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Giragossian C, Clark T, Piche-Nicholas N, Bowman CJ. Neonatal Fc receptor and its role in the absorption, distribution, metabolism and excretion of immunoglobulin G-based biotherapeutics. Curr Drug Metab. 2013;14:764–790. doi: 10.2174/13892002113149990099. [DOI] [PubMed] [Google Scholar]
- 210.Tzeng A, Kwan BH, Opel CF, Navaratna T, Wittrup KD. Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proc Natl Acad Sci U S A. 2015;112:3320–3325. doi: 10.1073/pnas.1416159112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.List T, Neri D. Immunocytokines: a review of molecules in clinical development for cancer therapy. Clinical Pharmacology. 2013;5:29–45. doi: 10.2147/CPAA.S49231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Becker JC, Pancook JD, Gillies SD, Mendelsohn J, Reisfeld RA. Eradication of human hepatic and pulmonary melanoma metastases in SCID mice by antibody-interleukin-2 fusion proteins. Proceedings Of The National Academy Of Sciences Of The United States Of America. 1996;93:2701–2707. doi: 10.1073/pnas.93.7.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wieckowski S, Hemmerle T, Prince SS, Schlienger BD, Hillinger S, Neri D, Zippelius A. Therapeutic efficacy of the F8-IL2 immunocytokine in a metastatic mouse model of lung adenocarcinoma. Lung cancer. 2015;88:9–15. doi: 10.1016/j.lungcan.2015.01.019. [DOI] [PubMed] [Google Scholar]
- 214.Epstein AL, Chen FM, Taylor CR. A novel method for detection of necrotic lesions in human cancers. Cancer Research. 1988;48:5842–5848. [PubMed] [Google Scholar]
- 215.Borsi L, Carnemolla B, Nicolo G, Spina B, Tanara G, Zardi L. Expression of different tenascin isoforms in normal, hyperplastic and neoplastic human breast tissues. International Journal of Cancer. 1992;52:688–692. doi: 10.1002/ijc.2910520504. [DOI] [PubMed] [Google Scholar]
- 216.Santimaria M, Moscatelli G, Viale GL, Giovannoni L, Neri G, Viti F, Leprini A, Borsi L, Castellani P, Zardi L, Neri D, Riva P. Immunoscintigraphic detection of the ED-B domain of fibronectin, a marker of angiogenesis, in patients with cancer. Clinical Cancer Research. 2003;9:571–579. [PubMed] [Google Scholar]
- 217.Neri D, Bicknell R. Tumour vascular targeting. Nature Reviews Cancer. 2005;5:436–446. doi: 10.1038/nrc1627. [DOI] [PubMed] [Google Scholar]
- 218.Carnemolla B, Balza E, Siri A, Zardi L, Nicotra MR, Bigotti A, Natali PG. A tumor-associated fibronectin isoform generated by alternative splicing of messenger RNA precursors. J Cell Biol. 1989;108:1139–1148. doi: 10.1083/jcb.108.3.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Castellani P, Viale G, Dorcaratto A, Nicolo G, Kaczmarek J, Querze G, Zardi L. The fibronectin isoform containing the ED-B oncofetal domain: a marker of angiogenesis. International Journal of Cancer. 1994;59:612–618. doi: 10.1002/ijc.2910590507. [DOI] [PubMed] [Google Scholar]
- 220.King DM, Albertini MR, Schalch H, Hank JA, Gan J, Surfus J, Mahvi D, Schiller JH, Warner T, Kim K, Eickhoff J, Kendra K, Reisfeld R, Gillies SD, Sondel P. Phase I clinical trial of the immunocytokine EMD 273063 in melanoma patients. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2004;22:4463–4473. doi: 10.1200/JCO.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Shusterman S, London WB, Gillies SD, Hank JA, Voss SD, Seeger RC, Reynolds CP, Kimball J, Albertini MR, Wagner B, Gan J, Eickhoff J, DeSantes KB, Cohn SL, Hecht T, Gadbaw B, Reisfeld RA, Maris JM, Sondel PM. Antitumor Activity of Hu14.18-IL2 in Patients With Relapsed/Refractory Neuroblastoma: A Children’s Oncology Group (COG) Phase II Study. Journal of Clinical Oncology. 