

Abstract
Mutations in genes encoding enzymes in the tricarboxylic acid cycle (TCA, also known as the Krebs cycle) have been implicated as causative genetic lesions in a number of human cancers, including renal cell cancers, glioblastomas, and pheochromocytomas. In recent studies, missense mutations in the Succinate dehydrogenase (SDH) complex have also been proposed to cause differentiated thyroid cancer. In order to gain mechanistic insight into this process, we generated mice lacking the SDH subunit D (SDHD) in the thyroid. We report that these mice develop enlarged thyroid glands with follicle hypercellularity and increased proliferation. In vitro, human thyroid cell lines with knockdown of SDHD exhibit an enhanced migratory capability, despite no change in proliferative capacity. Interestingly, these cells acquire stem-like features which are also observed in the mouse tumors. The stem-like characteristics are reversed by α-ketoglutarate, suggesting that SDH-associated tumorigenesis results from dedifferentiation driven by an imbalance in cellular metabolites of the TCA cycle. The results of this study reveal a metabolic vulnerability for potential future treatment of SDH-associated neoplasia.
Keywords: Succinate dehydrogenase, Thyroid cancer, Stem cells, Metabolism, Mouse models
INTRODUCTION
Thyroid cancer is the most common endocrine malignancy with a rapidly increasing incidence rate as well as a high degree of familiality for certain subtypes (Davies, et al. 2015; Leenhardt, et al. 2004). Thyroid cancer can be divided into the rare (5% of cases) medullary thyroid cancer (MTC), arising from parafollicular C-cells, and the more common (95%) epithelial thyroid cancer (also called non-medullary thyroid cancer, NMTC), arising from the thyrocytes themselves (Kitahara and Sosa 2016). The majority of patients with NMTC exhibit well differentiated pathologies of either papillary (PTC) or follicular (FTC) subtype, and almost all of the increase in the incidence is observed in PTC (Pringle, et al. 2014). The majority of PTC and FTC are sporadic, while the familial component is believed to be 5% of the cases (Morrison and Atkinson 2009; Nose 2011). Familial cases may occur as part of rare high penetrance genetic syndromes or from more prevalent low-penetrance susceptibility genes (Nagy, et al. 2004; Tomsic, et al. 2015). Cowden Syndrome (CS) is an autosomal dominant disorder that is characterized by high risk of breast cancer, thyroid neoplasms, uterine tumors, gastrointestinal hamartomas, and less frequent tumors in other solid organs (Hobert and Eng 2009; Marsh, et al. 1998). Most cases of CS and a small number of Cowden-like syndrome (CSL) are associated with inactivating mutations in the Phosphatase and tensin homolog (PTEN) tumor suppressor gene.
Recently, mutations in genes encoding the Succinate dehydrogenase (SDH) subunits were identified as additional susceptibility genes for CS, comprising about 10% of CS and CSL phenotypes (Ni, et al. 2008; Yu, et al. 2015). SDH is a heterotetrameric nuclear-encoded mitochondrial protein comprised of four subunits encoded by the four autosomal genes SDHA, SDHB, SDHC, and SDHD. These genes are known tumor suppressors owing to their established connection to the syndrome of inherited pheochromocytoma (PHEO) and paraganglioma (PGL) (Bardella, et al. 2011; Letouze, et al. 2013). Genetic variants in SDHB and SDHD have been identified in a subset of CS/CSL patients, and confer high risk of breast, thyroid and other cancers. Further, downregulation of SDH subunits has been observed in both PTC and FTC and has been shown to correlate with poorer prognosis. Interestingly, CS/CSL patients with variants in SDHD exhibit an increased risk for PTC, while individuals with mutations in PTEN have a predilection to both PTC and FTC (Ni, et al. 2015).
SDH is a component of the tricarboxylic acid cycle (TCA cycle, also known as the citric acid or Krebs cycle), as it oxidizes succinate to fumarate and leads to electron transport to ubiquinone in the electron transport chain (ETC). SDH was the first mitochondrial enzyme that proved that mutations in TCA cycle genes can in fact act as drivers of tumorigenesis. Later, mutations leading to dysfunction of other TCA cycle proteins such as SDH assembly factor 2 (SDHAF2), Fumarate hydratase (FH), and Isocitrate dehydrogenase (IDH) were found to be associated with multiple types of cancer formation (Cantor and Sabatini 2012). The spectrum of tumors caused by mutations in each of these genes differs, although there may be some overlap among the syndromes. Inherited or somatic mutations in any of the four subunits of SDH can lead to pheochromocytoma, paraganglioma, renal cell carcinoma, gastrointestinal stromal tumor, thyroid cancer and breast cancer (Bardella et al. 2011; Letouze et al. 2013; Millan-Ucles, et al. 2014; Williamson, et al. 2015; Xiao, et al. 2012).
Dysfunction of these metabolic enzymes have provided new insight into the observation that the majority, although not all cancers show a preference towards anaerobic glycolysis over oxidative phosphorylation. This phenomenon, known as the Warburg effect enables cells to grow under hypoxic conditions (Archetti 2015). This metabolic alteration was initially thought to be an adaptive mechanism to overcome the hypoxic conditions in tumors, but newer data suggest that metabolic dysfunction is itself a driver of tumorigenesis. Mechanisms that have been proposed to explain how loss of SDHD leads to thyroid tumorigenesis include 1) an increase in the production of reactive oxygen species (ROS), 2) activation of a hypoxia-like pathway under normoxic conditions (pseudo-hypoxia), and 3) genetic and epigenetics alterations due to the presence of oncometabolites (Jardim-Messeder, et al. 2015; MacKenzie, et al. 2007; Salminen, et al. 2014; Yu et al. 2015). Specifically, several lines of genetic and biochemical evidence suggest that citrate, succinate, fumarate and α-ketoglutarate (α-KG, also known as 2-oxoglutaric acid or 2-OG) may serve as oncometabolites to promote tumorigenesis by inhibiting a family of epigenetic modulator enzymes (Carey, et al. 2015; Letouze et al. 2013; Sciacovelli and Frezza 2016).
