

ABSTRACT
Bacterial membrane-associated NAD-independent d-lactate dehydrogenase (Fe-S d-iLDH) oxidizes d-lactate into pyruvate. A sequence analysis of the enzyme reveals that it contains an Fe-S oxidoreductase domain in addition to a flavin adenine dinucleotide (FAD)-containing dehydrogenase domain, which differs from other typical d-iLDHs. Fe-S d-iLDH from Pseudomonas putida KT2440 was purified as a His-tagged protein and characterized in detail. This monomeric enzyme exhibited activities with l-lactate and several d-2-hydroxyacids. Quinone was shown to be the preferred electron acceptor of the enzyme. The two domains of the enzyme were then heterologously expressed and purified separately. The Fe-S cluster-binding motifs predicted by sequence alignment were preliminarily verified by site-directed mutagenesis of the Fe-S oxidoreductase domain. The FAD-containing dehydrogenase domain retained 2-hydroxyacid-oxidizing activity, although it decreased compared to the full Fe-S d-iLDH. Compared to the intact enzyme, the FAD-containing dehydrogenase domain showed increased catalytic efficiency with cytochrome c as the electron acceptor, but it completely lost the ability to use coenzyme Q10. Additionally, the FAD-containing dehydrogenase domain was no longer associated with the cell membrane, and it could not support the utilization of d-lactate as a carbon source. Based on the results obtained, we conclude that the Fe-S oxidoreductase domain functions as an electron transfer component to facilitate the utilization of quinone as an electron acceptor by Fe-S d-iLDH, and it helps the enzyme associate with the cell membrane. These functions make the Fe-S oxidoreductase domain crucial for the in vivo d-lactate utilization function of Fe-S d-iLDH.
IMPORTANCE Lactate metabolism plays versatile roles in most domains of life. Lactate utilization processes depend on certain enzymes to oxidize lactate to pyruvate. In recent years, novel bacterial lactate-oxidizing enzymes have been continually reported, including the unique NAD-independent d-lactate dehydrogenase that contains an Fe-S oxidoreductase domain besides the typical flavin-containing domain (Fe-S d-iLDH). Although Fe-S d-iLDH is widely distributed among bacterial species, the investigation of it is insufficient. Fe-S d-iLDH from Pseudomonas putida KT2440, which is the major d-lactate-oxidizing enzyme for the strain, might be a representative of this type of enzyme. A study of it will be helpful in understanding the detailed mechanisms underlying the lactate utilization processes.
KEYWORDS: NAD-independent d-lactate dehydrogenase, flavoprotein, iron-sulfur protein, electron transfer, lactate utilization, Pseudomonas putida
INTRODUCTION
The utilization of lactate is an important process that occurs in most domains of life, including microorganisms, plants, and higher animals (1–4). Many microorganisms can utilize lactate as a source of carbon and energy for growth (3, 5–8). Microbial lactate utilization begins with its uptake into cells, and the first enzyme-catalyzed reaction is the oxidization of lactate to pyruvate. Various types of lactate dehydrogenases (LDHs) can participate in this process. Generally, these enzymes can be classified into NAD-dependent lactate dehydrogenases (nLDHs) and NAD-independent lactate dehydrogenases (iLDHs), and specific enzymes in each group may have specificity for one isomer of lactate, that is, l-lactate versus d-lactate (3). The nLDHs, which use NAD as a cofactor and primarily catalyze the reduction of pyruvate to lactate, require high pH and high substrate concentrations to catalyze the oxidization of lactate (9). For most lactate-utilizing microorganisms, iLDHs, also called respiratory lactate dehydrogenases, catalyze lactate oxidization reactions (3, 10). The current understanding of microbial iLDHs has been summarized recently (3). Most l-iLDHs belong to the family of α-hydroxyacid-oxidizing flavoproteins, use flavin mononucleotide (FMN) as a cofactor, and use O2, cytochrome c, or quinones as electron acceptors (11–13). Most d-iLDHs belong to the flavin adenine dinucleotide (FAD)-binding oxidoreductase/transferase type 4 family, use FAD instead of FMN as the cofactor, and use quinones or cytochrome c as electron acceptors (7, 14, 15). However, there have been continual reports of novel types of both l-iLDHs and d-iLDHs in recent years (16–19). The discovery of these novel enzymes further expands our knowledge of microbial lactate utilization, and detailed characterizations of the enzymes will be helpful in understanding the detailed mechanisms underlying these processes.
d-Lactate can be produced by many fermenting bacteria through glycolysis and pyruvate reduction. Also, it can be produced through methylglyoxal metabolism in many organisms (20, 21). Therefore, the ability to utilize d-lactate would be an advantage for microorganisms. However, compared to l-iLDHs, whose structures and catalytic mechanisms have been well studied over the years (22–26), there have been relatively few characterization studies of d-iLDHs. The d-iLDH from Escherichia coli, which uses quinone as an electron acceptor, is the most well-characterized d-iLDH with a resolved structure (15). Recently, two novel bacterial iLDHs responsible for l-lactate and d-lactate utilization, were identified in Shewanella oneidensis MR-1. The l-iLDH was annotated as a nonflavin iron-sulfur enzyme containing three subunits (denoted LldABC), while the d-iLDH was a multidomain enzyme (denoted Fe-S d-iLDH) (17). Compared with the typical quinone or cytochrome c-dependent d-iLDHs, the Fe-S d-iLDH has a clearly defined C-terminal Fe-S oxidoreductase domain and an N-terminal flavin-containing domain. Interestingly, a later report showed that in Campylobacter jejuni NCTC 11168, which possesses both of the two novel iLDHs, the homolog of Fe-S d-iLDH functioned as an l-lactate- but not d-lactate-oxidizing enzyme in vivo. This suggests that homologs of this enzyme cannot be assumed to be d-lactate specific (18).
Pseudomonas is a vast bacterial genus in which various species have the ability to utilize lactate for growth (27–29). Several Pseudomonas genes related to lactate utilization have been identified, and several of these enzymes have been characterized and used in biocatalysis processes (23, 30–33). Homologs of Fe-S d-iLDH seem to be present in all the identified lactate utilization operons of Pseudomonas (29). A recent report showed that in Pseudomonas putida KT2440, the Fe-S d-iLDH is not the only enzyme that supports d-lactate utilization and that a glycolate oxidase was also involved. Nevertheless, the two enzymes do not contribute equally to the d-lactate utilization process in P. putida KT2440, and the Fe-S d-iLDH plays the major role (34). Thus, as an important but poorly understood enzyme that is distributed widely in lactate-utilizing microbes, the Fe-S d-iLDH requires further characterization.
In this study, we characterized the Fe-S d-iLDH from P. putida KT2440 in detail. The enzyme was overexpressed in E. coli and purified. Its flavin cofactor was identified as FAD, and quinone was identified as its preferred electron acceptor. According to sequence analysis of the protein, it has two main parts: an FAD-containing dehydrogenase domain and an Fe-S oxidoreductase domain. Both of the domains were successfully overexpressed and purified independently. The isolated FAD-containing dehydrogenase domain was found to retain some substrate-oxidizing activity, but it preferentially used cytochrome c instead of quinone as an electron acceptor. We further found that the FAD-containing dehydrogenase domain no longer was associated with the cell membrane, and it could not support in vivo d-lactate utilization. Using the isolated Fe-S oxidoreductase domain, the Fe-S cluster-binding motifs were confirmed by mutagenesis. For this multidomain d-iLDH, the Fe-S oxidoreductase domain helps the FAD-containing catalytic center link to the cell membrane, as well as facilitating the electron transfer from the substrate to the appropriate acceptor within the Fe-S center.
RESULTS
Sequence analysis of Fe-S d-iLDH.
The Fe-S d-iLDH-encoding gene lldE (pp4737) is located in the four-gene lactate utilization operon pp4734 to pp4737 in the P. putida KT2440 genome (34). The protein sequence was analyzed using the Protein BLAST software (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome). The result revealed that this protein has an N-terminal FAD/FMN-containing dehydrogenase domain (coded as COG0277 in the cluster of orthologous groups [COGs] of proteins [35]) located at positions 10 to 519 and a C-terminal Fe-S oxidoreductase domain (COG0247) located at positions 531 to 936 (Fig. 1). Each of the two domains also contains several subdomains. The sequence of the FAD/FMN-containing dehydrogenase domain showed similarity to glycolate oxidase subunit D (GlcD) and mitochondrial d-lactate dehydrogenase (DLD) from various species. However, it has no appreciable sequence similarity to the well-characterized quinone-dependent d-iLDH from E. coli. Sequence alignment of this domain and its homologs shows that all of these proteins contain the characteristic sequence motif of the FAD-binding type 4 family (FAD-binding-4, coded as PF01565 in the Pfam database [36]) (Fig. 2). Although enzymes in this family exhibit minimal sequence conservation overall (37), a consensus sequence was determined based on sequence alignment and the resolved structures (37, 38). Most of the corresponding residues in the Fe-S d-iLDH from KT2440 are in accordance with the consensus sequence of this family, with two exceptions. The Thr-74 is not in accordance with the His residue at this position, and the Lys-138 is a positively charged but not a small amino acid (38). Besides, a C-terminal FAD-linked oxidase domain (FAD-oxidase-C, PF02913) was also observed in the sequence (Fig. 1).
FIG 1.
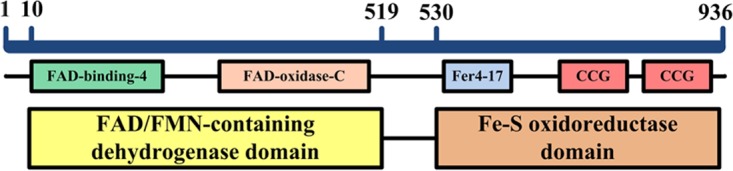
Outline of Fe-S d-iLDH primary structure.
FIG 2.