2010;28:4969–4975. doi: 10.1200/JCO.2009.27.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Schettini J, Kidiyoor A, Besmer DM, Tinder TL, Roy LD, Lustgarten J, Gendler SJ, Mukherjee P. Intratumoral delivery of CpG-conjugated anti-MUC1 antibody enhances NK cell anti-tumor activity. Cancer Immunology, Immunotherapy. 2012;61:2055–2065. doi: 10.1007/s00262-012-1264-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Li Z, Jang JK, Lechner MG, Hu P, Khawli L, Scannell CA, Epstein AL. Generation of tumor-targeted antibody–CpG conjugates. Journal of Immunological Methods. 2013;389:45–51. doi: 10.1016/j.jim.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 224.Schrand B, Berezhnoy A, Brenneman R, Williams A, Levay A, Kong LY, Rao G, Zhou S, Heimberger AB, Gilboa E. Targeting 4-1BB costimulation to the tumor stroma with bispecific aptamer conjugates enhances the therapeutic index of tumor immunotherapy. Cancer immunology research. 2014;2:867–877. doi: 10.1158/2326-6066.CIR-14-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Shir A, Ogris M, Roedl W, Wagner E, Levitzki A. EGFR-homing dsRNA activates cancer-targeted immune response and eliminates disseminated EGFR-overexpressing tumors in mice. Clinical Cancer Research. 2011;17:1033–1043. doi: 10.1158/1078-0432.CCR-10-1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Yu M, Lam J, Rada B, Leto TL, Levine SJ. Double-stranded RNA induces shedding of the 34-kDa soluble TNFR1 from human airway epithelial cells via TLR3–TRIF–RIP1-dependent signaling: Roles for dual oxidase 2-and caspase-dependent pathways. The Journal of Immunology. 2011;186:1180–1188. doi: 10.4049/jimmunol.1001499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Caster JM, Patel AN, Zhang T, Wang A. Investigational nanomedicines in 2016: a review of nanotherapeutics currently undergoing clinical trials. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017;9 doi: 10.1002/wnan.1416. [DOI] [PubMed] [Google Scholar]
- 228.Chow EK, Ho D. Cancer nanomedicine: from drug delivery to imaging. Sci Transl Med. 2013;5:216rv214. doi: 10.1126/scitranslmed.3005872. [DOI] [PubMed] [Google Scholar]
- 229.Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65:71–79. doi: 10.1016/j.addr.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 230.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- 231.Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1 [Google Scholar]
- 232.Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69:2506–2513. doi: 10.1158/0008-5472.CAN-08-4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Huo M, Zhao Y, Satterlee AB, Wang Y, Xu Y, Huang L. Tumor-targeted delivery of sunitinib base enhances vaccine therapy for advanced melanoma by remodeling the tumor microenvironment. J Control Release. 2017;245:81–94. doi: 10.1016/j.jconrel.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Xu Z, Wang Y, Zhang L, Huang L. Nanoparticle-delivered transforming growth factor-beta siRNA enhances vaccination against advanced melanoma by modifying tumor microenvironment. ACS Nano. 2014;8:3636–3645. doi: 10.1021/nn500216y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Li SY, Liu Y, Xu CF, Shen S, Sun R, Du XJ, Xia JX, Zhu YH, Wang J. Restoring anti-tumor functions of T cells via nanoparticle-mediated immune checkpoint modulation. Journal of Controlled Release. 2016;231:17–28. doi: 10.1016/j.jconrel.2016.01.044. [DOI] [PubMed] [Google Scholar]