To evaluate the contribution of Sdhd loss on thyroid tumorigenesis, we generated tissue-specific knockout of Sdhd in the mouse thyroid gland. These in vivo studies were complemented by in vitro analyses of human thyroid cancer cells with knockdown of SDHD. Together, these studies reveal the ability of SDHD/Sdhd loss in promoting thyroid neoplasia and provide new mechanistic insights for these observations.
METHODS
Animal Strains, Husbandry, and Maintenance
The use of animals was in compliance with federal and Ohio State University Laboratory Animal Resources regulations. SdhdloxP/loxP mice (Millan-Ucles et al. 2014) were crossed with Thyroid Peroxidase (TPO)–cre (Kusakabe, et al. 2004) to generate Sdhd-TpoKO. In addition, PtenloxP/loxP mice (Pten-TpoKO) were generated as described previously (Pringle, et al. 2012). Tpo-cre, Sdhd loxP/loxP and Pten loxP/loxP were mated to generate Tpo-Cre; Sdhd loxP/loxP Pten loxP/loxP (SP-TpoKO) mice. The experiments were performed using littermate mice from a mixed C57BL/6 and FVB genetic background.
Ultrasonography
All mice were imaged with a VisualSonics Vevo 2100 (VisualSonics, Toronto, CA) every 3 months up to 1 year age. MS550D transducer (22–55 MHz) was used with 3D-Mode imaging to determine size of the thyroid. The volume of both thyroid lobes was calculated using Vevolab 2.1 software in a blinded fashion.
Follicular area measurement
The H&E-stained sections were imaged at 20x magnification. The area of follicles was determined by measuring the luminal surface using the ImageJ software as described previously (Yeager, et al. 2007).
Cell culture and reagents
FTC133 (human follicular thyroid carcinoma) cells were maintained in DMEM media with 10% FBS and penicillin/streptomycin. NthyOri 3.1 (human follicular epithelial thyroid cells) were maintained in RPMI media with 10% FBS and penicillin/streptomycin. Octyl-α-ketoglutarate was purchased from Cayman Chemicals (Ann Arbor, MI). Lentiviral based ShRNA (sequences available on request) for SDHD knockdown was obtained from Sigma Aldrich (St. Louis, MO). Lentiviral supernatant was produced from phoenix cells transfected with Fugene (Promega, Madison WI), packaging mix (Sigma, St. Louis, MO) and viral vector. Stable SDHD-knockdown thyroid cells were generated by puromycin selection after transducing with ShRNA lentivirus.
Primary thyrocytes isolation
Primary thyroid cells were isolated from mouse tumors by enzymatic dissociation using Collagenase/Hyaluronidase solution, Dispase and Dnase using protocols adapted from manufacturer (StemCell Technologies, Vancouver, Canada). Briefly, the thyroid was dissected and chopped using automatic tissue chopper and placed in Collagenase/Hyaluronidase solution for 3 hours at 37o C with agitation. The sample was centrifuged at 350g for 5 mins and the pellet was resuspended in 0.25% trypsin-EDTA on ice for 1 hour. After centrifugation, the cell pellet was resuspended in ice-cold HBSS with 2% serum. After centrifugation, cells were dissociated with pre-warmed Dispase and Dnase I, resuspended ice cold HBSS with 2% serum and filtered through 40 uM cell strainer. Cells were kept on ice until use.
ALDEFLUOR assay
The ALDEFLUOR kit (StemCell Technologies, Vancouver, Canada) was used to isolate the cell population with a high ALDH activity. Cells were obtained from cultured cells or mouse thyroid tumors. Optimal conditions for the experiment were determined by titrating cell number and incubation time. Cultured cells and primary tumor cells were resuspended in ALDEFLUOR assay buffer containing ALDH substrate (BAAA, 5uL per 1×105 cells/mL) and incubated for 20 minutes at 37°C. An aliquot of each sample was treated with diethylaminobenzaldehyde (DEAB, 5uL per 1×105 cells/mL), a specific ALDH inhibitor as a negative control. Propidium iodide was used for cell viability. Cells were kept on ice after ALDEFLUOR reaction was completed until analyzed on BD LSR II flow cytometer. DEAB treated negative control sample was used as a gating control and gate was set to include 0.5% ALDHbright cells on DEAB treatment for each sample.
Western Blotting
The proteins were run on SDS-PAGE gel by standard procedure, and the membranes were probed with the indicated antibodies: Millipore-SDHD (ABT110), Cell Signaling Technology-GAPDH (2118). Signals were detected with SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher).
Mass Spectrometry
Organic acid and phosphorylated compounds were extracted from 1×106 cultured NthyOri 3.1 and FTC133 cells using boiling water and subjected to LC-MS/MS analysis using AS-11 column (Dionex, 2.1 × 250mm). 13C-fumarate was used as internal standard. Relative succinate levels were normalized by calculating the ratio of succinate to sum of all measured metabolites as described previously (Denkert, et al. 2008).
