

Abstract
Superhydrophobic surface simultaneously possessing exceptional stretchability, robustness, and non-fluorination is highly desirable in applications ranging from wearable devices to artificial skins. While conventional superhydrophobic surfaces typically feature stretchability, robustness, or non-fluorination individually, co-existence of all these features still remains a great challenge. Here we report a multi-performance superhydrophobic surface achieved through incorporating hydrophilic micro-sized particles with pre-stretched silicone elastomer. The commercial silicone elastomer (Ecoflex) endowed the resulting surface with high stretchability; the densely packed micro-sized particles in multi-layers contributed to the preservation of the large surface roughness even under large strains; and the physical encapsulation of the microparticles by silicone elastomer due to the capillary dragging effect and the chemical interaction between the hydrophilic silica and the elastomer gave rise to the robust and non-fluorinated superhydrophobicity. It was demonstrated that the as-prepared fluorine-free surface could preserve the superhydrophobicity under repeated stretching-relaxing cycles. Most importantly, the surface’s superhydrophobicity can be well maintained after severe rubbing process, indicating wear-resistance. Our novel superhydrophobic surface integrating multiple key properties, i.e. stretchability, robustness, and non-fluorination, is expected to provide unique advantages for a wide range of applications in biomedicine, energy, and electronics.
Keywords: superhydrophobic, stretchable, robust, non-fluorinated
Graphical Abstract
Stretchable, rubbing-proof superhydrophobic surface was realized by chemically bonding silicone elastomer network covering the surface of silica microparticles to form enhanced micro-scale surface roughness.
Introduction
Superhydrophobic surfaces that show extremely strong water repellency have attracted wide interest due to their extensive applications, especially in the fields including self-cleaning1–2 and anti-fouling surfaces,3–5moisture-proof electronics,6 and drag-reduction for marine vessels,7–10 among others.11 While the existing superhydrophobic materials are generally realized by combinations of delicate microscale and nanoscale heterogeneous structures, the resulting superhydrophobicity is vulnerable to scraping or abrasion due to the destruction of the brittle structures.12, 13 A possible solution to the vulnerability is to use elastic substrates instead of rigid ones. As an example, micro-sized pyramid silicon arrays covered with gold nanoparticles were used as a template to fabricate superhydrophobic polydimethylsiloxane-based surfaces.14 However, despite all the efforts devoted in micro/nanofabrication of the molds, there remains a severe challenge related with faithful replication in nanoscale of the elastic materials, which tends to either break or deform at nanoscale during peeling off from the molds. Consequently, the failure in nanostructural replication has made the elastomer superhydrophobicity unsuccessful. On the other hand, reducing surface free energy has been used to achieve superhydrophobicity on elastomers. In this aspect, poly- or perfluoroalkyl surface treatments (also termed fluorination) are frequently implemented to substantially reduce the surface free energy.15–18 Nevertheless, the use of fluorination has raised some environmental concerns regarding their potential threats to human health.19, 20 For example, it has been reported that the exposure of human to these substances may increase the chances of many intractable diseases such as cancer, immune disorder, and hormonal disturbance.21 Consequently, there is a strong need to develop a stable superhydrophobic surface without fluorination.
While non-fluorinated and robust superhydrophobic surfaces are highly desirable, those integrating another remarkable feature, stretchability, are showing even greater promise in areas ranging from devices related to liquid motion manipulation22–24 to modern biomedical applications such as artificial skin25, 26 and wearable devices.27, 28 The true value of elastic superhydrophobic materials lies in that they preserve the superhydrophobicity when being stretched. To achieve this goal, researchers have devised a number of different strategies. For example, by enabling crumpling of graphene on an elastomer film of polyacrylate adhesive (VHB, 3M), the surface showed obvious superhydrophobicity with water contact angle as high as 152° in the crumpled state.29, 30 Unfortunately, the superhydrophobicity disappeared when the substrate was stretched (with contact angle decreased to 103° at ~80% strain), due to the sharp reduction of the surface roughness during graphene unfolding. More recently, another attempt has been made by spraying the solution of carbon nanofiber/paraffin/toluene and depositing the blend onto a natural rubber surface followed by solvent evaporation.31 The resulting superhydrophobicity persisted under high stretch, but this surface might be vulnerable against rubbing or abrasion because of the brittleness of individual carbon nanofibers that protruded out of the surface, and the lack of chemical bonding between carbon nanofiber and natural rubber.
Here we introduce a simple and environment-friendly non-fluorination method to prepare a highly stretchable and robust superhydrophobic surface by combining physical encapsulation and chemical bonding. The substrate material we chose was a commercial elastomer Ecoflex (Smooth-On, Inc.), which is a silicone rubber possessing exceptional stretchability and known non-toxicity.32 In particular, hydrophilic micro-sized silica particles were used to generate the superhydrophobic surface without the need of further fluorination. The as-prepared surfaces proved to be able to maintain superhydrophobicity after at least 1000 cycles of stretching-relaxing. Moreover, surface’s superhydrophobicity could be well preserved after severe rubbing to the surfaces, which brings it exceptional value to practical applications.
