

ABSTRACT
Pulmonary infection by Streptococcus pneumoniae is characterized by a robust alveolar infiltration of neutrophils (polymorphonuclear cells [PMNs]) that can promote systemic spread of the infection if not resolved. We previously showed that 12-lipoxygenase (12-LOX), which is required to generate the PMN chemoattractant hepoxilin A3 (HXA3) from arachidonic acid (AA), promotes acute pulmonary inflammation and systemic infection after lung challenge with S. pneumoniae. As phospholipase A2 (PLA2) promotes the release of AA, we investigated the role of PLA2 in local and systemic disease during S. pneumoniae infection. The group IVA cytosolic isoform of PLA2 (cPLA2α) was activated upon S. pneumoniae infection of cultured lung epithelial cells and was critical for AA release from membrane phospholipids. Pharmacological inhibition of this enzyme blocked S. pneumoniae-induced PMN transepithelial migration in vitro. Genetic ablation of the cPLA2 isoform cPLA2α dramatically reduced lung inflammation in mice upon high-dose pulmonary challenge with S. pneumoniae. The cPLA2α-deficient mice also suffered no bacteremia and survived a pulmonary challenge that was lethal to wild-type mice. Our data suggest that cPLA2α plays a crucial role in eliciting pulmonary inflammation during pneumococcal infection and is required for lethal systemic infection following S. pneumoniae lung challenge.
KEYWORDS: Streptococcus pneumoniae, inflammation, neutrophils, phospholipase
INTRODUCTION
Streptococcus pneumoniae (the pneumococcus) is a Gram-positive pathogen that is capable of asymptomatically colonizing the nasopharynx of both children and adults. This organism can cause acute infections (sinusitis and otitis media) and is the most common cause of life-threatening community-acquired bacterial pneumonia (1, 2). Pneumococci are protected from clearance in the blood by an antiphagocytic polysaccharide capsule and other protective virulence factors; therefore, their entry into the circulation can lead to potentially lethal septicemia (3). Approximately 14.5 million cases of invasive pneumococcal disease occur annually worldwide, resulting in 0.5 to 1 million deaths of children less than 5 years old (4; http://worldpneumoniaday.org/wp-content/uploads/2014/10/Pneumococcal_factsheet.pdf).
A hallmark of pneumococcal pneumonia is a robust recruitment of neutrophils (polymorphonuclear cells [PMNs]) into the alveolar spaces (5, 6). Although PMN recruitment to the site of pathogenic insult is an integral part of innate immune defense, prolonged and robust PMN recruitment can contribute to disease and mortality (7–10). Although the potent antimicrobial activities of PMNs, which include production of reactive oxygen species, proteases, cationic peptides, and inflammatory mediators, help contain infection, the poorly regulated release of these factors after PMN accumulation in the lungs leads to tissue destruction and potentially to lung failure (8, 11, 12).
PMN recruitment to the alveolar mucosal surface is a complex multistep process involving interactions between PMNs and endothelial, interstitial, and epithelial cells, cytokines, and various PMN chemokines and chemoattractants (7, 13–23). Eicosanoids are bioactive lipids that play critical roles in this inflammatory process (14, 17, 22, 23). Arachidonic acid (AA), the precursor of eicosanoids, is acted upon by cyclooxygenases (COX) to produce prostaglandins and thromboxanes or by lipoxygenases (LOX) to produce leukotrienes, lipoxins, and hepoxilins (24–26). We previously showed that the 12-LOX pathway and its products are important for PMN transepithelial migration during S. pneumoniae infection (27). This observation is consistent with other studies that have shown the 12-LOX pathway to be critical for PMN transepithelial migration across pulmonary epithelia during Pseudomonas aeruginosa infection and across in vitro intestinal epithelia during infection by Salmonella enterica serovar Typhimurium, Shigella flexneri, or enteroaggregative Escherichia coli (28–33).
The bulk of AA in mammalian cells is generated from the fatty acyl chains of glycerophospholipids present in cell membranes (34–36). AA availability is a rate-limiting factor in the production of eicosanoids, because AA release from membrane phospholipids due to enhanced activity of phospholipase A2 (PLA2) results in the increased production of eicosanoids (14, 37). AA is generated in various cell types by the action of PLA2, which releases AA attached to the sn-2 position of membrane phospholipids, or by diacylglycerol (DAG) lipase, which generates AA from diacylglycerols (37–43). Several inflammatory stimuli, such as the extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (p38 MAPK), c-Jun-NH2-terminal kinase (JNK) and tumor necrosis factor alpha (TNF-α), enhance PLA2 activity, suggesting that PLA2 may play an important role at the initiation of inflammation (44, 45).
The PLA2 family includes at least three major subtypes: secretory PLA2 (sPLA2), calcium-independent PLA2 (iPLA2), and cytosolic PLA2 (cPLA2) (39, 42). The activity of sPLA2 depends on millimolar concentrations of calcium, while that of iPLA2, which is also cytosolic in nature, is calcium independent (39, 46). Of these subfamilies, the cPLA2s are thought to be required for AA generation, as they have a substrate preference for phospholipids with AA at the sn-2 position (14, 39, 40, 47). This enzyme is abundantly expressed in multiple tissue and cell types, including human lungs and alveolar epithelial cells, and has the highest transcript levels in the lungs, brain, kidneys, heart, and spleen (48, 49). Numerous inflammatory stimuli, such as interleukin 1β (IL-1β), gamma interferon (IFN-γ), and TNF-α, trigger the release of eicosanoids from human lung epithelial cells in a manner dependent on cPLA2 activation (45, 50, 51). cPLA2 has been implicated in PMN recruitment in vitro and promotes sepsis-induced acute lung injury (36, 52, 53), but its role in S. pneumoniae-induced lung inflammation and subsequent systemic spread of the pathogen has yet to be defined. Here we show that cPLA2α plays a critical role in S. pneumoniae-induced AA release from cultured human airway epithelial cells and subsequent PMN transepithelial migration and is required for pulmonary inflammation, bacteremia, and lethality upon pneumococcal lung infection of mice.
RESULTS
AA is released from membrane phospholipids during S. pneumoniae infection of cultured airway epithelial cells.
Robust recruitment of PMNs into alveolar spaces is a hallmark of pneumococcal pneumonia, and we previously showed that blocking the 12-LOX pathway abrogated PMN migration response across pulmonary epithelia (27). As arachidonic acid (AA) is the substrate for 12-LOX activity, we sought to determine whether pneumococcal infection leads to AA release from pulmonary epithelial cells. We incorporated [3H]AA into the membrane phospholipids of NCI-H292 (H292) cell monolayers, and following infection with the S. pneumoniae clinical isolate strains TIGR4, D39, and G54, we assessed AA release by scintillation counting of culture supernatants. Culture supernatants of monolayers treated with Hanks' balanced salt solution (HBSS) alone or the nonpathogenic Gram-positive bacterium Bacillus subtilis were used as negative controls, and the global signaling pathway activator phorbol myristate acetate (PMA), previously shown to induce AA release (54), was used as a positive control. Lactate dehydrogenase (LDH) release assays indicated that S. pneumoniae infection was not cytotoxic to H292 cells over the periods tested (see Table S1 in the supplemental material), but infection resulted in a significant increase in AA release from membrane phospholipids in a time-dependent manner, with no significant differences observed among the S. pneumoniae strains tested (Fig. 1A). Infection with B. subtilis resulted in significantly less release of AA than with the S. pneumoniae strains (2.5-fold lower than with strain TIGR4 and 2-fold lower than with strains D39 and G54; at 5 h postinfection, P < 0.005, P < 0.05, P < 0.05, respectively) and was statistically indistinguishable from treatment with HBSS. Strain TIGR4 (55) was previously used for monolayer and murine infection experiments (27, 56), including in investigations that revealed 12-LOX-dependent inflammation, and thus was utilized for the studies described below.
FIG 1.
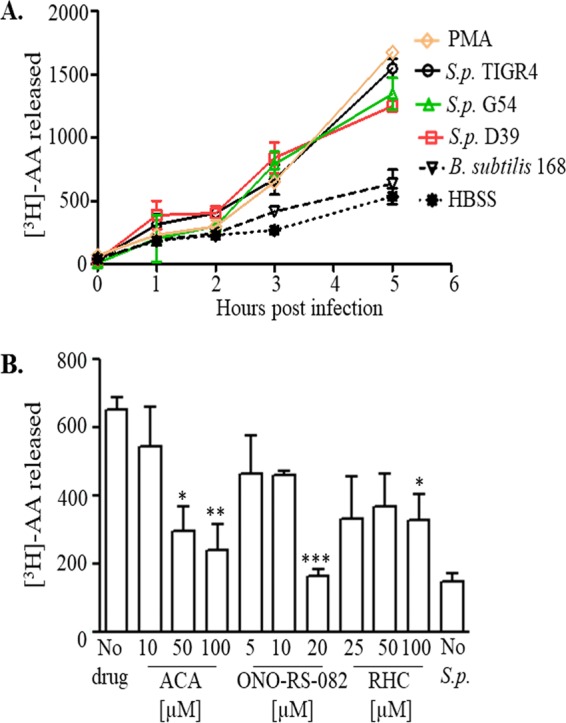
S. pneumoniae infection of airway epithelial cells triggers arachidonic acid (AA) release. (A) H292 cell monolayers were labeled with [3H]AA as detailed in Materials and Methods and infected with the indicated S. pneumoniae (S.p.) strains (each of a different capsular serotype) or with B. subtilis strain 168 at a multiplicity of infection (MOI) of 10 (1 ×107 CFU/monolayer). Supernatants were collected at the indicated times postinfection, and radioactive counts released into the supernatants were measured by scintillation counting. Labeled H292 cell monolayers treated with PMA and HBSS+Ca/Mg were used as positive and negative controls, respectively. Shown are results from a representative of two experiments. (B) H292 cell monolayers labeled with [3H]AA were treated with the indicated concentrations of pan-PLA2 inhibitors ACA or ONO-RS-082 or the DAG lipase inhibitor RHC-80267 (labeled as RHC) prior to infection with S. pneumoniae TIGR4, as detailed in Materials and Methods. Labeled H292 cell monolayers treated with HBSS+Ca/Mg were used as negative controls. Radioactive counts released into supernatants were determined by scintillation counting. Shown are results from a representative of three experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (compared with the no-drug control, using one-way ANOVA).