Oxygen consumption rate
FTC133 and NthyOri 3.1 cells were plated at a density of 20,000 cells per well (XF24 cell culture microplate; Seahorse Biosciences, North Billerica, MA). The cells were allowed to grow for 24 h, following which the cells were washed with XF Assay media (with 20mM glucose, 1mM sodium pyruvate, no sodium bicarbonate at pH 7.4). The cells were incubated for 1 h at 37 °C in a non-CO2 incubator. The assay was normalized to protein and analyzed using the XFe 2.3 software. Optimal seeding density and concentrations of oligomycin, rotenone and FCCP (purchased from Sigma) were determined for each cell line. For mitochondrial stress test, oxygen consumption rate (OCR) was measured with sequential addition of oligomycin, rotenone and FCCP. Spare reserve capacity was calculated as FCCP induced-maximum OCR relative to baseline OCR, whereas non-mitochondrial respiration was calculated from mitochondrial stress test based on residual respiration in response to rotenone.
Cell proliferation/growth
5000 cells were plated in 48 well plates for growth curve by crystal violet assay. At each time point, triplicate wells were stained with 0.05% crystal violet, 0.1% formalin for 20 mins and extracted with 10% acetic acid. Absorbance was measured at 590 nm.
Cell migration assay
Cell migration was assessed by scratch wound healing assay. For the measurement of cell migration, control and SDHD-knockdown cells were cultured in individual wells of a 6-well plate. After reaching a confluent state, cell layers were scratched with a 200 uL plastic micropipette tip. The medium was aspirated away and replaced by 1–2 ml of fresh serum-free medium. Cells were allowed to migrate in serum free medium for 24 hours. Images were obtained at 0 and 24 hours by phase contrast microscopy. For evaluation of scratch closure, the horizontal distance of migrating cells from the initial wound was measured at 2 points along each scratch using ImageJ software.
Immunohistochemistry
Dissected mouse thyroid tissues were fixed in 10% neutral-buffered formalin solution. Tissues were processed, embedded in paraffin, cut in 5 μm sections on positively charged slides, deparaffinized, rehydrated, and stained with hematoxylin and eosin (H&E). For immunohistochemistry, all sections were stained using a Bond Rx autostainer (Leica Biosystems, Richmond, IL) for Ki67 (Abcam, Cambridge, MA) antibody. For quantification of the DAB staining, the images were reviewed and analyzed using immunoRatio application.
Electron microscopy
Mouse thyroid tissue was fixed in 2.5 % Gluteraldehyde, 0.1 M Phosphate Buffer at pH 7.4 for TEM microscopy. The tissue sections were imaged on FEI Tecnai G2 Spirit TEM microscope.
RNA and Real-Time PCR
Total RNA from cells and tissue was isolated using Trizol using the Qiagen RNeasy kit. RNA quality was assessed by Nanodrop ND-1000 (Thermo Scientific, Waltham, MA). cDNA was prepared using an iScript cDNA Synthesis Kit (BioRad Laboratories, Hercules, CA) and subject to qRT-PCR using the iQ SYBR Green Supermix Kit (BioRad) as per manufacturer’s instructions. RT-PCR reactions were performed in triplicate. Primers sequences are available on request.
Genomic DNA isolation
Genomic DNA was isolated from control and SDHD-knockdown cells, and mouse thyroids using DNeasy Blood & Tissue Kit (Qiagen, Germantown, MD) according to the manufacturer’s protocol. The concentration and purity were determined by measuring the absorbance at 230, 260, and 280 nm using a Nanodrop ND-1000 (Thermo Scientific).
Methylation Analysis
Methylated DNA was quantified using 100 ng DNA using an ELISA assay for methylated DNA according to the manufacturer’s directions (Abcam). Absolute and relative methyl-cytosine content was then calculated using the supplied formula.
Statistics
All data was analyzed via student’s t-test, Mann Whitney test or ANOVA using GraphPad Prism software, p-values less than 0.05 were considered significant.
RESULTS
Sdhd knockout causes thyroid hyperplasia in mice
To gain insight into the role of Sdhd in thyroid tumorigenesis, we crossed mice carrying a conditional null allele of Sdhd (SdhdloxP) to mice expressing Cre recombinase under the control of the thyroid peroxidase (Tpo) promoter in order to generate thyroid specific Sdhd-KO mice (denoted as Sdhd-TpoKO). Sdhd-TpoKO mice were viable and fertile with normal lifespan. They also demonstrated normal thyroid function, with normal levels of serum TSH (WT: 71.8 ± 43.36 ng/ml vs. Sdhd-TpoKO: 57 ± 83.85 ng/ml, p value = 0.7) and no change in the expression of the sodium-iodine symporters (NIS) mRNA compared to controls (data not shown). Sdhd deletion resulted in a modest increase in thyroid volumes of Sdhd-TpoKO mice at 6 months compared to controls (Figure 1A), which was sustained up to 12 months (p value for the longitudinal analysis = 0.0075). Histopathologic analysis in a subset of mice revealed incidence of follicular adenoma as 18% (2/11) in Sdhd-TpoKO compared to 0% (0/7) in controls. However, the Sdhd-KO thyroids were more cellular, exhibiting both a decrease in follicular area and a 2-fold enhanced rate of proliferation as measured by Ki-67 staining (Figure 1B, C). Inflammatory component as well as apoptosis rate was low and unchanged in the Sdhd-TpoKO compared to littermate controls (data not shown). Ultrastructure analysis of the thyroids revealed severe degeneration of the mitochondria in Sdhd-TpoKO mice, similar to previous reports (Szarek, et al. 2015), suggesting altered bioenergetics (Figure 1D).