Multiple-sequence alignment of flavin binding domain of Fe-S d-iLDH and its homologs. PP Fe-S d-iLDH 1-519, predicted flavin binding domain of Fe-S d-iLDH from P. putida KT2440 (accession no. AAN70309.1); SO Fe-S d-iLDH 1-518, predicted flavin binding domain of Fe-S d-iLDH from S. oneidensis MR-1 (accession no. AAN54582.2); CJ Fe-S d-iLDH 1-513, predicted flavin binding domain of Fe-S d-iLDH from C. jejuni NCTC 11168 (accession no. CAL35682.1); SC DLD, d-lactate dehydrogenase from S. cerevisiae (accession no. CAA91571.1); EC GlcD, glycolate oxidase subunit D from E. coli (accession no. CAE85245.1). Blue boxes indicate sites that have high similarities among the sequences. White letters indicate identical residues; red letters indicate that residues are not identical but belong to the same group (alkaline, acidic, nonpolar, or nonionizing polar amino acids) or that vacancies occur in some sequences, with residues in other sequences being identical or similar. The underlined regions are the consensus sequence of FAD-binding site (s, small amino acids [G, A, S, and T]; h, hydrophobic amino acids [I, L, and V]; G, glycine; H, histidine) (38), and the asterisks indicate sites that do not accord with the consensus sequence.
Among the proteins showing similarity to the Fe-S oxidoreductase domain, subunit F of glycolate oxidase (GlcF) and subunit C of anaerobic glycerol-3-phosphate dehydrogenase were prominent. These enzymes are multimeric proteins with similar catalytic mechanisms, which oxidize molecules and transfer electrons through Fe-S subunits (39). There are 15 conserved cysteine residues in these sequences (Fig. 3). A Fer4-17 subdomain (PF13534) and two CCG subdomains (PF02754) were observed (Fig. 1). The Fer4-17 subdomain is a [4Fe-4S] dicluster-binding domain. However, one of the [4Fe-4S] cluster-binding motifs in the Fe-S d-iLDH from KT2440 lacks a cysteine, which is replaced by a threonine (T597), and such a motif might bind a [3Fe-4S] cluster instead (40). The cysteine-rich CCG subdomain has also been reported to bind to a [4Fe-4S] cluster (41, 42). Therefore, the protein is predicted to bind three [4Fe-4S] clusters and a [3Fe-4S] cluster overall.
FIG 3.

Multiple-sequence alignment of Fe-S oxidoreductase domain of Fe-S d-iLDH and its homologs. PP Fe-S d-iLDH 531-936, predicted Fe-S oxidoreductase domain of Fe-S d-iLDH from P. putida KT2440 (accession no. AAN70309.1); SO Fe-S d-iLDH 535-934, predicted Fe-S oxidoreductase domain of Fe-S d-iLDH from S. oneidensis MR-1 (accession no. AAN54582.2); CJ Fe-S d-iLDH 529-923, predicted Fe-S oxidoreductase domain of Fe-S d-iLDH from C. jejuni NCTC 11168 (accession no. CAL35682.1); EC GlpC, glycerol-3-phosphate dehydrogenase subunit C from E. coli (accession no. AAC75303.1); EC GlcF, glycolate oxidase subunit F from E. coli (accession no. AJE57532.1). Blue boxes indicate sites that have high similarities among the sequences. White letters indicate identical residues; red letters indicate that residues are not identical but belong to the same group (alkaline, acidic, nonpolar, or nonionizing polar amino acids) or that vacancies occur in some sequences, with residues in other sequences being identical or similar. The underlined regions are the consensus sequence of Fe-S cluster-binding sites (C, cysteine; P, proline; G, glycine), and the asterisks indicates the sites that do not accord with the consensus sequence.
Expression and purification of Fe-S d-iLDH.
Fe-S d-iLDH from P. putida KT2440 was heterologously expressed in E. coli C43(DE3) and purified as a His-tagged protein. The results of the purification procedure are summarized in Fig. 4A and Table S1. The specific activity after the final step was 44.5 U per mg of protein, which was a 35.6-fold increase over that of the crude cell extract. The molecular masses of the protein as estimated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. 4A) and gel filtration chromatography (Fig. 4B) were 100 kDa and 118 kDa, respectively, suggesting a monomeric form of Fe-S d-iLDH.
FIG 4.

Purification, molecular mass, and flavin cofactor analysis of recombinant Fe-S d-iLDH from P. putida KT2440. (A) SDS-PAGE analysis of the purified recombinant Fe-S d-iLDH. Lane M, molecular mass markers; lane 1, crude extract of E. coli C43(DE3) carrying the empty pETDuet-1 plasmid; lane 2, crude extract of E. coli C43(DE3) carrying the pET-lldE plasmid; lane 3, sample obtained from Ni2+-nitrilotriacetic acid (NTA) chromatography; lane 4, sample obtained from DEAE anion-exchange chromatography; lane 5, sample obtained from gel filtration chromatography. (B) Gel filtration chromatography of Fe-S d-iLDH on a Superdex 200 10/300 GL column. The standard molecular mass markers are thyroglobulin (660 kDa), ferritin (440 kDa), catalase (250 kDa), aldolase (158 kDa), lactic acid dehydrogenase (140 kDa), bovine serum albumin (67 kDa), chymotrypsinogen A (25 kDa), and RNase A (13.7 kDa). Ve/V0, ratio of elution volume to void volume of the column. (C) HPLC analysis of the flavin cofactor of Fe-S d-iLDH. (D) The fluorescence of authentic FAD before and after treatment with phosphodiesterase I. (E) The fluorescence of authentic FMN before and after treatment with phosphodiesterase I. (F) The fluorescence of flavin cofactor of Fe-S d-iLDH before and after treatment with phosphodiesterase I.
The flavin cofactor of Fe-S d-iLDH was investigated using heat denaturation of the protein, followed by high-performance liquid chromatography (HPLC) (Fig. 4C) and fluorescence spectroscopy (Fig. 4D to F). Both results demonstrated the presence of FAD as the cofactor of Fe-S d-iLDH. An analysis of the flavin contents of several preparations of homogeneous enzyme determined the ratio of Fe-S d-iLDH to FAD to be 0.92. Therefore, the native Fe-S d-iLDH protein should contain one FAD molecule.
2-Hydroxyacid oxidization activity of Fe-S d-iLDH.
The substrate specificity of the recombinant Fe-S d-iLDH was examined with 20 mM 2-hydroxyacids (d-lactate, l-lactate, glycolate, d-2-hydroxybutyrate, l-2-hydroxybutyrate, d-mandelate, l-mandelate, d-3-phenyllactate, d-glycerate, l-glycerate, dl-2-hydroxyisocaproate, and dl-2-hydroxyoctanoate), with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) as an electron acceptor (see Fig. S1A in the supplemental material). The Fe-S d-iLDH exhibited narrow substrate specificity. Only d-lactate, d-2-hydroxybutyrate, and d-glycerate were clearly oxidized by the enzyme. Notably, it also exhibited activity toward l-lactate. The effects of metal ions on the activity of the Fe-S d-iLDH were tested by adding metal salts (final concentration of 5 mM) to the assay buffer. As shown in Fig. S1B, K+ exhibited a slightly positive effect on Fe-S d-iLDH activity. Ni2+, Ba2+, Ca2+, Mg2+, and EDTA partially inhibited the activity, while Cu2+, Fe2+, Cd2+, Co2+, Mn2+, and Zn2+ completely inhibited its activity.
The rate of oxidization of the substrates catalyzed by recombinant Fe-S d-iLDH followed Michaelis-Menten kinetics. Double-reciprocal plots of the initial rates plotted against the concentrations of d-lactate, l-lactate, d-2-hydroxybutyrate, and d-glycerate yielded Km values of 57 ± 7.8, 1,040 ± 50, 100 ± 5.3, and 1,380 ± 140 μM, respectively. The kcat values were estimated to be 120 ± 19, 9.5 ± 1.1, 66 ± 5.0, and 8.0 ± 1.4 s−1, respectively, with MTT as the electron acceptor (Table 1).
TABLE 1.
Kinetic parameters of Fe-S d-iLDH and FAD-containing dehydrogenase domain toward different substratesa
| Substrate | Fe-S d-iLDH |
FAD-containing dehydrogenase domain |
||||
|---|---|---|---|---|---|---|
| kcat (s−1) | Km (μM) | kcat/Km (μM−1 · s−1) | kcat (s−1) | Km (μM) | kcat/Km (μM−1 · s−1) | |
| d-Lactate | 120 ± 19 | 57 ± 7.8 | 2.1 | 17 ± 1.5 | 94 ± 11 | 0.176 |
| l-Lactate | 9.5 ± 1.1 | 1,040 ± 50 | 9 × 10−3 | 1.5 ± 0.3 | 1,780 ± 270 | 0.84 × 10−3 |
| d-2-Hydroxybutyrate | 66 ± 5.0 | 100 ± 5.3 | 0.66 | 13 ± 0.65 | 170 ± 16 | 0.078 |
| d-Glycerate | 8.0 ± 1.4 | 1,380 ± 140 | 6 × 10−3 | 0.78 ± 0.1 | 1,670 ± 140 | 0.47 × 10−3 |
All experiments were carried out with 0.2 mM MTT as the electron acceptor. Values are the mean ± SD from three parallel replicates.
Electron acceptor of Fe-S d-iLDH.
The type of natural electron acceptor could be used as the basis of classification of iLDHs (3); thus, it was also investigated for Fe-S d-iLDH. As a respiratory lactate dehydrogenase, the electron transfer pathway via Fe-S d-iLDH could be analyzed by studying the inhibition effects of typical electron transport chain inhibitors. Whole cells of P. putida KT2440 ΔldhA ΔglcD, which uses Fe-S d-iLDH as the sole d-lactate-oxidizing enzyme (34), were used for these experiments. The biocatalysis rate from d-lactate to pyruvate was assayed to evaluate the electron transfer activity of Fe-S d-iLDH, with antimycin A or NaN3 added as an inhibitor. Antimycin A inhibits electron transfer from quinone to cytochrome c, while NaN3 inhibits electron transfer from cytochrome c to molecular oxygen. As shown in Table 2, compared to a control with no inhibitor, the d-lactate oxidization activity was reduced more than 70% when either antimycin A or NaN3 was added to the reaction system. This result suggested that both the electron transfer from quinone to cytochrome c and from cytochrome c to molecular oxygen could occur during Fe-S d-iLDH-mediated d-lactate oxidization in vivo. The kinetic parameters of Fe-S d-iLDH with different electron acceptors were also assayed (Table 3). Compare to the values obtained with cytochrome c, the Km with coenzyme Q10 was lower by an order of magnitude, while the kcat/Km ratio was higher by two orders of magnitude. These results suggest that Fe-S d-iLDH would be most likely to utilize quinone as the natural electron acceptor.