Results and Discussions
Fig. 1 depicts the possible structural basis for the robustness of the superhydrophobicity on our prepared surface, contrasting with that on a traditional elastic superhydrophobic surface lack of robustness. For the latter one that is simply covered with a dense layer of hydrophobic micro/nano-scaled particles, the superhydrophobic property may be easily lost by either stretching or rubbing the surface (shown in Fig. 1a). By stretching the surface, the distance between the particles is enlarged; by rubbing the surface, the particles on the surface tend to be scratched off due to the lack of stable particle/rubber adhesion, further giving rise to enlarged particle distance. Both processes will result in the decrease of the surface roughness, and thus the loss of superhydrophobicity. For our superhydrophobic surface taking advantage of the combination of the physical encapsulation and chemical bonding, by contrast, the superphydrophobicity can be well-preserved under much larger strains or after severe rubbing (Fig. 1b) due to the ultra-high density, multi-layered micro-sized particles chemically bonded to the substrate. It is worth to mention that the strategy of fabricating particle-composited superhydrophobic surface has been reported before. For example, multiple-step chemical reactions were designed to functionalize nanoparticles with water-repellent fluorinated groups, followed by spraying or spin-coating organic solution of the nanoparticles on glass substrates to fabricate superhydrophobic surfaces.33, 34 The as-prepared superhydrophobic surface showed remarkable abrasion resistance. Unfortunately, the lack of deformable substrates prevented the superhydrophobic surfaces from being stretchable. Moreover, the sophisticated procedures for functionalization of the nanoparticles further increased complexity of the preparation.
Fig. 1.
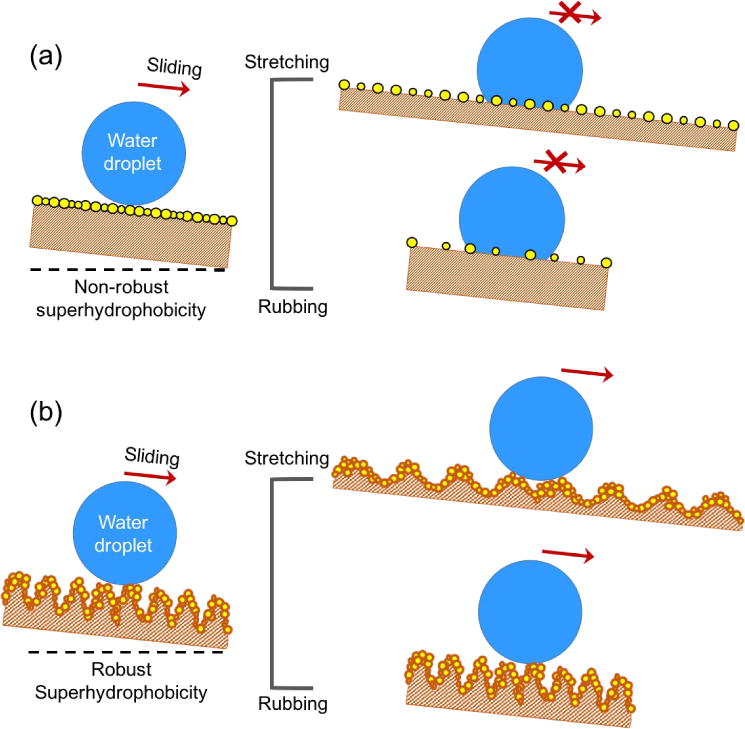
Structural comparison of the robust and conventional superhydrophobic surfaces. (a) Water droplet can slide off a lightly inclined freshly prepared non-robust superhydrophobic surface, while sticks to the surface if the surface receives stretching or rubbing due to increased particle distance and reduced surface roughness. (b) Water droplet slides off a lightly inclined robust superhydrophobic surface before and after stretching or rubbing process due to the preserved large surface roughness in both situations.
Fig. 2a schematically illustrates the typical fabrication process of such a robust superhydrophobic surface. A completely cured silicone elastomer membrane was firstly biaxially stretched to typical strains in the range of 50–200%. The stretched membrane was then spin-coated with a thin layer of the oligomer (with a weight ratio of part A to part B at 1:1), followed by deposition of micro-sized hydrophilic silica particles using a dumping method. The excessive particles were removed by gently shaking the membrane and the resulting surface was placed in a 100°C oven for at least 10 h, allowing the oligomer to cure completely and silica microparticles fixation. After the entire procedure, a highly stretchable and robust superhydrophobic surface can be obtained. Of note, the pre-stretch of the membrane before spin-coating of the oligomer is crucial for the surface to preserve superhydrophobicity under subsequent high stretch. Also, the hydrophilicity of the silica microparticles and the heating process were both indispensable to the final superior performance of the surface.
Fig. 2.
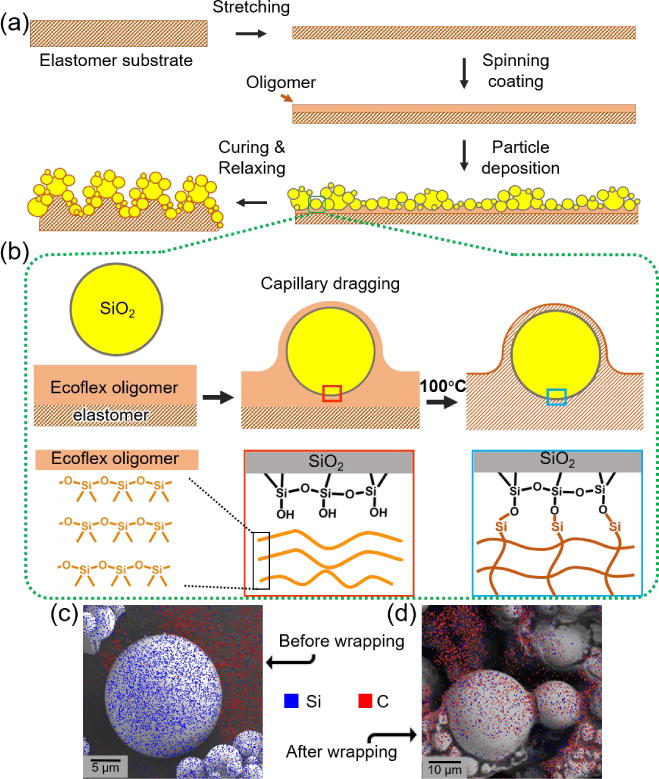
Schematics showing the fabrication and mechanism of the silicone elastomer/silica microparticle superhydrophobic surface. (a) A crosslinked silicone elastomer (Ecoflex) membrane is firstly stretched and spin-coated with a second layer of silicone oligomer. Silica microparticles are then deposited onto the silicone oligomer followed by curing at 100 °C. (b) As soon as the silica microparticles contact the silicone oligomers, the microparticles will be encapsulated by the oligomer via capillary dragging. At an elevated temperature of 100 °C, some Si-O backbones of the silicone oligomer break and bond with the hydroxyl group hanging outside of silica microparticles, forming stable chemical bonding. (c–d) EDX element mapping of the silica microparticles before and after encapsulated by the silicone elastomer. While large amount of carbon element could be detected on top of the silica microparticles after encapsulation (d), there were only negligible carbon signals on top of silica microparticles before the encapsulation(c).
