

Summary
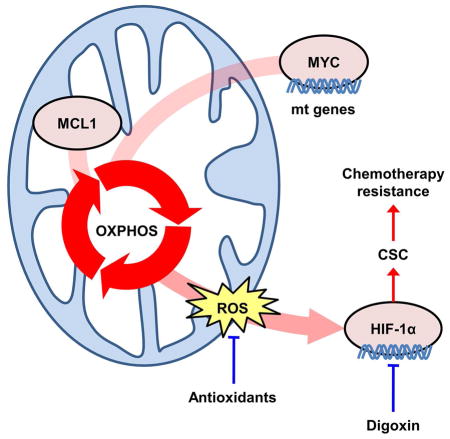
Most patients with advanced triple-negative breast cancer (TNBC) develop drug resistance. MYC and MCL1 are frequently co-amplified in drug-resistant TNBC after neoadjuvant chemotherapy. Herein, we demonstrate that MYC and MCL1 cooperate in the maintenance of chemotherapy-resistant cancer stem cells (CSCs) in TNBC. MYC and MCL1 increased mitochondrial oxidative phosphorylation (mtOXPHOS) and the generation of reactive oxygen species (ROS), processes involved in maintenance of CSCs. A mutant of MCL1 that cannot localize in mitochondria reduced mtOXPHOS, ROS levels and drug-resistant CSCs without affecting the anti-apoptotic function of MCL1. Increased levels of ROS, a by-product of activated mtOXPHOS, led to the accumulation of HIF-1α. Pharmacological inhibition of HIF-1α attenuated CSC enrichment and tumor initiation in vivo. These data suggest that 1) MYC and MCL1 confer resistance to chemotherapy by expanding CSCs via mtOXPHOS; and 2) targeting mitochondrial respiration and HIF-1α may reverse chemotherapy resistance in TNBC.
eTOC blurb
MYC and MCL1 are co-amplified in drug-resistant breast cancer. Lee et al. reveal that MYC and MCL1 cooperate to maintain cancer stem cells (CSCs) resistant to chemotherapy by increasing mitochondrial OXPHOS, ROS production and HIF-1α expression. Inhibition of HIF-1α blocks CSC expansion and restores chemotherapy sensitivity.
Introduction
Triple negative breast cancer (TNBC) comprises ~15% of all invasive breast cancers. TNBC lacks expression of the estrogen receptor (ER), progesterone receptor (PR), and amplification of ERBB2 (Carey et al., 2010). Due to the lack of known targetable molecular drivers in TNBC, cytotoxic chemotherapy is widely used in these patients. Many patients with TNBC develop resistance and relapse after adjuvant chemotherapy, ultimately succumbing to metastatic disease (Liedtke et al., 2008; Yu et al., 2013). Previous studies have proposed that a rare population of cancer cells, referred to as cancer stem-like cells (CSCs) or tumor-initiating cells (TICs), exhibit self-renewal capabilities and resistance to chemotherapy (Beck and Blanpain, 2013). This property of CSCs contributes to colonization of cancer cells at distant metastatic sites despite adjuvant chemotherapy (Clevers, 2011). Consistent with this notion, patients with TNBC whose tumors express CSC markers exhibit a worse outcome (Yu et al., 2013).
In a previous study, we demonstrated that TNBCs remaining in the breast following neoadjuvant chemotherapy (NAC) harbor amplification of MYC (54%) and MCL1 (35%) (Balko et al., 2014). In that study, 83% of MYC-amplified tumors exhibited co-amplification of MCL1, thus suggesting MYC and MCL1 may cooperatively contribute to chemotherapy resistance in TNBC. MYC is a proto-oncogene that encodes a transcription factor associated with cancer cell cycle progression, proliferation, apoptosis, and biosynthesis (Dang, 2012; Li et al., 2005a). Myeloid cell leukemia-1 (MCL1) is an anti-apoptotic Bcl-2 family protein which prevents apoptosis by suppressing cytochrome c release through association with pro-apoptotic Bcl-2 family proteins such as BID, BIM, PUMA and NOXA (Chen et al., 2005; Opferman et al., 2003; Shimazu et al., 2007).
Herein we show that MYC and MCL1 are overexpressed in TNBCs after chemotherapy and also in claudin-low TNBC cell lines where they contribute to tumor initiation and maintenance of CSCs. We also show that breast CSCs predominantly relied on mitochondrial oxidative phosphorylation (mtOXPHOS) whose activation is enhanced by both MYC and MCL1. This revealed a possible mechanism by which MYC and MCL1 promote CSC enrichment. Further, MYC- and MCL1-induced mtOXPHOS led to elevated production of reactive oxygen species (ROS) which, in turn, induced HIF-1α expression. Finally, knockdown of HIF-1α and use of a HIF-1α inhibitor, each in combination with anti-cancer chemotherapy markedly reduced drug-resistant CSCs, suggesting a novel therapeutic strategy for patients with this subtype of breast cancer.
Results
MYC and MCL1 are co-amplified in chemotherapy-resistant TNBC
We first performed targeted capture next-generation sequencing (NGS) on tumors from a small cohort of patients with TNBC treated with neoadjuvant chemotherapy (NAC). In 9 patients, tumor was available from the diagnostic pre-treatment biopsy, post-NAC mastectomy specimen, and a recurrent metastasis. In 9 additional patients, tumor was available from at least two of these sequential biopsies. In all tumors, a mutation in TP53 was detected. Overall, 8/18 (44%) cancers exhibited MYC and MCL1 co-amplification in at least one of the serial biopsies. MYC and MCL1 were co-amplified in 4/18 (22%) primary untreated tumors, 4/18 (22%) post-NAC mastectomies, and in 6/18 (33%) metastatic recurrences. Within the cohort with all three serial biopsies, 3/4 tumors with both genes amplified in the metastasis also contained the co-amplification in the original diagnostic biopsy. Overall, 17/18 (94%) TNBCs exhibited MYC and/or MCL1 amplification in at least one of the serial biopsies (Figure 1A). These data are consistent with and extend a previous report of ours (Balko et al., 2014) and further suggest an association of MYC and MCL1 co-amplification with drug-resistant TNBCs with a poor outcome as well as a higher frequency of each alteration than that reported by The Cancer Genome Atlas [TCGA; MYC: (14/18) 77.7% vs 44% in TCGA, p<0.0001; MCL1: (12/18) 66.6% vs 24% in TCGA, p<0.0001 both Fisher’s exact test].
Figure 1. MYC and MCL1 are amplified in post-NAC TNBC tumors and overexpressed in CSCs.

(A) Plot of genetic alterations as determined by targeted NGS in tumor DNA. X represents no biopsy was available. (B) ALDH+ cells were sorted and then subjected to intracellular labeling with MYC and MCL1 antibodies. (C) Cells were cultured in adherent conditions (ADH) or as mammospheres (MS) for 7 days. Cell lysates were subjected to immunoblot analysis with the indicated antibodies. (D) Relative levels of MYC and MCL1 protein in lysates from TNBC cell lines and quantified by Image J (*p<0.05). (E) MYC and MCL1 protein levels were plotted against the ratio of CD44:CD24 mRNA in the TNBC cell lines (Pearson’s correlation). (F) Levels of MYC mRNA in breast cancer biopsies before chemotherapy (Pre-T) and after chemotherapy (Post-T) were measured by NanoString analysis (n=17; paired t test, *p<0.005). (G) Left panel: H score of IHC analysis of MCL1 from tumor biopsies before chemotherapy and after chemotherapy (n=7; paired t test, *p<0.05). Right panel: Representative MCL1 IHC. (H) MYC, MCL1 and actin immunoblot analyses of lysates from paclitaxel resistant (PCTR) and parental (PAR) cells. Data are represented as mean ± SD.
A CSC phenotype is a known feature of many chemotherapy-resistant tumors (Beck and Blanpain, 2013). MDA-MB-436 and SUM159PT cells grown as mammospheres were overall less sensitive to chemotherapeutics, including paclitaxel, docetaxel, and doxorubicin, compared to those grown in adherent conditions, supporting an association of cells with tumor-initiating, stem-like capacity with drug tolerance and potential clinical resistance to anticancer chemotherapy (Figure S1A). Thus, we evaluated MYC and MCL1 protein levels in CSCs from TNBC cell lines sorted by aldehyde dehydrogenase (ALDH) activity, a typical marker of tumor-initiating cells (Croker et al., 2009). ALDH+ MDA-MB-436 and SUM159PT cells displayed enhanced mammosphere formation (Figure S1B) and higher MYC and MCL1 expression compared to ALDH− cells (Figure 1B). MYC and MCL1 expression were also increased in TNBC cells grown as mammospheres compared to adherent cells (Figure 1C). A CSC-derived gene expression signature is a feature of claudin-low TNBCs that persist following chemotherapy (Creighton et al., 2009; Hennessy et al., 2009). Hence, we next compared MYC and MCL1 protein levels between claudin-low and non-claudin-low TNBC subgroups (Figure S1C). Claudin-low TNBC cell lines exhibited higher expression of MYC and MCL1 compared to non-claudin-low TNBC lines (Figure 1D). Breast CSCs are also marked by CD44high/CD24low (Al-Hajj et al., 2003) and CD49+/EpCAM− (Visvader, 2009) expression. The ratio of mRNA levels of CD44 to CD24 and CD49 to EpCAM (Neve et al., 2006) also correlated with expression of MYC and MCL1 in TNBC cell lines (Figures 1E, S1D).