TABLE 2.
Effects of different inhibitors on the d-lactate-oxidizing activities of P. putida KT2440 ΔldhA ΔglcD
| Inhibitor | d-Lactate-oxidizing activity (U OD−1)a | Inhibition ratio (%) |
|---|---|---|
| No inhibitor | 0.072 ± 0.004 | 0 |
| Antimycin A | 0.016 ± 0.0003 | 77.6 |
| NaN3 | 0.019 ± 0.002 | 73.4 |
Values are the mean ± SD from three parallel replicates. OD, optical density.
TABLE 3.
Kinetics of Fe-S d-iLDH and FAD-containing dehydrogenase domain with different electron acceptorsa
| Electron acceptor | Fe-S d-iLDH |
FAD-containing dehydrogenase domain |
||||
|---|---|---|---|---|---|---|
| kcat (s−1) | Km (μM) | kcat/Km (μM−1 · s−1) | kcat (s−1) | Km (μM) | kcat/Km (μM−1 · s−1) | |
| MTT | 128 ± 5.7 | 48 ± 1.9 | 2.7 | 5.3 ± 0.97 | 31 ± 4.1 | 0.17 |
| Coenzyme Q10 | 2.4 ± 0.029 | 4.1 ± 0.35 | 0.59 | ND | ND | ND |
| Cytochrome c | 0.49 ± 0.04 | 51.5 ± 1.7 | 9.5 × 10−3 | 2.0 ± 0.07 | 9.5 ± 0.33 | 0.21 |
All experiments were carried out with 1.0 mM d-lactate as the substrate. ND, not detected. kcat and Km values are the mean ± SD from three parallel replicates.
Expression and purification of FAD-containing dehydrogenase domain.
Since the FAD-containing dehydrogenase domain of Fe-S d-iLDH is similar to that of many other integral flavoenzymes, we attempted to overexpress it separately to explore its properties. The amino acids from 1 to 530 of Fe-S d-iLDH (Fe-S d-iLDH 1-530) were expressed as the recombinant FAD-containing dehydrogenase domain with a His tag after trying to construct different C-terminal domains (Fig. S2). Detergent was found to be unnecessary for purification, suggesting that it may be not membrane associated. The protein appears to be in a monomeric form, since its molecular mass was estimated to be 60 kDa by SDS-PAGE and 64 kDa by gel filtration chromatography (Fig. 5A and B). The purified protein was yellow, and its absorption spectrum with two peaks at around 375 nm and 450 nm was in accordance with that of typical flavin-containing proteins (Fig. 5C). HPLC analysis indicated that 1 mol FAD-containing dehydrogenase domain contained 0.96 mol FAD, which is in accordance with the results using the full Fe-S d-iLDH. Additionally, the purified FAD-containing dehydrogenase domain contained no iron, as analyzed by a Ferene S method assay kit (data not shown).
FIG 5.

Purification of recombinant FAD-containing dehydrogenase domain (Fe-S d-iLDH 1-530). (A) SDS-PAGE analysis of the purified FAD-containing dehydrogenase domain. Lane M, molecular mass markers; lane 1, crude extract of E. coli C43(DE3) carrying the empty pETDuet-1 plasmid; lane 2, crude extract of E. coli C43(DE3) carrying the pET-lldEFAD2 plasmid; lane 3, sample obtained from Ni2+-NTA chromatography. (B) Gel filtration chromatography of FAD-containing dehydrogenase domain on a Superdex 200 10/300 GL column. The standard molecular mass markers are thyroglobulin (660 kDa), ferritin (440 kDa), catalase (250 kDa), aldolase (158 kDa), lactic acid dehydrogenase (140 kDa), bovine serum albumin (67 kDa), chymotrypsinogen A (25 kDa), and RNase A (13.7 kDa). Ve/V0, ratio of elution volume to void volume of the column. (C) The absorption spectrum of purified FAD-containing dehydrogenase domain.
Comparison of the FAD-containing dehydrogenase domain with the full Fe-S d-iLDH.
Activity assays showed that the recombinant FAD-containing dehydrogenase domain alone retained its oxidization activity with 2-hydroxyacids. These properties were compared with the full Fe-S d-iLDH. The activities and stabilities as a function of pH did not differ dramatically from those of the intact Fe-S d-iLDH (Fig. S3A and B). However, the FAD-containing dehydrogenase domain showed a higher optimal temperature and increased thermostability (Fig. S3C and D). The kinetic parameters of the FAD-containing dehydrogenase domain were also determined. As shown in Table 1, the Km values of the FAD-containing dehydrogenase domain with d-lactate, l-lactate, d-2-hydroxybutyrate, and d-glycerate were 94 ± 11, 1,780 ± 270, 170 ± 16, and 1,670 ± 140 μM, respectively, which were not substantially different from the values obtained with the full Fe-S d-iLDH. However, the kcat values with these substrates decreased to different extents, and the catalytic efficiencies (based on the kcat/Km values) decreased by 11.9-, 10.7-, 8.5-, and 12.8-fold with d-lactate, l-lactate, d-2-hydroxybutyrate, and d-glycerate, respectively.
As for the electron acceptors, the FAD-containing dehydrogenase domain exhibited a decreased kcat value with the artificial electron acceptor MTT, but the Km value did not change much. The truncated domain completely lost the ability to use coenzyme Q10 as an electron acceptor. However, a lower Km value and higher kcat value with cytochrome c were observed for the FAD-containing dehydrogenase domain compared to the full Fe-S d-iLDH (Table 3). These results suggest that in contrast to the intact Fe-S d-iLDH, the FAD-containing dehydrogenase domain preferentially uses cytochrome c rather than coenzyme Q10 as an electron acceptor. It is likely that individual expression of this domain exposes a site on the protein interacting with cytochrome c that is not physiologically necessary.
Expression and purification of the Fe-S oxidoreductase domain.
Several Fe-S cluster-binding motifs were predicted in Fe-S d-iLDH, but they still needed to be experimentally confirmed. Since the sequence of the Fe-S oxidoreductase domain covers all the predicted Fe-S cluster-binding motifs, we also tried to overexpress this domain separately to facilitate site-directed mutations. The amino acids from 531 to 936 of Fe-S d-iLDH (Fe-S d-iLDH 531-936) were expressed as the recombinant Fe-S oxidoreductase domain, with a His tag, after trying to construct different N-terminal domains (Fig. S2). The molecular mass of the purified Fe-S oxidoreductase domain detected by SDS-PAGE agreed with the theoretical value 46 kDa (Fig. 6A), and the solution exhibited an absorption spectrum characteristic of Fe-S proteins, with a shoulder at 330 nm and a distinctive peak at 420 nm (Fig. 6B) (43). Assays measuring the iron and protein concentrations of the purified protein solution showed that the iron-to-protein ratio of the Fe-S oxidoreductase domain was 2.8 (Table 4), suggesting that the Fe-S clusters might be incompletely assembled due to the protein overexpression process. Thus, reconstitution of the Fe-S clusters was then performed using the purified protein. The iron-to-protein ratio after reconstitution rose to 17.7 (Table 4), which is slightly higher than the theoretical value when binding three [4Fe-4S] clusters and a [3Fe-4S] cluster, which was predicted based on protein sequence.
FIG 6.

Purification of recombinant Fe-S oxidoreductase domain (Fe-S d-iLDH 531-936) and identification of Fe-S cluster-binding sites. (A) SDS-PAGE analysis of the purified Fe-S oxidoreductase domain. Lane M, molecular mass markers; lane 1, crude extract of E. coli C43(DE3) carrying the empty pETDuet-1 plasmid; lane 2, crude extract of E. coli C43(DE3) carrying the pET-lldEFeS3 plasmid; lane 3, sample obtained from Ni2+-NTA chromatography. (B) The absorption spectrum of purified Fe-S oxidoreductase domain and its derivates. (C) Absorption spectrum of Fe-S oxidoreductase domain and its derivates after Fe-S cluster reconstitution. (B and C) M-1, M-2, M-4, M-6, and M-7 stand for C540S, C540S-C594S, C540S-C594S-C751S-C752S, C540S-C594S-C751S-C752S-C540S-C594S, and C540S-C543S-C594S-C751S-C752S-C540S-C594S mutated Fe-S oxidoreductase domains, respectively.
TABLE 4.
Iron-to-protein ratio of purified Fe-S oxidoreductase domain and its derivates
| Proteina | Iron-to-protein ratiob |
|
|---|---|---|
| Before reconstitution | After reconstitution | |
| Fe-S oxidoreductase domain | 2.84 ± 0.035 | 17.71 ± 0.62 |
| M-1 | 2.80 ± 0.12 | 14.03 ± 0.31 |
| M-2 | 1.73 ± 0.12 | 13.58 ± 0.61 |
| M-4 | 0.79 ± 0.047 | 10.16 ± 0.17 |
| M-6 | 0.26 ± 0.017 | 8.07 ± 0.38 |
| M-7 | 0.033 ± 0.0024 | 4.51 ± 0.32 |
M-1, M-2, M-4, M-6, and M-7 stand for C540S, C540S-C594S, C540S-C594S-C751S-C752S, C540S-C594S-C751S-C752S-C540S-C594S, and C540S-C543S-C594S-C751S-C752S-C540S-C594S mutated Fe-S oxidoreductase domains, respectively.
Values are the mean ± SD from three parallel replicates.
Identification of Fe-S cluster-binding motifs.