cPLA2 has a high specificity for membrane phospholipids with AA at the sn-2 position, and its activation has been implicated in AA release and inflammation (54, 57). It is also known that diacylglycerol (DAG) lipase, in combination with monoacylglycerol lipase or fatty acid amidohydrolase, can result in AA release from DAG (43). To assess the potential role of PLA2 and DAG lipases in AA release during S. pneumoniae infection, we treated the [3H]AA-labeled H292 cell monolayers with various concentrations of the pan-PLA2 inhibitors ONO-RS-082 and N-(p-amylcinnamoyl) anthranilic acid (ACA) (50% inhibitory concentration [IC50s] = 9.3 and 10.6 μM, respectively) or the DAG lipase-specific inhibitor RHC-80267 (IC50 = 5 μM) prior to infecting the cells with S. pneumoniae TIGR4 (41, 58–60). RHC-80267 inhibited AA release 50% compared with that in untreated monolayers, but we detected no dose-response relationship and the maximal degree of inhibition was 50% (Fig. 1B). In contrast, the PLA2 inhibitors significantly (P < 0.05) reduced the amount of [3H]AA released from TIGR4-infected H292 cells in a dose-dependent manner, with a maximal reduction of 63 to 75% (Fig. 1B). Note that none of the inhibitors was cytotoxic to the H292 cells (see Table S3) or interfered with bacterial growth or binding to monolayers (see Fig. S1A and B, respectively).
cPLA2 is critical for eliciting PMN transepithelial migration during pneumococcal infection.
Since AA is the precursor of a number of proinflammatory eicosanoids, including the PMN chemoattractant HXA3 (17, 30), we inferred that increased levels of AA release may play a role in PMN transepithelial migration. To determine whether AA release by PLA2 or DAG lipases was required for TIGR4-induced PMN migration in vitro, we treated H292 cell monolayers with the pan-PLA2 or DAG lipase inhibitors prior to S. pneumoniae infection and measured subsequent PMN transepithelial migration. Pretreatment with the DAG lipase inhibitor did not significantly reduce PMN migration (Fig. 2). In contrast, the pan-PLA2 inhibitors resulted in a dose-dependent reduction in PMN transepithelial migration compared with that in untreated H292 cell monolayers, with a maximal inhibition of more than 80%. The specificity of this response was demonstrated by the observation that the pan-PLA2 inhibitors each failed to inhibit fMLP-induced PMN transepithelial migration (see Fig. S1C). Additionally, these inhibitors had no effect on H292 cell barrier properties at the doses used (see Table S4). This result suggests that the AA released during S. pneumoniae infection is derived from membrane phospholipids, a substrate of PLA2, rather than from diacylglycerols.
FIG 2.
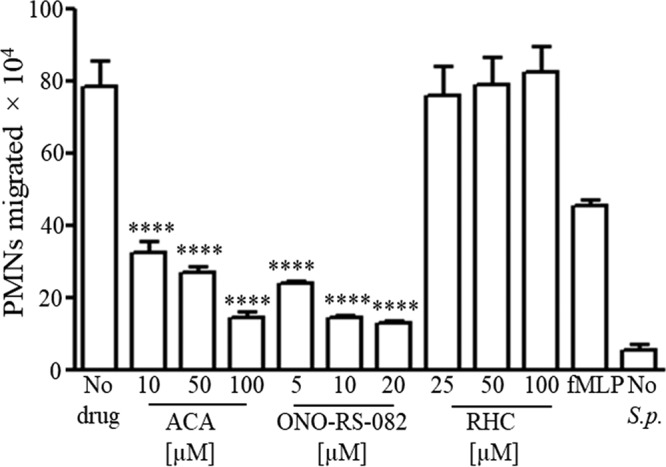
PLA2 is critical for S. pneumoniae-elicited PMN transepithelial migration. H292 cell monolayers were treated with different concentrations of pan-PLA2 inhibitors ACA and ONO-RS-082 or the DAG lipase inhibitor RHC-80267 prior to infection with S. pneumoniae TIGR4, as detailed in Materials and Methods. PMNs were added basolaterally, and the number of PMNs that migrated to the apical side was calculated using the myeloperoxidase (MPO) assay. Monolayers treated with fMLP and HBSS+Ca/Mg were used as positive and negative controls, respectively. Shown are results from a representative of three experiments. ****, P = 0.0001 compared to the no-drug control, using one-way ANOVA.
cPLA2 has a high specificity for membrane phospholipids with AA at the sn-2 position (39), and its activation has been implicated in the release of AA and inflammation (54, 57). This enzyme is activated upon serine phosphorylation by MAP kinases, leading to its translocation to the plasma membrane and catalysis of phospholipids (42, 50). To determine whether S. pneumoniae infection of lung epithelial cells leads to the phosphorylation and membrane localization of cPLA2, we infected H292 cell monolayers with S. pneumoniae and performed immunoblotting to determine both total amount of cPLA2 in cell lysates and the amount of phosphorylated cPLA2 in cell membrane preparations. PMA, a known activator of cPLA2 (61), was included as a positive control. We found that S. pneumoniae infection resulted in no change in total cPLA2 but also a dose-dependent activation of cPLA2, evidenced by increasing amounts of phosphorylated cPLA2 in membrane fractions (Fig. 3A).
FIG 3.
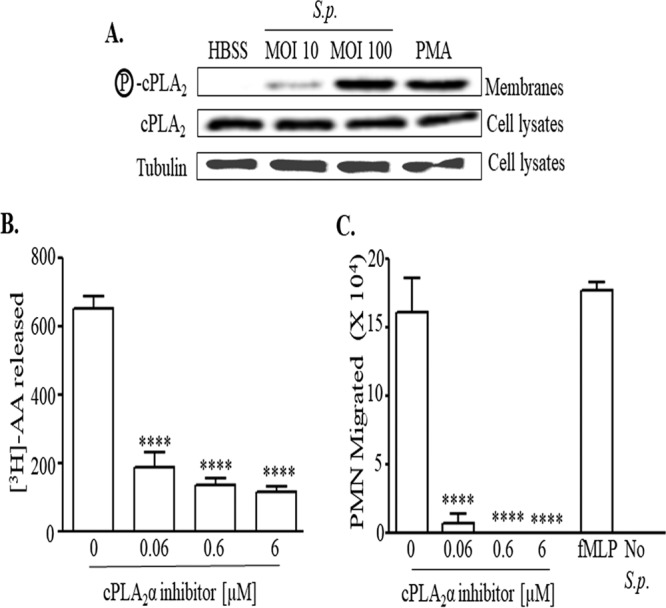
cPLA2 is activated upon pneumococcal infection of pulmonary epithelial monolayers and is critical for eliciting AA release and PMN transepithelial migration. (A) H292 cell monolayers were infected with S. pneumoniae TIGR4 at an MOI of 10 or 100. Monolayers treated with PMA and HBSS+Ca/Mg were used as positive and negative controls, respectively. Cell membranes were prepared as described in Materials and Methods and were probed with anti-phospho-cPLA2, anti-cPLA2, or antitubulin antibody. Shown are results from a representative of two independent experiments. (B) H292 cell monolayers labeled with [3H]AA were treated with the indicated concentrations of cPLA2α inhibitor prior to infection with S. pneumoniae TIGR4, as detailed in Materials and Methods. Radioactive counts in supernatants were measured by scintillation counting. Shown are results from a representative of three experiments. ****, P = 0.0001 compared to the untreated control, using one-way ANOVA. (C) H292 cell monolayers were treated with the indicated concentrations of the cPLA2α inhibitor prior to infection with S. pneumoniae TIGR4, as detailed in Materials and Methods. PMNs were added basolaterally, and the number of PMNs that migrated to the apical side was calculated using the MPO assay. Monolayers treated with fMLP and HBSS+Ca/Mg were used as positive and negative controls, respectively. Shown are results from a representative of three experiments. ****, P < 0.0001 compared to the respective no-drug controls, using one-way ANOVA.