In a previous report, we quantified expression of genes associated with chemotherapy resistance in matched biopsies from TNBCs before and after NAC using NanoString nCounter analysis (Bhola et al., 2013). Analysis of these data showed that MYC mRNA expression (Figure 1F) and MCL1 protein levels (Figure 1G) were statistically higher in TNBCs after NAC compared to before treatment. In line with these data from primary tumors, MYC and MCL1 protein levels were upregulated in SUM159PT and MDA-MB-436 cells made resistant to paclitaxel (PCTR) (Figures 1H and S1E). The PCTR cells exhibited enhanced mammosphere forming ability (Figure S1F) and an increased proportion of ALDH+ cells (Figure S1G), also suggesting chemotherapy expands drug-tolerant CSCs. Finally, following treatment with paclitaxel, ALDH activity was higher in live MDA-MB-436 cells than in apoptotic cells sorted by flow cytometry (Figure S1H). These results suggest an association between high expression of MYC and MCL1 with chemo-resistant CSCs.
MYC and MCL1 contribute to enrichment of CSCs
To investigate the role of MYC and MCL1 on maintenance of CSCs, we knocked down MYC or MCL1 using siRNAs in MDA-MB-436 and SUM159PT cells, which express high levels of both MYC and MCL1 (Figure S2A). Conversely, we stably expressed MCL1 or GFP (control) constructs, or doxycycline (DOX)-inducible MYC in MDA-MB-468 cells, which express low endogenous levels of MYC and MCL1 (Figure S2B). Knockdown of MYC or MCL1 attenuated mammosphere formation (Figure 2A) and reduced the CD44hi/CD24low and ALDH+ fraction in SUM159PT and MDA-MB-436 cells (Figure 2B), respectively. Conversely, DOX-mediated induction of MYC or transduction of MCL1 induced mammosphere formation and increased the proportion of CD44hi/CD24low MDA-MB-468 cells (Figures 2C, D). To assess the tumorigenic potential of MYC and MCL1, we injected SUM159PT cells stably transduced with shRNAs [non-targeting shRNA control (shCont) or shMYC or shMCL1, Figure S2C] in limiting dilutions into athymic mice and monitor them for tumor formation for 8 weeks. Both shMYC and shMCL1 significantly reduced tumor initiation in vivo (Figure 2E, F). These results suggest that MYC and MCL1 contribute to CSC enrichment and tumor-initiating capacity in TNBC.
Figure 2. MYC and MCL1 increase mammosphere formation and enrich for CSCs.

(A, C) SUM159PT and MDA-MB-436 cells were transfected with two different MYC or MCL1 siRNAs (A). MDA-MB-468 cells stably transduced with pINDUCER20-MYC (expression induced by 100 ng/mL DOX) or pLX302-GPF or -MCL1 were subjected to mammosphere assay (C). The indicated cells were seeded in mammosphere assays for 7 days (* p<0.05, **p<0.005); original magnification, x100. (B, D) ALDH+ or CD44hi/CD24low fractions in cells manipulated as in A and C were analyzed by FACS (*p<0.005, **p<0.0005). (E, F) SUM159PT cells stably transduced with MYC or MCL1 shRNA were serially diluted and then injected subcutaneously (s.c.) in the lateral dorsum of athymic female mice for ELDA. Data are represented as mean ± SD.
Breast CSCs exhibit enhanced mitochondrial respiration
Recent evidence suggests that functional mitochondria are crucial for the maintenance of CSCs in several tumor types (Lamb et al., 2014). Dormant pancreatic tumor cells, which exhibit CSC features, rely on mitochondrial oxidative phosphorylation (mtOXPHOS) for survival after oncogene ablation (Viale et al., 2014). Inhibition of mitochondrial function by metformin eliminates pancreatic CSCs (Sancho et al., 2015). However, evidence for a mitochondrial role in breast CSCs is inconclusive. Thus, we examined the mitochondrial respiratory capacity of breast CSCs. In SUM159PT and MDA-MB-436 cells, the basal and maximal respiratory capacities of their CSC fraction (ALDH+ and cells grown as mammospheres) were higher than in ALDH− and adherent cells, suggesting activated mtOXPHOS (Figure 3A). Consistent with this result, mitochondrial membrane potentials (mtMPs) were also upregulated in SUM159PT and MDA-MB-436 CSCs (Figure 3B).
Figure 3. Breast CSCs exhibit an enhanced mitochondrial respiratory phenotype.

(A) OCRs were determined in cells sorted by ALDH activity (upper panel) or grown as mammospheres or in adherent condition (lower panel). (B) MDA-MB-436 and SUM159PT cells sorted or grown as in A were stained with Mitotracekr Red CMXRos and then analyzed by flow cytometry (*p<0.005). (C) ROS levels were determined by ROS-Glo as described in Experimental Procedures (*p<0.05, **p<0.005). (D) Cells were stained with MitoSOX Red and analyzed by flow cytometry (*p<0.005, **p<0.0005). (E) Cells were sorted by flow cytometry upon intensity of Mitotracker Red CMXRos (upper) and then seeded in mammosphere assays for 7 days (* p<0.05). Data are represented as mean ± SD.
Next, we measured levels of reactive oxygen species (ROS), which can be elevated through activation of the electron transport chain (ETC) in mitochondria. Hydrogen peroxide (H2O2) and mitochondrial superoxide levels were elevated in SUM159PT and MDA-MB-436 CSCs compared to non-CSCs, in line with a hyperactive mtOXPHOS (Figures 3C, D). SUM159PT and MDA-MB-436 cells were then sorted for high vs. low mtMPs by the intensity of Mitotracker Red CMXRos and plated as mammospheres. Mammosphere formation was markedly higher in cells with high mtMPs compared to low mtMPs (Figure 3E). Paclitaxel-resistant SUM159PT cells, which are enriched with CSCs (Figures S1F, G), also exhibited elevated mtOXPHOS capacity, mtMPs, ROS levels, and mitochondrial superoxide levels compared to parental drug-sensitive cells (Figures S3A–D). A previous report showed that targeting mitochondria with oligomycin A, an ATP synthase inhibitor, can attenuate mammosphere formation by pancreatic cancer cells (Viale et al., 2014). Treatment of TNBCs with oligomycin A reduced the oxygen consumption rate (OCR) and mammosphere formation by SUM159PT and MDA-MB-436 cells (Figures S3E, F). Similarly, metformin, a mitochondrial complex I inhibitor, reduced mammosphere formation and limited tumor initiation (Figure S3G, S7L). Moreover, PCTR cells and SUM159PT cells grown as mammospheres exhibited a statistically higher inhibition of OCR when treated with etomoxir, an inhibitor of fatty acid oxidation, compared to cells treated with BPTES and UK5099, suggesting a higher bioenergetic reliance on fatty acid oxidation in TNBC CSCs (Figures S3H, I). These results suggest that TNBC CSCs exhibit hyperactive mtOXPHOS which, in turn, maintains their self-renewal capacity.
MYC induces mtOXPHOS through mitochondrial biogenesis
Previous studies have shown that MYC directly regulates expression of genes involved in mitochondrial biogenesis (Li et al., 2005b) and affects metabolic processes required for mitochondrial respiration (Stine et al., 2015). To examine whether MYC contributes to mtOXPHOS in TNBC, we determined the OCR in MDA-MB-468 cells stably expressing DOX-inducible MYC. Induction of MYC by DOX increased basal and maximal mitochondrial respiratory capacities in MDA-MB-468 cells (Figure 4A, left). Conversely, knockdown of MYC by siRNA reduced mitochondrial respiratory capacities in MDA-MB-436 and SUM159PT cells (Figure 4A, middle and right). Ablation of MYC with siRNA also inhibited mitochondrial biogenesis in MDA-MB-468 cells as assessed by the number of mitochondria measured by transmission electron microscopy (TEM; Figure 4B). Both the mitochondrial DNA content and mass were also reduced upon MYC knockdown in MDA-MB-436 and SUM159PT cells. Conversely, expression of DOX-inducible MYC increased the mitochondrial DNA content and mass in MDA-MB-468 cells (Figures 4C, D). Further, induction of MYC upregulated ROS production in TNBC cells (Figure 4E). Finally, the inhibitory effect of oligomycin A on mammosphere formation was much more potent in MDA-MB-468 cells expressing DOX-inducible MYC compared to uninduced cells, suggesting that a MYC-dependent tumor-initiating capacity relies on mtOXPHOS (Figure 4F). These results imply that MYC potentiates mtOXPHOS and the subsequent enrichment of CSCs through its role in mitochondrial biogenesis.