A proposed mechanism is illustrated in Fig. 2b. Upon contact with the Ecoflex oligomer, the hydrophilic silica microparticles are rapidly encapsulated by the oligomer under capillary dragging, which is driven by the difference of the surface free energy between the silica microparticle and the silicone elastomer, Δγ. The viscosity η of the elastomer retards the encapsulation. By dimensional analysis, the relaxation time (τ) needed for the encapsulation process scales following the equation below,
where L is the diameter of the silica microparticle. Specific to the experiment here, the silica microparticles used had a diameter distribution from 1 µm to 24 µm, with approximately 80% falling in the range of 2 µm to 5 µm (see Fig. S1). From the rheology measurement shown in Fig. S2, η was calculated to be around 5 Pa·s for freshly prepared silicone oligomer. Taking Δγ ~ 55 mN/m (surface free energy of the hydrophilic silica microparticles and the silicone oligomer is ~ 75 mN/m and 20 mN/m, respecitvely,35, 36) and L ~ 5 µm, it was derived that the relaxation time τ equaled to roughly 0.5 ms. Indeed, the result suggested that all silica microparticles were well encapsulated by the silicone oligomer almost immediately after deposition on the surface. Therefore, this physical encapsulation can firmly immobilize the microparticles on the surface of silicone elastomer. In addition, the high-temperature incubation during the elastomer curing process further facilitated the creation of chemical bonding between the silica microparticles and the silicone matrix. As shown in Fig. 1b, the reactive component in the silicone oligomer is polydimethylsiloxane. Upon heating to 100 °C, some Si-O groups in the backbone of polydimethylsiloxane break and bond with the hydroxyl groups hanging outside of silica microparticles, forming covalent bonding between the surface of the silica microparticle and silicone.37–39 In the meantime, the silicone oligomer is crosslinked by crosslinker through hydrosilylation reaction40 and firmly adheres to the pre-existing underlying elastomer substrate (see Fig. S3 for detailed reaction between silicone oligomer and silica microparticles, as well as crosslinking of silicone oligomer). The combined physical encapsulation and covalent chemical bonding of the silica microparticles with the elastomer matrix ensured the robustness of the fabricated stretchable superhydrophobic surface.
The encapsulation of the silica microparticles by the silicone elastomer was verified by the element distribution on the surface of silica microparticles before and after the wrapping process, using energy dissipative x-ray spectroscopy (EDX) element mapping method, as shown in Fig. 2c and 2d. While the silicon element could be detected on the silica microparticles in both situations (shown in blue), the carbon element only appeared on top of those after wrapping (shown in red). Since the elastomer layer that covered the outer surface of the silica microparticle was the only source of carbon signal gained during the survey, the presence of the carbon element on top of silica microparticles clearly indicated their wrapping by the elastomer. In addition, the encapsulation was also evidenced from appearance of the wrinkles on the otherwise smooth silica microparticles, as shown in the Fig. S4, where the red arrows denote the wrinkles formed during the curing process of the oligomer. This wrapping of elastomer on the surface of the silica microparticles simultaneously endowed the surface with roughness and low surface free energy, which led to the superhydrophobicity.
Fig. 3a shows the appearance of the pristine and superhydrophobic elastomer surfaces. The translucent membrane became totally opaque after silica microparticle deposition. This uniform opaque surface differed much from the surface prepared by depositing hydrophobic silica microparticles onto the same silicone oligomer, which could only result in scattered non-uniform islands due to the inability of the oligomer to wrap the hydrophobic silica microparticles that already have low surface free energy (Fig. S5a). Fig. 3b shows a scanning electron microscopy (SEM) image of the cross-section of the as-prepared elastomeric superhydrophobic material. An elastomer/silica microparticle composite layer was observed with a clear boundary to the underlying smooth elastomer substrate. The thickness of the composite layer was approximately 70 µm, which was formed under a spin-coating speed of 4500 rpm and silica microparticles were deposited onto the oligomer surface immediately after the spin-coating. Top-view SEM images in Fig. 3c and 3e also indicate the microscale structural changes before and after the superhydrophobic treatment. The pristine elastomer surface was smooth (Fig. 3c) and the water contact angle was approximately 99.6° (Fig. 3d), exhibiting intrinsic hydrophobicity. After superhydrophobic treatment, the surface became enormously rough at microscale, characterizing multi-layer hierarchical structures (Fig. 3e). Water contact angle on the resulting surface was 151.2° (Fig. 3f), much higher than that of 114.5° on the elastomer surface with hydrophobic silica microparticle deposition (Fig. S5b).