Of the six isoforms of cPLA2 (39), cPLA2α is crucial for eicosanoid production in the airway epithelium (42, 45, 50, 62), and we took advantage of a potent (IC50 = 0.05 μM) and highly specific cPLA2α inhibitor (63) to further investigate the role of cPLA2α in generating AA during pneumococcal infection. This inhibitor was not cytotoxic to H292 cells (see Table S3) and did not affect bacterial growth and binding to monolayers (Fig. S1A and B, respectively). Nevertheless, treatment of [3H]AA-labeled H292 cell monolayers with as little as a 0.06 μM concentration of this inhibitor prior to S. pneumoniae infection reduced AA release 3.5-fold (P = 0.001 [Fig. 3B]) and almost completely inhibited PMN transepithelial migration compared with that in untreated H292 monolayers (P < 0.005 [Fig. 3C]), strongly suggesting that cPLA2α plays a critical role in inflammatory signaling during S. pneumoniae infection.
cPLA2α deficiency diminishes pulmonary inflammation in mice challenged with S. pneumoniae.
We cannot rule out off-target effects of the cPLA2 inhibitors utilized as described above or that the immortalized H292 cells fully reflect the response of bona fide pulmonary epithelium. Hence, to test the role of cPLA2α in pneumococcal disease, we utilized wild-type (WT) or cPLA2α−/− BALB/c mice (64). Basal levels of pulmonary dendritic cells, macrophages, and T cells, measured by flow cytometry of single-cell lung suspensions, were significantly lower in cPLA2α-deficient mice than in WT mice (Fig. 4A, “DC,” “M-phage,” and “T cell”), consistent with the lower baseline levels of eicosanoids, such as the proinflammatory leukotriene B4 (LTB4), associated with cPLA2α deficiency (52).
FIG 4.
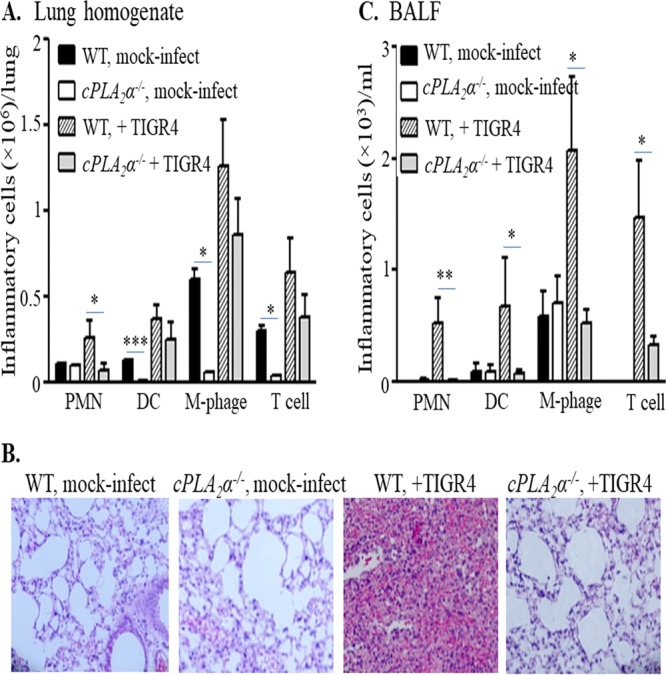
cPLA2α promotes pulmonary inflammation in S. pneumoniae-infected mice. Mice (cPLA2α−/− or their WT littermates) were mock infected with PBS (n = 4 for each strain, per experiment) or infected intratracheally with S. pneumoniae TIGR4 (n = 5 for each strain, per experiment), as detailed in Materials and Methods. Lungs and bronchoalveolar lavage fluid (BALF) were collected from the mock-infected or TIGR4-infected mice. Cells present in the digested lungs and BALF were stained with relevant MAbs, and the fluorescence intensities of the stained cells were determined by flow cytometry. Collected data were analyzed to determine the numbers of PMNs, dendritic cells (DC), macrophages (M-phage), or T cells in the lungs (A) and in the BALF (C). Statistical significance was analyzed by one-way ANOVA followed by individual Student's t test analyses. *, P < 0.05; **, P < 0.005; ***, P < 0.0005. For histological analyses (B), H&E-stained lung sections were prepared and examined by light microscopy (original magnification, ×20). Shown are results from a representative of two independent experiments.
Notably, striking differences between WT and cPLA2α−/− mice were observed upon intratracheal challenge with 2× 105 CFU of S. pneumoniae TIGR4. Similar to our previous observation in C57BL/6 mice (27), wild-type BALB/c mice exhibited a robust acute inflammatory response at 48 h postinfection, with vascular congestion and massive cellular infiltrates (Fig. 4B, “WT, mock-infect” versus “WT, +TIGR4”). Flow cytometry of lung homogenates and/or bronchoalveolar lavage fluid (BALF) revealed greater numbers of PMNs, dendritic cells, macrophages, and T cells in infected mice than in uninfected controls (Fig. 4A and C). In contrast, infection of cPLA2α−/− littermates resulted in dramatically lower levels of pulmonary inflammation upon S. pneumoniae challenge (Fig. 4B, “WT, +TIGR4” versus “cPLA2−/−, +TIGR4”). Flow cytometric analyses of pulmonary homogenates showed that PMN recruitment to the lung was 2.5-fold lower than in WT mice, i.e., diminished to background (uninfected) levels (Fig. 4A). PMN recruitment to the airways was also dramatically affected, as virtually no PMNs were detected in BALF (Fig. 4C). Quantitation of PMNs in lung homogenates or BALF by myeloperoxidase (MPO), a lysosomal peroxidase present in the azurophilic granules of PMNs, also indicated an essential role for cPLA2α in promoting acute inflammation following pulmonary challenge with S. pneumoniae TIGR4 (see Fig. S2A). Finally, the recruitment of dendritic cells, macrophages, and T cells to airway spaces, as assessed by flow cytometric analysis of BALF, was also significantly diminished by ablation of cPLA2α (Fig. 4C), consistent with the lower levels of a variety of eicosanoids in cPLA2α-deficient mice in other lung injury models (52). Interestingly, the numbers of bacteria in either lung homogenates or BALF at 48 h postinfection were not significantly different between WT and cPLA2α−/− mice (see Fig. S2B), consistent with previous findings that genetic ablation or chemical inhibition of 12-LOX, which is required for the production of HXA3, did not result in an increase in bacterial load in the lung (27). These data indicate that cPLA2α plays a critical role in the recruitment of PMNs and other inflammatory cells following S. pneumoniae lung infection.
Ablation of cPLA2α leads to decreased bacteremia and increased survival in an otherwise lethal S. pneumoniae lung challenge.
Acute pulmonary inflammation during S. pneumoniae lung infection may disrupt epithelial barrier function and enhance tissue damage (10, 27, 65, 66), and we previously found that genetic ablation or chemical inhibition of 12-LOX mitigated bacteremia and lethality following intratracheal challenge with S. pneumoniae (27). To determine whether cPLA2α is important for invasive pneumococcal disease, we infected cPLA2α−/− mice and littermate BALB/c controls intratracheally with S. pneumoniae and monitored bacteremia, illness, and survival. At 48 h postinfection, ∼105 CFU/ml were present in the tail vein blood of WT mice, and at 72 h postinfection, at which time bacteremia reached ∼106 CFU/ml, all the WT mice succumbed to infection or were moribund and euthanized (Fig. 5). In contrast, infected cPLA2α−/− mice suffered no detectable bacteremia and uniformly survived (Fig. 5).
FIG 5.
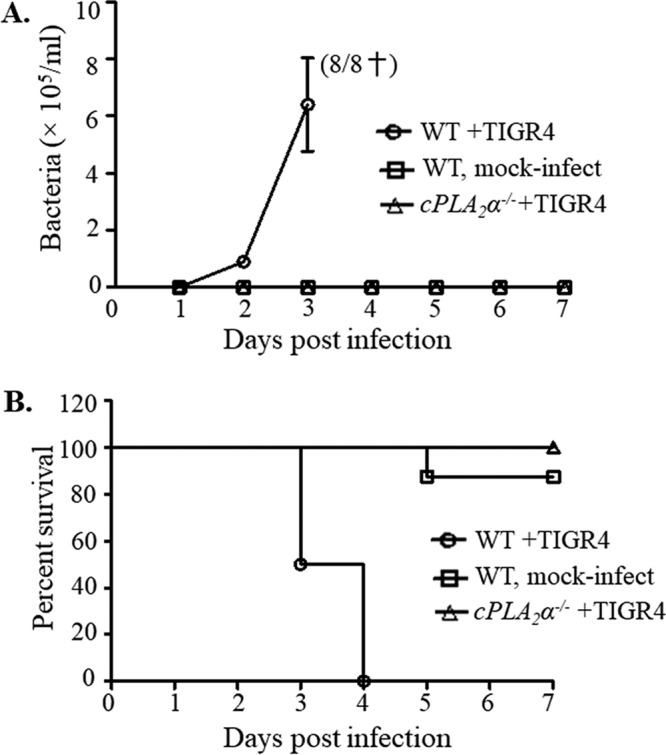
Ablation of cPLA2α leads to decreased bacteremia and increased survival in an otherwise lethal S. pneumoniae lung challenge. cPLA2α−/− mice or their littermate WT controls were intratracheally inoculated with TIGR4 (see Materials and Methods). A control group of WT mice received PBS. (A) Bacteremia was determined by plating tail vein blood at the specified time points. Shown are results from a combination of two experiments. Death of all infected WT mice is indicated by a dagger. (B) Survival of animals was monitored over a 7-day postinfection period. The log rank (Mantel-Cox) test was performed for survival curve analysis. The experiment was performed twice, with similar results. Shown are results from a combination of two experiments. A total of 8 mice were used per group; panels A and B represent the same cohorts of mice.