Figure 4. MYC drives CSC enrichment via mitochondrial biogenesis.

(A) OCRs were determined by Seahorse XFe96 extracellular flux analyzer. (B) Number of mitochondria in cells transduced with MYC siRNA was measured as described in Experimental Procedures (*p<0.05). The yellow arrows point to each mitochondrion. (C) Levels of mitochondrial DNA were measured by real time quantitative PCR using the Human Mitochondrial DNA Monitoring Primer Set (*p<0.05, **p<0.005). (D) Fluorescent intensity of cells stained with NAO was determined by flow cytometry. (E) ROS levels were determined by ROS-Glo (*p<0.05, **p<0.005). (F) Cells transduced with pINDUCER20-MYC were seeded in mammosphere assays and treated with DMSO or 0.1 μM oligomycin A ± 100 ng/mL DOX for 7 days (*p<0.05). Data are represented as mean ± SD.
MCL1-mediated induction of CSCs is driven by mtOXPHOS
Despite the established anti-apoptotic role of MCL1, knockdown of MCL1 with siRNA did not induce apoptosis in MDA-MB-436 and SUM159PT cells (Figure S4A). Thus, we speculated that the anti-CSC effect of MCL1 knockdown (Figure 2) might be due to a mechanism independent of its anti-apoptotic function. When associated with the outer mitochondrial membrane (OMM), MCL1 serves as an anti-apoptotic protein, whereas inside the mitochondrial matrix, MCL1 stimulates mitochondrial respiration (Perciavalle et al., 2012). Stable overexpression of MCL1 in MDA-MB-468 cells upregulated OCR, mtMPs, and ROS levels. Conversely, transfection of MCL1 siRNA into MDA-MB-436 and SUM159PT cells reduced OCR, mtMPs, and ROS levels (Figures 5A–C). During the respiratory phase, mitochondria appear elongated and their cristae compartments are enlarged; in the glycolytic phase, however, the cristae appear round and fragmented (Alirol and Martinou, 2006). Knockdown of MCL1 with siRNA modulated mitochondria from an elongated shape to a round shape, indicative of an inactive respiratory phenotype (Figure 5D). In addition, the inhibitory effect of oligomycin A on mammosphere formation was more potent against MDA-MB-468 cells stably overexpressing MCL1 compared to control cells, suggesting that self-renewal capacity potentiated by MCL1 also relies on mtOXPHOS (Figure 5E). Metabolome analysis of SUM159PT cells showed ablation of MCL1 with siRNA reduced level of TCA cycle metabolites, including citrate, isocitrate, succinate, fumarate and malate, compared to control siRNA, suggesting a role of MCL1 in the aerobic oxidation of mitochondrial fuels (Figure 5F).
Figure 5. MCL1-induced increase in CSCs is mediated by mtOXPHOS.

(A–C) OCRs were determined by Seahorse XFe96 extracellular flux analyzer (A). Cells were stained with Mitotracker Red CMXRos and then were analyzed by flow cytometry (B; *p<0.0005) ROS levels were determined by ROS-Glo (C; *p<0.05, **p<0.005). (D) Cells were transduced with MCL1 siRNA and then prepared for transmission electron microscope imaging. Yellow dots outline each individual mitochondrion. (E) Cells transduced with MCL1 were seeded in mammosphere assays and treated with DMSO or 0.1 μM oligomycin A for 7 days (*p<0.05). (F) Cells were transduced with MCL1 siRNA and then lysed and subjected to LC-MS/MS. TCA cycle metabolites were analyzed as described in Methods (Multiple t-tests, *q<0.05, **q<0.0005). (G–I) OCRs and the proportion of CD44hi/CD24low cells were determined by Seahorse XFe96 extracellular flux analyzer (G) and flow cytometry (H), respectively (*p<0.005, **p<0.0005). Cells were seeded in mammosphere assays for 7 days (I; *p<0.05). (J) Cells were transduced with MCL1 siRNA or treated with VU0659158 for 4 days and then seeded in mammosphere assays for 7 days (*p<0.05, ** p <0.005). Data are represented as mean ± SD.
The localization of murine MCL1 within the mitochondrial matrix is mediated by its N-terminal mitochondrial targeting sequence (MTS) (Huang and Yang-Yen, 2010) and this localization is causally associated with its role in mitochondrial respiration (Perciavalle et al., 2012). Hence, we next conducted an in silico analysis of the human MCL1 protein sequence to examine a probability of mitochondrial export (Claros and Vincens, 1996). In this analysis, amino acids 1-44 of human MCL1 were highly predicted to play a role in mitochondrial export (0.8364; Figure S4B). The role of N-terminal domain of murine MCL1 in its proper mitochondrial localization was previously reported (Perciavalle et al., 2012). To test whether the localization of MCL1 in the mitochondrial matrix is required to induce CSCs, we designed a human MCL1 mutant without the N-terminal MTS (MCL1-ΔMTS; Figure S4C). Deletion of the MCL1 MTS impaired its ability to localize in the mitochondrial matrix (Figures S4D, E), resulting in reduced mitochondrial respiratory capacities in MDA-MB-468 cells (Figure 5G). Similarly, expression of MCL1-ΔMTS diminished mtMPs and ROS production, suggesting mitochondrial localization is required for the ability of MCL1 to regulate mtOXPHOS (Figures S4F, G). Further, CD44hi/CD24low cells and mammosphere formation were reduced in MDA-MB-468 cells expressing MCL1-ΔMTS compared to wild-type MCL1 (Figures 5H, I). Finally, deletion of the MTS attenuated enrichment of CSCs but did not affect the anti-apoptotic activity of MCL1 (Figure S4H).
To further investigate the discordance between the mitochondrial and anti-apoptotic actions of MCL1, we used the BH3-mimetic small molecule MCL1 inhibitor VU0659158 (Lee et al., 2017). Since this drug specifically binds the BH3-binding groove and mimics the binding of pro-apoptotic BH3 family members, we hypothesized that VU0659158 would not affect MCL1 action in the mitochondrial matrix where multi-BH domain cell death effectors, such as BAX and BAK, are absent. Unlike MCL1 siRNA, treatment with VU0659158 did not attenuate mammosphere formation or the OCR in MDA-MB-436 and SUM159PT cells (Figures 5J, S4I) but still induced apoptosis as measured by cleaved caspase 3/7 activity (Figure S4J). Collectively, these data suggest that mitochondrial MCL1 contributes to CSC enrichment through enhancement of mtOXPHOS, independent of its BH3 domain-mediated anti-apoptotic function.
MYC and MCL1 cooperate to induce expansion of triple negative breast cancer stem cells
Since we observed that ~44% of post-treatment, drug-resistant TNBCs harbor co-amplification of MYC and MCL1 (Figure 1A), we next explored whether MYC and MCL1 cooperate to enrich CSCs. Knockdown of both MYC and MCL1 with siRNA reduced expression of both MYC and MCL1 (Figure S5A). Simultaneous knockdown of MYC and MCL1 reduced ALDH+ cells and mammosphere formation in MDA-MB-436 and SUM159PT cells more potently than knockdown of MYC or MCL1 alone (Figures 6A, B). A reduction in mammosphere formation and the CD44hi/CD24low fraction was also observed in PCTR SUM159PT cells transfected with both siRNAs (Figures 6C, D). Conversely, we stably expressed MCL1 or GFP in MDA-MB-468 cells and then transduced them with DOX-inducible MYC (Figure S5B). Induction of MYC in MDA-MB-468 cells overexpressing MCL1 stimulated mammosphere formation and CD44hi/CD24low cells to a larger degree than in control cells expressing GFP (Figures 6E, F). Subsequently, simultaneous knockdown of MYC and MCL1 resulted in a more potent reduction in OCR and ROS levels compared to knockdown of MYC or MCL1 alone (Figures 6G, H).
Figure 6. MYC and MCL1 cooperate to expand CSCs.