Fig. 3.

Photoimage, morphology, and water contact angles of the pristine and superhydrophobic elastomer. (a) The superhydrophobic elastomer was opaque due to the presence of silica particles embedded. (b) The typical thickness of the composite layer is around 70 µm. (c) and (e) SEM images of the pristine and superhydrophobic elastomer. The superhydrophobic elastomer surface show homogeneous hierarchical microscale roughness. (d) and (f) The water contact angles on the pristine and superhydrophobic elastomer surfaces are 99.6° and 151.2°, respectively.
In fact, the thickness of the composite layer can be tuned by varying the spin-coating speed of the silicone oligomers (Fig. S6). The thicknesses of the composite layer corresponding to spin-coating speeds of 3000 rpm, 4500 rpm, and 6000 rpm, were 80 µm, 70 µm, and 60 µm, respectively. Despite of the difference in thickness, the resulting surfaces were all superhydrophobic, with the water contact angles on those surfaces being greater than 150°. However, the water sliding angles on these surfaces varied with the different thicknesses arisen from different spin-coating speeds. As shown in Fig. S6, the surface with a thickness of 70 µm prepared at 4500 rpm gave the lowest sliding angle of approximately 10°. It was believed that when the spin-coating speed was exceedingly slow (e.g. 3000 rpm), the thickness of the silicone oligomer layer was much larger than the diameter of the silica microparticles. During deposition, the silica microparticles (especially those small in size) were mostly immersed into the viscous liquid. As a result, the hierarchical surface roughness due to the different sizes of the silica microparticles could not be fully realized with most parts of the surface showing only bulk microscale roughness. Consequently, water droplet was prone to get pinned as it slid upon tilting. On the other hand, although for the surface obtained under a high spin-coating speed (e.g. 6000 rpm), the hierarchical structures could still be formed, since the thickness of the silicone oligomer layer is small, it was hard to achieve a homogeneous hierarchical structure on the surface (i.e., generation of clusters), leaving surface defected. In this situation, the water sliding angle was also increased. Surfaces prepared at 4500 rpm with a medium thickness of silicone oligomer showed superiority on both the generation of hierarchical structure and homogeneous structural distribution, resulting in the smallest water sliding angle.
In addition to the spin-coating speed, pre-curing time of the silicone oligomer prior to silica microparticle deposition is another parameter that would affect the ultimate performance of the surfaces. As shown in Fig. S7, with the pre-curing time varied from 0 min to 80 min (i.e. maximal time before the silicone Ecoflex oligomer completely cures) at room temperature, the resulting thickness of the composite layer monotonically decreased from approximately 70 µm to 10 µm. This reduction in thickness was believed to correlate with the increased viscosity of the oligomer and thus the wrapping hysteresis when pre-curing time was increased. Similar to the effect of spin-coating speed, the resulting change in the thickness did not significantly alter the water contact angles on these surfaces, with all being around 150° due to the large roughness and complete coverage of silica microparticles by silicone elastomer. Despite of the similarity of the water contact angles, sliding angles on these surfaces were quite distinct. From pre-curing time of 0 min to 20 min, the sliding angle increased from approximately 10° to nearly 30°, which was caused by the reduction of hierarchical assembly of the structures as abovementioned. Consequently, the spin-coating speed of 4500 rpm and 0 min pre-curing that gave rise to the largest water contact angle and smallest water sliding angle were determined to be the optimal conditions for preparing superhydrophobic elastomeric composite material, which were used for surface fabrication in following experiments unless otherwise noted.
As practical applications call for surface’s superhydrophobicity to remain stable under large strains, we tested the stability of the superhydrophobicity of our surface in different stretching states. Fig. 4a and 4b show the water droplets sliding on the relaxed and stretched surfaces, respectively. When an 8-µL water droplet was dispensed onto the slightly tilted surface with zero strain, the droplet could slide off the surface with ease (Movie S1). After receiving a uniaxial strain of 200%, water droplet with the same volume could still slide off the surface at the same level of tilting (Movie S2). Interestingly, the sliding speed of water droplet on the stretched surface was even larger than that on the relaxed surface, with further moving distance within 133 ms on the superhydrophobic surface at 200% strain (approximate 17.6 mm) than that at 0% strain (approximate 10.8 mm). In fact, this superhydrophobicity could be maintained at even more stretched states of beyond 200% strain. As shown in Fig. 4c, the water contact angles maintained at around 150º at the strain < 500%, with inset images showing the uniaxial stretch under different strains. Beyond the strain of 500%, the superhydrophobic surface would break abruptly. However, the large water contact angle as well as the highly roughed microstructures were preserved on the broken surface after it was relaxed (Fig. S8). The large surface roughness stemming from deposition of silica microparticles on a pre-stretch surface contributed to this stable superhydrophobicity.
Fig. 4.