Figure 1. Sdhd deletion leads to mitochondrial defects and enhanced cellularity in vivo.

A. Thyroid volumes of WT (n=12) and Sdhd-TpoKO (n=16) mice analyzed by 3D ultrasonography at 6 months age. B. Representative images of follicles in WT, Sdhd-TpoKO, Pten-TpoKO and SP-TpoKO mice (200x magnification) at 1 year age. Quantification of the average follicular size of WT thyroids compared to Sdhd-TpoKO , and Pten-TpoKO thyroids compared to SP-TpoKO is on right. C. Ki-67 staining by immunohistochemistry in formalin fixed thyroid sections of WT, Sdhd-TpoKO , Pten-TpoKO and SP-TpoKO mice (100x magnification). Quantification of Ki-67 positive nuclei is on right. D. Mitochondrial ultrastructure (17,000x magnification) of WT and Sdhd-TpoKO mice (n=3 each) at 6 months age. Representative TEM images are shown and mitochondria are indicated by the asterisk. Error bars represents standard deviation (SD). Statistical analyses were performed by two-tailed Student’s t-test (*P value ≤ 0.05, **P value ≤ 0.01).
Similarly, to test whether Sdhd deletion enhances tumorigenesis of the thyroid in cooperation with loss of Pten, we generated Sdhd Pten-TpoKO double knockout mice (denoted as SP-TpoKO) and compared them to mice lacking Pten in their thyroids. We have previously shown that tissue-specific ablation of Pten alone generates mice with enlarged thyroids and follicular adenomas without cancer (Pringle et al. 2014). At 1 year age, mice with both Sdhd and Pten deletion in the thyroid showed no significant difference from Pten-only deletion in terms of thyroid size or the absence of follicular carcinoma (data not shown). At the cellular level, SP-TpoKO thyroids also showed markedly enhanced proliferation and a reduced follicle size, leading to overall enhanced cellularity in the thyroid, although gross size was not altered (Figure 1B, C). However when aged up to 18 months, SP-TpoKO mice demonstrated 81% penetrance of follicular carcinomas, whereas Pten-TpoKO did not undergo malignant transformation. Lung metastases were also observed in SP-TpoKO mice. Pten-TpoKO mice were euthyroid (mean TSH 39.2 ± 67.98 ng/mL), and SP-TpoKO mice also exhibited no change in thyroid function (mean TSH 42.2 ± 23.31 ng/mL) (Yeager et al. 2007). Together, these data suggest that Sdhd deletion causes thyroid hyperproliferation as an early sign of tumor initiation, and when combined with Pten KO, has the potential to progress to advanced disease over time.
SDHD deficiency leads to metabolic defects
Because mouse thyroid primary cells are difficult to manipulate in primary culture, we selected human thyroid cell lines and used lentiviral shRNAs to generate stable SDHD knockdown using Pten-null FTC133 follicular thyroid cancer cell line and the non-malignant NthyOri 3.1 cells as model systems. The efficiency of SDHD knockdown was tested by quantitative RT-PCR and verified by Western blot (Figure 2 A–C). Interestingly, SDHD-deficient cells showed normal expression levels of genes encoding the other SDH subunits (data not shown), indicating a lack of co-regulation of the genes encoding this multi subunit protein. To assess the metabolic effects of SDHD knockdown, we performed quantitative mass spectrometry of small organic acids from the cells. This analysis demonstrated that SDHD downregulation led to an accumulation of succinate in both cell lines following transfection (Figure 2 D, E). Changes in other metabolites of the TCA cycle were inconsistent in the two cell lines (Supplemental figure 1).
Figure 2. SDHD depletion leads to succinate accumulation in vitro.

A. Quantification of SDHD knockdown in FTC133 by RT-PCR. Gene expression is normalized to B2M. B. Quantification of SDHD knockdown in NthyOri 3.1 by RT-PCR. Gene expression is normalized to B2M. C. Western blot showing SDHD knockdown in lentivirus transfected thyroid cell lines in FTC133 and NthyOri 3.1. D. Quantification of relative succinate levels in FTC133 cells analyzed by mass spectrometry. Data represents average of four biological replicates. E. Quantification of relative succinate levels in NthyOri 3.1 cells analyzed by mass spectrometry. Data represents average of four biological replicates. Error bars represents standard deviation (SD). Statistical analyses were performed by two-tailed Student’s t-test (*P value ≤ 0.05, **P value ≤ 0.01).
As SDHD is involved in metabolism through its role in TCA cycle and associated ETC, we investigated whether SDHD dysfunction results in defects in cellular respiration and metabolism by Seahorse assay. Interestingly, SDHD knockdown did not completely abrogate the basal oxygen consumption rate (OCR) in either cell lines. However, SDHD knockdown cell lines responded differently to FCCP treatment (Figure 3 A, B). SDHD depletion thus caused decreased in spare capacity and rendered cells more sensitive to energy stress (Figure 3 E, F). Moreover, SDHD deficient cells showed less sensitivity in response to Oligomycin treatment, indicating a greater reliance on non-mitochondrial respiration. SDHD knockdown cells had increased non-mitochondrial respiration than control cells in both cell lines (Figure 3 C, D). Overall, knockdown of SDHD decreased mitochondrial reserve respiratory capacity.
