

ABSTRACT
Activation of the innate immune receptor NLRP1B leads to the formation of an inflammasome, which induces autoproteolytic processing of pro-caspase-1, and ultimately to the release of inflammatory cytokines and to the execution of pyroptosis. One of the signals to which NLRP1B responds is metabolic stress that occurs in cells deprived of glucose or treated with metabolic inhibitors. NLRP1B might therefore sense microbial infection, as intracellular pathogens such as Listeria monocytogenes and Shigella flexneri cause metabolic stress as a result of nutrient scavenging and host cell damage. Here we addressed whether these pathogens activate the NLRP1B inflammasome. We found that Listeria infection activated the NLRP1B inflammasome in a reconstituted fibroblast model. Activation of NLRP1B by Listeria was diminished in an NLRP1B mutant shown previously to be defective at detecting energy stress and was dependent on the expression of listeriolysin O (LLO), a protein required for vacuolar escape. Infections of either Listeria or Shigella activated NLRP1B in the RAW264.7 murine macrophage line, which expresses endogenous NLRP1B. We conclude that NLRP1B senses cellular infection by distinct invasive pathogens.
KEYWORDS: Listeria monocytogenes, NLRP1B, Shigella, caspase-1, inflammasome
INTRODUCTION
NLRP1B is a pattern recognition receptor that forms a multiprotein complex termed an inflammasome after it detects an activating signal (1). The NLRP1B inflammasome complex consists of multiple copies of NLRP1B and pro-caspase-1. The assembly of the complex leads to autoproteolysis of pro-caspase-1 and, consequently, to processing of inflammatory cytokines interleukin-1β (IL-1β) and IL-18 and to a type of cell death called pyroptosis (2, 3).
NLRP1B has four domains (2) (Fig. 1A). The N-terminal NACHT domain (a domain present in NAIP, CIITA, HET-E, and TP-1) is a nucleotide-binding domain that self-associates. The leucine-rich repeat (LRR) domain is involved in autoinhibition (4, 5), and the function to find domain (FIIND) undergoes autoproteolytic processing, which facilitates inflammasome assembly (6–8). The C-terminal caspase-activating and recruitment domain (CARD) binds the CARD of pro-caspase-1 (4).
FIG 1.

Listeria monocytogenes activates the NLRP1B inflammasome in a reconstituted system. (A) Schematic of NLRP1B. NACHT domain (residues 87 to 435), LRR domain (residues 627 to 719), FIIND (residues 720 to 1140; repeated sequences indicated as R1 and R2), and CARD (residues 1140 to 1233) are shown. (B) HT1080 cells expressing pro-caspase-1-T7, pro-IL-1β-HA, and wild-type NLRP1B were infected with Listeria monocytogenes at an MOI of 50 for the indicated times. Cell lysates were assayed for ATP. (C) Supernatants of cells as described for panel B were assayed for LDH activity. (D) Supernatants of cells as described for panel B were immunoprecipitated with anti-HA antibodies and then probed for HA-tagged IL-1β by immunoblotting. (E) HT1080 cells expressing pro-caspase-1-T7, pro-IL-1β-HA, and either wild-type (WT) NLRP1B or CARD deletion mutant NLRP1B1–1140, were treated with 10 mM 2DG in glucose-free, serum-free DMEM for 3 h or were infected with Listeria monocytogenes (Lis) for 3 h at an MOI of 50. Cell lysates were assayed for ATP. (F) Supernatants of cells as described for panel E were assayed for LDH activity. (G) Supernatants of cells as described for panel E were immunoprecipitated with anti-HA antibodies and then probed for HA-tagged IL-1β by immunoblotting. Blots are representative of three independent experiments. Cross-reacting bands were detected between 25 and 40 kDa. Graphed data represent means ± standard deviations from three independent experiments.
Anthrax lethal toxin is the only known direct activator of murine NLRP1B (1). The proteolytic component of the toxin cleaves NLRP1B near its N terminus; this cleavage is sufficient to relieve autoinhibition and allow oligomerization (9–11). Depletion of intracellular ATP is another activator of NLRP1B but one that probably triggers inflammasome assembly indirectly (12). The N-terminal region of NLRP1B is not cleaved after depletion of ATP, and the FIIND of NLRP1B facilitated the detection of this signal instead (5). Thus, activation of NLRP1B occurs through at least two distinct mechanisms.
The intracellular parasite Toxoplasma gondii is also detected by NLRP1B (13, 14), although the direct signal has not been determined. It is possible that Toxoplasma infection causes a reduction in cytosolic ATP. Notably, the parasite cannot synthesize its own purines and must import them from the host cell (15, 16). We thought that it was possible that intracellular bacterial pathogens might also be detected by NLRP1B. Listeria monocytogenes and Shigella flexneri have developed strategies that allow the bacteria to escape from the phagocytic vacuole, move intracellularly, and replicate in the cytosol (17, 18). These processes are likely to cause energy stress in the host cell. In addition, Listeria and Shigella infections have been shown to cause fragmentation of the mitochondrial network, resulting in a decrease of membrane potential and ultimately to a decrease in intracellular ATP (19–21).