(A, C) Cells were transduced with MYC and/or MCL1. After 4 days, cells were seeded in mammosphere assays for 7 days (*p<0.05, **p<0.005, ***p<0.0005). (B, D) Proportion of ALDH+ or CD44hi/CD24low cells was determined by flow cytometry (*p<0.05, **p<0.005, ***p<0.0005). (E, F) MDA-MB-468 cells transduced with pLX302-GFP or -MCL1 were re-transduced with pINDUCER20-MYC. Cells were then seeded in a mammosphere assay for 7 days ± 100 ng/mL DOX (E). Proportion of CD44hi/CD24low cells was determined by flow cytometry after 4 days of treatment with DOX (F). (G) OCRs were determined by Seahorse XFe96 extracellular flux analyzer. (H) ROS levels were examined by ROS-Glo (*p<0.05, **p<0.005, ***p<0.0005). (I) GSVA score was examined with a gene set [Reactome_Pyruvate metabolism and Citric Acid (TCA) cycle] in breast cancers in TCGA (*p<0.0005). Data are represented as mean ± SD.
To generate correlative data in human tumors, we performed Gene Set Variation Analysis (GSVA) of TCGA data and found that both OXPHOS and ROS pathways are significantly upregulated in breast tumors with MYC and/or MCL1 amplifications or mRNA upregulation (Table S1 and Figure S6A). We confirmed that levels of MYC or MCL1 mRNA were elevated in those tumors with MYC and MCL1 alterations (Figure S6B). Further, the GSVA score determined using a gene set involved in OXPHOS was higher in breast tumors with MYC and MCL1 amplifications or mRNA upregulation compared to tumors with overexpression of MYC or MCL1 alone (Figure 6I). These results suggest that MYC and MCL1 cooperate to enhance mitochondrial respiration which, in turn, maintains cancer cell stemness.
CSCs are augmented by MYC and MCL1 through the hypoxia pathway
Hypoxia is a micro-environmental state that activates hypoxia inducible factor (HIF) signaling, promoting CSCs and tumorigenesis (Lee and Simon, 2012; Rankin and Giaccia, 2016). As a byproduct of mtOXPHOS, ROS stabilize HIF proteins, resulting in activation of hypoxia signaling (Hwang and Lee, 2011; Mathieu et al., 2011). ROS levels are elevated in CSC-enriched tumor populations (Viale et al., 2014), which we confirmed in TNBC cells (Figure 3C). Similarly, HIF-1α protein was upregulated in MDA-MB-436 and SUM159PT cells cultured as mammospheres compared to adherent cells (Figure S7A). Analysis of expression data from the Cancer Cell Line Encyclopedia (CCLE) using Gene Set Enrichment Analysis (GSEA) found that epithelial-to-mesenchymal transition (EMT), ROS, and hypoxia pathways are enriched in claudin-low TNBC cell lines (Figure 7A). Moreover, we interrogated gene expression data in primary human TNBCs in the METABRIC database- and found that -EMT, ROS and Hypoxia pathways are statistically higher in claudin-low TNBCs compared to non-claudin-low TNBCs (Figures 7B, S7B). Therefore, we next asked whether ROS, stimulated by MYC and MCL1, contribute to hypoxia and, as a result, CSCs enrichment in TNBC.
Figure 7. Enrichment of CSCs driven by MYC and MCL1 is mediated by ROS-induced HIF-1α stabilization.

(A) mRNA expression data of TNBC cell lines (n=20) in the CCLE was analyzed using GSEA. Plots indicate hallmark gene sets enriched in claudin-low TNBC cell lines. (B) TNBCs in the METABRIC database (n=320) were divided as claudin-low and non-claudin low based on PAM50 analysis. GSVA scores were plotted accordingly. (C, D) Cells treated with H2O2 for 4 days were subjected to ALDH assay (C, *p<0.005, **p<0.0005) or seeded in mammosphere assays for 7 days (D, *p<0.05, **p<0.005). (E, I) Lysates were subjected to immunoblot analysis with HIF-1α and actin antibodies. (F) GSVA scores were generated with a gene set (Hallmark_Hypoxia) in TCGA breast tumors (*p<0.05, **p<0.005, ***p<0.0005). (G) Hallmark gene sets enriched in TNBCs after chemotherapy were determined using GSVA. Heatmap shows significantly enriched gene sets (q<0.05). (H) IHC analysis of HIF-1α in FFPE sections from TNBC biopsies before and after chemotherapy (n = 7; paired t tests, *p<0.05). (J, K) Cells were transduced with HIF1A siRNA (J, left and middle panels) or treated with 50 nM digoxin (K, left and middle panels) in presence of 200 μM H2O2 for 4 days. Cells were then subjected to ALDH assay (*p<0.05, **p<0.005, ***p<0.0005). (L, M) Cells manipulated as in J and K were seeded in mammosphere assays for 7 days (*p<0.05, **p<0.0005). (N) PCTR SUM159PT cells were treated with 50 nM paclitaxel ± 100 nM digoxin for 10 days (*p<0.0005). Data are represented as mean ± SD.
Treatment of MDA-MB-436 and SUM159PT cells with H2O2 increased HIF-1α protein expression, the ALDH+ fraction, and mammosphere formation (Figures S7C and 7C, D). Next, we determined whether MYC and MCL1 affect expression of HIF-1α. Knockdown of MYC or MCL1 reduced HIF-1α protein levels and expression of CA9 and ADM mRNAs, both HIF-1α target genes, in SUM159PT cells (Figure 7E, S7D). Conversely, induction of MYC and overexpression of MCL1 increased HIF-1α expression in MDA-MB-468 cells (Figure 7E). Consistent with upregulated ROS and OXPHOS pathways in breast tumors with MYC and MCL1 amplifications or mRNA upregulation, the hypoxia pathway was also found upregulated in those tumors (Figure S6A and Table S1). Moreover, the GSVA score of the hypoxia pathway was significantly higher in breast tumors with alterations in both MYC and MCL1 compared to tumors with amplification and overexpression of only MYC or MCL1 (Figure 7F), suggesting MYC and MCL1 cooperatively induce a hypoxia phenotype in breast cancer. To assess whether the hypoxia pathway is also activated in drug-resistant breast tumors, we conducted GSVA on the Nanostring nCounter pre- and post-chemotherapy expression data from a previously reported cohort of TNBCs at our institution (Bhola et al., 2013). Similar to the findings of higher MYC and MCL1 expression in drug-resistant treated cancers, the hypoxia and EMT pathways were upregulated in tumor mRNA after chemotherapy compared to before treatment (Figure 7G and Table S2). Protein levels of HIF-1α as measured by immunohistochemistry (IHC) were also elevated in TNBCs after chemotherapy (Figure 7H). Finally, HIF-1α protein measured by immunoblot was also upregulated in PCTR MDA-MB-436 and PCTR SUM159PT cells compared to their respective parental drug-sensitive cells (Figure 7I).
To test if HIF-1α is required for ROS-mediated induction of CSCs, we measured ALDH+ cells and mammosphere formation in cells transfected with HIF1A siRNA and treated with H2O2. Indeed, knockdown of HIF1A in MDA-MB-436 and SUM159PT cells abrogated induction of CSCs upon exposure to H2O2. The ALDH+ fraction and mammosphere formation were also diminished by HIF1A siRNA in PCTR SUM159PT cells (Figures 7J, L and S7E, F).
To complement these studies with a pharmacological approach, we used digoxin, which inhibits translation of HIF-1α (Zhang et al., 2008). Treatment with digoxin reduced the ALDH+ fraction and mammosphere formation in MDA-MB-436 and SUM159PT cells exposed to H2O2 and in PCTR SUM159PT cells (Figures 7K, M and S7G, H). Similarly, digoxin resensitized PCTR SUM159PT cells to paclitaxel (Figure 7N). Pretreatment of PCTR SUM159PT cells with digoxin before inoculation into mice reduced the rate of tumor formation (Figure S7L), further suggesting TNBCs enriched with CSCs can be reduced upon HIF-1α inhibition in vivo. Another inhibitor of HIF-1α, the antioxidant N-acetylcysteine (N-AC), also reduced the ALDH+ fraction and mammosphere formation (Figures S7I, J). Moreover, S3QEL2, complex III ROS scavenger, reduced HIF-1α protein levels in and limited tumor initiation by PCTR SUM159PT cells (Figure S7K, L). Finally, suggesting an association of HIF-1α with more virulent breast cancer biology, overexpression of HIF1A correlated with a shorter relapse-free survival in patients with TNBC (Figure S7M). Taken together, these data suggest that MYC and MCL1 promote ROS production which, in turn, activates the hypoxia pathway and subsequent enrichment of CSCs in TNBC. These results also provide a rationale for the therapeutic targeting of MCL1, MYC and HIF-1α in TNBC.