Figure 3. SDHD-KD leads to lower respiration and decreased spare capacity.
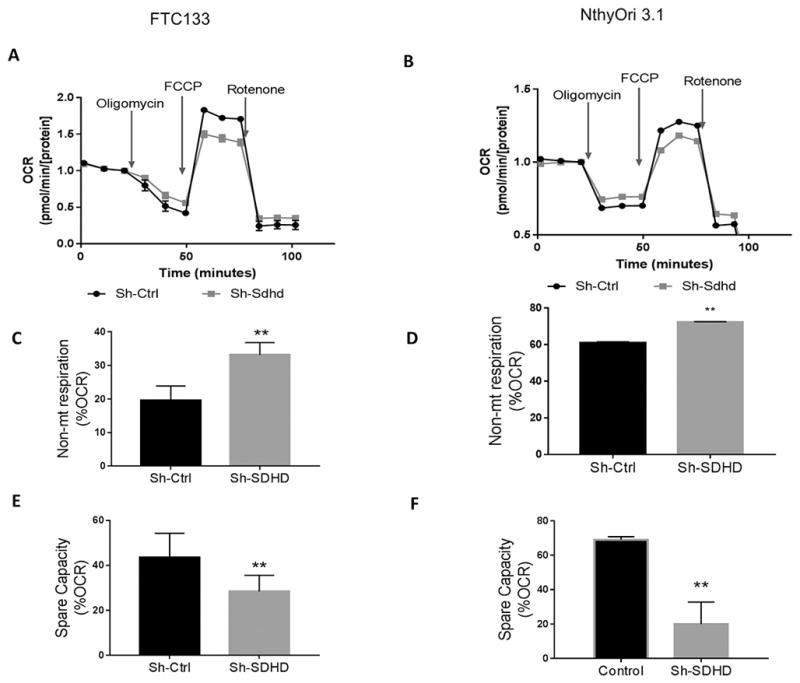
A. Seahorse respiratory profile analysis of SDHD-deficient FTC133 cells. B. Seahorse respiratory profile analysis of SDHD-deficient NthyOri 3.1 cells. C–F. SDHD knockdown cells show a corresponding increase in non-mitochondrial respiration and decrease in spare capacity. Data represents one of three representative experiments. Error bars represent standard deviation (SD). Statistical analyses were performed by two-tailed Student’s t-test (*P value ≤ 0.05, **P value ≤ 0.01).
SDHD depletion leads to increased migration in thyroid cancer cell lines
To assess the effect of SDHD knockdown on cellular markers of cancer behavior, we first measured cell proliferation. Over the course of 4 days of growth, there was no difference in cell numbers between WT and SDHD-KD cells for either cell line (Figure 4 A,B). Next, we measured migratory ability using a 2-D scratch assay. In both cell lines, SDHD-KD cells demonstrated an increased migratory capacity compared to control cells (Figure 4 C,D). This migratory phenotype was also confirmed by Boyden chamber assay (Figure 4 E,F).
Figure 4. SDHD depletion does not affect proliferation but increases migratory capacity of thyroid derived cell lines.

A,B) Proliferation of WT or Sdhd-knockdown FTC133 and NthyOri 3.1 cells analyzed by crystal violet assay. C,D) Effect of Sdhd-knockdown on FTC133 and NthyOri 3.1 cell migration at 24 hours as measured by wound healing assays. E,F) Effect of Sdhd-knockdown on FTC133 and NthyOri 3.1 cell migration at 24 hours as measured by a Boyden chamber assay. Each graph represents data from three independent experiments. Error bars represents standard deviation (SD). Statistical analyses were performed by two-tailed Student’s t-test (*P value ≤ 0.05, **P value ≤ 0.01, ns=non-significant).
SDHD depletion promotes stemness in thyrocytes
Collectively, our data suggest that loss of SDHD leads to increased cellularity in vivo, and a migratory phenotype in vitro. SDHD dysfunction also lead to altered metabolic properties, mainly reduced oxidative phosphorylation (OXPHOS) in vitro. These characteristics are associated with a so-called ‘stemness phenotype’ (Burgess, et al. 2014; Hermann, et al. 2007; Li, et al. 2017; Pardal, et al. 2005). We further tested if SDHD dysfunction had any impact on cellular differentiation. We utilized our SDHD knockdown human thyroid cancer cell lines to test the expression of stem cell transcription factors. This analysis demonstrated that transcription factors Nanog and Oct-4 were upregulated in SDHD knockdown NthyOri 3.1 cells relative to the control cells (Figure 5A). We next analyzed the cells for expression of intracellular aldehyde dehydrogenases (ALDH) activity, which has been proposed to play a key role in stem/progenitor cell expansion and differentiation as well as tumor initiation and progression (Rodriguez-Torres and Allan 2016). In agreement with RT-PCR data, we observed a marked increase in the fraction of ALDH positive cells in SDHD depleted NthyOri 3.1 cells (Figure 5B). Unlike NthyOri 3.1 cells, FTC133 cells showed no ALDH activity nor increased “stem” gene expression as these cells are known to lack ALDH activity (Nagayama, et al. 2016).
Figure 5. SDHD-KD leads to increased stem-like response in vitro.