DISCUSSION
Robust pulmonary infiltration of PMNs is a characteristic of pneumococcal lung infection (5, 6). Eicosanoids, which are formed from AA by the action of lipoxygenases, cyclooxygenases, and/or cytochrome P450, are widely implicated in lung inflammation by S. pneumoniae and other bacteria (27, 31, 67). We previously showed that pneumococcus-mediated induction of 12-LOX, an enzyme essential for the conversion of AA to the neutrophil chemoattractant HXA3, promotes basolateral-to-apical transepithelial recruitment of PMNs in vitro, as well as the corresponding apical-to-basolateral migration of bacteria (27). 12-LOX expression is increased during pneumococcal infection, and chemical inhibition or genetic ablation of 12-LOX protected mice from pulmonary inflammation and lethal systemic infection following intratracheal challenge with S. pneumoniae (27).
Pneumococcal infection triggers the release of AA and its metabolites in the middle ear during otitis media (68).We showed in this study that pneumococcal infection of respiratory epithelial monolayers also results in the release of AA. Both phospholipases and DAG lipases have been implicated in the generation of AA (41), and we found that a DAG inhibitor, at the highest concentration tested, decreased AA production 2-fold. However, PLA2 has been shown to play a more important role in releasing AA in a variety of other physiologic and pathophysiologic processes (64), and two pan-PLA2 inhibitors resulted in greater (3- to 4-fold) decreases in AA release. In addition, both PLA2 inhibitors diminished PMN transepithelial migration by >80% during pneumococcal infection of respiratory epithelial monolayers, whereas the DAG lipase inhibitor had no effect. Although we cannot rule out a role for DAG in the production of eicosanoid PMN chemoattractants, PLA2 has been previously shown to be activated by pneumococcal virulence factors, such as the pore-forming toxin pneumolysin, in pulmonary artery endothelial cells and human neutrophils (69, 70).
The PLA2 superfamily, which cleaves the ester bond of phospholipids at the sn-2 position to generate free fatty acids, includes at least three major subfamilies: sPLA2s, iPLA2s and cPLA2s (39, 42). Of these, cPLA2 is highly specific for phospholipids with AA at the sn-2 position and thus plays an important role in the generation of free AA (39, 50, 62). cPLA2 has been implicated in airway inflammation both in humans and mice (37, 71, 72). For example, lung epithelial cells of cystic fibrosis (CF) patients have elevated cPLA2 activity and produce more eicosanoids than do normal epithelial cells (73). Activation of cPLA2 via phosphorylation is imperative for the production of eicosanoids and is dependent on MAP kinases and protein kinase C (PKC) (42, 50, 74), and we found that phosphorylated cPLA2 accumulated in the membrane fraction of lung epithelial cells after pneumococcal infection.
The cPLA2 family consists of six members (cPLA2α, -β, -γ, -δ, -ε, and -ζ) (62). cPLA2γ is induced in eosinophils and pulmonary epithelial cells upon sensitization of mice with Aspergillus fumigatus extracts (75), but overall the role of the non-cPLA2α members of the cPLA2 family remains poorly characterized. In contrast, cPLA2α has been shown to be crucial for eicosanoid production in a variety of tissues (45, 50, 62), including the human intestine (76, 77), and has been implicated in pulmonary disorders such as acute respiratory distress syndrome (ARDS), CF, and allergy (71, 78). For example, during ARDS, cPLA2α−/− mice display lower baseline levels of eicosanoids in BALF than do littermate control WT mice, as well as lower levels of neutrophil infiltration (52). We found that an inhibitor of cPLA2α dramatically diminished AA release and virtually eliminated PMN migration across epithelial monolayers in vitro. Given the inherent limitations of in vitro chemical inhibitor studies, we utilized a murine model to assess the role of cPLA2α and found that cPLA2α deficiency (64) entirely blocked pneumococcus-induced PMN recruitment to the lung. cPLA2α was also required for maximal recruitment of macrophages, dendritic cells, and T cells in the lung and airways upon infection, suggesting a more general role for cPLA2α in pulmonary inflammation. The cPLA2α-dependent extravasation of these other inflammatory cell types may in part be a consequence of generalized compromise of integrity of the airway epithelial barrier resulting from transepithelial PMN migration (27). In addition, other AA metabolites function as inflammatory mediators for a variety of cells (79–81) such that the release of AA by cPLA2α may promote the migration of several inflammatory cell types into airway spaces.
Previous reports have shown that in several models, PMNs are required for pneumococcal control early after pulmonary challenge (82, 83) but may not promote bacterial clearance at later phases of infection (10, 27). We also did not observe an effect of cPLA2α-mediated inflammatory signaling on bacterial numbers in the lungs or BALF of mice at 48 h postinfection with pneumococcal doses included in this study, consistent with our previous observations that 12-LOX-deficient mice also suffered no greater pneumococcal loads in the lung and airways than did WT C57BL/6 mice (27; unpublished data).
Finally, pulmonary inflammation during pneumococcal infection contributes to epithelial barrier compromise and promotes systemic disease (65, 84, 85). We found that not only did cPLA2α fail to promote bacterial clearance in the lung, but also this enzyme actively promoted lethal systemic disease: cPLA2α-deficient mice, like 12-LOX-deficient mice (27), were protected from S. pneumoniae bacteremia after pulmonary challenge and uniformly survived an otherwise lethal challenge. Our study supports the possibility that inhibition of cPLA2-mediated PMN migration may provide a strategy to combat invasive pneumococcal infection. Given that cPLA2-mediated inflammation appears central to other pulmonary disorders, such as ARDS, CF, and allergy (71, 78), these inhibitors may have broad therapeutic value for noninfectious disorders as well.
MATERIALS AND METHODS
Bacterial strains.
S. pneumoniae strains TIGR4 (serotype 4 [55]), D39 (serotype 2 [86]), and G54 (serotype 19F [87]) were grown in Todd-Hewitt broth (BD Biosciences, San Jose, CA) supplemented with yeast extract at 37°C and 5% CO2 and used in mid-log phase to late log phase. Bacillus subtilis strain 168 was grown overnight at 37°C in Luria broth. S. pneumoniae strain TIGR4 is highly virulent in mice and has been used in our previous murine infection studies, including those on 12-LOX-dependent pulmonary inflammation (27, 56). For the murine infection experiments, bacteria (5 ×108/ml) were stored in medium supplemented with 25% (vol/vol) glycerol at −80°C. Prior to use, the frozen cultures were thawed and diluted in phosphate-buffered saline (PBS). Bacterial stock titers were confirmed by plating serial dilutions on blood agar at 37°C and 5% CO2. When required, S. pneumoniae strains were heat killed at 65°C for 1 h (27).
Growth, maintenance, and infection of epithelial cells.
Human mucoepidermoid pulmonary carcinoma cell line NCI-H292 (H292) has been used in a number of studies of pulmonary inflammation and/or eicosanoid production (88–92), as well as in studies of pulmonary infection by bacteria (93–95). These cells were cultured in RPMI 1640 medium (American Type Culture Collection, Manassas, VA) supplemented with 10% fetal bovine serum, 2 mM l-glutamine, and penicillin-streptomycin. Polarized H292 cell monolayers were grown on the underside of collagen (rat tail, type IV)-coated 0.33-cm2 membrane filters (Corning Life Sciences, Tewksbury, MA). Cells were infected with bacteria resuspended in Hanks' balanced salt solution (HBSS) supplemented with Ca2+ and Mg2+ (HBSS+Ca/Mg), at 1 × 107 bacteria/monolayer, a multiplicity of infection of 10. The cytotoxicity of H292 cell monolayers in response to S. pneumoniae TIGR4 infection as measured by lactate dehydrogenase (LDH) release (30) is shown in Table S1 in the supplemental material. As the transepithelial resistance of lung epithelial monolayers is typically very low (96), permeability to horseradish peroxidase (HRP) was used to assess monolayer barrier integrity (30) and is shown in Table S2.
Animals.
cPLA2α knockout (cPLA2α−/−) mice were generated in a BALB/c background using gene targeting in mouse embryonic stem cells to disrupt the exon containing Ser228, thus generating a null allele (64). The cPLA2α−/−mice were backcrossed for 10 generations and genotyped using tail DNA. All animal experiments were performed with cPLA2α−/− mice and their wild-type BALB/c littermate controls in accordance with Tufts University Animal Care and Use Committee-approved protocols.
PMN isolation and transmigration assay.
PMNs were isolated from blood drawn from healthy human volunteers according to previously described protocols (30, 97). The PMN isolation protocol was approved by the Tufts University Institutional Review Board, and informed written consent was obtained from all volunteers. Basolateral-to-apical PMN transmigration assays were performed as described previously (27). Formyl-methionyl-leucyl-phenylalanine (fMLP) at 10−11 M or buffer alone (HBSS+Ca/Mg) was used as the positive or negative control, respectively, for induction of PMN transmigration.
AA release assay.