Using a reconstituted system in which fibroblasts were transfected with plasmids encoding murine NLRP1B, pro-caspase-1, and pro-IL-1β, we found that infection with Listeria monocytogenes caused metabolic stress, as indicated by lowered cytosolic ATP levels, and induced NLRP1B-dependent pro-IL-1β processing. The N-terminal region of NLRP1B was dispensable for Listeria-mediated activation, whereas mutations in FIIND diminished its activation, which is consistent with the notion that metabolic stress caused by Listeria is the signal that triggers inflammasome assembly. We next used the macrophage cell line RAW264.7 to determine whether endogenously expressed NLRP1B was activated by ATP depletion and infection; we found that infection with either Listeria monocytogenes or Shigella flexneri reduced cytosolic ATP levels and induced pro-caspase-1 processing that was partially dependent on NLRP1B.
RESULTS
Listeria monocytogenes lowers cytosolic ATP levels and activates the NLRP1B inflammasome in transfected fibroblasts.
To determine whether cellular infection with Listeria monocytogenes reduces cytosolic ATP and activates NLRP1B, we used a transfected fibroblast model in which HT1080 cells are transfected with plasmids encoding NLRP1B, pro-caspase-1, and pro-IL-1β; inflammasome activation is then assessed by measuring IL-1β in cell supernatants. Human HT1080 cells were used because they are easily transfected and lack murine inflammasome components. Cells infected with Listeria at a multiplicity of infection (MOI) of 50 caused a 50% reduction in ATP after 0.5 h of infection (Fig. 1B). The ATP levels recovered slightly to 60 to 70% at 1 h, 2 h, and 3 h postinfection. Lactate dehydrogenase (LDH) release, used to measure the integrity of the plasma membrane, was approximately 15% of total LDH at 0.5 h and increased to 30% by 3 h (Fig. 1C). We next monitored caspase-1 activation by measuring the processing and release of IL-1β, which was detected in cell supernatants as early as 0.5 h postinfection; a moderately higher level of IL-1β was observed at 1 to 3 h postinfection (Fig. 1D).
To assess whether IL-1β release in response to Listeria was dependent upon expression of NLRP1B, we transfected HT1080 cells with either wild-type NLRP1B (NLRP1B1–1233) or a mutant NLRP1B lacking CARD (NLRP1B1–1140) (4). The cells were then infected with Listeria, and cytosolic ATP levels, LDH release, and IL-1β release were measured 3 h after infection. Cytosolic ATP levels and LDH release were similar in cells transfected with either wild-type (WT) NLRP1B or NLRP1B1–1140 (Fig. 1E and F). Because NLRP1B1–1140 (used as a negative control) lacks a CARD and is therefore unable to activate pro-caspase-1, we interpret these data to mean that LDH release results from either necrosis or membrane damage caused by the infection. IL-1β release was detected only from cells transfected with WT NLRP1B (Fig. 1G), however, demonstrating that IL-1β release in response to Listeria infection was dependent on the NLRP1B inflammasome. These results were similar to those in which cells were treated with the glycolysis inhibitor 2-deoxyglucose (2DG) (Fig. 1E to G).
NLRP1B FIIND is involved in NLRP1B inflammasome activation in response to Listeria infection.
Different regions of NLRP1B are involved in the response to lethal toxin and to metabolic stress (5). The N terminus of NLRP1B is the target of lethal toxin, and the repeated sequences in FIIND facilitate the response to ATP depletion. We next assessed whether these regions were important for activation in response to Listeria infection. HT1080 cells were first transfected with either WT NLRP1B or a mutant (NLRP1B80–1233) lacking 80 amino acids from the N terminus, and then the cells either were infected with Listeria or were treated with either lethal toxin or 2DG (Fig. 2A). As previously reported, cells that express NLRP1B80–1233 release similar amounts of IL-1β either when left untreated or when treated with lethal toxin, which is consistent with the notion that the N-terminal region of the protein is involved in autoinhibition and is the target of the toxin. In contrast, the amount of IL-1β released from cells treated with 2DG exceeds the amount released from untreated cells, indicating that the deletion does not fully activate NLRP1B and that metabolic stress is sensed outside the N-terminal region. Infection of cells expressing NLRP1B80–1233 with Listeria led to a small increase in the amount of IL-1β released compared to untreated cells, demonstrating that the N-terminal region is not essential for detection of the infection.
FIG 2.