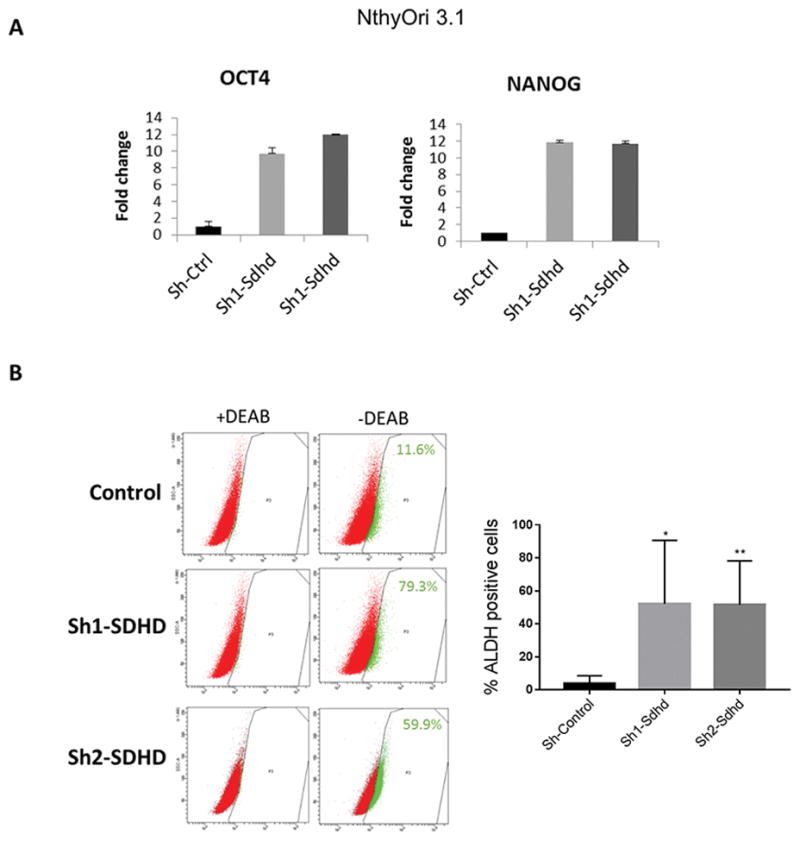
A. Quantitative gene expression analysis in NthyOri 3.1 cells. Gene expression is normalized to B2M. Graph represents one of three representative experiments. B. ALDH activity in SDHD depleted NthyOri 3.1 cells by ALDHFLUORTM assay. The percentage of ALDHhigh cells from single experiment is shown in each panel. On right, mean of four independent experiments measuring % ALDH positive cells gated according to DEAB treated negative controls is quantified. Error bars represents standard deviation (SD). Statistical analyses were performed by two-tailed student’s t-test (*P value ≤ 0.05, **P value ≤ 0.01).
With the identification of stem-like features in the cell lines, we wanted to see if the same observations were applicable to murine tumors in vivo, as acquisition of this stem-like phenotype would be a significant indicator of tumor initiation and neoplasia. To test this hypothesis, we isolated P and SP thyrocytes for measurement of their ALDH activity. As shown in Figure 6, the population of ALDH positive cells in SP thyroid tumors was significantly elevated compared to Pten-only mice. These data confirm that the ability of Sdhd ablation to promote tumorigenesis is significantly reliant on promotion of this stem-like phenotype.
Figure 6. SDHD-KD leads to increased stemness response in vivo.

A. ALDH activity in primary mouse thyrocytes by ALDHFLUORTM assay. The percentage of ALDHhigh cells from single experiment is shown in each panel. B. Mean of three independent experiment measuring % ALDH positive cells gated according to DEAB treated negative control. Error bars represents standard deviation (SD). Statistical analyses were performed by two-tailed student’s t-test (*P value ≤ 0.05).
SDHD deficiency increases global DNA methylation in thyroid cells
As described above, three mechanisms have been proposed to account for the tumorigenic effect of mutation of genes in the TCA cycle: generation of free radicals, hypoxia/pseudohypoxia, and epigenetic alterations caused by metabolites excess. To assess free radical generation, we measured ROS in control and SDHD-deficient cells; while ROS levels could be manipulated by altering the oxidizing environment of the cells, no differences were detected between control and KD cells at baseline or under stimulation (Supplemental Figure 2). To produce a pseudohypoxic state, it has been suggested that succinate accumulation can inhibit prolyl hydroxylase (PHD) leading to a hypoxic response (Briere, et al. 2005). However, we did not observe induction of HIF1α (data not shown), suggesting that this pathway is not operative in these cells. We also examined the ERK, PI3K, and mTOR pathways to evaluate activation in SDHD-deficient cells. While p-AKT and p-ERK levels remained unchanged both in vivo and in vitro (data not shown), we observed mildly increased levels of p-mTOR in Sdhd-TpoKO and SP-TpoKO mouse thyroids. However, the ratio of p-mTOR to total mTOR was unaltered in vitro (Supplemental Figure 3).
It has been shown that TCA cycle intermediates fumarate and succinate can inhibit the histone demethylases of the Jumonji C class in a dose dependent manner (Xiao et al. 2012). Also, SDHC mutations give rise to hypermethylator phenotype in PGL (Letouze et al. 2013). Thus, we tested whether SDHD depletion directly affected DNA methylation in thyroid cell lines and mouse tumors. In NthyOri 3.1 cells, there was an approximately 3-fold increase in 5-methylcytosine (5-mC) levels after depletion of SDHD (Figure 7A). In contrast, DNA methylation levels in FTC133 cells seemed to be unaffected by SDHD loss. It is unclear if the lack of change is similar to (or caused by) the same defect that prevents ALDH expression, or if this is a limitation of the sensitivity of the assay (Figure 7B). In mouse tumors, there was a clear trend towards an increase in 5-mC levels between SP tumors and Pten-only tumors although these results did not reach significance, perhaps due to high variability observed between mice (Figure 7C).