Discussion
We report herein a series of studies supporting the role of MYC and MCL1 in chemotherapy resistance of triple negative breast cancer (TNBC). We found MYC and MCL1 to be overexpressed in cancer stem cells (CSCs) in TNBCs. MYC and MCL1 cooperated in the promotion of mitochondrial oxidative phosphorylation (mtOXPHOS) that, in turn, activated the hypoxia pathway to further potentiate CSC enrichment, altogether revealing novel mechanism(s) by which MYC and MCL1 induce anticancer drug resistance.
CSCs exhibit a constellation of malignant traits, including increased motility, invasiveness, and self-renewal capacity, in line with their correlation with poor patient outcome (Chaffer and Weinberg, 2011). Other studies have suggested that CSCs represent a subpopulation of tumor cells that drive tumor recurrence following adjuvant therapy (Visvader and Lindeman, 2008). Gene expression analyses of residual drug-refractory breast tumors following chemotherapy have revealed an enrichment of CSC-like gene expression signatures (Creighton et al., 2009), also suggesting a causal association between CSCs and drug resistance. Therefore, strategies to prevent the re-emergence of CSCs have been devised to augment the impact of anticancer therapy.
Metabolic reprogramming is essential for cancer cells to survive and grow by using conventional metabolic pathways involved in energy production, biosynthesis, and redox balance. Oncogenic signaling pathways and transcriptional networks involving HIF, MYC and sterol regulatory element binding protein-1 (SERBP-1) have been shown to drive this metabolic reprogramming (DeBerardinis and Chandel, 2016). Cancer cells without stem-like features predominantly exhibit an aerobic glycolytic phenotype to produce energy, known as the Warburg effect (Koppenol et al., 2011), whereas CSCs display distinct metabolic features. For example, in pancreatic ductal adenocarcinoma, a subpopulation of dormant tumor cells that resist oncogene ablation exhibit CSC-like features and rely on mtOXPHOS for survival (Viale et al., 2014). Furthermore, mammospheres obtained from hormone-dependent, estrogen receptor (ER) positive breast cancer cell lines exhibit upregulation of key mitochondrial enzymes involved in beta-oxidation and ketone metabolism (Lamb et al., 2014), suggesting that CSC metabolism relies on mitochondrial respiration. In this report, we show that mitochondrial metabolic markers and activities, such as mtOXPHOS, mitochondrial membrane potential, and levels of mitochondrial superoxide and ROS, are elevated in TNBC stem-like cells and tumors.
Previous reports have shown that MYC activates a stem cell-like potential in several cancer types such as leukemia (Schubbert et al., 2014), hepatocellular carcinoma (Akita et al., 2014), prostate cancer (Civenni et al., 2013) and breast cancer (Terunuma et al., 2014). MYC is also known to participate in mitochondrial metabolic regulation processes, including mitochondrial biogenesis (Li et al., 2005b) and glutamine metabolism (Gao et al., 2009). Consistent with these data, we found that MYC positively regulates mitochondrial biogenesis and mtOXPHOS, which subsequently contributes to CSC maintenance in TNBC.
MCL1 is an anti-apoptotic Bcl-2 family member known to sequester pro-apoptotic Bcl-2 family proteins, such as BIM, BID, PUMA, and NOXA (Akgul, 2009). Through these actions, MCL1 has been proposed to promote chemotherapy resistance and cancer relapses (Perciavalle and Opferman, 2013). However, siRNA-based knockdown of MCL1 was found not to induce apoptosis in the claudin-low subgroup of TNBC (Goodwin et al., 2015), which we confirmed in claudin-low SUM159PT and MDA-MB-436 cells in this study (Figure S4A). Some reports have suggested that MCL1 serves as an anti-apoptotic protein when associated with the outer mitochondrial membrane (OMM), but improves mitochondrial respiration when located inside the mitochondrial matrix (Perciavalle and Opferman, 2013; Perciavalle et al., 2012). Indeed, we showed that, like MYC, MCL1 also facilitates mitochondrial respiration and promotes CSCs in TNBC. Thus, using a BH3 mimetic small molecule inhibitor of MCL1, we next asked whether MCL1 contributes to chemotherapy resistance independent of its anti-apoptotic function. Treatment with the MCL1 inhibitor VUI0659158 induced tumor cell apoptosis but did not abolish the effect of MCL1 on CSC maintenance, supporting the concept that this activity is independent of MCL1 binding with and disabling pro-apoptotic Bcl-2 family proteins. Considering the role of CSCs on drug resistance and cancer relapses, this result would also suggest that the sparing of CSCs by small molecule MCL1 inhibitors in clinical development could be a limitation to the overall anticancer efficacy of drugs relying exclusively on blockade of the BH3 domain-dependent anti-apoptotic function of MCL1.
We recognize that other mechanisms for MYC and MCL1 cooperativity in transformation have been proposed. For example, MCL1 has a protective role to delay apoptosis induced by MYC overexpression (Reynolds et al., 1994). Conversely, MYC directly regulates transcription of MCL1 (Labisso et al., 2012). Herein, we showed that MYC and MCL1 cooperate in stimulating mtOXPHOS and ROS production, resulting in activation of hypoxia signaling. Consistent with previous reports showing that HIFs are required for chemotherapy resistance mediated by breast CSCs (Samanta et al., 2014), we demonstrated that inhibition of HIF-1α reduces cancer cell stemness, chemotherapy resistance, and tumor initiation in TNBC. In addition, knockdown of MYC or MCL1 reduced expression of Nanog (Figure S7N), a major target of HIF-1α transcription (Mathieu et al., 2011), further suggesting that MYC and MCL1 enhance the stem cell-like potential of breast tumors via the hypoxia pathway. These data also suggest that a combination of a HIF-1α inhibitor and a BH3 mimetic MCL1 inhibitor may be required for complete blockade of MCL1 anti-apoptotic and mitochondrial functions.
In summary, we have shown a cooperative role of MYC and MCL1 on CSC enrichment that, in turn, leads to chemotherapy resistance in MYC and MCL1 co-amplified TNBC cells and tumors. This effect is due at least in part to an increase in mtOXPHOS, ROS production and HIF-1α expression. Genetic and pharmacological inhibition of HIF-1α reduced CSCs and restored chemotherapy action. Taken together, these results suggest HIF-1α as a therapeutic target in chemotherapy-resistant TNBC harboring MYC and/or MCL1 amplification.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Carlos L. Arteaga (carlos.arteaga@vanderbilt.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients and tumor specimens
Diagnostic core biopsies, surgically resected mastectomy specimens and metastatic biopsies were from patients with TNBC diagnosed and treated with neoadjuvant chemotherapy at the Vanderbilt-Ingram Cancer Center, the Instituto Nacional de Enfermedades Neoplásicas (INEN) in Lima, Perú, or the Hospital Clínico Universitario in Valencia, Spain. Female patients with triple negative breast cancer (TNBC) with ages between 24 and 78 (stage IIb to IIIc). Clinical and pathologic data were retrieved from medical records under institutionally approved protocols (IRB#030747, INEN 10-018, and 2012/189, respectively, at each clinical site). Breast tumors were determined to be triple-negative if they were negative for ER, PR, and HER2 overexpression measured by IHC and/or FISH in the diagnostic biopsy following clinical guidelines.
Cell lines
SUM159PT, MDA-MB-436, and MDA-MB-468 human breast cancer cell lines are derived from female Homo sapiens, were kindly provided from Dr. Jennifer A. Pietenpol (Lehmann et al., 2011). SUM159PT cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM; GIBCO) supplemented with 5% FBS and 0.5 μg/mL hydrocortisone. MDA-MB-436 and MDA-MB-468 cells were cultured in DMEM supplemented with 10% FBS. All cell lines were maintained at 37°C in 5% CO2. MDA-MB-436 and SUM159PT cells were treated with increasing concentrations of paclitaxel for 6 months to establish paclitaxel resistant cells.
Mice
Hsd:Athymic Nude-Foxn1nu female mice (6 weeks of age) for xenograft experiments were purchased from Envigo. Mice were housed in a controlled environment (12-h light/12-h dark cycle, light intensity ~350 lux) with ad libitum access to water and standard food (13.5% kcal from fat, LabDiet 5L0D). All animal experiments were performed in compliance with the NIH Guide for the Care and Use of Laboratory Animals and Animal Welfare Act. All animal studies were approved and performed in accordance with the Vanderbilt Institutional Animal Care and Use Committee.