Variations of the conserved cysteine residues to serines were made in the Fe-S oxidoreductase domain to confirm the Fe-S cluster-binding motifs. Sequence alignment shows that C540, C543, C546, and C550 could form a conserved [4Fe-4S] cluster-binding motif; C594, C600, and C604 could form a [3Fe-4S] cluster-binding motif; C709, C751, C752, and C794 could form a CCG subdomain; and C839, C869, C870, and C908 form another CCG subdomain. Based on these predictions, C540S, C540S-C594S, C540S-C594S-C751S-C752S, and C540S-C594S-C751S-C752S-C540S-C594S variant proteins of Fe-S oxidoreductase domain were constructed in a step-by-step manner to disrupt the binding of each Fe-S cluster (Fig. S4). However, the UV-visible absorption spectrum of the sextuple-variant protein still exhibited a weak peak at 420 nm (Fig. 6B). It has been reported that a mutation of one of the four cysteine residues that binds a [4Fe-4S] cluster would lead to a conformation change, leading the protein to bind to a [3Fe-4S] cluster instead (40). Thus, the C543 was further changed to serine to construct the septuple variant C540S-C543S-C594S-C751S-C752S-C540S-C594S Fe-S oxidoreductase domain. This protein lost the characteristic Fe-S protein absorption spectrum completely (Fig. 6B). Like the wild-type Fe-S oxidoreductase domain, the iron-to-protein ratios of these variants (except the septuple variant) were all dramatically lower than the theoretical values; hence, Fe-S cluster reconstitution was also performed with these proteins. Although the multiple variations of amino acids might affect the protein conformation, all of the proteins exhibited increased absorption at 330 nm and 420 nm after reconstitution, and the septuple-variant protein lacked the characteristic spectrum of Fe-S proteins (Fig. 6C). As a result, these variations preliminarily confirmed our predicted Fe-S cluster-binding sites in Fe-S d-iLDH. However, like the wild-type protein, the iron-to-protein ratios of these proteins after reconstitution were all close to but higher than the theoretical values, and the septuple-variant protein still had an iron-to-protein ratio of 4.51 (Table 4). This may be caused by the binding of iron to the His tag of recombinant proteins, since histidine exhibits strong interaction with metal ions (44).
The Fe-S oxidoreductase domain is indispensable for the in vivo function of Fe-S d-iLDH.
Since the Fe-S oxidoreductase domain has been demonstrated to be unnecessary for substrate oxidization by Fe-S d-iLDH, its function within the enzyme was further investigated. As many Fe-S proteins are involved in electron transfer systems, it was speculated that the Fe-S oxidoreductase domain may play the same role in Fe-S d-iLDH, and thus, the lack of this domain may impair the utilization of d-lactate in P. putida KT2440. To test this hypothesis, the FAD-containing dehydrogenase domain was expressed in the P. putida KT2440 ΔlldE ΔglcD strain, in which d-lactate utilization was completely eliminated (34). The resulting strain was investigated for its ability to use d-lactate as a carbon source. The results showed that this strain could not grow on minimal medium supplied with d-lactate as the sole carbon source. In contrast, P. putida KT2440 ΔlldE ΔglcD complemented with the lldE gene encoding the full Fe-S d-iLDH grew similarly to the wild-type strain (Fig. S5). This result demonstrates that deletion of the Fe-S oxidoreductase domain of Fe-S d-iLDH impairs its d-lactate utilization function in vivo.
The Fe-S oxidoreductase domain helps Fe-S d-iLDH associate with the cell membrane.
Like with many enzymes involved in the respiratory chain, Fe-S d-iLDH is associated with cell membranes (34), and a detergent was needed for the purification of Fe-S d-iLDH. However, it was noted that there was no need for detergent for the purification of the FAD-containing dehydrogenase domain, which suggested that it may not be a membrane-associated protein. To identify if the Fe-S oxidoreductase domain is necessary for membrane binding of Fe-S d-iLDH, the distribution of d-lactate oxidization activities in E. coli C43(DE3) strains expressing Fe-S d-iLDH or the FAD-containing dehydrogenase domain was analyzed. The membrane fraction of the cell extract was separated using ultracentrifugation, and the MTT-related d-lactate oxidization activity of each fraction was assayed (Fig. 7). Since E. coli has a constitutive membrane-associated d-iLDH (15), E. coli C43(DE3) carrying an empty pETDuet-1 plasmid was also assayed as a control. Compared to the control strain, the specific activity increased significantly in the crude cell extract and supernatant but decreased slightly in the precipitate of the strain expressing the FAD-containing dehydrogenase domain. This suggests that the heterogeneously expressed protein remains in the soluble fraction. For the strain expressing Fe-S d-iLDH, the activity increased in both fractions. However, the increased activity in the supernatant might be due to some of the overexpressed Fe-S d-iLDH protein not binding tightly to the membrane. These results confirm that the FAD-containing dehydrogenase domain is no longer associated with the cell membrane.
FIG 7.
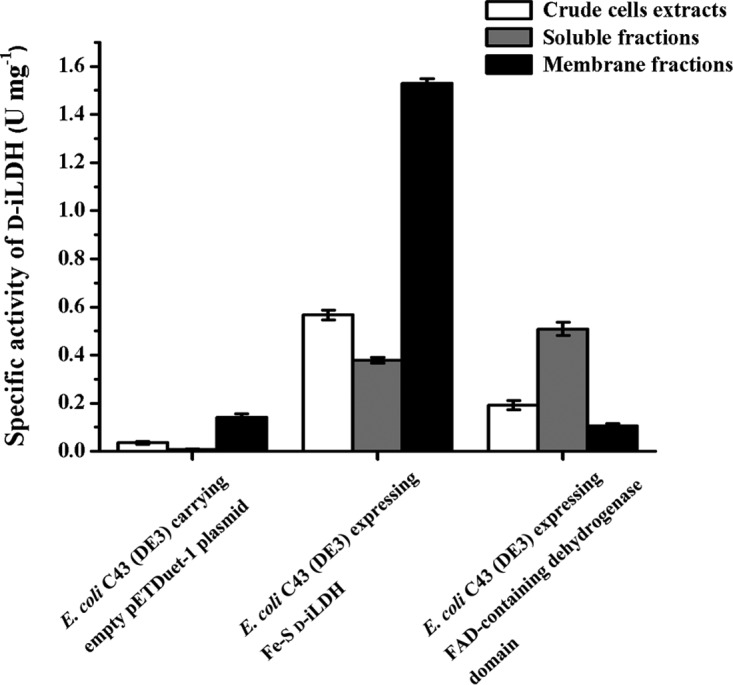
Distribution of d-lactate oxidization activities in E. coli C43(DE3) strains expressing Fe-S d-iLDH or FAD-containing dehydrogenase domain. Values are the mean ± standard deviation (SD) from three parallel replicates.
DISCUSSION
Microbial iLDHs give microbes the ability to use lactate as a source of carbon and energy for growth, which could be an advantage for microbes dwelling in habitats that are rich in organic acids, including lactate (34). These enzymes exhibit different substrate specificities, utilize different cofactors and electron acceptors, and have different catalytic mechanisms. Hence, we have grouped them in a previous review (3). In general, the d-lactate-specific iLDHs are not as well understood as l-iLDHs (22–26). Most microbial d-iLDHs belong to the FAD-binding type 4 family, and previously, they have been classified into three groups: quinone-dependent d-iLDH (Q d-iLDH), cytochrome c-dependent d-iLDH (Cyt d-iLDH), and Fe-S d-iLDH (3). The structure of the Q d-iLDH from E. coli has been resolved (15). A membrane-binding subdomain (PF09330) at the C terminus is a distinctive feature of this type of d-iLDH (Fig. S6). The Cyt d-iLDH, which are distributed more widely, have a specific FAD oxidase C subdomain (PF02913). Interestingly, a recently characterized d-iLDH from Gluconobacter oxydans (denoted DLOX), which has similarity to cytochrome c-dependent d-iLDH, was demonstrated to use molecular oxygen as an electron acceptor (19). The phylogenetic relationships between these d-iLDH enzymes were analyzed with two other proteins that have sequence features resembling Cyt d-iLDH (Fig. 8).
FIG 8.

Evolutionary relationships of d-iLDHs and other related FAD-binding-4 family proteins from various bacterial genera. The phylogenetic tree was constructed using neighbor-joining method with Mega 5 software. Bootstrap values (percent) are for 500 replicates. The scale at the bottom indicates sequence divergence. The accession numbers (from the National Center for Biotechnology Information [NCBI]) are given in parentheses. The triangle indicates the Fe-S d-iLDH characterized in this work.
The first additional protein is GlcD, the flavin-containing subunit of a multisubunit bacterial glycolate dehydrogenase GlcDEF (encoded by the glcDEF gene cluster). It was reported that in some cases, glycolate dehydrogenase catalyzes the oxidization of both d-lactate and glycolate or even d-lactate only (34, 45). The second additional protein is d-2-hydroxyglutarate dehydrogenase (D2HGDH), which is phylogenetically related to d-iLDH (14). As shown in the phylogenetic tree, the Q d-iLDHs have the most distant relationship with the other enzymes, indicating that the alterations of the FAD oxidase C subdomain and membrane-binding subdomain should be important events during evolution. Among the five other types of enzymes, the Fe-S d-iLDHs are clustered in a separate clade. It is possible that the divergence of these proteins began with the retention or loss of the Fe-S cluster-containing domain, while the divergence in substrates or electron acceptors occurred later. The GlcF subunit of GlcDEF has similar sequence features to the Fe-S oxidoreductase domain of Fe-S d-iLDH (Fig. S6). Thus, it appears that Fe-S d-iLDH is a merged form of GlcDEF without GlcE (another flavin-containing subunit). Interestingly, it was found that the glcD genes in several bacterial species were not clustered with both glcE and glcF but with only glcF. The phylogenetic relationship of these GlcD proteins (denoted DF-GlcD), the typical GlcDs (whose genes are clustered with both glcE and glcF), and the predicted FAD-containing dehydrogenase domains of Fe-S d-iLDHs were further analyzed (Fig. S7). It can be seen that DF-GlcDs and typical GlcDs are clustered in different clades, suggesting that divergence occurred in GlcD proteins accompanied by the retention or loss of GlcE. Therefore, GlcDF might have existed as an intermediate form during the transformation between GlcDEF and Fe-S d-iLDH. Nevertheless, the hypothesis needs to be confirmed by further studies.