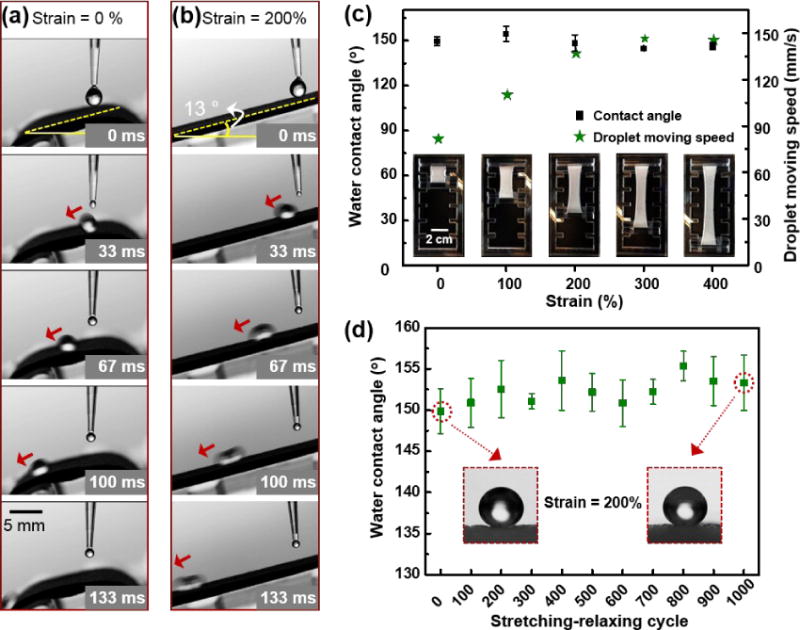
Stability of the superhydrophobicity of the surface under different strain. (a) and (b) Water droplet with volume of 8 µL sliding down the inclined superhydrophobic surface under strain = 0% and strain = 200%. (c) The water contact angles remain larger than 150° under different strain of the superhydrophobic surface, and the water sliding velocity increases with increasing strain. (d) The hydrophobicity of the surface remains after 1000 cycles of stretching-relaxing, with the strain=200%.
In addition, from Fig. 4c, we can also derive that the droplet moving speed on the surfaces with the same tilted angle of 13° but with different strains increased with increasing strains, which was consist with the results on the movement of water droplet in Fig. 4a and 4b. This increase of the moving speed with increasing strain could have arisen from the more obvious anisotropy of the surface due to the uniaxial stretch (Fig. S9), where anisotropic surfaces are more beneficial for directional movement of the droplets along the aligned direction comparing with isotropic surfaces.41, 42 Fig. 4d further demonstrated the stability of the superhydrophobicity under cyclic stretching between 0% and 200%. The superhydrophobicity of the surface could be well maintained after 1000 cycles. The inset images of water droplet on the surface before any stretch and after 1000 cycles of stretch-relax showed little difference with regard to the contact angle. Moreover, after 1000 cycles of stretching-relaxing at a strain of 200%, the superhydrophobic elastomer could still give a water sliding angle of 8°, similar to the value (9–10°) for that before stretching. This unchanged sliding angle was consistent with the well-preserved rough microstructures of the surface after cyclic stretching-relaxing, as shown in Fig. S10. These results indicated that the superhydrophobic surface was able to maintain stability in highly stretch states, which is promising in applications requiring superhydrophobicity under different strains. Of note, the method we show here can be extended to preparation of highly stretchable superhydrophobic surfaces for a broad range of elastomeric materials. It was also believed that with larger pre-stretch ratio of the elastomer substrate before the spin-coating process, the resulting surface should be able to maintain its superhydrophobicity at even larger strains. In other words, in our approach the stretch limit for maintain stable superhydrophobicity on a given elastomer surface is only limited by its own stretchability.
While stable superhydrophobicity of surfaces upon stretching are highly desired in certain applications, robustness of the superhydrophobicity under rubbing processes is another crucial advantage in applications related with surface wettability.42 To assess the robustness of our superhydrophobic surface, droplet adhesion or moving behavior on the superhydrophobic surface before and after receiving rubbing was recorded (Movie S3). As shown in Fig. 5a, the as prepared silicone elastomer-based superhydrophobic surface (white) was first wrapped around a finger. A water droplet of 8-µL staining red dye was dispensed on the superhydrophobic elastomeric surface. The water droplet slid down off the surface within 170 ms, as indicated by the yellow arrows showing the positions of the droplet at different time points. This non-adhesive down-moving behavior is typical for a superhydrophobic surface. After the initial test, the surface was rubbed with two fingers and then wrapped on the finger again, shown in Fig. 5b. Again, stained water droplets were deposited on the post-rubbing surface. As shown in Fig. 5c, the water droplets slid down off the surface fluently (170 ms) in the same manner with those on the surface before rubbing. Moreover, we also measured the water contact angle and the water sliding angle of the post-rubbing surface. As shown in Fig. S11, a contact angle of 148.5 ±0.4° and a sliding angle of approximately 11° could be achieved on the surface, suggesting the robustness of the surface superhydrophobicity.
Fig. 5.
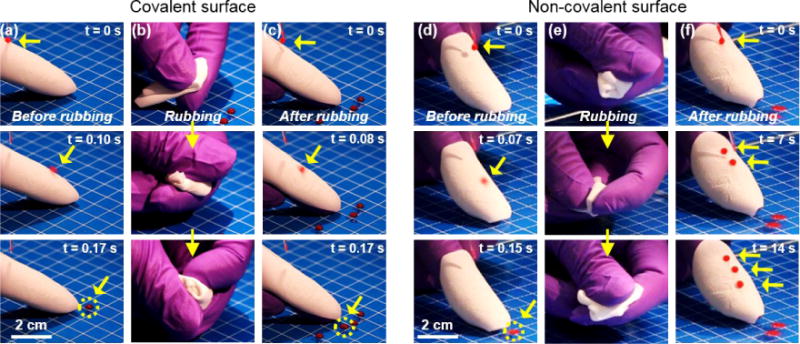
Robustness of the superhydrophobicity of the surface with covalent bonding and vulnerability of the superhydrophobicity of the surface without covalent bonding. (a) Before any rubbing, a water droplet could easily slide down off the covalent bonded superhydrophobic silicone elastomer membrane wrapping a finger, as indicated by the yellow arrows in different frames and the yellow circle in the last frame. (b) Rubbing was performed, making sure that the superhydrophobic elastomer received large amount of shear stress. (c) After the rubbing process, the superhydrophobicity of the elastomer membrane was kept, as shown the fast sliding of water droplet dispensed on the membrane. (d) Before rubbing, water droplet could slide down off the non-covalent bonded superhydrophobic elastomer membrane. (e) A same rubbing process to that in (b) was conducted. (f) After the rubbing process, water droplets with the same volume to that in (d) stuck firmly to the post-rubbed surface, indicating compromised superhydrophobicity.