- 236.Park J, Wrzesinski SH, Stern E, Look M, Criscione J, Ragheb R, Jay SM, Demento SL, Agawu A, Limon P Licona, Ferrandino AF, Gonzalez D, Habermann A, Flavell RA, Fahmy TM. Combination delivery of TGF-beta inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat Mater. 2012;11:895–905. doi: 10.1038/nmat3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Chen Y, Xia R, Huang Y, Zhao W, Li J, Zhang X, Wang P, Venkataramanan R, Fan J, Xie W, Ma X, Lu B, Li S. An immunostimulatory dual-functional nanocarrier that improves cancer immunochemotherapy. Nature communications. 2016;7 doi: 10.1038/ncomms13443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, Kohler RH, Iwamoto Y, Yang KS, Askevold B, Kolishetti N, Pittet M, Lippard SJ, Farokhzad OC, Weissleder R. Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nature communications. 2015;6:8692. doi: 10.1038/ncomms9692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Jeanbart L, Kourtis IC, van der Vlies AJ, Swartz MA, Hubbell JA. 6-Thioguanine-loaded polymeric micelles deplete myeloid-derived suppressor cells and enhance the efficacy of T cell immunotherapy in tumor-bearing mice. Cancer Immunology, Immunotherapy. 2015;64:1–14. doi: 10.1007/s00262-015-1702-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016;37:208–220. doi: 10.1016/j.it.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Lu M, Cohen MH, Rieves D, Pazdur R. FDA report: Ferumoxytol for intravenous iron therapy in adult patients with chronic kidney disease. American Journal of Hematology. 2010;85:315–319. doi: 10.1002/ajh.21656. [DOI] [PubMed] [Google Scholar]
- 242.Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, Pajarinen JS, Nejadnik H, Goodman S, Moseley M, Coussens LM, Daldrup-Link HE. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nature Nanotechnology. 2016;11:986–994. doi: 10.1038/nnano.2016.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Sindrilaru A, Peters T, Wieschalka S, Baican C, Baican A, Peter H, Hainzl A, Schatz S, Qi Y, Schlecht A, Weiss JM, Wlaschek M, Sunderkotter C, Scharffetter-Kochanek K. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. J Clin Invest. 2011;121:985–997. doi: 10.1172/JCI44490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Laskar A, Eilertsen J, Li W, Yuan XM. SPION primes THP1 derived M2 macrophages towards M1-like macrophages. Biochemical and Biophysical Research Communications. 2013;441:737–742. doi: 10.1016/j.bbrc.2013.10.115. [DOI] [PubMed] [Google Scholar]
- 245.Huang Z, Zhang Z, Jiang Y, Zhang D, Chen J, Dong L, Zhang J. Targeted delivery of oligonucleotides into tumor-associated macrophages for cancer immunotherapy. J Control Release. 2012;158:286–292. doi: 10.1016/j.jconrel.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 246.Zhu S, Niu M, O’Mary H, Cui Z. Targeting of tumor-associated macrophages made possible by PEG-sheddable, mannose-modified nanoparticles. Mol Pharm. 2013;10:3525–3530. doi: 10.1021/mp400216r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Chen Q, Xu L, Liang C, Wang C, Peng R, Liu Z. Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nature communications. 2016;7:13193. doi: 10.1038/ncomms13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, Sheng D, Liu X, Sims PW, VanderVeen LA, Ali CF, K CT, Konakova M, Pena RL, Kanhere RS, Kirksey YM, Ji C, Wang Y, Huang J, Sweeney TD, Kantak SS, Doberstein SK. NKTR-214, an Engineered Cytokine with Biased IL2 Receptor Binding, Increased Tumor Exposure, and Marked Efficacy in Mouse Tumor Models. Clinical Cancer Research. 2016;22:680–690. doi: 10.1158/1078-0432.CCR-15-1631. [DOI] [PubMed] [Google Scholar]