Figure 7. DNA methylation analysis of SDHD depleted cells.

A. 5-mC levels in NthyOri 3.1 cells DNA analyzed by ELISA assay. Graph represents mean ± SD of three biological replicates. B. 5-mC levels in FTC133 cells DNA analyzed by ELISA assay. Graphs represents mean ± SD of three biological replicates. C. 5-mC levels in DNA isolated from SP-TpoKO and Pten-TpoKO tumors analyzed by ELISA assay. Graphs represents mean ± SD of five mice. Statistical analyses were performed by two-tailed student’s t-test (*P value ≤ 0.05, ns=non-significant).
α-ketoglutarate treatment reverses stem cell-like phenotype caused by mutant SDHD
It is increasingly appreciated that DNA methylation has important role in cancer development and it helps maintain transcriptional silencing of genes (Baylin 2005). Epigenetic modifications are reversible and can be therapeutically targeted. The phenotype caused by SDHD loss can be explained by possibly altered αKG/succinate ratio, as we have reported increased succinate levels in our model. If an alteration of the relative amounts of TCA cycle intermediates is responsible for this effect, then reestablishing the balance should revert this effect. To test this possibility, we treated cells with cell permeable α-KG and measured the phenotypic characteristics as described above. Indeed, treatment with low dose of α-KG for three days fully reversed the migratory phenotype as well as stem-like response evidenced by ALDH assay and reduced the ALDH levels back to that of control cells (Figure 8A, C). To examine whether unavailability of α-KG is also responsible for increased levels of DNA methylation observed with SDHD deficiency, we measured 5-mC levels in NthyOri 3.1 cells treated with α-KG. Indeed, treatment with high dose of α-KG could rescue the hypermethylation phenotype in NthyOri cells (Figure 8B).
Figure 8. α-KG reverses stem-like phenotype in vitro.

A. SDHD knockdown NthyOri 3.1 cells with aKG (0.1 mM) treatment for 3 days were subjected to FACS analysis for measurement of ALDH activity. The percentage of ALDHhigh cells from single experiment is shown in each panel. Quantification of three independent experiment measuring % ALDH positive cells gated according to DEAB treated negative control is shown on right (statistical comparison is to the untreated Sh-Ctrl). B. DNA methylation levels of NthyOri 3.1 cells after treatment with 1mM α-KG for 3 days. Graphs represents mean ± SD of five biological replicates. C. Migration in thyroid cell lines treated with aKG (0.1 mM) at 12 hours performed by wound healing assays. Graph represents data from three independent experiments. Error bars represents standard deviation (SD). Statistical analyses were performed by two-tailed student’s t-test for ALDH assay and migration, and by Mann Whitney test in case of DNA methylation. (*P value ≤ 0.05, **P value ≤ 0.01).
DISCUSSION
Although mutations in SDH genes are known to cause human cancers, analysis of the mechanism by which this occurs has been hampered by a lack of robust model systems. Previous efforts to study tumorigenesis in vivo did not identify an effect of mutant IDH, FH, or SDH genes, including in tissues clearly affected in human patients (Lu, et al. 2013; Millan-Ucles et al. 2014; Szarek et al. 2015). In this report, we address the role of SDHD in thyroid cancer by ablating Sdhd in the thyroid gland. Because it has been suggested that there may be an interaction between loss of the PTEN tumor suppressor and mutations in SDH genes, we also generated mice lacking both of these genes and compared them to Pten-KO thyroids, which only develop thyroid adenomas. Studying the role of tumor suppressors and oncogenes in the thyroid provides an advantageous model system to study pre-neoplastic and neoplastic changes because of the unique architecture of the thyroid follicle, as well as the opportunity to study the differentiated function of thyroid hormone secretion.
In contrast to the prior studies looking at the role of SDHD in neural crest-derived tissue (including the adrenal medulla), we observed that Sdhd null thyroids show enlarged glands, a potential indicator of early neoplasia. Examination of the tissue revealed hypercellularity caused by enhanced proliferation, further supporting the impression that Sdhd ablation promotes tumor initiation in the thyroid gland. In fact, when combined with Pten KO, these mice developed FTC with metastasis at advanced age. Although Sdhd ablation from the thyroid causes enhanced proliferation in vivo, we did not observe this same effect in vitro. We attribute the differences either to the fact that in vitro studies were carried out in permanent cell lines, which may already have altered proliferation properties, or to the lack of a normal 3D tissue environment which exists in vivo.
One of the most striking observations from this study is that KO of Sdhd from the thyroid in vivo or knockdown of the gene in vitro promotes a shift to a stem-like phenotype, including expression of both stem cell transcription factors and cellular production of the thyroid stem cell marker aldehyde dehydrogenase (ALDH). Although the tumor cells retained sufficient differentiated function to maintain normal thyroid function, the acquisition of stem-like features is likely to play an important role in the ability of the hyperplastic glands to eventually progress to thyroid cancer.