In addition, to indirectly prove the formation of the covalent bonding between the hydrophilic silica particles and the silicone elastomer in our surface as well as its critical role to the robustness of the superhydrophobicity, we compared the droplet sliding behavior on a superhydrophobic surface with the same physical encapsulation but without the chemical bonding. The procedure for preparing such a surface was similar to the strategy used to prepare robust superhydrophobic surfaces with the difference that the curing process of the silicone oligomer after deposition of micro-sized silica particles was conducted at ambient temperature, making sure little or no chemical bonding was formed.37, 39 As shown in Fig. 5d–f, an 8-µL stained water droplet could slide down off the inclined superhydrophobic surface due to the presence of surface roughness; however, after a similar rubbing process, water droplets readily adhered to the surface, indicating damage to its superhydrophobicity. Indeed, changes of the microstructures of the surface before and after the rubbing process were observed (Fig. S12), showing loss of the silica microparticles from the surface. The distinct result from the comparison clearly shows that the chemical bonding between the silica particles and the silicone elastomer is critical in making the superhydrophobicity of the surface durable by preventing the silica microparticles from detaching the surface during harsh treatments such as rubbing.
Moreover, the robustness of the chemically bonded superhydrophobic surface was further verified by a sand paper abrasion test (Fig. S13), where the superhydrophobic surface was pressed against a 500-grit sandpaper surface by a 100-g weight placed on top. A horizontal force was used to draw the copper wire tethered to the weight, making the superhydrophobic surface move steadily on the sandpaper for 10 cm. Water droplets deposited on the resulting surface could still easily slide off the surface afterwards (Fig. S13a and 13b). Upon further observation of the fine structures of the superhydrophobic surface after the abrasion against sandpaper, there were only negligible differences comparing with that before abrasion treatment (Fig. S13c and 13d). Of note, the water contact angle and microstructure of the superhydrophobic elastomer surface were able to maintain even after 10 cycles of such test (Fig. S14). The preserved behavior of water droplets on the superhydrophobic surface with unchanged microstructures indicated robust superhydrophobicity against abrasion. Therefore, the combination of physical encapsulation and chemical bonding between silica microparticles and silicone elastomer should have accounted for the robustness of the superhydrophobicity.
Conclusions
In conclusion, we demonstrated a simple and environment-friendly method to fabricate superhydrophobic, non-fluorinated composite elastomer surfaces with multiple exceptional properties, such as stability under extensive and cyclic stretching, as well as robustness after severe rubbing and abrasion. The combination of the physical encapsulation and chemical bonding attributed to this superior performance. Difference between the relatively low surface tension of silicone oligomers and the high surface free energy of the silica microparticles was the driving force for the physical encapsulation; reaction between hydroxyl groups on the surface of silica microparticles and the Si-O groups at an elevated temperature resulted in the formation of covalent chemical bonding between silica and silicone elastomer. This highly stretchable and robust non-fluorinated superhydrophobic surface is expected to provide unique advantages for a wide range of applications in biomedicine, energy management, and electronics. In addition, the preparation method is simple and readily scalable, and can be applied as a general paradigm for fabricating composite elastomeric materials with versatile surface functions. Functional micro/nano-objects that are intrinsically rich of hydroxyl groups or can be easily treated to possess hydroxyl groups will be able to serve as the embedding materials for the fabrication of superhydrophobic surfaces while maintaining their own functionalities, such as magnetic ferroferric oxide particles.44
Experimental Section
Preparation of silicone elastomer/silica microparticle composite surface
The composite surface was fabricated in two steps. First, commercial silicone elastomer Ecoflex sheets (with typical thickness of 1 mm) was prepared using a mixture of part A and part B at a weight ratio of 1:1 and allowed to cure completely at room temperature for 24 h. Second, the elastomer sheet was pre-stretched at a strain of 200% using a fixture, and silicone oligomers with same mixing ratio was deposited on the stretched silicone elastomer sheet by spin-coating. The spin speed was varied. After spin-coating, the substrate was maintained horizontally for a certain period of time (from 0 min to 80 min). Then a large amount of silica microparticles was directly deposited onto the substrate. After 1 min, excessive microparticles were physically shaken off. The surface was then incubated in an oven at 100 °C for 10 h.
Water contact angle and sliding angel measurement
The contact angle was measured and analyzed by OCA 20 (Dataphysics). A 5-µL water droplet was carefully deposited on the composite elastomer surface. A digital camera (Canon 7D) was used to take close-up photos in parallel to the elastomer surface. Optical fiber light source was applied from the background to provide a bright field. At least five points on a single surface were measured to obtain mean value of contact angle. For testing the sliding angle, the sample were fixed on a custom-made device. The device was gradually raised from horizontal plane to a certain tilting degree. The lowest tilting angle that allowed an 8-µL water droplet to slide from the surface was recorded as the slide angle.