While there have been characterizations of similar enzymes, such as DLOX (19), Cyt d-iLDH (7), Q d-iLDH (15, 46), GlcD (39), and D2HGDH (47), the Fe-S d-iLDH characterized in this study is more unusual, although comparative genome analysis shows that it is widely distributed among bacterial species (17). The homolog of Fe-S d-iLDH from S. oneidensis or C. jejuni was identified to be responsible for d-lactate or l-lactate utilization, respectively, while the purified enzyme from C. jejuni (Cj1585c) exhibited higher activity with d-lactate than with l-lactate (17, 18). In the case of P. putida KT2440, the Fe-S d-iLDH has been confirmed to be the major d-lactate-oxidizing enzyme, and it does not participate in l-lactate utilization (34). In this study, we determined the substrate spectrum of purified Fe-S d-iLDH. In accordance with the results using Cj1585c, Fe-S d-iLDH from P. putida KT2440 exhibits activity with both d- and l-lactate, although the activity with l-lactate is relatively low. Compared with the l-iLDHs that are usually strict in chiral specificity (6, 48), both the characterized Q d-iLDH and Fe-S d-iLDH exhibit a relatively loose selectivity in the chirality of lactate (46, 49). These facts suggest that the substrate specificity of Fe-S d-iLDH does not determine its physiological function, and the ability of Cj1585c in C. jejuni to utilize l-lactate requires further investigation.
Based on the protein sequence analysis, the most distinctive feature of Fe-S d-iLDH is that it has an Fe-S oxidoreductase domain besides the FAD-containing dehydrogenase domain. The two domains are clearly separated, and each of them shows similarity to a certain group of proteins. The unusual form of this enzyme leaves some questions, like if the additional domain is necessary for its catalytic activity or physiological function, what role the additional domain plays in its catalytic process, and what is the connection between the two domains. As a member of the α-hydroxyacid oxidase family, Fe-S d-iLDH was identified as a monomeric protein. In contrast, several other members of this protein family, which do not have cofactors other than flavin, have been confirmed to be homotetramers (22, 24, 50). This difference leads to a speculation that it is the presence of the Fe-S oxidoreductase domain that affects the multimerization of Fe-S d-iLDH. However, the FAD-containing dehydrogenase domain expressed separately was also a monomeric protein, which refutes this hypothesis. As a part of the whole enzyme, the FAD-containing dehydrogenase domain retained the ability to bind to the same amount of FAD cofactor and still showed substrate oxidization activity. The substrate binding capacities of the FAD-containing dehydrogenase domain also did not change substantially from that of the intact Fe-S d-iLDH. However, the absence of the Fe-S oxidoreductase domain should have an effect on the conformation of the FAD-containing dehydrogenase domain. First, decreased MTT-related catalytic efficiencies were observed with the FAD-containing dehydrogenase domain, and its kinetic parameters with MTT exclude issues with MTT binding. Second, the purified FAD-containing dehydrogenase domain showed more resistance to heat than the intact Fe-S d-iLDH. Based on these facts, we can speculate that the FAD-containing dehydrogenase domain expressed separately should have a changed conformation compared to when it is linked with the Fe-S oxidoreductase domain. This change would not affect the binding of FAD, 2-hydroxyacids, or MTT; although it makes the protein more stable, it impairs the substrate-oxidizing process to some extent.
The C-terminal domain of Fe-S d-iLDH is rich in Fe-S cluster-binding motifs. Fe-S clusters can accept or donate electrons and, thus, are involved as cofactors in electron transfer systems (51). In this study, the Fe-S cluster-binding motifs predicted by sequence alignment were preliminarily validated by mutagenesis. Nevertheless, further quantitative or qualitative analysis with spectroscopic techniques, like variable-temperature magnetic circular dichroism (VTMCD) and electron paramagnetic resonance (EPR), will provide more accurate information regarding these Fe-S clusters. Some Fe-S clusters containing electron transfer components, such as cytochromes, have been reported to exist in either an independent or a merged form (52–54). One of the most well-characterized cytochrome fused proteins is flavocytochrome b2, an l-iLDHs from Saccharomyces cerevisiae. The cytochrome domain mediates electron transfer from FMN to cytochrome c. Interruption of the intraprotein electron transfer in flavocytochrome b2 does not affect the lactate dehydrogenase function of the enzyme, suggesting that there is a clear-cut separation of functions in these two domains (53). In contrast, in the case of an ortho-nitrophenol 2-monooxygenase with a fused cytochrome b5, the cytochrome b5 domain was identified to impose a significant effect on the activity of the enzyme (54). In this study, it was demonstrated that, although the lack of Fe-S oxidoreductase domain does not abolish substrate oxidization activities, it does cause a complete loss of d-lactate utilization by Fe-S d-iLDH. This result suggests that the Fe-S oxidoreductase domain plays an indispensable role in the process of electron transfer from d-lactate to the natural acceptors. Meanwhile, we have shown that Fe-S d-iLDH prefers to utilize quinone as an electron acceptor. Therefore, it can be speculated that it is the Fe-S oxidoreductase domain that accommodates the quinone molecule. We also showed that the FAD-containing dehydrogenase domain alone could not utilize quinone but had increased catalytic efficiency with cytochrome c as an electron acceptor. Considering that the sequence features of cytochrome c-dependent d-iLDHs are similar to those of the FAD-containing dehydrogenase domain, it could be speculated that the two types of d-iLDHs are phylogenetically related. The Fe-S oxidoreductase domain functions as an accessory to facilitate the utilization of quinone as an electron acceptor, while it also hinders the utilization of cytochrome c.
Another feature of Fe-S proteins is that they are often linked to biomembranes (55–57). Fe-S d-iLDH has been previously identified as a membrane-associated enzyme (34), but no possible transmembrane helix was predicted for the whole Fe-S d-iLDH (http://www.ch.embnet.org/software/TMPRED_form.html). In this study, we show that the FAD-containing dehydrogenase domain alone is a soluble protein. Therefore, it is possible that the Fe-S oxidoreductase domain also functions as the membrane-binding region. It has been demonstrated that some peripheral membrane proteins associate with membranes by a positively charged surface that interacts with the negatively charged phospholipid head groups of the membrane (15, 58). Interestingly, the theoretical isoelectric points of intact Fe-S d-iLDH, FAD-containing dehydrogenase domain, and Fe-S oxidoreductase domain were predicted to be 6.38, 5.33, and 8.62, respectively (http://web.expasy.org/compute_pi/). The high isoelectric point of the Fe-S oxidoreductase domain suggests that it is positively charged under physiological conditions. Therefore, although the specific region has not been identified, Fe-S d-iLDH may also use electrostatic forces in the Fe-S oxidoreductase domain to associate with membranes. Nevertheless, another possibility is that a loss of the Fe-S oxidoreductase domain induces a conformational change in the FAD-containing dehydrogenase domain, impeding its association with the cell membrane. As a part of the respiratory chain, the link between the enzyme and cell membrane might facilitate electron transfer to enhance catalytic efficiency in vivo.
In summary, the investigation of Fe-S d-iLDH in this study provides a better understanding of this novel enzyme. A model for d-lactate utilization by the Fe-S d-iLDH in P. putida KT2440 is proposed based on these results (Fig. 9). The enzyme can accommodate d-lactate, l-lactate, d-2-hydroxybutyrate, and d-glycerate as substrates and likely utilizes quinone as an electron acceptor. Compared to other typical d-iLDH, this enzyme's most distinctive feature is the additional Fe-S oxidoreductase domain, which is distinct from the FAD-containing dehydrogenase domain where substrate oxidization occurs. This domain binds several Fe-S clusters as electron transfer cofactors, and it is crucial for the in vivo d-lactate utilization function of Fe-S d-iLDH. In addition to being an electron transfer component, it also helps the enzyme bind to the cell membrane. Furthermore, this domain could be the key factor that determines the type of electron acceptor utilized by the enzyme. To further understand the catalytic mechanism and the role of the Fe-S oxidoreductase domain, detailed information, especially in regard to the protein structure, is needed. Moreover, iLDHs are good candidates for developing biosensors and biocatalysts. Thus, a better understanding of the novel d-iLDH could facilitate its application in the development of these techniques.
FIG 9.

Proposed model for d-lactate utilization via the Fe-S d-iLDH in P. putida KT2440. The gray area indicates cell membrane. The Fe-S d-iLDH could be a phospholipid-anchored membrane protein. The − symbols indicate the negatively charged phospholipid head groups of the membrane, and + symbols indicate the supposed positively charged surface of Fe-S d-iLDH. Gray arrows indicate the electron transfer pathway. The LldP is the lactate permease encoded by pp4735 (34).
MATERIALS AND METHODS
Chemicals.
l-Lactate, glycerate, d-2-hydroxybutyrate, l-2-hydroxybutyrate, d-mandelate, l-mandelate, d-3-phenyllactate, d-glycerate, l-glycerate, dl-2-hydroxyisocaproate, dl-2-hydroxyoctanoate, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), isopropyl-β-d-1-thiogalactopyranoside (IPTG), dithiothreitol (DTT), sodium lauroyl sarcosine (SLS), phosphodiesterase I, antimycin A, coenzyme Q10, and cytochrome c were purchased from Sigma. d-Lactate was purchased from Fluka. All other chemicals were of reagent grade.
Bacteria and culture conditions.
The bacterial strains used in this study are listed in Table S2. The P. putida KT2440 was cultured in minimal salt medium (MSM) supplemented with d-lactate (6 g · liter−1) as the sole carbon source, at 30°C (59). E. coli strains were grown in lysogenic broth (LB) medium at 37°C. Antibiotics were used, when appropriate, at the following concentrations: ampicillin, 100 μg · ml−1; kanamycin, 50 μg · ml−1, and gentamicin sulfate, 15 μg · ml−1.
Expression and purification of Fe-S d-iLDH in E. coli.