- 249.Puskas J, Skrombolas D, Sedlacek A, Lord E, Sullivan M, Frelinger J. Development of an attenuated interleukin-2 fusion protein that can be activated by tumour-expressed proteases. Immunology. 2011;133:206–220. doi: 10.1111/j.1365-2567.2011.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Huang B, Abraham WD, Zheng Y, Lopez SC Bustamante, Luo SS, Irvine DJ. Active targeting of chemotherapy to disseminated tumors using nanoparticle-carrying T cells. Sci Transl Med. 2015;7:291ra294. doi: 10.1126/scitranslmed.aaa5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14:642–662. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AG, Wandless TJ. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell. 2006;126:995–1004. doi: 10.1016/j.cell.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.June C, Rosenberg SA, Sadelain M, Weber JS. T-cell therapy at the threshold. Nat Biotechnol. 2012;30:611–614. doi: 10.1038/nbt.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Sakamoto N, Ishikawa T, Kokura S, Okayama T, Oka K, Ideno M, Sakai F, Kato A, Tanabe M, Enoki T, Mineno J, Naito Y, Itoh Y, Yoshikawa T. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J Transl Med. 2015;13:277. doi: 10.1186/s12967-015-0632-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Burke B, Sumner S, Maitland N, Lewis CE. Macrophages in gene therapy: cellular delivery vehicles and in vivo targets. J Leukoc Biol. 2002;72:417–428. [PubMed] [Google Scholar]
- 256.Stephan MT, Irvine DJ. Enhancing Cell therapies from the Outside In: Cell Surface Engineering Using Synthetic Nanomaterials. Nano Today. 2011;6:309–325. doi: 10.1016/j.nantod.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73:2412–2417. doi: 10.1158/0008-5472.CAN-12-4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Eisenstein S, Coakley BA, Briley-Saebo K, Ma G, Chen HM, Meseck M, Ward S, Divino C, Woo S, Chen SH, Pan PY. Myeloid-derived suppressor cells as a vehicle for tumor-specific oncolytic viral therapy. Cancer Res. 2013;73:5003–5015. doi: 10.1158/0008-5472.CAN-12-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Qiao J, Kottke T, Willmon C, Galivo F, Wongthida P, Diaz RM, Thompson J, Ryno P, Barber GN, Chester J, Selby P, Harrington K, Melcher A, Vile RG. Purging metastases in lymphoid organs using a combination of antigen-nonspecific adoptive T cell therapy, oncolytic virotherapy and immunotherapy. Nat Med. 2008;14:37–44. doi: 10.1038/nm1681. [DOI] [PubMed] [Google Scholar]
- 261.Muthana M, Kennerley AJ, Hughes R, Fagnano E, Richardson J, Paul M, Murdoch C, Wright F, Payne C, Lythgoe MF, Farrow N, Dobson J, Conner J, Wild JM, Lewis C. Directing cell therapy to anatomic target sites in vivo with magnetic resonance targeting. Nat Commun. 2015;6:8009. doi: 10.1038/ncomms9009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Yang JC. Toxicities Associated With Adoptive T-Cell Transfer for Cancer. Cancer J. 2015;21:506–509. doi: 10.1097/PPO.0000000000000157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Tey SK. Adoptive T-cell therapy: adverse events and safety switches. Clin Transl Immunology. 2014;3:e17. doi: 10.1038/cti.2014.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat Med. 2010;16:1035–1041. doi: 10.1038/nm.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Stephan MT, Stephan SB, Bak P, Chen J, Irvine DJ. Synapse-directed delivery of immunomodulators using T-cell-conjugated nanoparticles. Biomaterials. 2012;33:5776–5787. doi: 10.1016/j.biomaterials.