Arachidonic acid (AA) release in epithelial cell culture was assayed using previously described protocols (37, 98). Briefly, H292 cells grown on 24-well tissue culture inserts were incubated in medium containing 0.2 μCi/ml of [3H]AA (PerkinElmer, Waltham, MA) for 18 to 24 h prior to infection, to allow for the incorporation of [3H]AA into membrane phospholipids. Cells were then washed in PBS and infected with 1 × 107 S. pneumoniae organisms resuspended in 0.5 ml of HBSS+Ca/Mg. Supernatants were collected at different time points and measured for radioactivity using a scintillation counter (Becton Dickinson). At the end of the experiment, H292 cells were lysed in 1% SDS and 1% Triton X-100 to determine total radioactivity.
Drug treatment.
H292 cell monolayers were incubated with different doses of the pan-PLA2 inhibitor ONO-RS-082 or N-(p-amylcinnamoyl) anthranilic acid (ACA) or the DAG lipase inhibitor RHC-80267 (Enzo Life Sciences, Farmington, NY) (38, 58). cPLA2α was specifically inhibited in H292 cell monolayers by incubation with the cPLA2α inhibitor (N-{(2S,4R)-4-(biphenyl-2-ylmethyl-isobutyl-amino)-1-[2-(2,4-difluorobenzoyl)-benzoyl]-pyrrolidin-2-ylmethyl}-3-[4-(2,4-dioxothiazolidin-5-ylidenemethyl)-phenyl]acrylamide), HCl (EMD Millipore, Billerica, MA), diluted to 0.06, 0.6, and 6 μM from a stock concentration of 6 mM in dimethyl sulfoxide (DMSO). Working dilutions were made in HBSS+Ca/Mg from the stock solutions. Monolayers were incubated with inhibitors for 2 h at 37°C and then washed to remove the residual drug prior to infection. Vehicle controls were used to rule out nonspecific effects. The inhibitors did not affect cell viability or H292 cell barrier integrity, as assessed by LDH and HRP migration assays, and had no effect on bacterial growth or adherence to H292 cell monolayers.
Cell viability and bacterial binding assays.
Cell viability in response to bacterial or drug treatment was measured by LDH assays as previously described (30). Bacterial binding was measured after incubating the bacteria for 40 min with H292 cell monolayers, using a previously described protocol (27).
Detection of cPLA2 activation.
H292 cell monolayers were infected with S. pneumoniae TIGR4 for 2.5 h. Control monolayers received buffer (HBSS+Ca/Mg) alone as a negative control or 1 μM phorbol myristate acetate (PMA) as a positive control. Membrane fractions were prepared as previously described (99). Equal amounts of protein were run on a 4 to 20% gradient polyacrylamide gel (Bio-Rad, Hercules, CA) and then transferred onto nitrocellulose membranes. Western blots were probed with anti-cPLA2 or anti-phospho-cPLA2 antibodies (Cell Signaling Technologies, Danvers, MA) and detected with ECL reagent. To verify equal sample loading, blots were probed with antitubulin antibody.
Murine infection studies.
cPLA2α−/− mice (64) and their littermate wild-type (WT) BALB/c controls were challenged with 2 × 105 S. pneumoniae TIGR4 organisms in 50 μl of PBS via intratracheal inoculation, whereas control mice received only PBS. For histological study of pulmonary inflammation, mice were euthanized at 48 h postinfection, and whole lungs were excised. Lungs were fixed in 10% buffered formalin (Sigma-Aldrich, St. Louis, MO) and embedded in paraffin. Lung blocks were sectioned at 5 μM and adhered to silanized slides. Three mice per group were analyzed following hematoxylin and eosin (H&E) staining, and lungs were visualized using a Nikon TMS microscope. PMN migration into the lung and airways was measured by myeloperoxidase (MPO) assay using a previously described protocol (100). To determine the bacterial load in the lungs at 48 h, lungs were isolated and homogenized in PBS, and serial dilutions were plated on blood agar. Serial dilutions of bronchoalveolar lavage fluid (BALF) were plated on blood agar to enumerate bacteria in the airways. For bacteremia, tail blood was collected and dilutions were plated on blood agar plates every 24 h postinfection. Mice were monitored for bacteremia, sickness, and survival over 7 days following infection, and moribund animals were euthanized as per the protocol approved by the Tufts University Animal Care and Use Committee.
Flow cytometry.
Anti-Ly-6G-phycoerythrin (PE) (clone 1A8), anti-CD11c-fluorescein isothiocyanate (FITC) (clone N418), anti-F4-80-PE-Cy7 (clone BM8), and anti-T cell receptor β (TCRβ)-allophycocyanin (clone H57-597) were obtained from BD Biosciences, San Jose, CA. Murine lungs were collected at 48 h postinfection, and excised lung tissue was digested with type II collagenase and DNase (1 mg/ml and 50 U/ml, respectively; Worthington, Lakewood, NJ) to obtain a single-cell suspension, as described previously (101). For collection of BALF, mice were sacrificed at 48 h postinfection and lungs were washed twice with 1 ml of PBS via a cannula. Cells in the pulmonary single-cell suspension or in the BALF were stained with relevant monoclonal antibodies (MAbs) on ice, washed, and run through a FACSCalibur flow cytometer (BD Biosciences). Fluorescence intensities of the stained cells were determined. Data were analyzed using FlowJo software (Tree Star) to determine the numbers of PMNs (Ly6G+), dendritic cells (F4/80− CD11c+), macrophages (F4/80+), and T cells (TCRβ+) in both the lung single-cell suspensions and BALF.
Statistical analyses.
Statistical analysis was performed using the GraphPad Prism program (GraphPad Software). The log rank (Mantel-Cox) test was performed for survival curve analysis. All quantitative results were analyzed by one-way analysis of variance (ANOVA). In cases where data sets contained more than two groups, one-way ANOVA was followed by individual Student's t test analyses. P values of <0.05 were considered significant in all cases.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by American Lung Association Senior Research Training Fellowship RT 194942 N (to R.B.). B.A.M. was supported by NIH grants DK056754 and DK109677. J.V.B. was supported by NIH grants R37 DK039773 and RO1 DK072381.
We thank Eileen O'Brien and Yushuan Lai for technical assistance, Bryan Hurley for helpful discussion, and Marcia Osburne, Elsa Bou Ghanem, Walter Adams, Sara Roggensack, and Dakshina Murthy Jandhyala for critical review of the manuscript. Bacillus subtilis strain 168 was a kind gift from Linc Sonenshein.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00280-17.
REFERENCES
- 1.Gillespie SH, Balakrishnan I. 2000. Pathogenesis of pneumococcal infection. J Med Microbiol 49:1057–1067. doi: 10.1099/0022-1317-49-12-1057. [DOI] [PubMed] [Google Scholar]
- 2.McCullers JA, Tuomanen EI. 2001. Molecular pathogenesis of pneumococcal pneumonia. Front Biosci 6:D877–D889. [DOI] [PubMed] [Google Scholar]
- 3.Lynch JP III, Zhanel GG. 2010. Streptococcus pneumoniae: epidemiology and risk factors, evolution of antimicrobial resistance, and impact of vaccines. Curr Opin Pulm Med 16:217–225. [DOI] [PubMed] [Google Scholar]
- 4.O'Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, Lee E, Mulholland K, Levine OS, Cherian T. 2009. Hib and Pneumococcal Global Burden of Disease Study Team: burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374:893–902. doi: 10.1016/S0140-6736(09)61204-6. [DOI] [PubMed] [Google Scholar]
- 5.Doerschuk CM, Markos J, Coxson HO, English D, Hogg JC. 1994. Quantitation of neutrophil migration in acute bacterial pneumonia in rabbits. J Appl Physiol 77:2593–2599. [DOI] [PubMed] [Google Scholar]
- 6.Moreland JG, Bailey G, Nauseef WM, Weiss JP. 2004. Organism-specific neutrophil-endothelial cell interactions in response to Escherichia coli, Streptococcus pneumoniae, and Staphylococcus aureus. J Immunol 172:426–432. doi: 10.4049/jimmunol.172.1.426. [DOI] [PubMed] [Google Scholar]