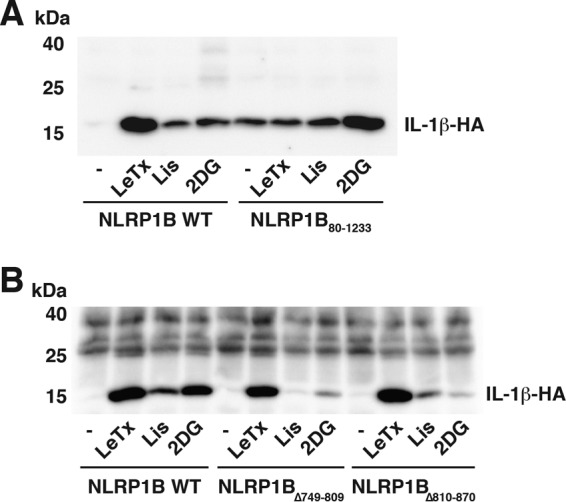
NLRP1B FIIND domain facilitates NLRP1B inflammasome activation in response to Listeria infection. (A) HT1080 cells expressing pro-caspase-1-T7, pro-IL-1β-HA, and either wild-type NLRP1B or N-terminal deletion mutant NLRP1B80–1233 were treated with lethal toxin (LeTx, consisting of 1 × 10−8 M PA and 1 × 10−8 M LF), or infected with Listeria (Lis) at an MOI of 50, or incubated with glucose-free, serum-free DMEM supplemented with 10 mM 2DG for 3 h. Cell supernatants were immunoprecipitated with anti-HA antibodies and then probed for HA-tagged IL-1β by immunoblotting. (B) HT1080 cells expressing pro-caspase-1-T7, pro-IL-1β-HA, and either wild-type NLRP1B or the indicated FIIND deletion mutant were treated with lethal toxin, or infected with Listeria (Lis) at an MOI of 50, or incubated with glucose-free, serum-free DMEM supplemented with 10 mM 2DG for 3 h. Cell supernatants were immunoprecipitated with anti-HA antibodies and then probed for HA-tagged IL-1β by immunoblotting. Blots are representative of three independent experiments.
We then transfected cells either with WT NLRP1B or with mutants lacking one of the duplicated sequences in FIIND, NLRP1BΔ749–809, and NLRP1BΔ810–870 (Fig. 2B). Cells expressing these mutants released an amount of IL-1β after treatment with lethal toxin similar to the amount released by cells expressing WT NLRP1B. In contrast, cells expressing either NLRP1BΔ749–809 or NLRP1BΔ810–870 secreted less IL-1β after treatment with 2DG than did cells expressing the wild-type protein (Fig. 2B). Similar results were observed when the cells were infected with Listeria. Together, our data suggest that activation of NLRP1B by Listeria occurs through a mechanism more closely related to that initiated by metabolic inhibition than by intoxication.
NLRP1B inflammasome activation in response to Listeria infection is dependent on LLO.
We next addressed whether Listeria Δhly or ΔactA mutant is able to activate the NLRP1B inflammasome. hly encodes listeriolysin O (LLO), a pore-forming toxin essential for endosomal escape (22). ActA is a protein on the bacterial cell surface that initiates actin-based motility and consequently promotes spreading of bacteria to neighboring cells (18, 23). The Listeria Δhly mutant failed to reduce cytosolic ATP or induce LDH release or IL-β release (Fig. 3A to C). The ΔactA mutant caused a reduction in ATP levels and release of LDH and IL-β that was comparable to WT Listeria (Fig. 3D to F). These results demonstrate that Listeria must express LLO, but not ActA, in order to reduce cytosolic ATP levels and activate the NLRP1B inflammasome.
FIG 3.

NLRP1B inflammasome activation in response to Listeria infection is dependent on listeriolysin O (LLO). (A) HT1080 cells expressing pro-caspase-1-T7, pro-IL-1β-HA, and NLRP1B were infected with either wild-type (WT) Listeria or Listeria Δhly mutant at an MOI of 50 or were incubated with glucose-free, serum-free DMEM supplemented with 10 mM 2DG for 3 h. The cell lysates were then assayed for ATP. (B) Supernatants of cells as described for panel A were assayed for LDH activity. (C) Supernatants of cells as described for panel A were immunoprecipitated with anti-HA antibodies and then probed for HA-tagged IL-1β by immunoblotting. (D) HT1080 cells expressing pro-caspase-1-T7, pro-IL-1β-HA, and NLRP1B were infected with either wild-type (WT) Listeria or Listeria ΔactA mutant at an MOI of 50 or were incubated with glucose-free, serum-free DMEM supplemented with 10 mM 2DG for 3 h. The cell lysates were then assayed for ATP. (E) Supernatants of cells as described for panel D were assayed for LDH activity. (F) Supernatants of cells as described for panel D were immunoprecipitated with anti-HA antibodies and then probed for HA-tagged IL-1β by immunoblotting. Blots are representative of three independent experiments. Cross-reacting bands were detected between 25 and 40 kDa. Graphed data represent means ± standard deviations from three independent experiments.
Metabolic inhibition activates the NLRP1B inflammasome in a murine macrophage cell line.
We sought to determine whether metabolic stress activates the inflammasome in the murine macrophage cell line RAW264.7, which expresses NLRP1B endogenously. RAW264.7 cells were treated with lipopolysaccharide (LPS) (commonly used to prime inflammasomes), 2DG, or a combination of the two compounds. Processed caspase-1 was detected in lysates derived from cells treated with the combination of LPS and 2DG (Fig. 4A; see also Fig. S2 in the supplemental material).
FIG 4.