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Burga RA, Patel S, Bollard CM, CR YC, Fernandes R. Conjugating Prussian blue nanoparticles onto antigen-specific T cells as a combined nanoimmunotherapy. Nanomedicine (Lond) 2016;11:1759–1767. doi: 10.2217/nnm-2016-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Wayteck L, Dewitte H, De Backer L, Breckpot K, Demeester J, De Smedt SC, Raemdonck K. Hitchhiking nanoparticles: Reversible coupling of lipid-based nanoparticles to cytotoxic T lymphocytes. Biomaterials. 2016;77:243–254. doi: 10.1016/j.biomaterials.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 268.Kaasgaard T, Andresen TL. Liposomal cancer therapy: exploiting tumor characteristics. Expert Opin Drug Deliv. 2010;7:225–243. doi: 10.1517/17425240903427940. [DOI] [PubMed] [Google Scholar]
- 269.Cole C, Qiao J, Kottke T, Diaz RM, Ahmed A, Sanchez-Perez L, Brunn G, Thompson J, Chester J, Vile RG. Tumor-targeted, systemic delivery of therapeutic viral vectors using hitchhiking on antigen-specific T cells. Nat Med. 2005;11:1073–1081. doi: 10.1038/nm1297. [DOI] [PubMed] [Google Scholar]
- 270.Spangler JB, Moraga I, Mendoza JL, Garcia KC. Insights into cytokine-receptor interactions from cytokine engineering. Annu Rev Immunol. 2015;33:139–167. doi: 10.1146/annurev-immunol-032713-120211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Krieg C, Létourneau S, Pantaleo G, Boyman O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proceedings Of The National Academy Of Sciences Of The United States Of America. 2010;107:11906–11911. doi: 10.1073/pnas.1002569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, Moraga I, Raeber ME, Bowman GR, Novick P, Pande VS, Fathman CG, Boyman O, Garcia KC. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature. 2012;484:529–533. doi: 10.1038/nature10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Berezhnoy A, Castro I, Levay A, Malek TR, Gilboa E. Aptamer-targeted inhibition of mTOR in T cells enhances antitumor immunity. J Clin Invest. 2014;124:188–197. doi: 10.1172/JCI69856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Sato K, Sato N, Xu B, Nakamura Y, Nagaya T, Choyke PL, Hasegawa Y, Kobayashi H. Spatially selective depletion of tumor-associated regulatory T cells with near-infrared photoimmunotherapy. Sci Transl Med. 2016;8:352ra110. doi: 10.1126/scitranslmed.aaf6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, DuPage M, Tammela T, Kerper NR, Farago AF, Robbins R, Crowley DM, Bronson RT, Jacks T. Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses. Immunity. 2015;43:579–590. doi: 10.1016/j.immuni.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Goebeler ME, Bargou R. Blinatumomab: a CD19/CD3 bispecific T cell engager (BiTE) with unique anti-tumor efficacy. Leuk Lymphoma. 2016;57:1021–1032. doi: 10.3109/10428194.2016.1161185. [DOI] [PubMed] [Google Scholar]
- 277.Huehls AM, Coupet TA, Sentman CL. Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol. 2015;93:290–296. doi: 10.1038/icb.2014.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Shen J, Vallera DA, Wagner CR. Prosthetic Antigen Receptors. J Am Chem Soc. 2015;137:10108–10111. doi: 10.1021/jacs.5b06166. [DOI] [PubMed] [Google Scholar]
- 279.Schutz C, Varela JC, Perica K, Haupt C, Oelke M, Schneck JP. Antigen-specific T cell Redirectors: a nanoparticle based approach for redirecting T cells. Oncotarget. 2016;7:68503–68512. doi: 10.18632/oncotarget.11785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Zheng Y, Stephan MT, Gai SA, Abraham W, Shearer A, Irvine DJ. In vivo targeting of adoptively transferred T-cells with antibody- and cytokine-conjugated liposomes. J Control Release. 2013 doi: 10.1016/j.jconrel.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Frick SU, Domogalla MP, Baier G, Wurm FR, Mailaender V, Landfester K, Steinbrink K. Interleukin-2 Functionalized Nanocapsules for T Cell-Based Immunotherapy. ACS Nano. 2016 doi: 10.1021/acsnano.5b07973. [DOI] [PubMed] [Google Scholar]