- 7.Craig A, Mai J, Cai S, Jeyaseelan S. 2009. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect Immun 77:568–575. doi: 10.1128/IAI.00832-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss SJ. 1989. Tissue destruction by neutrophils. N Engl J Med 320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 9.Bou Ghanem EN, Clark S, Du X, Wu D, Camilli A, Leong JM, Meydani SN. 2015. The alpha-tocopherol form of vitamin E reverses age-associated susceptibility to Streptococcus pneumoniae lung infection by modulating pulmonary neutrophil recruitment. J Immunol 194:1090–1099. doi: 10.4049/jimmunol.1402401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bou Ghanem EN, Clark S, Roggensack SE, McIver SR, Alcaide P, Haydon PG, Leong JM. 2015. Extracellular adenosine protects against Streptococcus pneumoniae lung infection by regulating pulmonary neutrophil recruitment. PLoS Pathog 11(8):e1005126. doi: 10.1371/journal.ppat.1005126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grommes J, Soehnlein O. 2011. Contribution of neutrophils to acute lung injury. Mol Med 17:293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li LF, Lai YT, Chang CH, Lin MC, Liu YY, Kao KC, Tsai YH. 2014. Neutrophil elastase inhibitor reduces ventilation-induced lung injury via nuclear factor-kappaB and NF-kappaB repressing factor in mice. Exp Biol Med 239:1045–1057. doi: 10.1177/1535370214529393. [DOI] [PubMed] [Google Scholar]
- 13.Burns AR, Smith CW, Walker DC. 2003. Unique structural features that influence neutrophil emigration into the lung. Physiol Rev 83:309–336. doi: 10.1152/physrev.00023.2002. [DOI] [PubMed] [Google Scholar]
- 14.Diaz BL, Arm JP. 2003. Phospholipase A(2). Prostaglandins Leukot Essent Fatty Acids 69:87–97. [DOI] [PubMed] [Google Scholar]
- 15.Jeyaseelan S, Manzer R, Young SK, Yamamoto M, Akira S, Mason RJ, Worthen GS. 2005. Induction of CXCL5 during inflammation in the rodent lung involves activation of alveolar epithelium. Am J Respir Cell Mol Biol 32:531–539. doi: 10.1165/rcmb.2005-0063OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koh AY, Priebe GP, Ray C, Van Rooijen N, Pier GB. 2009. Inescapable need for neutrophils as mediators of cellular innate immunity to acute Pseudomonas aeruginosa pneumonia. Infect Immun 77:5300–5310. doi: 10.1128/IAI.00501-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCormick BA. 2007. Bacterial-induced hepoxilin A3 secretion as a pro-inflammatory mediator. FEBS J 274:3513–3518. doi: 10.1111/j.1742-4658.2007.05911.x. [DOI] [PubMed] [Google Scholar]
- 18.Prince AS, Mizgerd JP, Wiener-Kronish J, Bhattacharya J. 2006. Cell signaling underlying the pathophysiology of pneumonia. Am J Physiol Lung Cell Mol Physiol 291:L297–L300. doi: 10.1152/ajplung.00138.2006. [DOI] [PubMed] [Google Scholar]
- 19.Schultz MJ, Rijneveld AW, Florquin S, Edwards CK, Dinarello CA, van der Poll T. 2002. Role of interleukin-1 in the pulmonary immune response during Pseudomonas aeruginosa pneumonia. Am J Physiol Lung Cell Mol Physiol 282:L285–L290. doi: 10.1152/ajplung.00461.2000. [DOI] [PubMed] [Google Scholar]
- 20.Smart SJ, Casale TB. 1994. Pulmonary epithelial cells facilitate TNF-alpha-induced neutrophil chemotaxis. A role for cytokine networking. J Immunol 152:4087–4094. [PubMed] [Google Scholar]
- 21.Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G, Nijkamp FP, Blalock JE. 2006. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med 12:317–323. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- 22.Kubala SA, Patil SU, Shreffler WG, Hurley BP. 2014. Pathogen induced chemo-attractant hepoxilin A3 drives neutrophils, but not eosinophils across epithelial barriers. Prostaglandins Other Lipid Mediat 108:1–8. doi: 10.1016/j.prostaglandins.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pazos MA, Pirzai W, Yonker LM, Morisseau C, Gronert K, Hurley BP. 2015. Distinct cellular sources of hepoxilin A3 and leukotriene B4 are used to coordinate bacterial-induced neutrophil transepithelial migration. J Immunol 194:1304–1315. doi: 10.4049/jimmunol.1402489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pace-Asciak CR, Reynaud D, Demin P, Nigam S. 1999. The hepoxilins. A review. Adv Exp Med Biol 447:123–132. doi: 10.1007/978-1-4615-4861-4_12. [DOI] [PubMed] [Google Scholar]
- 25.Serhan CN, Savill J. 2005. Resolution of inflammation: the beginning programs the end. Nat Immunol 6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 26.Williams KI, Higgs GA. 1988. Eicosanoids and inflammation. J Pathol 156:101–110. doi: 10.1002/path.1711560204. [DOI] [PubMed] [Google Scholar]
- 27.Bhowmick R, Tin Maung NH, Hurley BP, Ghanem EB, Gronert K, McCormick BA, Leong JM. 2013. Systemic disease during Streptococcus pneumoniae acute lung infection requires 12-lipoxygenase-dependent inflammation. J Immunol 191:5115–5123. doi: 10.4049/jimmunol.1300522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boll EJ, Struve C, Sander A, Demma Z, Krogfelt KA, McCormick BA. 2012. Enteroaggregative Escherichia coli promotes transepithelial migration of neutrophils through a conserved 12-lipoxygenase pathway. Cell Microbiol 14:120–132. doi: 10.1111/j.1462-5822.2011.01706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boll EJ, Struve C, Sander A, Demma Z, Nataro JP, McCormick BA, Krogfelt KA. 2012. The fimbriae of enteroaggregative Escherichia coli induce epithelial inflammation in vitro and in a human intestinal xenograft model. J Infect Dis 206:714–722. doi: 10.1093/infdis/jis417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hurley BP, Siccardi D, Mrsny RJ, McCormick BA. 2004. Polymorphonuclear cell transmigration induced by Pseudomonas aeruginosa requires the eicosanoid hepoxilin A3. J Immunol 173:5712–5720. doi: 10.4049/jimmunol.173.9.5712. [DOI] [PubMed] [Google Scholar]
- 31.Mrsny RJ, Gewirtz AT, Siccardi D, Savidge T, Hurley BP, Madara JL, McCormick BA. 2004. Identification of hepoxilin A3 in inflammatory events: a required role in neutrophil migration across intestinal epithelia. Proc Natl Acad Sci U S A 101:7421–7426. doi: 10.1073/pnas.0400832101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mumy KL, Bien JD, Pazos MA, Gronert K, Hurley BP, McCormick BA. 2008. Distinct isoforms of phospholipase A2 mediate the ability of Salmonella enterica serotype Typhimurium and Shigella flexneri to induce the transepithelial migration of neutrophils. Infect Immun 76:3614–3627. doi: 10.1128/IAI.00407-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tamang DL, Pirzai W, Priebe GP, Traficante DC, Pier GB, Falck JR, Morisseau C, Hammock BD, McCormick BA, Gronert K, Hurley BP. 2012. Hepoxilin A(3) facilitates neutrophilic breach of lipoxygenase-expressing airway epithelial barriers. J Immunol 189:4960–4969. doi: 10.4049/jimmunol.1201922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gil-de-Gómez L, Astudillo AM, Meana C, Rubio JM, Guijas C, Balboa MA, Balsinde J. 2013. A phosphatidylinositol species acutely generated by activated macrophages regulates innate immune responses. J Immunol 190:5169–5177. doi: 10.4049/jimmunol.1203494. [DOI] [PubMed] [Google Scholar]
- 35.Hurley BP, Pirzai W, Mumy KL, Gronert K, McCormick BA. 2011. Selective eicosanoid-generating capacity of cytoplasmic phospholipase A2 in Pseudomonas aeruginosa-infected epithelial cells. Am J Physiol Lung Cell Mol Physiol 300:L286–L294. doi: 10.1152/ajplung.00147.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang CM, Lee IT, Chi PL, Cheng SE, Hsiao LD, Hsu CK. 2014. TNF-alpha induces cytosolic phospholipase A2 expression via Jak2/PDGFR-dependent Elk-1/p300 activation in human lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 306:L543–L551. doi: 10.1152/ajplung.00320.2013. [DOI] [PubMed] [Google Scholar]
- 37.Hurley BP, Williams NL, McCormick BA. 2006. Involvement of phospholipase A2 in Pseudomonas aeruginosa-mediated PMN transepithelial migration. Am J Physiol Lung Cell Mol Physiol 29:L703–L709. doi: 10.1152/ajplung.00390.2005. [DOI] [PubMed] [Google Scholar]
- 38.Burgoyne RD, Morgan A. 1990. The control of free arachidonic acid levels. Trends Biochem Sci 15:365–366. doi: 10.1016/0968-0004(90)90227-3. [DOI] [PubMed] [Google Scholar]
- 39.Hurley BP, McCormick BA. 2008. Multiple roles of phospholipase A2 during lung infection and inflammation. Infect Immun 76:2259–2272. doi: 10.1128/IAI.00059-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kudo I, Murakami M. 2002. Phospholipase A2 enzymes. Prostaglandins Other Lipid Mediat 68-69:3–58. [DOI] [PubMed] [Google Scholar]
- 41.Liu L. 1999. Regulation of lung surfactant secretion by phospholipase A2. J Cell Biochem 72:103–110. [PubMed] [Google Scholar]
- 42.Schaloske RH, Dennis EA. 2006. The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta 1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 43.Tang X, Edwards EM, Holmes BB, Falck JR, Campbell WB. 2006. Role of phospholipase C and diacylglyceride lipase pathway in arachidonic acid release and acetylcholine-induced vascular relaxation in rabbit aorta. Am J Physiol Heart Circ Physiol 290:H37–H45. doi: 10.1152/ajpheart.00491.2005. [DOI] [PubMed] [Google Scholar]