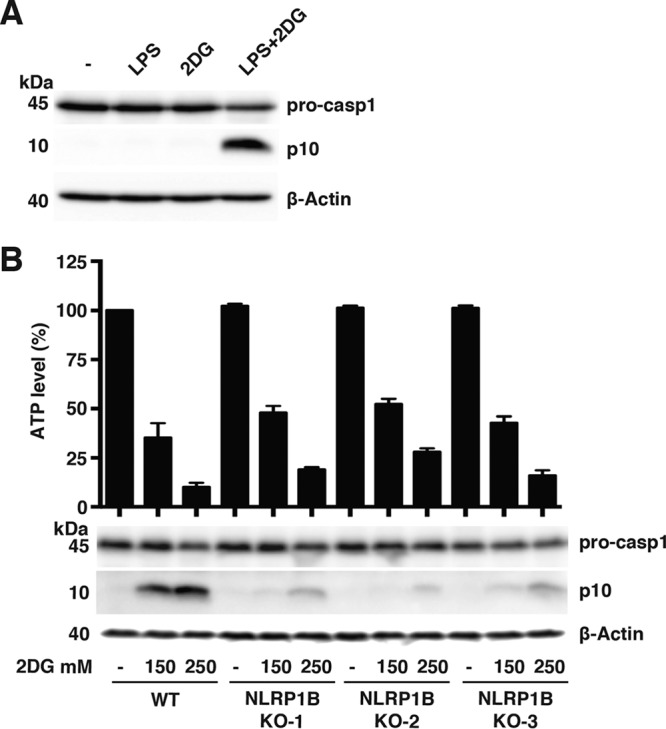
Metabolic inhibition activates the NLRP1B inflammasome in murine macrophage cell line RAW264.7. (A) WT RAW264.7 cells were treated for 4 h with either medium alone, 10 μg/ml LPS, 250 mM 2DG, or a combination of LPS and 2DG. The cell lysates were probed for pro-caspase-1, caspase-1 p10, and β-actin by immunoblotting. (B) WT RAW264.7 cells and three NLRP1B KO clones from independent CRISPR constructs were treated with 10 μg/ml LPS and the indicated concentrations of 2DG for 4 h. The cell lysates were assayed for ATP (top) and were probed for pro-caspase-1, caspase-1 p10, and β-actin by immunoblotting (bottom). Blots are representative of three independent experiments. Graphed data represent means ± standard deviations from three independent experiments.
To address whether the processing of pro-caspase-1 was dependent on NLRP1B, we used clustered regularly interspaced short palindromic repeat(s) (CRISPR)/Cas9 to knock out the gene. Three independent CRISPR constructs were used to generate NLRP1B knockout (KO) cell lines; each was tested for cytosolic ATP levels and pro-caspase-1 processing in response to LPS and different concentrations of 2DG. Cytosolic ATP levels were similar for WT RAW264.7 cells and all three NLRP1B KO cell lines under the different conditions (Fig. 4B); however, NLRP1B KO cells exhibited reduced levels of processed caspase-1, demonstrating that pro-caspase-1 processing in response to 2DG is partially dependent on NLRP1B (Fig. 4B).
Listeria monocytogenes and Shigella flexneri reduce cytosolic ATP levels and activate the NLRP1B inflammasome in RAW264.7 cells.
RAW264.7 cells were treated with LPS and infected with Listeria for 4 h to address whether Listeria monocytogenes can activate the inflammasome in this murine macrophage cell line. Infection with Listeria lowered ATP levels and induced pro-caspase-1 processing in wild-type RAW264.7 cells (Fig. 5A and B; see also Fig. S3 in the supplemental material). Levels of processed caspase-1 were reduced in Listeria-infected NLRP1B KO cells compared to wild-type cells (Fig. 5B), indicating that Listeria infection activates NLRP1B. Similar to what was observed with transfected HT1080 cells, the ΔactA mutant reduced intracellular ATP and caused processing of pro-caspase-1, but the Δhly mutant did neither (Fig. 5C and D).
FIG 5.

Listeria monocytogenes reduces cytosolic ATP levels and activates the NLRP1B inflammasome in RAW264.7 cells. (A) WT RAW264.7 or NLRP1B KO-1 cells were treated with LPS and were either uninfected (black bars) or infected with Listeria monocytogenes (white bars) for 4 h; cell lysates were assayed for ATP. (B) LPS-treated WT RAW264.7 or NLRP1B KO-1 cells were either uninfected (−) or infected with Listeria monocytogenes (Lis) for 4 h. Cell lysates were probed for pro-caspase-1, caspase-1 p10, and β-actin by immunoblotting. (C, D) WT RAW264.7 cells were treated with LPS and infected with either wild-type Listeria monocytogenes, the Δhly mutant, or the ΔactA mutant; cell lysates were assayed for ATP (C) or probed for pro-caspase-1, caspase-1 p10, and β-actin (D). Blots are representative of three independent experiments. Graphed data represent means ± standard deviations from three independent experiments; for each cell line, the infected sample was normalized to its uninfected control.
To test whether a different intracellular pathogen, Shigella flexneri, is able to activate the NLRP1B inflammasome, WT RAW264.7 and NLRP1B KO cells were treated with LPS and infected with Shigella for 1 h; ATP levels and pro-caspase-1 processing were measured. Shigella was able to lower cytosolic ATP and induce pro-caspase-1 processing in WT RAW264.7 cells (Fig. 6A and B; see also Fig. S4 in the supplemental material), while NLRP1B KO cells were impaired in their ability to process pro-caspase-1 (Fig. 6B).