The Fe-S d-iLDH-encoding gene lldE was PCR amplified from the P. putida KT2440 genome with the high-fidelity DNA polymerase TransStart FastPfu (TransGen) using primers lldE-F and lldE-R (see Table S3 in the supplemental material). The PCR product was doubly digested with EcoRI and HindIII and then ligated into similarly treated pETDuet-1 to form the expression construct pET-lldE, in which a six-His tag-encoding sequence was fused into lldE. Its sequence was verified by DNA sequencing.
E. coli C43(DE3) carrying the pET-lldE plasmid was grown in LB medium (100 μg · ml−1 ampicillin) at 37°C to an optical density at 620 nm of 0.5. Then, 1 mM IPTG was added to induce the expression of Fe-S d-iLDH. Cells were harvested by centrifugation at 14,000 × g for 5 min at 4°C and washed with 0.85% (wt/vol) NaCl solution. The cell pellets were subsequently suspended in the binding buffer (20 mM sodium phosphate, 20 mM imidazole, 500 mM NaCl, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1 mM DTT, 0.1% Triton X-100, and 10% glycerol [pH 7.4]). Then, the cells were sonicated with a Sonics (USA) sonicator (500 W/20 kHz) in an ice bath. The sonicate was then centrifuged at 14,000 × g for 20 min at 4°C. The supernatant was loaded onto a HisTrap HP column (5 ml) and eluted with 50% binding buffer and 50% elution buffer (20 mM sodium phosphate, 500 mM imidazole, 500 mM NaCl, 1 mM PMSF, 1 mM DTT, 0.1% Triton X-100, and 10% glycerol [pH 7.4]). The fractions containing Fe-S d-iLDH were desalted with Sephadex G-25 using buffer A (50 mM Tris-HCl containing 30 mM NaCl, 0.1% Triton X-100, and 1 mM DTT [pH 8.0]) and then applied to a column of DEAE-Sepharose Fast Flow equilibrated with buffer A. The column was washed with 60% buffer A and 40% buffer B (50 mM Tris-HCl containing 500 mM NaCl, 0.1% Triton X-100, and 1 mM DTT [pH 8.0]). The fractions containing Fe-S d-iLDH were then loaded onto a Superdex 200 10/300 GL column equilibrated with buffer A for desalting and further purification. The fractions containing Fe-S d-iLDH were concentrated by ultrafiltration. Glycerol was added to the purified Fe-S d-iLDH solution at a concentration of 20%, and then it was stored at −20°C.
Expression and purification of FAD-containing dehydrogenase domain and Fe-S oxidoreductase domain in E. coli.
To express the FAD-containing dehydrogenase domain of Fe-S d-iLDH separately, the predicted region for the FAD-containing dehydrogenase domain of lldE was amplified by PCR. The region was cloned with 3 different 3′ termini using primers lldE-F/lldEFAD1-R, lldE-F/lldEFAD2-R, and lldE-F/lldEFAD3-R (Table S3). lldEFAD1-R, lldEFAD2-R, and lldEFAD3-R each introduced a termination codon. The resulting genes, lldEFAD1, lldEFAD2, and lldEFAD3, encode proteins Fe-S d-iLDH 1-519, Fe-S d-iLDH 1-530, and Fe-S d-iLDH 1-538 with different C termini. Fe-S d-iLDH 1-530 was chosen to be further studied as the FAD-containing dehydrogenase domain (Fig. S2). The expression procedure of His-tagged FAD-containing dehydrogenase domain was the same as the expression procedure of Fe-S d-iLDH. While in the purification procedure, a relative high purity of protein could be obtained just by His tag purification. Except for desalting, the following steps (with DEAE-Sepharose Fast Flow and Superdex 200 10/300 GL columns) in the Fe-S d-iLDH purification procedure were omitted when purifying the FAD-containing dehydrogenase domain. Triton X-100 was omitted in the corresponding buffers applied.
The predicted region for the Fe-S oxidoreductase domain of lldE was cloned with 3 different 5′ termini using primers lldEFeS1-F/lldE-R, lldEFeS2-F/lldE-R, and lldEFeS3-F/lldE-R (Table S3), and lldEFeS1-R, lldEFeS2-R, and lldEFeS3-R each introduced a start codon. The resulting genes, lldEFeS1, lldEFeS2, and lldEFeS3, encode proteins Fe-S d-iLDH 511-936, Fe-S d-iLDH 520-936, and Fe-S d-iLDH 531-936, with different N termini. Fe-S d-iLDH 531-936 was chosen to be further studied as an Fe-S oxidoreductase domain (Fig. S2). The expression procedure for the His-tagged Fe-S oxidoreductase domain was the same as the expression procedure for Fe-S d-iLDH. In the purification procedure, the cell pellets were subsequently suspended in the DB buffer (20 mM sodium phosphate, 500 mM NaCl, 1 mM PMSF, and 10% glycerol [pH 7.4]) and sonicated. The sonicate was ultracentrifuged at 46,000 × g for 60 min at 4°C. The pellet containing the membrane fraction was resuspended in the SLS binding buffer (20 mM sodium phosphate, 500 mM NaCl, 1 mM PMSF, 1 mM DTT, 0.5% SLS, and 10% glycerol [pH 7.4]) and centrifuged at 14,000 × g for 20 min at 4°C. The supernatant was loaded onto a HisTrap HP column (5 ml) and eluted with 50% SLS binding buffer and 50% SLS elution buffer (20 mM sodium phosphate, 200 mM imidazole, 500 mM NaCl, 1 mM PMSF, 1 mM DTT, 0.1% SLS, and 10% glycerol [pH 7.4]). The fractions containing the Fe-S oxidoreductase domain were desalted with Sephadex G-25 using SLS buffer (50 mM Tris-HCl containing 20 mM NaCl, 0.1% SLS, and 1 mM DTT [pH 8.0]).
Cofactor analysis.
The flavin cofactor of Fe-S d-iLDH was extracted by heat treatment. The protein solution was heated to 100°C for 5 min and then centrifuged at 10,000 × g for 10 min to remove denatured protein. The fluorescence of the released cofactor was measured before and after treatment with 0.1 mg/ml phosphodiesterase I (25°C, 60 min) using an FP-4500 fluorescence spectrophotometer (Jasco, Japan) at 450 nm excitation, and the emission between 480 nm and 650 nm was recorded.
HPLC analysis of flavin cofactor was carried out with an Agilent 1100 series HPLC (Hewlett-Packard, USA) using an ODS C18 column (4.6 by 150 mm; particle size, 5 μm). The eluent was 82% 100 mM ammonium bicarbonate in 18% methanol (60). Standard FAD solutions of 0.05, 0.1, 0.15, 0.2, 0.25, and 0.3 mM were used for quantitative analysis and detected at 450 nm.
The iron contents of proteins were analyzed using an iron colorimetric assay kit (BioVision, USA), and experiments were carried out according to the instructions in the manual.
Site-directed mutagenesis.
Site-directed mutagenesis was performed using the MutanBEST kit (TaKaRa). The lldEFeS3 gene was subcloned into the pEASY-Blunt vector (TransGen) to construct the pEASY-Blunt-lldEFeS3 vector. The C540S mutation of the Fe-S oxidoreductase domain was introduced using the primers 540F/540R to amplify the entire sequence of pEASY-Blunt-lldEFeS3. The linear PCR products were then cyclized using the ligase from the MutanBEST kit. The C540S-C594S, C540S-C594S-C751S-C752S, C540S-C594S-C751S-C752S-C540S-C594S, and C540S-C543S-C594S-C751S-C752S-C540S-C594S mutated proteins were constructed step by step by repeating the procedure using corresponding primers (Table S3). The mutant lldEFeS3 genes were confirmed by DNA sequencing.
Fe-S cluster reconstitution.
Protein solution (25 μM), 20 mM Na2S, 20 mM FeCl3, and 100 mM DTT were deoxygenated in separate tubes under a stream of nitrogen for 1 h. After this time, DTT was added to the protein solution to a final concentration of 10 mM, and the mixture was incubated for 30 min. Then, FeCl3 was added to the mixture drop by drop to a final concentration of 2 mM. When the observed color change to red was stable, Na2S was added to the mixture drop by drop to a final concentration of 2 mM. The reaction was allowed to proceed for 60 min, and then the mixture was desalted with Superdex 200 10/300 GL column using SLS buffer.
Complementation of lldEFAD2 gene in P. putida KT2440 ΔlldE ΔglcD.
For the complementation of lldEFAD2, fragment lldEFAD2 was PCR amplified by using lldE-pBBR-F/lldEFAD2-pBBR-R as primers. The resulting PCR product was cloned into pBBR1MCS-5, a broad-host-range plasmid (61). The resulting plasmid was conjugated into P. putida KT2440 ΔlldE ΔglcD via a triparental mating method, as previously described (6).
Analytical procedures.
The activities of purified recombinant Fe-S d-iLDH and FAD-containing dehydrogenase domain were determined at 30°C in 1 ml of 50 mM Tris-HCl (pH 7.4) and various electron acceptors (MTT, coenzyme Q10, or cytochrome c). The reaction was started by the addition of substrate. For the kinetic measurements of enzymes toward different substrates, 0.005 to 0.35 mM d-lactate, 0.30 to 6.25 mM l-lactate, 0.015 to 1.25 mM d-2-hydroxybutyrate, and 0.30 to 6.25 mM d-glycerate were used with 0.2 mM MTT as an electron acceptor. For the kinetic measurements of enzymes toward different electron acceptors, 0.003 to 0.25 mM MTT, 0.003 to 0.03 mM coenzyme Q10, and 0.003 to 0.10 mM cytochrome c were used with 1.0 mM d-lactate as the substrate. The rate of MTT, coenzyme Q10, or cytochrome c reduction was determined by measuring the changes in absorbance at 578 nm, 340 nm, or 550 nm, respectively. The ε values of these electron acceptors are 1.78 × 104 (mol/liter)−1 · cm−1, 6.22 × 103 (mol/liter)−1 · cm−1, and 2.0 × 104 (mol/liter)−1 · cm−1, respectively (62–64). One unit of d-iLDH activity was defined as the amount reducing 1.0 μmol electron acceptor per minute under the test conditions. Protein concentrations were determined by the Lowry method, with bovine serum albumin (BSA) as the standard.