Material stretch apparatus
The as-prepared composite elastomer was clamped at both ends on an Instron Materials testing machine (Instron, 5960 dual column). The material was then subjected to single stretch at different strains and cyclic stretch with strain = 200%. For the cyclic stretching, different samples were stretched from 100 to 1000 cycles at an increment of 100 cycles. Upon finishing the cyclic stretch test, the composite elastomer was released for contact angle measurement.
Surface structure characterization
All the microstructures of the as-prepared composite surfaces and the silica microparticles were imaged on a Field-Emission SEM (FESEM, Zeiss ultra 55) after coated with 10 nm-thickness of Pt/Pd conductive layer on a sputter coater (EMS 300T D Dual Head Sputter Coater). The EDX mapping was conducted on the same SEM.
Rheology of Ecoflex
The viscosity of the silicone oligomers with prolonged time of curing was monitored using a Rheology (Anton-Paar MCR501) under constant shear mode.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge funding from the Office of Naval Research Young National Investigator Award, the National Institutes of Health (AR057837, DE021468, D005865, AR068258, AR066193, EB022403, EB021148), and the Presidential Early Career Award for Scientists and Engineers (PECASE). Y.S.Z. acknowledges the National Cancer Institute of the National Institutes of Health Pathway to Independence Award (K99CA201603). X.H. acknowledges the support of Recruitment Program for Young Professionals, China, and National Natural Science Foundation (21673197), China and Research Institute for Biomimetics and Soft Matter, Fujian Provincial Key Laboratory for Soft Functional Materials Research, Xiamen University, supported by the 111 Project (B16029).
Footnotes
Supporting Information
Supporting Information is available online from Wiley Online Library or from the author.
Contributor Information
Jie Ju, Biomaterials Innovation Research Center, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139; Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
Xi Yao, School of Engineering and Applied Sciences, Kavli Institute for Bionano Science and Technology, Harvard University, Cambridge, MA 02138.
Xu Hou, Biomaterials Innovation Research Center, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139; Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139, USACollege of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China.
Qihan Liu, School of Engineering and Applied Sciences, Kavli Institute for Bionano Science and Technology, Harvard University, Cambridge, MA 02138.
Yu Shrike Zhang, Biomaterials Innovation Research Center, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139; Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139, USAWyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA.
Ali Khademhosseini, Biomaterials Innovation Research Center, Division of Engineering in Medicine, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Cambridge, MA 02139; Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA; Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, MA 02115, USA; Department of Bioindustrial Technologies, College of Animal Bioscience and Technology, Konkuk University, Hwayang-dong, Gwangjin-gu, Seoul 143-701, Republic of Korea; Department of Physics, King Abdulaziz University, Jeddah 21569, Saudi Arabia.
References
- 1.Feng L, Jiang L, Zhu D. Adv Mater. 2002;14:1857–1860. [Google Scholar]
- 2.Neinhuis C, Barthlott W. Annals of Botany. 1997;79 [Google Scholar]
- 3.Tuteja A, Choi W, Ma M, Mabry JM, Mazzella SA, Rutledge GC, McKinley GH, Cohen RE. Science. 2007;318:1618–1622. doi: 10.1126/science.1148326. [DOI] [PubMed] [Google Scholar]
- 4.Magin CM, Cooper SP, Brennan AB. Materials Today. 2010;13:36–44. [Google Scholar]
- 5.Liu M, Zheng Y, Zhai J, Jiang L. Accounts of Chemical Research. 2010;43:368–377. doi: 10.1021/ar900205g. [DOI] [PubMed] [Google Scholar]
- 6.Lee S, Kim W, Yong K. Adv Mater. 2011;23:4398–4402. doi: 10.1002/adma.201101580. [DOI] [PubMed] [Google Scholar]
- 7.Truesdell R, Mammoli A, Vorobieff P, van Swol F, Brinker CJ. Phys Rev Lett. 2006;97:044504. doi: 10.1103/PhysRevLett.97.044504. [DOI] [PubMed] [Google Scholar]
- 8.Daniello RJ, Waterhouse NE, Rothstein JP. Physics of Fluids. 2009;21:085103. [Google Scholar]
- 9.Lee C, Kim CJ. Phys Rev Lett. 2011;106:014502. doi: 10.1103/PhysRevLett.106.014502. [DOI] [PubMed] [Google Scholar]
- 10.Zhang S, Ouyang X, Li J, Gao S, Han S, Liu L, Wei H. Langmuir. 2015;31:587–593. doi: 10.1021/la504451k. [DOI] [PubMed] [Google Scholar]
- 11.Hou Y, Wang Z, Guo J, Shen H, Zhang H, Zhao N, Zhao Y, Chen L, Liang S, Jin Y, Xu J. J Mater Chem A. 2015;3:23252. [Google Scholar]