FIG 6.
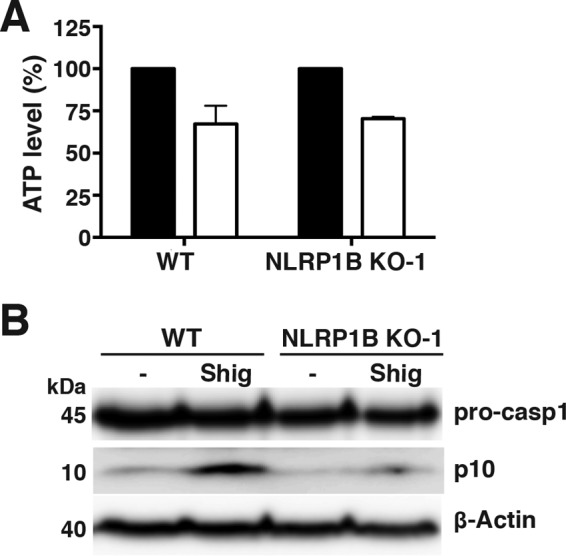
Shigella flexneri reduces cytosolic ATP levels and activates the NLRP1B inflammasome in RAW264.7 cells. (A) WT RAW264.7 or NLRP1B KO-1 cells were treated with LPS and were uninfected (black bars) or infected with Shigella flexneri (white bars) for 1 h. The cell lysates were then assayed for ATP. (B) LPS-treated WT RAW264.7 or NLRP1B KO-1 cells were either uninfected (−) or infected with Shigella flexneri (Shig) for 1 h. The cell lysates were probed for pro-caspase-1, caspase-1 p10, and β-actin by immunoblotting. Blots are representative of three independent experiments. Graphed data represent means ± standard deviations from three independent experiments; for each cell line, the infected sample was normalized to its uninfected control.
Taken together, these results demonstrate that the two intracellular pathogens reduce cytosolic ATP levels in the infected cell and activate the NLRP1B inflammasome.
DISCUSSION
We previously found that NLRP1B was activated in cells treated with metabolic inhibitors or subjected to glucose starvation—conditions that led to decreased cytosolic ATP (12). This led to the hypothesis that the sensing of energy stress by NLRP1B might allow it to detect a variety of intracellular pathogens. We demonstrate here that NLRP1B detects cellular infection by two pathogens, Listeria monocytogenes and Shigella flexneri, that reduce host cell ATP.
The mechanisms by which invasive pathogens deplete host cell ATP have not been well characterized. We found that depletion of ATP by Listeria was dependent on LLO. This pore-forming toxin is required for disruption of the vacuolar membrane, an event that may reduce ATP levels: membrane damage by Shigella and Listeria causes amino acid starvation and may likewise affect ATP levels (24, 25). In addition, LLO-dependent entry of Listeria into the cytosol might contribute to ATP depletion, as Listeria expresses a translocase that takes up host hexose phosphate, which might decrease glycolysis in the host cell (26). A third possibility is that LLO contributes to energy stress through disruption of mitochondrial function. Cells treated with purified LLO or infected with Listeria exhibit fragmented mitochondria (19, 27).
Shigella does not use a pore-forming toxin to exit the vacuole (17). Instead, a type three secretion system translocon protein mediates escape (28); this process is facilitated by an injected effector protein that recruits Rab11 vesicles to the vacuole (29, 30). Membrane damage or scavenging of host nutrients might cause the observed reduction of ATP. It is perhaps less likely that actin-based motility is responsible for the drop in ATP since loss of ActA had no effect on ATP levels during Listeria infection and Shigella has a mechanistically similar system (23).
We used two in vitro cell models to assess whether an invasive bacterium could activate NLRP1B. Fibroblasts that ectopically express NLRP1B, pro-caspase-1, and pro-IL-1β secreted IL-1β after being infected with Listeria. Because cells expressing NLRP1B FIIND mutants secreted less IL-1β, we believe that NLRP1B is detecting a signal generated by metabolic stress rather than by a host or microbial protease that cleaves its N terminus.
The second cell model employed to study NLRP1B was the RAW264.7 macrophage cell line, which has been used extensively to study anthrax lethal toxin. RAW264.7 cells do not express the apoptosis-associated speck-like (ASC) adaptor protein that is an essential component of the NLRP3, AIM2, and pyrin inflammasomes but is not required for the NLRP1B inflammasome (3). Furthermore, the expression level of the NAIP/NLRC4 inflammasome in RAW264.7 cells is very low and does not become activated under our experimental conditions (31–33). We observed processing of pro-caspase-1 in wild-type cells treated with 150 or 250 mM 2DG but little processing in the NLRP1B KO line. Infection of RAW264.7 cells by Listeria or Shigella also led to the activation of pro-caspase-1 that was in part a consequence of the NLRP1B inflammasome. It is unclear how pro-caspase-1 becomes activated by metabolic inhibitors or by infection in the NLRP1B KO cells. There are three NLRP1 paralogs in mice (NLRP1A, -B, and -C) (1). NLRP1C encodes a truncated protein that lacks CARD, so it is unlikely that NLRP1C is a functional protein. The domain structures of NLRP1A and NLRP1B proteins are similar. The activation signal of NLRP1A is still unknown and may function similarly to NLRP1B as a sensor of metabolic stress. We were unable, however, to detect NLRP1A mRNA in RAW264.7 cells by real-time quantitative PCR (qPCR) (data not shown).