The d-lactate-oxidizing activities of P. putida KT2440 ΔldhA ΔglcD were determined at 30°C in 1 ml of 50 mM Tris-HCl (pH 7.4) containing 20 mM EDTA to prevent the degradation of pyruvate (65). Whole cells of P. putida KT2440 ΔldhA ΔglcD were added at a final optical density of 1.0. d-Lactate and inhibitors (antimycin A or NaN3) were added at final concentrations of 20 mM and 2 mM, respectively. After 30 min of reaction in shaking bath, the reaction solution was centrifuged at 8,000 × g for 5 min to remove the cells. The pyruvate concentration in the supernatant was then assayed by the dynamic nuclear polarization (DNP) method. One unit of d-lactate-oxidizing activity was defined as the amount producing 1.0 μmol pyruvate per minute under the test conditions.
The concentrations of lactate and pyruvate were measured by HPLC (Agilent 1100 series) equipped with an Aminex HPX-87H column (300 mm by 7.8 mm, 9 μm) and a differential refractive index detector (RID) at 55°C. The mobile phase was 10 mM H2SO4 at a flow rate of 0.4 ml · min−1.
Supplementary Material
ACKNOWLEDGMENTS
The work was supported by National Natural Science Foundation of China (grants 31470199, 31670041, and 31400033), the Young Scholars Program of Shandong University (grant 2015WLJH25), and the Chinese National Program for High Technology Research and Development (grant 2014AA021206).
We declare no conflicts of interest with the contents of this article.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00342-17.
REFERENCES
- 1.Gruber PJ, Frederick SE, Tolbert NE. 1974. Enzymes related to lactate metabolism in green algae and lower land plants. Plant Physiol 53:167–170. doi: 10.1104/pp.53.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Medina JM, Tabernero A. 2005. Lactate utilization by brain cells and its role in CNS development. J Neurosci Res 79:2–10. doi: 10.1002/jnr.20336. [DOI] [PubMed] [Google Scholar]
- 3.Jiang T, Gao C, Ma C, Xu P. 2014. Microbial lactate utilization: enzymes, pathogenesis, and regulation. Trends Microbiol 22:589–599. doi: 10.1016/j.tim.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 4.Mächler P, Wyss MT, Elsayed M, Stobart J, Gutierrez R, von Faber-Castell A, Kaelin V, Zuend M, San Martin A, Romero-Gomez I, Baeza-Lehnert F, Lengacher S, Schneider BL, Aebischer P, Magistretti PJ, Barros LF, Weber B. 2016. In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab 23:94–102. doi: 10.1016/j.cmet.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 5.Aguilera L, Campos E, Gimenez R, Badia J, Aguilar J, Baldoma L. 2008. Dual role of LldR in regulation of the lldPRD operon, involved in l-lactate metabolism in Escherichia coli. J Bacteriol 190:2997–3005. doi: 10.1128/JB.02013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao C, Jiang T, Dou P, Ma C, Li L, Kong J, Xu P. 2012. NAD-independent l-lactate dehydrogenase is required for l-lactate utilization in Pseudomonas stutzeri SDM. PLoS One 7:e36519. doi: 10.1371/journal.pone.0036519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reed DW, Hartzell PL. 1999. The Archaeoglobus fulgidus d-lactate dehydrogenase is a Zn(2+) flavoprotein. J Bacteriol 181:7580–7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richardson AR, Libby SJ, Fang FC. 2008. A nitric oxide-inducible lactate dehydrogenase enables Staphylococcus aureus to resist innate immunity. Science 319:1672–1676. doi: 10.1126/science.1155207. [DOI] [PubMed] [Google Scholar]
- 9.Goffin P, Lorquet F, Kleerebezem M, Hols P. 2004. Major role of NAD-dependent lactate dehydrogenases in aerobic lactate utilization in Lactobacillus plantarum during early stationary phase. J Bacteriol 186:6661–6666. doi: 10.1128/JB.186.19.6661-6666.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garvie EI. 1980. Bacterial lactate dehydrogenases. Microbiol Rev 44:106–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lederer F. 2011. Another look at the interaction between mitochondrial cytochrome c and flavocytochrome b2. Eur Biophys J 40:1283–1299. doi: 10.1007/s00249-011-0697-0. [DOI] [PubMed] [Google Scholar]
- 12.Maeda-Yorita K, Aki K, Sagai H, Misaki H, Massey V. 1995. l-Lactate oxidase and l-lactate monooxygenase: mechanistic variations on a common structural theme. Biochimie 77:631–642. doi: 10.1016/0300-9084(96)88178-8. [DOI] [PubMed] [Google Scholar]
- 13.Stansen C, Uy D, Delaunay S, Eggeling L, Goergen JL, Wendisch VF. 2005. Characterization of a Corynebacterium glutamicum lactate utilization operon induced during temperature-triggered glutamate production. Appl Environ Microbiol 71:5920–5928. doi: 10.1128/AEM.71.10.5920-5928.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cristescu ME, Egbosimba EE. 2009. Evolutionary history of d-lactate dehydrogenases: a phylogenomic perspective on functional diversity in the FAD binding oxidoreductase/transferase type 4 family. J Mol Evol 69:276–287. doi: 10.1007/s00239-009-9274-x. [DOI] [PubMed] [Google Scholar]
- 15.Dym O, Pratt EA, Ho C, Eisenberg D. 2000. The crystal structure of d-lactate dehydrogenase, a peripheral membrane respiratory enzyme. Proc Natl Acad Sci U S A 97:9413–9418. doi: 10.1073/pnas.97.17.9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuller JR, Vitko NP, Perkowski EF, Scott E, Khatri D, Spontak JS, Thurlow LR, Richardson AR. 2011. Identification of a lactate-quinone oxidoreductase in Staphylococcus aureus that is essential for virulence. Front Cell Infect Microbiol 1:19. doi: 10.3389/fcimb.2011.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pinchuk GE, Rodionov DA, Yang C, Li X, Osterman AL, Dervyn E, Geydebrekht OV, Reed SB, Romine MF, Collart FR, Scott JH, Fredrickson JK, Beliaev AS. 2009. Genomic reconstruction of Shewanella oneidensis MR-1 metabolism reveals a previously uncharacterized machinery for lactate utilization. Proc Natl Acad Sci U S A 106:2874–2879. doi: 10.1073/pnas.0806798106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas MT, Shepherd M, Poole RK, van Vliet AH, Kelly DJ, Pearson BM. 2011. Two respiratory enzyme systems in Campylobacter jejuni NCTC 11168 contribute to growth on l-lactate. Environ Microbiol 13:48–61. doi: 10.1111/j.1462-2920.2010.02307.x. [DOI] [PubMed] [Google Scholar]
- 19.Sheng B, Xu J, Zhang Y, Jiang T, Deng S, Kong J, Gao C, Ma C, Xu P. 2015. Utilization of d-lactate as an energy source supports the growth of Gluconobacter oxydans. Appl Environ Microbiol 81:4098–4110. doi: 10.1128/AEM.00527-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phillips SA, Thornalley PJ. 1993. Formation of methylglyoxal and d-lactate in human red blood cells in vitro. Biochem Soc Trans 21:163S. doi: 10.1042/bst021163s. [DOI] [PubMed] [Google Scholar]
- 21.Inoue Y, Kimura A. 1995. Methylglyoxal and regulation of its metabolism in microorganisms. Adv Microb Physiol 37:177–227. doi: 10.1016/S0065-2911(08)60146-0. [DOI] [PubMed] [Google Scholar]
- 22.Furuichi M, Suzuki N, Dhakshnamoorhty B, Minagawa H, Yamagishi R, Watanabe Y, Goto Y, Kaneko H, Yoshida Y, Yagi H, Waga I, Kumar PK, Mizuno H. 2008. X-ray structures of Aerococcus viridans lactate oxidase and its complex with d-lactate at pH 4.5 show an alpha-hydroxyacid oxidation mechanism. J Mol Biol 378:436–446. doi: 10.1016/j.jmb.2008.02.062. [DOI] [PubMed] [Google Scholar]
- 23.Jiang T, Gao C, Dou P, Ma C, Kong J, Xu P. 2012. Rationally re-designed mutation of NAD-independent l-lactate dehydrogenase: high optical resolution of racemic mandelic acid by the engineered Escherichia coli. Microb Cell Fact 11:151. doi: 10.1186/1475-2859-11-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tegoni M, Begotti S, Cambillau C. 1995. X-ray structure of two complexes of the Y143F flavocytochrome b2 mutant crystallized in the presence of lactate or phenyl lactate. Biochemistry 34:9840–9850. doi: 10.1021/bi00031a004. [DOI] [PubMed] [Google Scholar]
- 25.Tegoni M, Cambillau C. 1994. The 2.6-Å refined structure of the Escherichia coli recombinant Saccharomyces cerevisiae flavocytochrome b2-sulfite complex. Protein Sci 3:303–313. doi: 10.1002/pro.5560030214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsai CL, Gokulan K, Sobrado P, Sacchettini JC, Fitzpatrick PF. 2007. Mechanistic and structural studies of H373Q flavocytochrome b2: effects of mutating the active site base. Biochemistry 46:7844–7851. doi: 10.1021/bi7005543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang T, Gao C, Su F, Zhang W, Hu C, Dou P, Zheng Z, Tao F, Ma C, Xu P. 2012. Genome sequence of Pseudomonas stutzeri SDM-LAC, a typical strain for studying the molecular mechanism of lactate utilization. J Bacteriol 194:894–895. doi: 10.1128/JB.06478-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao C, Hu C, Ma C, Su F, Yu H, Jiang T, Dou P, Wang Y, Qin T, Lv M, Xu P. 2012. Genome sequence of the lactate-utilizing Pseudomonas aeruginosa strain XMG. J Bacteriol 194:4751–4752. doi: 10.1128/JB.00943-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao C, Wang Y, Zhang Y, Lv M, Dou P, Xu P, Ma C. 2015. NAD-independent l-lactate dehydrogenase required for l-lactate utilization in Pseudomonas stutzeri A1501. J Bacteriol 197:2239–2247. doi: 10.1128/JB.00017-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao C, Ma C, Xu P. 2011. Biotechnological routes based on lactic acid production from biomass. Biotechnol Adv 29:930–939. doi: 10.1016/j.biotechadv.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 31.Gao C, Qiu J, Li J, Ma C, Tang H, Xu P. 2009. Enantioselective oxidation of racemic lactic acid to d-lactic acid and pyruvic acid by Pseudomonas stutzeri SDM. Bioresour Technol 100:1878–1880. doi: 10.1016/j.biortech.2008.09.053. [DOI] [PubMed] [Google Scholar]