- 282.Zheng Y, Tang L, Mabardi L, Kumari S, Irvine D. Enhancing adoptive cell therapy of cancer through targeted delivery of small-molecule immunomodulators to internalizing or non- internalizing receptors. ACS Nano. doi: 10.1021/acsnano.7b00078. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Sacchetti C, Rapini N, Magrini A, Cirelli E, Bellucci S, Mattei M, Rosato N, Bottini N, Bottini M. In vivo targeting of intratumor regulatory T cells using PEG-modified single-walled carbon nanotubes. Bioconjug Chem. 2013;24:852–858. doi: 10.1021/bc400070q. [DOI] [PubMed] [Google Scholar]
- 284.Hellerstein M, Xu Y, Marino T, Lu S, Yi H, Wright ER, Robinson HL. Co-expression of HIV-1 virus-like particles and granulocyte-macrophage colony stimulating factor by GEO-D03 DNA vaccine. Hum Vaccin Immunother. 2012;8:1654–1658. doi: 10.4161/hv.21978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Iyer SS, Amara RR. DNA/MVA Vaccines for HIV/AIDS. Vaccines (Basel) 2014;2:160–178. doi: 10.3390/vaccines2010160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Ilett E, Kottke T, Donnelly O, Thompson J, Willmon C, Diaz R, Zaidi S, Coffey M, Selby P, Harrington K, Pandha H, Melcher A, Vile R. Cytokine conditioning enhances systemic delivery and therapy of an oncolytic virus. Mol Ther. 2014;22:1851–1863. doi: 10.1038/mt.2014.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Chu D, Zhao Q, Yu J, Zhang F, Zhang H, Zhenjia W. Nanoparticle targeting of neutrophils for improved cancer immunotherapy. Advanced Healthcare Materials. 2016;5:1088–1093. doi: 10.1002/adhm.201500998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Smith BR, Ghosn EE, Rallapalli H, Prescher JA, Larson T, Herzenberg LA, Gambhir SS. Selective uptake of single-walled carbon nanotubes by circulating monocytes for enhanced tumour delivery. Nat Nanotechnol. 2014;9:481–487. doi: 10.1038/nnano.2014.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Kullberg M, Martinson H, Mann K, Anchordoquy TJ. Complement C3 mediated targeting of liposomes to granulocytic myeloid derived suppressor cells. Nanomedicine. 2015;11:1355–1363. doi: 10.1016/j.nano.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. 2012;12:786–798. doi: 10.1038/nri3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Liu J, Blake SJ, Harjunpaa H, Fairfax KA, Yong MC, Allen S, Kohrt HE, Takeda K, Smyth MJ, Teng MW. Assessing Immune-Related Adverse Events of Efficacious Combination Immunotherapies in Preclinical Models of Cancer. Cancer Res. 2016;76:5288–5301. doi: 10.1158/0008-5472.CAN-16-0194. [DOI] [PubMed] [Google Scholar]
- 292.Martino MM, Hubbell JA. The 12th-14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 2010;24:4711–4721. doi: 10.1096/fj.09-151282. [DOI] [PubMed] [Google Scholar]
- 293.Martino MM, Tortelli F, Mochizuki M, Traub S, Ben-David D, Kuhn GA, Müller R, Livne E, Eming SA, Hubbell JA. Engineering the growth factor microenvironment with fibronectin domains to promote wound and bone tissue healing. Science Translational Medicine. 2011;3:100ra189. doi: 10.1126/scitranslmed.3002614. [DOI] [PubMed] [Google Scholar]
- 294.Martino MM, Briquez PS, Guc E, Tortelli F, Kilarski WW, Metzger S, Rice JJ, Kuhn GA, Muller R, Swartz MA, Hubbell JA. Growth factors engineered for super-affinity to the extracellular matrix enhance tissue healing. Science. 2014;343:885–888. doi: 10.1126/science.1247663. [DOI] [PubMed] [Google Scholar]