We conclude from this work that NLRP1B functions as a sensor of metabolic stress induced by intracellular pathogens. The direct activator of NLRP1B in these experiments remains unclear and still needs to be elucidated.
MATERIALS AND METHODS
Reagents.
Protective antigen (PA) and lethal factor (LF) were purified as described previously and applied to cells at a final concentration of 10−8 M (34). LPS and 2DG were purchased from Sigma-Aldrich and used at the indicated concentrations.
Cell culture and bacterial strains.
HT1080 cells (ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. RAW264.7 cells (ATCC) were cultured in RPMI supplemented with 5% fetal bovine serum and 1% penicillin-streptomycin. Listeria monocytogenes WT strain 10403S and the isogenic Δhly-DPL2161 (35) and ΔactA-DPL3078 (36) strains were grown in brain heart infusion broth (Wisent). Shigella flexneri strain M90T was grown in tryptic soy broth (Wisent).
Plasmids and CRISPR/Cas9-mediated genome editing.
Plasmids encoding NTAP-NLRP1B allele 1, NTAP-NLRP1B1–1140, NTAP-NLRP1B80–1233, NTAP-NLRP1BΔ749–809, NTAP-NLRP1BΔ810–870, pro-caspase-1-T7, and pro-IL-1β-hemagglutinin (HA) have been described previously (5).
CRISPR guides targeting NLRP1B allele 1 were selected using the CRISPR design tool from MIT (http://crispr.mit.edu), and annealed oligonucleotides were cloned into pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene plasmid number 42230; a gift from Feng Zhang) (37): gNLRP1B-1, 5′-CACCGAGGATTCAGTGGGATTGAAC-3′; gNLRP1B-2, 5′-CACCGTGGAAGTCCTAGGATTCAGT-3′; gNLRP1B-3, 5′-CACCGTGGAAGTCCTAGGATTCAG-3′. RAW264.7 NLRP1B KO clones were selected following lethal toxin treatment in 96-well plates and were validated by sequencing. Each NLRP1B KO clone contained two mutations. For NLRP1B KO-1, the first mutation was a 55-bp deletion from nucleotides 834 to 888 of the NLRP1B allele 1 coding region (GenBank DQ117583.1), and the second was the insertion of a G at nucleotide 848. For NLRP1B KO-2, the first mutation was the deletion of the A at nucleotide 838, while the second was a 2-nucleotide (AG) deletion at nucleotides 838 and 839. For NLRP1B KO-3, the first mutation was the deletion of the C at nucleotide 837, and the second mutation was the deletion of the T at position 836. Each insertion or deletion resulted in a frameshift mutation and premature stop codon.
Listeria infection.
Cells cultured in antibiotic-free medium were infected with exponentially growing Listeria strains with a multiplicity of infection (MOI) of 50 for HT1080 cells and an MOI of 10 for RAW264.7 cells. Bacteria and cells were centrifuged at 1,000 × g for 10 min at room temperature and incubated at 37°C and 5% CO2 for 30 min. HT1080 cells were washed with phosphate-buffered saline (PBS) and treated with fresh gentamicin-containing (50 μg/ml) complete medium. RAW264.7 cells were incubated with fresh gentamicin-containing (30 μg/ml) complete medium supplemented with LPS (10 μg/ml).
Shigella infection.
RAW264.7 cells cultured in antibiotic-free medium were infected with exponentially growing Shigella such that the multiplicity of infection was 1. Bacteria and cells were centrifuged at 1,000 × g for 15 min at room temperature and incubated at 37°C and 5% CO2 for 30 min. Cells were incubated with fresh gentamicin-containing (30 μg/ml) complete medium supplemented with LPS (10 μg/ml).
IL-1β release assay in HT1080 cells.
The IL-1β release assay was performed as described previously (5). Sixteen hours prior to transfection, 1 × 106 HT0180 cells were seeded into 10-cm dishes. The cells were washed in PBS and transfected with 1 μg each of pNTAP-NLRP1B or the indicated mutant, pcDNA3-pro-caspase-1-T7, and pcDNA3-pro-IL-1β-HA. Polyethyleneimine adjusted to pH 7.2 was used as the transfection reagent. Cells were washed in PBS 24 h after transfection and treated with 10 mM 2DG in serum-free, glucose-free DMEM or with lethal toxin (LeTx, consisting of 1 × 10−8 M PA and 1 × 10−8 M LF) or infected with Listeria for 3 h. Cell supernatants were collected and incubated with α-HA antibody (12CA5) at 4°C for 2 h. Protein A Sepharose beads (RepliGEN) were then added to the supernatants and incubated overnight at 4°C. Protein A Sepharose beads were washed in EBC lysis buffer (50 mM Tris [pH 7.6], 150 mM NaCl, 0.5% [vol/vol] NP-40, 1 mM phenylmethylsulfonyl fluoride), and proteins were eluted from the beads with SDS loading dye and subjected to immunoblotting using a polyclonal anti-α-HA antibody (Santa Cruz sc805). Cross-reacting bands were detected between 25 kDa and 40 kDa, and pro-IL-1β was sometimes detected in the supernatants (Fig. S1).