- 44.Mizumura K, Hashimoto S, Maruoka S, Gon Y, Kitamura N, Matsumoto K, Hayashi S, Shimizu K, Horie T. 2003. Role of mitogen-activated protein kinases in influenza virus induction of prostaglandin E2 from arachidonic acid in bronchial epithelial cells. Clin Exp Allergy 33:1244–1251. doi: 10.1046/j.1365-2222.2003.01750.x. [DOI] [PubMed] [Google Scholar]
- 45.Wu T, Ikezono T, Angus CW, Shelhamer JH. 1996. Tumor necrosis factor-alpha induces the 85-kDa cytosolic phospholipase A2 gene expression in human bronchial epithelial cells. Biochim Biophys Acta 1310:175–184. doi: 10.1016/0167-4889(95)00143-3. [DOI] [PubMed] [Google Scholar]
- 46.Triggiani M, Granata F, Giannattasio G, Marone G. 2005. Secretory phospholipases A2 in inflammatory and allergic diseases: not just enzymes. J Allergy Clin Immunol 116:1000–1006. doi: 10.1016/j.jaci.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 47.Clark JD, Schievella AR, Nalefski EA, Lin LL. 1995. Cytosolic phospholipase A2. J Lipid Mediat Cell Signal 12:83–117. [DOI] [PubMed] [Google Scholar]
- 48.Kramer RM, Sharp JD. 1997. Structure, function and regulation of Ca2+-sensitive cytosolic phospholipase A2 (cPLA2). FEBS Lett 410:49–53. doi: 10.1016/S0014-5793(97)00322-0. [DOI] [PubMed] [Google Scholar]
- 49.Leslie CC. 1997. Properties and regulation of cytosolic phospholipase A2. J Biol Chem 272:16709–16712. doi: 10.1074/jbc.272.27.16709. [DOI] [PubMed] [Google Scholar]
- 50.Ghosh M, Tucker DE, Burchett SA, Leslie CC. 2006. Properties of the group IV phospholipase A2 family. Prog Lipid Res 45:487–510. doi: 10.1016/j.plipres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 51.Tokumoto H, Croxtall JD, Flower RJ. 1994. Differential role of extra- and intracellular calcium in bradykinin and interleukin 1 alpha stimulation of arachidonic acid release from A549 cells. Biochim Biophys Acta 1211:301–309. doi: 10.1016/0005-2760(94)90154-6. [DOI] [PubMed] [Google Scholar]
- 52.Nagase T, Uozumi N, Ishii S, Kume K, Izumi T, Ouchi Y, Shimizu T. 2000. Acute lung injury by sepsis and acid aspiration: a key role for cytosolic phospholipase A2. Nat Immunol 1:42–46. doi: 10.1038/76897. [DOI] [PubMed] [Google Scholar]
- 53.Hadad N, Burgazliev O, Elgazar-Carmon V, Solomonov Y, Wueest S, Item F, Konrad D, Rudich A, Levy R. 2013. Induction of cytosolic phospholipase a2alpha is required for adipose neutrophil infiltration and hepatic insulin resistance early in the course of high-fat feeding. Diabetes 62:3053–3063. doi: 10.2337/db12-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gijón MA, Leslie CC. 1999. Regulation of arachidonic acid release and cytosolic phospholipase A2 activation. J Leukoc Biol 65:330–336. [DOI] [PubMed] [Google Scholar]
- 55.Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, Heidelberg J, DeBoy RT, Haft DH, Dodson RJ, Durkin AS, Gwinn M, Kolonay JF, Nelson WC, Peterson JD, Umayam LA, White O, Salzberg SL, Lewis MR, Radune D, Holtzapple E, Khouri H, Wolf AM, Utterback TR, Hansen CL, McDonald LA, Feldblyum TV, Angiuoli S, Dickinson T, Hickey EK, Holt IE, Loftus BJ, Yang F, Smith HO, Venter JC, Dougherty BA, Morrison DA, Hollingshead SK, Fraser CM. 2001. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293:498–506. doi: 10.1126/science.1061217. [DOI] [PubMed] [Google Scholar]
- 56.Bou Ghanem EN, Lee JN, Joma BH, Meydani SN, Leong JM, Panda A. 2017. The alpha-tocopherol form of vitamin E boosts elastase activity of human PMNs and their ability to kill Streptococcus pneumoniae. Front Cell Infect Microbiol 7:161. doi: 10.3389/fcimb.2017.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park JY, Pillinger MH, Abramson SB. 2006. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin Immunol 119:229–240. doi: 10.1016/j.clim.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 58.Pietrobon EO, Soria M, Dominguez LA, Monclus Mde L, Fornes MW. 2005. Simultaneous activation of PLA2 and PLC are required to promote acrosomal reaction stimulated by progesterone via G-proteins. Mol Reprod Dev 70:58–63. doi: 10.1002/mrd.20190. [DOI] [PubMed] [Google Scholar]
- 59.Moriyama T, Urade R, Kito M. 1999. Purification and characterization of diacylglycerol lipase from human platelets. J Biochem 125:1077–1085. doi: 10.1093/oxfordjournals.jbchem.a022389. [DOI] [PubMed] [Google Scholar]
- 60.Pajor AM, Randolph KM. 2007. Inhibition of the Na+/dicarboxylate cotransporter by anthranilic acid derivatives. Mol Pharmacol 72:1330–1336. doi: 10.1124/mol.107.035352. [DOI] [PubMed] [Google Scholar]
- 61.Maxwell AP, Goldberg HJ, Tay AH, Li ZG, Arbus GS, Skorecki KL. 1993. Epidermal growth factor and phorbol myristate acetate increase expression of the mRNA for cytosolic phospholipase A2 in glomerular mesangial cells. Biochem J 295(Part 3):763–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leslie CC. 2015. Cytosolic phospholipase A(2): physiological function and role in disease. J Lipid Res 56:1386–1402. doi: 10.1194/jlr.R057588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moon SH, Jenkins CM, Mancuso DJ, Turk J, Gross RW. 2008. Smooth muscle cell arachidonic acid release, migration, and proliferation are markedly attenuated in mice null for calcium-independent phospholipase A2beta. J Biol Chem 283:33975–33987. doi: 10.1074/jbc.M805817200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bonventre JV. 1999. The 85-kD cytosolic phospholipase A2 knockout mouse: a new tool for physiology and cell biology. J Am Soc Nephrol 10:404–412. [DOI] [PubMed] [Google Scholar]
- 65.Clarke TB, Francella N, Huegel A, Weiser JN. 2011. Invasive bacterial pathogens exploit TLR-mediated downregulation of tight junction components to facilitate translocation across the epithelium. Cell Host Microbe 9:404–414. doi: 10.1016/j.chom.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marks M, Burns T, Abadi M, Seyoum B, Thornton J, Tuomanen E, Pirofski LA. 2007. Influence of neutropenia on the course of serotype 8 pneumococcal pneumonia in mice. Infect Immun 75:1586–1597. doi: 10.1128/IAI.01579-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pace-Asciak CR. 2015. Pathophysiology of the hepoxilins. Biochim Biophys Acta 1851:383–396. doi: 10.1016/j.bbalip.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 68.Nonomura N, Giebink GS, Zelterman D, Harada T, Juhn SK. 1991. Early biochemical events in pneumococcal otitis media: arachidonic acid metabolites in middle ear fluid. Ann Otol Rhinol Laryngol 100(5 Part 1):385–388. doi: 10.1177/000348949110000507. [DOI] [PubMed] [Google Scholar]
- 69.Rubins JB, Mitchell TJ, Andrew PW, Niewoehner DE. 1994. Pneumolysin activates phospholipase A in pulmonary artery endothelial cells. Infect Immun 62:3829–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cockeran R, Theron AJ, Steel HC, Matlola NM, Mitchell TJ, Feldman C, Anderson R. 2001. Proinflammatory interactions of pneumolysin with human neutrophils. J Infect Dis 183:604–611. doi: 10.1086/318536. [DOI] [PubMed] [Google Scholar]
- 71.Nagase T, Uozumi N, Aoki-Nagase T, Terawaki K, Ishii S, Tomita T, Yamamoto H, Hashizume K, Ouchi Y, Shimizu T. 2003. A potent inhibitor of cytosolic phospholipase A2, arachidonyl trifluoromethyl ketone, attenuates LPS-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol 284:L720–L726. doi: 10.1152/ajplung.00396.2002. [DOI] [PubMed] [Google Scholar]
- 72.Rubin BB, Downey GP, Koh A, Degousee N, Ghomashchi F, Nallan L, Stefanski E, Harkin DW, Sun C, Smart BP, Lindsay TF, Cherepanov V, Vachon E, Kelvin D, Sadilek M, Brown GE, Yaffe MB, Plumb J, Grinstein S, Glogauer M, Gelb MH. 2005. Cytosolic phospholipase A2-alpha is necessary for platelet-activating factor biosynthesis, efficient neutrophil-mediated bacterial killing, and the innate immune response to pulmonary infection: cPLA2-alpha does not regulate neutrophil NADPH oxidase activity. J Biol Chem 280:7519–7529. doi: 10.1074/jbc.M407438200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Berguerand M, Klapisz E, Thomas G, Humbert L, Jouniaux AM, Olivier JL, Bereziat G, Masliah J. 1997. Differential stimulation of cytosolic phospholipase A2 by bradykinin in human cystic fibrosis cell lines. Am J Respir Cell Mol Biol 17:481–490. doi: 10.1165/ajrcmb.17.4.2734. [DOI] [PubMed] [Google Scholar]