METHOD DETAILS
Targeted next generation sequencing analysis
Before targeted profiling, histopathological diagnoses were independently confirmed on hematoxylin and eosin (H&E)-stained slides. Genomic profiling was performed in a CLIA-certified, CAP-accredited reference laboratory (Foundation Medicine). At least 50 ng of DNA per sample were extracted from 40 μm of unstained formalin-fixed paraffin embedded (FFPE) tumor sections with the Promega Maxwell 16 Tissue LEV DNA Kit and analyzed by hybridization capture of all coding exons from 236 cancer-related genes and selected introns of 19 genes commonly rearranged in cancer. Specimens were sequenced to high, uniform coverage on Illumina HiSeq instruments. Genomic base substitutions and indels (short insertions and deletions) were detected using custom tools optimized for mutation calling based on statistical modeling of sequence quality scores and local sequence assembly. Variations were filtered using dbSNP, then annotated for known and likely somatic mutations using COSMIC. Copy number alterations were detected by comparing targeted genomic DNA sequence coverage with a process-matched normal control sample. Genomic rearrangements were detected by clustering chimeric reads mapping to targeted introns.
Xenograft studies
SUM159PT cells stably expressing MYC shRNA or MCL1 shRNA were mixed with matrigel (1:1) and then injected subcutaneously (s.c.) in the lateral dorsum of athymic female mice in limiting dilution (100,000; 10,000; 1,000 cells). Paclitaxel-resistant and parental SUM159PT cells pretreated with 100 nM digoxin, 3 mM Metformin, or 2.5 μM S3QEL2 for 7 days before inoculation were mixed with matrigel (1:1) and then injected s.c. in the lateral dorsum of athymic female mice in limiting dilution (100,000; 10,000; 1,000 cells). Mice (n=10 per tumor cell dilution) were followed for tumor formation for 8 weeks. Proportion of tumor-initiating cells was estimated using extreme limiting dilution analysis (ELDA) (Hu and Smyth, 2009).
Immunohistochemistry
Sections of formalin-fixed paraffin-embedded tumor blocks obtained before and after neoadjuvant chemotherapy (n=7) from a previous study were subjected to IHC. The following primary antibodies were used: MCL1 (Santa Cruz Biotechnology), HIF-1α (Abcam). Antigen retrieval for MCL1 and HIF-1α was performed in citrate buffer (pH 6.0) under pressure for 15 minutes; endogenous peroxidase activity was blocked by incubating with 3% H2O2 for 10 minutes. The sections were incubated with the antibodies at 1:800 dilution at 4°C overnight and developed by DAB substrate (Vector Laboratories). Stained tumor regions were scored by an expert breast pathologist (M.V.E) by calculating the product of the percentage of cells staining at each intensity level and the intensity level (1+ to 3+ intensity). An H score was then calculated by summing the individual intensity level scores.
Immunoblot analysis
Cells were lysed with RIPA buffer (Sigma) containing 10 mM sodium fluoride, 1 mM sodium orthovanadate, and 1x protease inhibitor cocktail (Roche). Lysates (40 μg) were subjected to SDS-PAGE and transferred to nitrocellulose membranes. The nitrocellulose membranes loading the lysates were incubated with primary antibodies at 4°C overnight, followed by incubation with HRP-conjugated anti-rabbit or anti-mouse secondary antibodies for 2 hours at room temperature. Immunoreactive bands were visualized by enhanced chemiluminescence (Thermo SCIENTIFIC).
Lentiviral transductions
pLX302-GFP, -MCL1, and pINDUCER20-MYC plasmids were designed and cloned previously (Balko et al., 2014). Control, MYC and MCL1 shRNA plasmids were purchased from Santa Cruz Biotechnology. The MCL1 open reading frame without the N-terminal MTS was obtained through PCR selection from pLX302-MCL1 and was recombined into pLX302 using the LR clonase (Invitrogen) resulting in pLX302-MCL1-ΔMTS. Purified plasmids were transfected into 293FT cells along with pΔ8.9 and pVSVG to generate lentiviruses. Conditioned medium was applied to target cells (SUM159PT and MDA-MB-468) in the presence of Polybrene for 2 days prior to puromycin or G418 selection.
siRNA transfections
MYC, MCL1, HIF1A, and control siRNAs were purchased from Ambion. MDA-MB-436 and SUM159PT cells were seeded in 60 mm dishes 24 hours prior to transfection. Medium containing Lipofectamine RNAiMAX (Thermo SCIENTIFIC) was incubated for 5 minutes at room temperature and then mixed with medium containing 2 μg siRNA for 15 minutes at room temperature.
XF Cell Mito Stress analysis
The mitochondrial respiratory capacity was determined using XF Cell Mito Stress Test Kit (Agilent Technologies). Cells seeded in the XF Cell Culture Microplate were incubated for 24 hours at 37°C. Next day, cells were incubated with the base medium containing 2 mM L-glutamine, 1 mM sodium pyruvate, and 10 mM glucose for 1 hour prior to assay. The oxygen consumption rate (OCR) was measured by XFe96 extracellular flux analyzer (Agilent Technologies) with sequential injection of 1 μM oligomycin A, 0.5 μM FCCP, and 0.5 μM rotenone/antimycin A.
XF Mito Fuel Flex Test analysis
Mitochondrial fuel usage in live cells was measured by XF Mito Fuel Flex Test Kit (Agilent Technologies). Cells seeded in the XF Cell Culture Microplate were incubated for 24 hours at 37°C. The next day, cells were incubated with the base medium containing 2 mM L-glutamine, 1 mM sodium pyruvate, and 10 mM glucose for 1 hour prior to the assay. The oxygen consumption rate (OCR) was measured by XFe96 extracellular flux analyzer after injection of 3 μM BPTES, 4 μM etomoxir, or 2 μM UK5099. Each inhibitor was injected in a different order in order to determine the cell dependence on each fuel oxidation pathway. For example, to determine glucose dependence, UK5099 was injected first followed by measurement of the OCR. After 40 minutes, BPTES and etoximor were injected followed by a second OCR measurement. The dependence (D) for each mitochondrial fuel was calculated with the following formula:
Transmission electron microscopy (TEM)
Cells transfected with MYC or MCL1 siRNA for 72 hours were processed for TEM and imaged at the Vanderbilt Cell Imaging Shared Resource-Research Electron Microscopy facility.
Gene expression analysis
Nornalized RNA-seq data of breast tumors (n=816) and gene expression data of TNBCs (n=320) obtained from TCGA (Cerami et al., 2012; Gao et al., 2013) and METABRIC (Pereira et al., 2016), respectively, were analyzed with GSVA (Hanzelmann et al., 2013) using the gene sets provided from Molecular Signatures Database (DSigDB) (Subramanian et al., 2005). Gene expression data determined by NanoString nCounter analysis of pre- and post-chemotherapy TNBCs in a recently reported cohort (n=34) (Bhola et al., 2013) were also analyzed with GSVA. Gene expression data of TNBC cell lines (n=20) from CCLE (Barretina et al., 2012) was analyzed with GSEA (Subramanian et al., 2005) using h.all.v5.2.symbols gene sets.
Flow cytometry
The ALDEFLUOR assay (Stemcell Technologies) was performed to identify cells with high aldehyde dehydrogenase (ALDH) activity according to the manufacturer’s protocol. For cell surface protein labeling, cells were incubated with anti-CD44-APC (BD Biosciences), anti-CD24-PE (BD Biosciences) for 30 minutes at 37°C. For intracellular labeling, cells were fixed with 4% paraformaldehyde and then permeabilized with 0.1% Triton X-100. Cells were incubated with anti-c-Myc-PE (R&D Systems) and anti-MCL1-Alexa647 (Abcam) for 30 minutes at room temperature. For mitochondrial staining, cells were incubated with 50 nM Mitotracker Red CMXRos or 5 μM Mitosox™ for 30 minutes at 37°C in 5% CO2. Cells were stained with 0.5 μM 10-N-nonyl acridine orange (NAO) for 15 minutes at room temperature. Mitotracker Red CMXRos, Mitosox™ and NAO were purchased from Thermo SCIENTIFIC. All assays were performed using BD LSRFortessa X-20 Cell Analyzer (BD Biosciences).
Mammosphere assays
Single cell suspensions were seeded in six-well ultra-low attachment plates (Corning) in serum-free DMEM/F12 with 20 ng/mL EGF (R&D Systems) and 1X B27 (Invitrogen). The number of mammosphere was determined using the Gelcount mammalian cell colony counter (Oxford Optronix).
Cell viability assays
Cells treated with paclitaxel for 3 days were incubated with 10% trichloroacetic acid (TCA; Sigma) for 30 minutes at 4°C. The next day, fixed cells were stained with 0.4% sulforhodamine B for 10 minutes at room temperature and then washed with 1% acetic acid. Cells stained with sulforhodamine B were solubilized by 10 mM Tris-Cl and optical densities of the cell monolayers were determined by GloMax®-Multi Detection System (Promega).