Detection of caspase-1 p10.
RAW264.7 cells infected with Listeria or treated with LPS and 2DG were lysed after 4 h; Shigella-infected cells were lysed after 1 h. Cells were lysed in EBC buffer by shaking at 4°C for 1 h or by sonicating twice for 3 s. Lysates were clarified by centrifugation, and protein concentrations were determined using the Bradford assay. Equivalent amounts of cell lysate protein were subjected to SDS-polyacrylamide gel electrophoresis and immunoblotted with antibodies against caspase-1 p10 (Santa Cruz sc-514) and β-actin.
ATP level assay.
Intracellular ATP levels were measured by a CellTiter-Glo luminescent cell viability assay (Promega G7571) in accordance with the manufacturer's instructions. HT1080 cells were treated 24 h after transfection with glucose-free, serum-free DMEM and 2DG or infected with Listeria, as indicated. Cells were lysed and assayed for intracellular ATP. RAW264.7 cells were treated with LPS and 2DG for 4 h or infected with Listeria (4 h) or Shigella (1 h) and then lysed and assayed for intracellular ATP.
LDH assay.
Release of cytoplasmic LDH into the cell supernatant was measured by a CytoTox 96 nonradioactive cytotoxicity assay (Promega G1780) in accordance with the manufacturer's instructions. The percentage of LDH released was calculated as 100 × (experimental LDH − spontaneous LDH)/(maximum LDH − spontaneous LDH).
Supplementary Material
ACKNOWLEDGMENTS
We thank Dan Portnoy (U.C. Berkeley) for the generous gift of the Listeria strains.
This work was supported by grants from the Canadian Institutes of Health Research to S.E.G. and J.M.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00338-17.
REFERENCES
- 1.Boyden ED, Dietrich WF. 2006. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 2.Chavarria-Smith J, Vance RE. 2015. The NLRP1 inflammasomes. Immunol Rev 265:22–34. doi: 10.1111/imr.12283. [DOI] [PubMed] [Google Scholar]
- 3.Man SM, Kanneganti TD. 2015. Regulation of inflammasome activation. Immunol Rev 265:6–21. doi: 10.1111/imr.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liao KC, Mogridge J. 2009. Expression of Nlrp1b inflammasome components in human fibroblasts confers susceptibility to anthrax lethal toxin. Infect Immun 77:4455–4462. doi: 10.1128/IAI.00276-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neiman-Zenevich J, Liao KC, Mogridge J. 2014. Distinct regions of NLRP1B are required to respond to anthrax lethal toxin and metabolic inhibition. Infect Immun 82:3697–3703. doi: 10.1128/IAI.02167-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finger JN, Lich JD, Dare LC, Cook MN, Brown KK, Duraiswami C, Bertin JJ, Gough PJ. 2012. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J Biol Chem 287:25030–25037. doi: 10.1074/jbc.M112.378323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frew BC, Joag VR, Mogridge J. 2012. Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog 8:e1002659. doi: 10.1371/journal.ppat.1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D'Osualdo A, Weichenberger CX, Wagner RN, Godzik A, Wooley J, Reed JC. 2011. CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5-like domain. PLoS One 6:e27396. doi: 10.1371/journal.pone.0027396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chavarria-Smith J, Vance RE. 2013. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog 9:e1003452. doi: 10.1371/journal.ppat.1003452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hellmich KA, Levinsohn JL, Fattah R, Newman ZL, Maier N, Sastalla I, Liu S, Leppla SH, Moayeri M. 2012. Anthrax lethal factor cleaves mouse nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PLoS One 7:e49741. doi: 10.1371/journal.pone.0049741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH, Moayeri M. 2012. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog 8:e1002638. doi: 10.1371/journal.ppat.1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liao KC, Mogridge J. 2013. Activation of the Nlrp1b inflammasome by reduction of cytosolic ATP. Infect Immun 81:570–579. doi: 10.1128/IAI.01003-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ewald SE, Chavarria-Smith J, Boothroyd JC. 2013. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect Immun 82:460–468. doi: 10.1128/IAI.01170-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorfu G, Cirelli KM, Melo MB, Mayer-Barber K, Crown D, Koller BH, Masters S, Sher A, Leppla SH, Moayeri M, Saeij JP, Grigg ME. 2014. Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. mBio 5(1):e01117-13. doi: 10.1128/mBio.01117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sibley LD. 2011. Invasion and intracellular survival by protozoan parasites. Immunol Rev 240:72–91. doi: 10.1111/j.1600-065X.2010.00990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker DM, Oghumu S, Gupta G, McGwire BS, Drew ME, Satoskar AR. 2014. Mechanisms of cellular invasion by intracellular parasites. Cell Mol Life Sci 71:1245–1263. doi: 10.1007/s00018-013-1491-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Philpott DJ, Edgeworth JD, Sansonetti PJ. 2000. The pathogenesis of Shigella flexneri infection: lessons from in vitro and in vivo studies. Philos Trans R Soc Lond B Biol Sci 355:575–586. doi: 10.1098/rstb.2000.0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vazquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Dominguez-Bernal G, Goebel W, Gonzalez-Zorn B, Wehland J, Kreft J. 2001. Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev 14:584–640. doi: 10.1128/CMR.14.3.584-640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stavru F, Bouillaud F, Sartori A, Ricquier D, Cossart P. 2011. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc Natl Acad Sci U S A 108:3612–3617. doi: 10.1073/pnas.1100126108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stavru F, Palmer AE, Wang C, Youle RJ, Cossart P. 2013. Atypical mitochondrial fission upon bacterial infection. Proc Natl Acad Sci U S A 110:16003–16008. doi: 10.1073/pnas.1315784110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carneiro LA, Travassos LH, Soares F, Tattoli I, Magalhaes JG, Bozza MT, Plotkowski MC, Sansonetti PJ, Molkentin JD, Philpott DJ, Girardin SE. 2009. Shigella induces mitochondrial dysfunction and cell death in nonmyleoid cells. Cell Host Microbe 5:123–136. doi: 10.1016/j.chom.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 22.Seveau S. 2014. Multifaceted activity of listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Subcell Biochem 80:161–195. doi: 10.1007/978-94-017-8881-6_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldberg MB. 2001. Actin-based motility of intracellular microbial pathogens. Microbiol Mol Biol Rev 65:595–626. doi: 10.1128/MMBR.65.4.595-626.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LA, Yang C, Emili A, Philpott DJ, Girardin SE. 2012. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11:563–575. doi: 10.1016/j.chom.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 25.Tattoli I, Sorbara MT, Yang C, Tooze SA, Philpott DJ, Girardin SE. 2013. Listeria phospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures. EMBO J 32:3066–3078. doi: 10.1038/emboj.2013.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chico-Calero I, Suarez M, Gonzalez-Zorn B, Scortti M, Slaghuis J, Goebel W, Vazquez-Boland JA, European Listeria Genome Consortium. 2002. Hpt, a bacterial homolog of the microsomal glucose-6-phosphate translocase, mediates rapid intracellular proliferation in Listeria. Proc Natl Acad Sci U S A 99:431–436. doi: 10.1073/pnas.012363899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stavru F, Cossart P. 2011. Listeria infection modulates mitochondrial dynamics. Commun Integr Biol 4:364–366. doi: 10.4161/cib.4.3.15506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du J, Reeves AZ, Klein JA, Twedt DJ, Knodler LA, Lesser CF. 2016. The type III secretion system apparatus determines the intracellular niche of bacterial pathogens. Proc Natl Acad Sci U S A 113:4794–4799. doi: 10.1073/pnas.1520699113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mellouk N, Weiner A, Aulner N, Schmitt C, Elbaum M, Shorte SL, Danckaert A, Enninga J. 2014. Shigella subverts the host recycling compartment to rupture its vacuole. Cell Host Microbe 16:517–530. doi: 10.1016/j.chom.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 30.Weiner A, Mellouk N, Lopez-Montero N, Chang YY, Souque C, Schmitt C, Enninga J. 2016. Macropinosomes are key players in early Shigella invasion and vacuolar escape in epithelial cells. PLoS Pathog 12:e1005602. doi: 10.1371/journal.ppat.1005602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffmann C, Galle M, Dilling S, Kappeli R, Muller AJ, Songhet P, Beyaert R, Hardt WD. 2010. In macrophages, caspase-1 activation by SopE and the type III secretion system-1 of S. Typhimurium can proceed in the absence of flagellin. PLoS One 5:e12477. doi: 10.1371/journal.pone.0012477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katagiri N, Shobuike T, Chang B, Kukita A, Miyamoto H. 2012. The human apoptosis inhibitor NAIP induces pyroptosis in macrophages infected with Legionella pneumophila. Microbes Infect 14:1123–1132. doi: 10.1016/j.micinf.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 33.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. 2006. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol 7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 34.Kassam A, Der SD, Mogridge J. 2005. Differentiation of human monocytic cell lines confers susceptibility to Bacillus anthracis lethal toxin. Cell Microbiol 7:281–292. doi: 10.1111/j.1462-5822.2004.00458.x. [DOI] [PubMed] [Google Scholar]
- 35.Jones S, Portnoy DA. 1994. Characterization of Listeria monocytogenes pathogenesis in a strain expressing perfringolysin O in place of listeriolysin O. Infect Immun 62:5608–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Skoble J, Portnoy DA, Welch MD. 2000. Three regions within ActA promote Arp2/3 complex-mediated actin nucleation and Listeria monocytogenes motility. J Cell Biol 150:527–538. doi: 10.1083/jcb.150.3.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.