- 74.Chen HM, Yang CM, Chang JF, Wu CS, Sia KC, Lin WN. 2016. AdipoR-increased intracellular ROS promotes cPLA2 and COX-2 expressions via activation of PKC and p300 in adiponectin-stimulated human alveolar type II cells. Am J Physiol Lung Cell Mol Physiol 311:L255–L269. [DOI] [PubMed] [Google Scholar]
- 75.Bickford JS, Newsom KJ, Herlihy JD, Mueller C, Keeler B, Qiu X, Walters JN, Su N, Wallet SM, Flotte TR, Nick HS. 2012. Induction of group IVC phospholipase A2 in allergic asthma: transcriptional regulation by TNFalpha in bronchoepithelial cells. Biochem J 442:127–137. doi: 10.1042/BJ20111269. [DOI] [PubMed] [Google Scholar]
- 76.Adler DH, Cogan JD, Phillips JA III, Schnetz-Boutaud N, Milne GL, Iverson T, Stein JA, Brenner DA, Morrow JD, Boutaud O, Oates JA. 2008. Inherited human cPLA(2alpha) deficiency is associated with impaired eicosanoid biosynthesis, small intestinal ulceration, and platelet dysfunction. J Clin Invest 118:2121–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Faioni EM, Razzari C, Zulueta A, Femia EA, Fenu L, Trinchera M, Podda GM, Pugliano M, Marongiu F, Cattaneo M. 2014. Bleeding diathesis and gastro-duodenal ulcers in inherited cytosolic phospholipase-A2 alpha deficiency. Thromb Haemost 112:1182–1189. doi: 10.1160/TH14-04-0352. [DOI] [PubMed] [Google Scholar]
- 78.Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, Miyazaki J, Shimizu T. 1997. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature 390:618–622. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 79.Miyahara N, Takeda K, Miyahara S, Taube C, Joetham A, Koya T, Matsubara S, Dakhama A, Tager AM, Luster AD, Gelfand EW. 2005. Leukotriene B4 receptor-1 is essential for allergen-mediated recruitment of CD8+ T cells and airway hyperresponsiveness. J Immunol 174:4979–4984. doi: 10.4049/jimmunol.174.8.4979. [DOI] [PubMed] [Google Scholar]
- 80.Wen Z, Liu H, Li M, Li B, Gao W, Shao Q, Fan B, Zhao F, Wang Q, Xie Q, Yang Y, Yu J, Qu X. 2015. Increased metabolites of 5-lipoxygenase from hypoxic ovarian cancer cells promote tumor-associated macrophage infiltration. Oncogene 34:1241–1252. doi: 10.1038/onc.2014.85. [DOI] [PubMed] [Google Scholar]
- 81.Dehle FC, Mukaro VR, Jurisevic C, Moffat D, Ahern J, Hodge G, Jersmann H, Reynolds PN, Hodge S. 2013. Defective lung macrophage function in lung cancer +/− chronic obstructive pulmonary disease (COPD/emphysema)-mediated by cancer cell production of PGE2? PLoS One 8(4):e61573. doi: 10.1371/journal.pone.0061573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hsu A, Aronoff DM, Phipps J, Goel D, Mancuso P. 2007. Leptin improves pulmonary bacterial clearance and survival in ob/ob mice during pneumococcal pneumonia. Clin Exp Immunol 150:332–339. doi: 10.1111/j.1365-2249.2007.03491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jutila CK, Jutila MA, Crowell RE, Chick TW, Van Epps DE, Reed WP. 1992. Neutrophil responses to intravascular pneumococcal sonicate. Inflammation 16:135–146. doi: 10.1007/BF00918953. [DOI] [PubMed] [Google Scholar]
- 84.Attali C, Durmort C, Vernet T, Di Guilmi AM. 2008. The interaction of Streptococcus pneumoniae with plasmin mediates transmigration across endothelial and epithelial monolayers by intercellular junction cleavage. Infect Immun 76:5350–5356. doi: 10.1128/IAI.00184-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Beisswenger C, Coyne CB, Shchepetov M, Weiser JN. 2007. Role of p38 MAP kinase and transforming growth factor-beta signaling in transepithelial migration of invasive bacterial pathogens. J Biol Chem 282:28700–28708. doi: 10.1074/jbc.M703576200. [DOI] [PubMed] [Google Scholar]
- 86.Lanie JA, Ng WL, Kazmierczak KM, Andrzejewski TM, Davidsen TM, Wayne KJ, Tettelin H, Glass JI, Winkler ME. 2007. Genome sequence of Avery's virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J Bacteriol 189:38–51. doi: 10.1128/JB.01148-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dopazo J, Mendoza A, Herrero J, Caldara F, Humbert Y, Friedli L, Guerrier M, Grand-Schenk E, Gandin C, de Francesco M, Polissi A, Buell G, Feger G, García E, Peitsch M, García-Bustos JF. 2001. Annotated draft genomic sequence from a Streptococcus pneumoniae type 19F clinical isolate. Microb Drug Resist 7:99–125. doi: 10.1089/10766290152044995. [DOI] [PubMed] [Google Scholar]
- 88.Chen CC, Sun YT, Chen JJ, Chang YJ. 2001. Tumor necrosis factor-alpha-induced cyclooxygenase-2 expression via sequential activation of ceramide-dependent mitogen-activated protein kinases, and IkappaB kinase 1/2 in human alveolar epithelial cells. Mol Pharmacol 59:493–500. [DOI] [PubMed] [Google Scholar]
- 89.Chokki M, Mitsuhashi H, Kamimura T. 2006. Metalloprotease-dependent amphiregulin release mediates tumor necrosis factor-alpha-induced IL-8 secretion in the human airway epithelial cell line NCI-H292. Life Sc 78:3051–3057. doi: 10.1016/j.lfs.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 90.Lee JW, Park JW, Kwon OK, Lee HJ, Jeong HG, Kim JH, Oh SR, Ahn KS. 2017. NPS2143 inhibits MUC5AC and proinflammatory mediators in cigarette smoke extract (CSE)-stimulated human airway epithelial cells. Inflammation 40:184–194. doi: 10.1007/s10753-016-0468-2. [DOI] [PubMed] [Google Scholar]
- 91.Shin IS, Shin NR, Park JW, Jeon CM, Hong JM, Kwon OK, Kim JS, Lee IC, Kim JC, Oh SR, Ahn KS. 2015. Melatonin attenuates neutrophil inflammation and mucus secretion in cigarette smoke-induced chronic obstructive pulmonary diseases via the suppression of Erk-Sp1 signaling. J Pineal Res 58:50–60. doi: 10.1111/jpi.12192. [DOI] [PubMed] [Google Scholar]
- 92.Yang T, Luo F, Shen Y, An J, Li X, Liu X, Ying B, Liao Z, Dong J, Guo L, Wang T, Xu D, Chen L, Wen F. 2012. Quercetin attenuates airway inflammation and mucus production induced by cigarette smoke in rats. Int Immunopharmacol 13:73–81. doi: 10.1016/j.intimp.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 93.Hao Y, Kuang Z, Jing J, Miao J, Mei LY, Lee RJ, Kim S, Choe S, Krause DC, Lau GW. 2014. Mycoplasma pneumoniae modulates STAT3-STAT6/EGFR-FOXA2 signaling to induce overexpression of airway mucins. Infect Immun 82:5246–5255. doi: 10.1128/IAI.01989-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kawamoto Y, Morinaga Y, Kimura Y, Kaku N, Kosai K, Uno N, Hasegawa H, Yanagihara K. 2017. TNF-alpha inhibits the growth of Legionella pneumophila in airway epithelial cells by inducing apoptosis. J Infect Chemother 23:51–55. doi: 10.1016/j.jiac.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 95.Park JW, Kim YJ, Shin IS, Kwon OK, Hong JM, Shin NR, Oh SR, Ha UH, Kim JH, Ahn KS. 2016. Type III secretion system of Pseudomonas aeruginosa affects matrix metalloproteinase 12 (MMP-12) and MMP-13 expression via nuclear factor kappaB signaling in human carcinoma epithelial cells and a pneumonia mouse model. J Infect Dis 214:962–969. doi: 10.1093/infdis/jiw278. [DOI] [PubMed] [Google Scholar]
- 96.Mason RJ, Williams MC. 1980. Phospholipid composition and ultrastructure of A549 cells and other cultured pulmonary epithelial cells of presumed type II cell origin. Biochim Biophys Acta 617:36–50. doi: 10.1016/0005-2760(80)90222-2. [DOI] [PubMed] [Google Scholar]
- 97.McCormick BA, Miller SI, Carnes D, Madara JL. 1995. Transepithelial signaling to neutrophils by salmonellae: a novel virulence mechanism for gastroenteritis. Infect Immun 63:2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pawliczak R, Huang XL, Nanavaty UB, Lawrence M, Madara P, Shelhamer JH. 2002. Oxidative stress induces arachidonate release from human lung cells through the epithelial growth factor receptor pathway. Am J Respir Cell Mol Biol 27:722–731. doi: 10.1165/rcmb.2002-0033OC. [DOI] [PubMed] [Google Scholar]
- 99.Sakaguchi T, Kohler H, Gu X, McCormick BA, Reinecker HC. 2002. Shigella flexneri regulates tight junction-associated proteins in human intestinal epithelial cells. Cell Microbiol 4:367–381. doi: 10.1046/j.1462-5822.2002.00197.x. [DOI] [PubMed] [Google Scholar]
- 100.Bergeron Y, Ouellet N, Deslauriers AM, Simard M, Olivier M, Bergeron MG. 1998. Cytokine kinetics and other host factors in response to pneumococcal pulmonary infection in mice. Infect Immun 66:912–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hollifield M, Bou Ghanem E, de Villiers WJ, Garvy BA. 2007. Scavenger receptor A dampens induction of inflammation in response to the fungal pathogen Pneumocystis carinii. Infect Immun 75:3999–4005. doi: 10.1128/IAI.00393-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.