- 32.Gao C, Zhang W, Lv C, Li L, Ma C, Hu C, Xu P. 2010. Efficient production of 2-oxobutyrate from 2-hydroxybutyrate by using whole cells of Pseudomonas stutzeri strain SDM. Appl Environ Microbiol 76:1679–1682. doi: 10.1128/AEM.02470-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao C, Zhang W, Ma C, Liu P, Xu P. 2011. Kinetic resolution of 2-hydroxybutanoate racemic mixtures by NAD-independent l-lactate dehydrogenase. Bioresour Technol 102:4595–4599. doi: 10.1016/j.biortech.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, Jiang T, Sheng B, Long Y, Gao C, Ma C, Xu P. 2016. Coexistence of two d-lactate-utilizing systems in Pseudomonas putida KT2440. Environ Microbiol Rep 8:699–707. doi: 10.1111/1758-2229.12429. [DOI] [PubMed] [Google Scholar]
- 35.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. 2003. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res 40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dym O, Eisenberg D. 2001. Sequence-structure analysis of FAD-containing proteins. Protein Sci 10:1712–1728. doi: 10.1110/ps.12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fraaije MW, Van Berkel WJ, Benen JA, Visser J, Mattevi A. 1998. A novel oxidoreductase family sharing a conserved FAD-binding domain. Trends Biochem Sci 23:206–207. doi: 10.1016/S0968-0004(98)01210-9. [DOI] [PubMed] [Google Scholar]
- 39.Pellicer MT, Badia J, Aguilar J, Baldoma L. 1996. glc locus of Escherichia coli: characterization of genes encoding the subunits of glycolate oxidase and the glc regulator protein. J Bacteriol 178:2051–2059. doi: 10.1128/jb.178.7.2051-2059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bingemann R, Klein A. 2000. Conversion of the central [4Fe-4S] cluster into a [3Fe-4S] cluster leads to reduced hydrogen-uptake activity of the F420-reducing hydrogenase of Methanococcus voltae. Eur J Biochem 267:6612–6618. doi: 10.1046/j.1432-1327.2000.01755.x. [DOI] [PubMed] [Google Scholar]
- 41.Hamann N, Mander GJ, Shokes JE, Scott RA, Bennati M, Hedderich R. 2007. A cysteine-rich CCG domain contains a novel [4Fe-4S] cluster binding motif as deduced from studies with subunit B of heterodisulfide reductase from Methanothermobacter marburgensis. Biochemistry 46:12875–12885. doi: 10.1021/bi700679u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamann N, Bill E, Shokes JE, Scott RA, Bennati M, Hedderich R. 2009. The CCG-domain-containing subunit SdhE of succinate:quinone oxidoreductase from Sulfolobus solfataricus P2 binds a [4Fe-4S] cluster. J Biol Inorg Chem 14:457–470. doi: 10.1007/s00775-008-0462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McIver L, Baxter RL, Campopiano DJ. 2000. Identification of the [Fe-S] cluster-binding residues of Escherichia coli biotin synthase. J Biol Chem 275:13888–13894. doi: 10.1074/jbc.275.18.13888. [DOI] [PubMed] [Google Scholar]
- 44.Terpe K. 2003. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol 60:523–533. doi: 10.1007/s00253-002-1158-6. [DOI] [PubMed] [Google Scholar]
- 45.Aboelmy MH, Peterhansel C. 2014. Enzymatic characterization of Chlamydomonas reinhardtii glycolate dehydrogenase and its nearest proteobacterial homologue. Plant Physiol Biochem 79:25–30. doi: 10.1016/j.plaphy.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 46.Futai M. 1973. Membrane d-lactate dehydrogenase from Escherichia coli. Purification and properties. Biochemistry 12:2468–2474. doi: 10.1021/bi00737a016. [DOI] [PubMed] [Google Scholar]
- 47.Zhang W, Zhang M, Gao C, Zhang Y, Ge Y, Guo S, Guo X, Zhou Z, Liu Q, Zhang Y, Ma C, Tao F, Xu P. 2017. Coupling between d-3-phosphoglycerate dehydrogenase and d-2-hydroxyglutarate dehydrogenase drives bacterial l-serine synthesis. Proc Natl Acad Sci U S A 114:E7574–E7582. doi: 10.1073/pnas.1619034114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Futai M, Kimura H. 1977. Inducible membrane-bound l-lactate dehydrogenase from Escherichia coli. Purification and properties. J Biol Chem 252:5820–5827. [PubMed] [Google Scholar]
- 49.Kato O, Youn JW, Stansen KC, Matsui D, Oikawa T, Wendisch VF. 2010. Quinone-dependent d-lactate dehydrogenase Dld (Cg1027) is essential for growth of Corynebacterium glutamicum on d-lactate. BMC Microbiol 10:321. doi: 10.1186/1471-2180-10-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lindqvist Y. 1989. Refined structure of spinach glycolate oxidase at 2 Å resolution. J Mol Biol 209:151–166. doi: 10.1016/0022-2836(89)90178-2. [DOI] [PubMed] [Google Scholar]
- 51.Loiseau L, Gerez C, Bekker M, Ollagnier-de Choudens S, Py B, Sanakis Y, Teixeira de Mattos J, Fontecave M, Barras F. 2007. ErpA, an iron sulfur (Fe S) protein of the A-type essential for respiratory metabolism in Escherichia coli. Proc Natl Acad Sci U S A 104:13626–13631. doi: 10.1073/pnas.0705829104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schenkman JB, Jansson I. 2003. The many roles of cytochrome b5. Pharmacol Ther 97:139–152. doi: 10.1016/S0163-7258(02)00327-3. [DOI] [PubMed] [Google Scholar]
- 53.Sharp RE, White P, Chapman SK, Reid GA. 1994. Role of the interdomain hinge of flavocytochrome b2 in intra- and inter-protein electron transfer. Biochemistry 33:5115–5120. doi: 10.1021/bi00183a015. [DOI] [PubMed] [Google Scholar]
- 54.Xiao Y, Liu TT, Dai H, Zhang JJ, Liu H, Tang H, Leak DJ, Zhou NY. 2012. OnpA, an unusual flavin-dependent monooxygenase containing a cytochrome b5 domain. J Bacteriol 194:1342–1349. doi: 10.1128/JB.06411-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu D, Liu X, Zhao J, Zhao J. 2005. FesM, a membrane iron-sulfur protein, is required for cyclic electron flow around photosystem I and photoheterotrophic growth of the cyanobacterium Synechococcus sp. PCC 7002. Plant Physiol 138:1586–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karnauchov I, Herrmann RG, Klosgen RB. 1997. Transmembrane topology of the Rieske Fe/S protein of the cytochrome b6/f complex from spinach chloroplasts. FEBS Lett 408:206–210. doi: 10.1016/S0014-5793(97)00427-4. [DOI] [PubMed] [Google Scholar]
- 57.Cole ST, Eiglmeier K, Ahmed S, Honore N, Elmes L, Anderson WF, Weiner JH. 1988. Nucleotide sequence and gene-polypeptide relationships of the glpABC operon encoding the anaerobic sn-glycerol-3-phosphate dehydrogenase of Escherichia coli K-12. J Bacteriol 170:2448–2456. doi: 10.1128/jb.170.6.2448-2456.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yeh JI, Chinte U, Du S. 2008. Structure of glycerol-3-phosphate dehydrogenase, an essential monotopic membrane enzyme involved in respiration and metabolism. Proc Natl Acad Sci U S A 105:3280–3285. doi: 10.1073/pnas.0712331105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ma C, Gao C, Qiu J, Hao J, Liu W, Wang A, Zhang Y, Wang M, Xu P. 2007. Membrane-bound l- and d-lactate dehydrogenase activities of a newly isolated Pseudomonas stutzeri strain. Appl Microbiol Biotechnol 77:91–98. doi: 10.1007/s00253-007-1132-4. [DOI] [PubMed] [Google Scholar]
- 60.Eppink MH, Boeren SA, Vervoort J, van Berkel WJ. 1997. Purification and properties of 4-hydroxybenzoate 1-hydroxylase (decarboxylating), a novel flavin adenine dinucleotide-dependent monooxygenase from Candida parapsilosis CBS604. J Bacteriol 179:6680–6687. doi: 10.1128/jb.179.21.6680-6687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kallnik V, Meyer M, Deppenmeier U, Schweiger P. 2010. Construction of expression vectors for protein production in Gluconobacter oxydans. J Biotechnol 150:460–465. doi: 10.1016/j.jbiotec.2010.10.069. [DOI] [PubMed] [Google Scholar]
- 62.Nagel M, Andreesen JR. 1990. Purification and characterization of the molybdoenzymes nicotinate dehydrogenase and 6-hydroxynicotinate dehydrogenase from Bacillus niacini. Arch Microbiol 154:605–613. doi: 10.1007/BF00248844. [DOI] [Google Scholar]
- 63.Petitdemange H, Marczak R, Raval G, Gay R. 1980. Menadione reductase from Clostridium tyrobutyricum. Can J Microbiol 26:324–329. doi: 10.1139/m80-053. [DOI] [PubMed] [Google Scholar]
- 64.Lechner MC. 1994. Cytochrome P450: biochemistry, biophysics, and molecular biology, p 24–28, 8th International Conference, 24 to 28 October 1993, Lisbon, Portugal. [Google Scholar]
- 65.Ma C, Xu P, Dou Y, Qu Y. 2003. Highly efficient conversion of lactate to pyruvate using whole cells of Acinetobacter sp. Biotechnol Prog 19:1672–1676. doi: 10.1021/bp0341242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.