Submitochondrial fractionation
Mitochondria were isolated using Mitochondrial Isolation Kit (Thermo SCIENTIFIC) and then swollen in 10 mM KH2PO4 (pH 7.4) for 20 minutes at 4°C. Equal volume of 10 mM Hepes (pH 7.4), 1.8 M sucrose, 10 mM MgCl2 was added to shrink the swollen mitochondria for 15 minutes at 4°C. Mitochondria were sonicated and then centrifuged at 12,000 g to obtain mitoplast fraction in pellet. Supernatant was centrifuged at 100,000 g to obtain outer mitochondrial membrane (OMM) fraction in pellet. Mitoplast fraction was sonicated and centrifuged at 12,000 g to remove unbroken mitoplast. Anti-SOD2 (#13141) was used as the OMM marker and anti-Bcl-xL (#7264) was used as the mitoplast marker. Both antibodies were purchased from Cell Signaling Technology.
Annexin V apoptotic cell staining
MDA-MB-436 and SUM159PT cells were transfected with MCL1 siRNA for 72 hours. Cells were then trypsinized and incubated with 1X Annexin V binding buffer containing Annexin V-alexa488 or –pacific blue™ (Invitrogen) for 30 minutes at room temperature in the dark. After this period of time, cells were incubated with propidium iodide (PI; Sigma) for 10 minutes at room temperature in the dark. Fraction of apoptotic cells was measured using flow cytometry.
ROS-Glo H2O2 Assays
The ROS-Glo H2O2 Assay was performed according to the manufacturer’s protocol (Promega). Cells were incubated with H2O2 substrate solution for 6 hours. The media was then incubated with the ROS-Glo detection solution containing D-Cysteine and the signal enhancer solution for 20 minutes at room temperature. Luminescence intensities from the mixture were measured by the GloMax®-Multi Detection System (Promega).
Quantification of mitochondrial DNA
Mitochondrial DNA extracted from the cells was quantified using the Human Mitochondrial DNA (mtDNA) Monitoring Primer Set (Takara) according to manufacturer’s guidelines. Genomic DNA was extracted from MDA-MB-436 and SUM159PT cells which were transfected with MYC siRNA for 72 hours. Primer sets for ND1 and ND5 were used for the detection of mtDNA and those of SLCO2B1 and SERPINA1 were used for the detection of nuclear DNA.
Immunocytochemistry
Cells were stained with 50 nM Mitotracker Red CMXRos for 30 minutes at 37°C in 5% CO2, fixed with complete media (DMEM + 10% FBS) containing 4% paraformaldehyde for 10 minutes at room temperature, and then permeabilized with 0.1% Triton X-100 for 10 minutes also at room temperature. Fixed cells were blocked with PBS containing 5% skimmed milk for 1 hour at room temperature. Finally, cells were stained with anti-MCL1 (#5453) and TOPRO3 (Invitrogen) for 1 hour at room temperature and then imaged using the Zeiss LSM510 Meta confocal microscope.
Caspase-Glo 3/7 assays
Caspase-Glo 3/7 assay was performed according to manufacturer’s instruction (Promega). Cells were treated with paclitaxel for 72 hours at 37°C in 5% CO2 and then incubated with Caspase-Glo reagent for 1 hour at room temperature. Luminescence intensities were measured by GloMax®-Multi Detection System.
Real time quantitative PCR
RNA was extracted from cells using the RNAeasy Kit (Qiagen) and synthesized to cDNA using iSCRPIT cDNA synthesis Kit (Bio-Rad) according to the manufacturer’s instructions. PCR reaction mixtures containing 1 μg of cDNA were prepared using SYBR Green Master Mix (Bio-Rad) with HIF1A, BNIP3, CA9, ADM, NANOG, and GAPDH primers (Qiagen). PCR reaction was performed using CFX96 thermal cycler (Bio-Rad).
Clonogenic assays
PCTR SUM159PT cells (5×102) were seeded on 6 well plates and treated with 50 nM paclitaxel ± 100 nM digoxin for 10 days. Cells were fixed with methanol for 10 minutes and then stained with 0.5% crystal violet for 10 minutes at room temperature. Stained cells were resolved with 10% acetic acid and the optical densities from the cells were measured by GloMax®-Multi Detection System.
LC-MS/MS
The quantification of TCA cycle intermediates was performed as described (Tan et al., 2014). Cells washed with ice-cold saline were sonicated. Samples were precipitated with ice-cold methanol overnight and then supernatants were obtained and dried. The dried samples corresponding to metabolites from 50 μg of cellular protein were reconstituted in 100 μl H2O containing 50 ng 3-13C-lactate (Cambridge Isotope Laboratories) and 50 ng pivalic acid (Sigma-Aldrich) for internal standard control and calibration. Following derivatization the samples were dried under a nitrogen stream and reconstituted in 100 μl mobile phase buffer A (water/MeCN (95:5) with 0.1% formic acid). The derivatized samples (10 μl) were injected onto an LC–triple quadrupole mass spectrometer (TSQ Quantum Ultra, Thermo SCIENTIFIC), coupled with an Acquity UPLC system (Waters). Samples were separated by UPLC using a BEH C8 column (1.7 μm particle size, 2.1 mm × 150 mm, Waters) with a two-solvent system [A: water/MeCN (95:5) with 0.1% formic acid; B: MeCN/water (95:5) with 0.1% formic acid] and a flow rate of 0.3 ml min–1. The gradient program was as follows: 0 to 1.0 min, 0% B; 1.0 to 8.0 min, 0 to 100% B; 8.0 to 10.0 min, 100% B; 10.0 to 10.5 min, 100 to 0% B; 10.5 to 15 min, 0% B. The injection volume was 10 μl. The MS acquisition methods used positive electrospray ionization (ESI) and analysis in the multiple reaction monitoring (MRM) mode. The source parameters were: N2 sheath gas 30 psi, N2 auxiliary gas 45 psi, Ar Collision cell pressure 1.5 mTorr and spray voltage was 5.0 kV. The ion source temperature was maintained at 300°C. The data analysis was performed using LCQuan 2.7 software (Thermo SCIENTIFIC). For metabolite quantification, calibration curves for peak areas of standards of known concentration were calculated using least-squares linear regression in Prism 7 (GraphPad software).
Kaplan-Meier plotter
Relapse free survival (RFS) was determined using the Kaplan-Meier Plotter (Gyorffy et al., 2013) with 200989_at as Affy id for HIF1A in ER−, PR−, and HER2− breast cancers.
QUANTIFICATION AND STATISTICAL ANALYSIS
All analyses were performed with GraphPad Prism 7. Data were presented as mean ± SD. All experiments were conducted at least three times with representative data shown. Statistically significant differences were determined using the Student’s t test. Paired t test were used for analyses of IHC (n=7) and NanoString data from matched tumor pairs (n=17, before and after chemotherapy). Correlations were evaluated using the Pearson r method. A value of p<0.05 was considered statistically significant. For the analyses of metabolites, comparisons between groups were made using Multiple t-tests with a False Discovery Rate of 0.05 using the two-stage setup method of Benjamini, Krieger and Yekutieli in Prism 7. A value of q (adjusted p value) <0.05 was considered statistically significant in GSVA with TCGA data (n=816), METABRIC data (n=320), and Nanostring data from matched tumor pairs (n=17). For the comparison among multiple groups in GSVA, one-way ANOVA were used to determine statistical significance. In GSEA for comparison between claudin-low and non-claudin-low cell lines (n=20), a value of p<0.05 was considered statistically significant. In the xenograft experiments, the number of tumors that formed in mice (n=10 per tumor cell dilution) was subjected to the ELDA and a value of p<0.05 was considered statistically significant.
Supplementary Material
Table S1, related to Figure S6. Enriched hallmarks in breast tumors harboring MYC and/or MCL1 amplifications
Table S2, related to Figure 7G. Enriched hallmarks in TNBCs following chemotherapy
Highlights.
MYC and MCL1 increase cancer stem cells in chemotherapy resistant TNBC
MYC and MCL1 cooperatively promote mtOXPHOS which, in turn, induces HIF-1α
MCL1 induces CSCs independent of its BH3-dependent, anti-apoptotic function
Inhibition of HIF1-α abolishes CSC enrichment in chemotherapy resistant TNBC
Acknowledgments
This work was supported by Susan G. Komen for the Cure Foundation grant SAC100013 (CLA), a grant from the Breast Cancer Research Foundation, NIH Breast Cancer SPORE grant P50 CA98131; and Vanderbilt-Ingram Cancer Center Support Grant P30 CA68485. The Agilent Seahorse Extracellular Flux Analyzer is housed and managed within the Vanderbilt High-throughput Screening Core Facility, an institutionally supported core, and was funded by NIH Shared Instrumentation Grant 1S10OD018015. J.M.B. received funding and support for this work from the IBC Network Foundation, Komen Foundation Career Catalyst Research grant 14299052 and NIH/NCI grant R00-CA181491. S.W.F. was supported by the NIH Director’s Pioneer Award DP1OD006933/DP1CA174419, and NCI Experimental Therapeutics (NExT) Program BOA29XS129TO22 under the Leidos Biomed Prime Contract No. HHSN261200800001E. A.L. received funding from Institute of Health Carlos III for the RD12/0036/0070.