- 12.Zimmermann J, Reifler FA, Fortunato G, Gerhardt LC, Seeger S. Advanced Functional Materials. 2008;18:3662–3669. [Google Scholar]
- 13.Verho T, Bower C, Andrew P, Franssila S, Ikkala O, Ras RH. Adv Mater. 2011;23:673–678. doi: 10.1002/adma.201003129. [DOI] [PubMed] [Google Scholar]
- 14.Dong Z, Wu L, Wang J, Ma J, Jiang L. Adv Mater. 2015;27:1745. doi: 10.1002/adma.201405387. [DOI] [PubMed] [Google Scholar]
- 15.Cho WK, Park S, Jon S, Choi IS. Nanotechnology. 2007;18:395602. doi: 10.1088/0957-4484/18/39/395602. [DOI] [PubMed] [Google Scholar]
- 16.Privett BJ, Youn J, Hong SA, Lee J, Han J, Shin JH, Schoenfisch MH. Langmuir. 2011;27:9597–9601. doi: 10.1021/la201801e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, Xue Y, Ding J, Feng L, Wang X, Lin T. Angew Chem Int Ed Engl. 2011;50:11433–11436. doi: 10.1002/anie.201105069. [DOI] [PubMed] [Google Scholar]
- 18.Ganesh VA, Nair AS, Raut HK, Yuan Tan TT, He C, Ramakrishna S, Xu J. Journal of Materials Chemistry. 2012;22:18479. [Google Scholar]
- 19.Lassen C, Jensen AA, Potrykus A, Christensen F, Kjølholt J, Jeppesen CN, Mikkelsen SH, Innanen S. Journal. 2013 [Google Scholar]
- 20.Hu XC, Andrews DQ, Lindstrom AB, Bruton TA, Schaider LA, Grandjean P, Lohmann R, Carignan CC, Blum A, Balan SA, Higgins CP, Sunderland EM. Environ Sci Technol Lett. 2016;3:344–350. doi: 10.1021/acs.estlett.6b00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin JW, Whittle DM, G DC, Mabury SA. Environ Sci Technol. 2004;38:5379–5385. doi: 10.1021/es049331s. [DOI] [PubMed] [Google Scholar]
- 22.Yao X, Ju J, Yang S, Wang J, Jiang L. Adv Mater. 2014;26:1895–1900. doi: 10.1002/adma.201304798. [DOI] [PubMed] [Google Scholar]
- 23.Seo J, Lee SK, Lee J, Seung Lee J, Kwon H, Cho SW, Ahn JH, Lee T. Sci Rep. 2015;5:12326. doi: 10.1038/srep12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hao C, Liu Y, Chen X, Li J, Zhang M, Zhao Y, Wang Z. Small. 2016;12:1825–1839. doi: 10.1002/smll.201503060. [DOI] [PubMed] [Google Scholar]
- 25.Sun JY, Keplinger C, Whitesides GM, Suo Z. Adv Mater. 2014;26:7608–7614. doi: 10.1002/adma.201403441. [DOI] [PubMed] [Google Scholar]
- 26.Yu B, Kang SY, Akthakul A, Ramadurai N, Pilkenton M, Patel A, Nashat A, Anderson DG, Sakamoto FH, Gilchrest BA, Anderson RR, Langer R. Nat Mater. 2016;15:911–918. doi: 10.1038/nmat4635. [DOI] [PubMed] [Google Scholar]
- 27.Billinghurst M, Starner T. Computer. 1999;32:57–64. [Google Scholar]
- 28.Ig Mo K, Kwangmok J, Ja Choon K, Jae-Do N, Young Kwan L, Hyouk Ryeol C. IEEE Transactions on Robotics. 2008;24:549–558. [Google Scholar]
- 29.Zang J, Ryu S, Pugno N, Wang Q, Tu Q, Buehler MJ, Zhao X. Nat Mater. 2013;12:321–325. doi: 10.1038/nmat3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zang J, Cao C, Feng Y, Liu J, Zhao X. Sci Rep. 2014;4:6492. doi: 10.1038/srep06492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mates JE, Bayer IS, Palumbo JM, Carroll PJ, Megaridis CM. Nat Commun. 2015;6:8874. doi: 10.1038/ncomms9874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amjadi M, Yoon YJ, Park I. Nanotechnology. 2015;26:375501. doi: 10.1088/0957-4484/26/37/375501. [DOI] [PubMed] [Google Scholar]
- 33.Fu Y, Jiang J, Zhang Q, Zhan X, Chen F. J Mater Chem A. 2017;5:275. [Google Scholar]
- 34.Zhan X, Yan Y, Zhang Q, Chen F. J Mater Chem A. 2014;2:9390. [Google Scholar]
- 35.Babik A, Mistrik J, Zemek J, Cech V. J Adhe Sci Tech. 2012;26:2543. [Google Scholar]
- 36.Roth J, Albrecht V, Nitschke M, Bellmann C, Simon F, Zschoche S, Michel S, Luhmann C, Grundke K, Voit B. Langmuir. 2008;24:12603. doi: 10.1021/la801970s. [DOI] [PubMed] [Google Scholar]
- 37.Krumpfer JW, McCarthy TJ. Langmuir. 2011;27:11514–11519. doi: 10.1021/la202583w. [DOI] [PubMed] [Google Scholar]
- 38.Krumpfer JW, McCarthy TJ. Faraday Discussion. 2010;146:103–111. doi: 10.1039/b925045j. [DOI] [PubMed] [Google Scholar]
- 39.Wooh S, Vollmer D. Angew Chem Int Ed. 2016;55:6822–6824. doi: 10.1002/anie.201511895. [DOI] [PubMed] [Google Scholar]
- 40.Nakajima Y, Shimada S. RSC Adv. 2015;5:20603. [Google Scholar]
- 41.Zhang P, Liu H, Meng J, Yang G, Liu X, Wang S, Jiang L. Adv Mater. 2014;26:3131–3135. doi: 10.1002/adma.201305914. [DOI] [PubMed] [Google Scholar]
- 42.Yoshimitsu Z, Nakajima A, Watanabe T, Hashimoto K. Langmuir. 2002;18:5818–5822. [Google Scholar]
- 43.Lu Y, Sathasivam S, Song J, Crick CR, Carmalt CJ, Parkin IP. Science. 2015;347:1132–1135. doi: 10.1126/science.aaa0946. [DOI] [PubMed] [Google Scholar]
- 44.Costa AMS, Dencheva NV, Caridade SG, Denchev ZZ, Mano JF. Advanced Materials Interfaces. 2016;3:1600074. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.