Footnotes
Author Contributions
K.M.L. and C.L.A. designed the studies and wrote the manuscript. K.E.H. edited the manuscript. J.M.G. analyzed genetic alterations in TNBC tumor specimens. J.M.B. designed and produced lentiviral plasmids. L.J.S. helped animal experiments. A.L.G. contributed to bioinformatics analyses. M.J.N., M.V.E., V.S. and M.E.S. conducted the IHC analysis. T.L. and S.W.F. supplied VU0659158. H.G., A.L. and J.A.P.F. provided tumor specimens from patient with TNBC. M.S.W., G.A. and J.C.R. contributed to metabolites analysis. All coauthors reviewed the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akgul C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol Life Sci. 2009;66:1326–1336. doi: 10.1007/s00018-008-8637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akita H, Marquardt JU, Durkin ME, Kitade M, Seo D, Conner EA, Andersen JB, Factor VM, Thorgeirsson SS. MYC activates stem-like cell potential in hepatocarcinoma by a p53-dependent mechanism. Cancer research. 2014;74:5903–5913. doi: 10.1158/0008-5472.CAN-14-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alirol E, Martinou JC. Mitochondria and cancer: is there a morphological connection? Oncogene. 2006;25:4706–4716. doi: 10.1038/sj.onc.1209600. [DOI] [PubMed] [Google Scholar]
- Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, Owens P, Sanders ME, Kuba MG, Sanchez V, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014;4:232–245. doi: 10.1158/2159-8290.CD-13-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13:727–738. doi: 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- Bhola NE, Balko JM, Dugger TC, Kuba MG, Sanchez V, Sanders M, Stanford J, Cook RS, Arteaga CL. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest. 2013;123:1348–1358. doi: 10.1172/JCI65416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol. 2010;7:683–692. doi: 10.1038/nrclinonc.2010.154. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Molecular cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Civenni G, Malek A, Albino D, Garcia-Escudero R, Napoli S, Di Marco S, Pinton S, Sarti M, Carbone GM, Catapano CV. RNAi-mediated silencing of Myc transcription inhibits stem-like cell maintenance and tumorigenicity in prostate cancer. Cancer research. 2013;73:6816–6827. doi: 10.1158/0008-5472.CAN-13-0615. [DOI] [PubMed] [Google Scholar]
- Claros MG, Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313–319. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croker AK, Goodale D, Chu J, Postenka C, Hedley BD, Hess DA, Allan AL. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J Cell Mol Med. 2009;13:2236–2252. doi: 10.1111/j.1582-4934.2008.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin CM, Rossanese OW, Olejniczak ET, Fesik SW. Myeloid cell leukemia-1 is an important apoptotic survival factor in triple-negative breast cancer. Cell Death Differ. 2015;22:2098–2106. doi: 10.1038/cdd.2015.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013;8:e82241. doi: 10.1371/journal.pone.0082241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J, Sahin A, Agarwal R, Joy C, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer research. 2009;69:4116–4124. doi: 10.1158/0008-5472.CAN-08-3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Huang CR, Yang-Yen HF. The fast-mobility isoform of mouse Mcl-1 is a mitochondrial matrix-localized protein with attenuated anti-apoptotic activity. FEBS Lett. 2010;584:3323–3330. doi: 10.1016/j.febslet.2010.07.013. [DOI] [PubMed] [Google Scholar]
- Hwang AB, Lee SJ. Regulation of life span by mitochondrial respiration: the HIF-1 and ROS connection. Aging (Albany NY) 2011;3:304–310. doi: 10.18632/aging.100292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Labisso WL, Wirth M, Stojanovic N, Stauber RH, Schnieke A, Schmid RM, Kramer OH, Saur D, Schneider G. MYC directs transcription of MCL1 and eIF4E genes to control sensitivity of gastric cancer cells toward HDAC inhibitors. Cell Cycle. 2012;11:1593–1602. doi: 10.4161/cc.20008. [DOI] [PubMed] [Google Scholar]
- Lamb R, Harrison H, Hulit J, Smith DL, Lisanti MP, Sotgia F. Mitochondria as new therapeutic targets for eradicating cancer stem cells: Quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget. 2014;5:11029–11037. doi: 10.18632/oncotarget.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KE, Simon MC. From stem cells to cancer stem cells: HIF takes the stage. Curr Opin Cell Biol. 2012;24:232–235. doi: 10.1016/j.ceb.2012.01.005. [DOI] [PubMed] [Google Scholar]
- Lee T, Bian Z, Zhao B, Hogdal LJ, Sensintaffar JL, Goodwin CM, Belmar J, Shaw S, Tarr JC, Veerasamy N, et al. Discovery and biological characterization of potent myeloid cell leukemia-1 inhibitors. FEBS Lett. 2017;591:240–251. doi: 10.1002/1873-3468.12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA, Kim JW, Yustein JT, Lee LA, Dang CV. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol. 2005a;25:6225–6234. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Wang YY, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA, Kim JW, Yustein JT, Lee LA, Dang CV. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Molecular and cellular biology. 2005b;25:6225–6234. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:1275–1281. doi: 10.1200/JCO.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CM, Hubaud A, Stadler B, Choi M, Bar M, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011;71:4640–4652. doi: 10.1158/0008-5472.CAN-10-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- Perciavalle RM, Opferman JT. Delving deeper: MCL-1’s contributions to normal and cancer biology. Trends Cell Biol. 2013;23:22–29. doi: 10.1016/j.tcb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, Temirov J, Cleland MM, Pelletier S, Schuetz JD, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012;14:575–583. doi: 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016;7:11479. doi: 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. 2016;352:175–180. doi: 10.1126/science.aaf4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JE, Yang T, Qian L, Jenkinson JD, Zhou P, Eastman A, Craig RW. Mcl-1, a member of the Bcl-2 family, delays apoptosis induced by c-Myc overexpression in Chinese hamster ovary cells. Cancer Res. 1994;54:6348–6352. [PubMed] [Google Scholar]
- Samanta D, Gilkes DM, Chaturvedi P, Xiang L, Semenza GL. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci U S A. 2014;111:E5429–5438. doi: 10.1073/pnas.1421438111. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, Barneda D, Sellers K, Campos-Olivas R, Grana O, et al. MYC/PGC-1alpha Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015;22:590–605. doi: 10.1016/j.cmet.2015.08.015. [DOI] [PubMed] [Google Scholar]
- Schubbert S, Cardenas A, Chen H, Garcia C, Guo W, Bradner J, Wu H. Targeting the MYC and PI3K pathways eliminates leukemia-initiating cells in T-cell acute lymphoblastic leukemia. Cancer research. 2014;74:7048–7059. doi: 10.1158/0008-5472.CAN-14-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu T, Degenhardt K, Nur EKA, Zhang J, Yoshida T, Zhang Y, Mathew R, White E, Inouye M. NBK/BIK antagonizes MCL-1 and BCL-XL and activates BAK-mediated apoptosis in response to protein synthesis inhibition. Genes & development. 2007;21:929–941. doi: 10.1101/gad.1522007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, Metabolism, and Cancer. Cancer discovery. 2015;5:1024–1039. doi: 10.1158/2159-8290.CD-15-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan B, Lu Z, Dong S, Zhao G, Kuo MS. Derivatization of the tricarboxylic acid intermediates with O-benzylhydroxylamine for liquid chromatography-tandem mass spectrometry detection. Anal Biochem. 2014;465:134–147. doi: 10.1016/j.ab.2014.07.027. [DOI] [PubMed] [Google Scholar]
- Terunuma A, Putluri N, Mishra P, Mathe EA, Dorsey TH, Yi M, Wallace TA, Issaq HJ, Zhou M, Killian JK, et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. The Journal of clinical investigation. 2014;124:398–412. doi: 10.1172/JCI71180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–632. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE. Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes & development. 2009;23:2563–2577. doi: 10.1101/gad.1849509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- Yu KD, Zhu R, Zhan M, Rodriguez AA, Yang W, Wong S, Makris A, Lehmann BD, Chen X, Mayer I, et al. Identification of prognosis-relevant subgroups in patients with chemoresistant triple-negative breast cancer. Clin Cancer Res. 2013;19:2723–2733. doi: 10.1158/1078-0432.CCR-12-2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, Rey S, Hammers H, Chang D, Pili R, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008;105:19579–19586. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, related to Figure S6. Enriched hallmarks in breast tumors harboring MYC and/or MCL1 amplifications
Table S2, related to Figure 7G. Enriched hallmarks in TNBCs following chemotherapy