Three mechanisms have been proposed to account of the mechanism by which mutations in SDH (or other TCA cycle genes) may account for the acquisition of a malignant phenotype. In the thyroid gland, we could find no evidence for an excess of reactive oxygen species nor for significant induction of a pseudohypoxia signature. These results differ from a number of studies on TCA cycle tumor suppressors that have resulted in inconsistent reports on HIF and ROS involvement, casting doubts on pseudo-hypoxia and free-radical mechanisms (Aspuria, et al. 2014; Briere et al. 2005; Edalat, et al. 2015; Millan-Ucles et al. 2014). Based on our observations, we focused on the role of succinate as an oncometabolite. Levels of succinate were elevated in SDHD-KD cells, although there were minimal effects on other TCA cycle intermediates. Succinate has been proposed to affect DNA demethylases, and, indeed, knockdown cells exhibited increased DNA methylation, an observation which was seen in vivo but which did not reach statistical significance due to high variability. The in vivo studies may reflect not only variability in the thyrocytes themselves, but also contributions from stromal cells, which would be expected to have normal SDH function and thus reduce the magnitude of any alterations restricted to thyroid epithelial cells.
It has been proposed that the effects of oncometabolites are mediated by the imbalance, which alters the ratio of various allosteric enzyme modulators. In the case of succinate, it has been suggested that the effects are mediated by an imbalance in the ratio of succinate to its precursor, alpha-ketoglutarate (α-KG). We tested this experimentally by treating cells with α-KG and demonstrated a nearly complete reversal of phenotypes with this treatment. The fact that SDH-associated changes can be reversed by excess α-KG suggests that altered α-KG/succinate ratio, rather than absolute levels likely contributes to tumorigenesis by enzymatic inhibition of α-KG-dependent dioxygenases. The differential response of SDH dysfunction in different cell systems may be attributed to the strength of epigenetic effects which often varies between cell-types. This observation may have clinical implications, although one would need to be cautious about manipulation of these metabolites, lest other intracellular processes were disturbed.
As expected, Sdhd-KO thyroids exhibited aberrant mitochondria, and studies in vitro demonstrated altered respiratory function, consistent with the metabolic remodeling. In the case of SDHD mutations, our data is consistent with the notion that the metabolic abnormalities can drive tumorigenesis, rather than occur as a secondary effect. The thyroid gland is a unique environment, in that it is a tissue that is highly reliant on intracellular oxidation (through the action of thyroid peroxidase) for its biological function of generating thyroid hormone. This fact may render thyrocytes either susceptible or more resistant to changes that affect reactive oxygen species. In our hands, we did not observe a noticeable effect on reactive oxygen species.
In summary, Sdhd knockout from the thyroid is sufficient to cause excess thyroid cell growth in mice, which may play a role in tumor initiation. In addition, the present results unveil a role for an aberrant TCA cycle in the generation of a stem-like phenotype in vivo and in vitro. Together, our data tie metabolic dysfunction with tumorigenic response, and warrant further studies for in-depth analysis of regulation of gene expression due to epigenetic changes in each model. These data also suggest that therapeutic reversal of DNA methylation may arise as an attractive approach to add to existing treatments of SDHx mutated tumors. The identification of a hypermethylator phenotype, albeit observed in single non-tumor derived NthyOri cell line helps in explaining both the tumor-suppressive role of SDH and the context-specific phenotypic characteristics. It warrants further studies of the impact of these mutations on specific genomic regions which will help us identify signaling pathways that play a role in the SDH related oncogenesis.
Supplementary Material
Metabolite levels in SDHD depleted cells. A. Mass spectrometry analysis of TCA cycle metabolites in SDHD depleted FTC133 cells. B. Mass spectrometry analysis of TCA cycle metabolites in SDHD depleted NthyOri 3.1 cells. C. Relative levels of ATP in FTC133 cells as analyzed by mass spectrometry. D. Relative levels of ATP in NthyOri 3.1 cells as analyzed by mass spectrometry. Graphs represent mean± SD of four biological replicates. Statistical analyses were performed by two-tailed student’s t-test. (*P value ≤ 0.05, **P value ≤ 0.01, ns= non significant).
SDHD depletion does not induce ROS in vitro. A. ROS levels in FTC133 cells as analyzed by flow cytometry. B. ROS levels in NthyOri 3.1 cells as analyzed by flow cytometry. C. Graph represents mean of percent FITC positive cells calculated from three independent experiments gated at 5% of untreated Sh-control cells. NAC (1mM) treatment was used as a negative control and H2O2 (1mM) treatment was used as a positive control. Statistical analyses were performed by two-tailed student’s t-test. (P value = non significant).
SDHD deletion activates mTOR pathway in vivo. A. p-mTOR staining by immunohistochemistry in formalin fixed thyroid sections of WT, Sdhd-TpoKO, Pten-TpoKO and SP-TpoKO mice (200x magnification). B. Western blot of mTOR in SDHD-KD cell lines.
Acknowledgments
We would like to thank Dr Jean-Christophe Cocuron and the OSU Targeted Metabolomics Laboratory for the mass spectrometry analysis of cellular metabolites. We would also like to acknowledge Dr. Xuguang Zhu and Shueu-yann Cheng (Laboratory of Molecular Biology, National Cancer Institute, Bethesda, MD) for performing TSH measurements on the mouse serum. Dr Matthew Ringel and Dr Charis Eng provided insightful comment on this work during its development. This work was supported by NIH grant P01CA124570 (to LSK) and P30CA0168058 (OSU Comprehensive Cancer Center). AA, DH, and AM were supported in part by individual Pelotonia Fellowship Program grants.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- Archetti M. Heterogeneity and proliferation of invasive cancer subclones in game theory models of the Warburg effect. Cell Prolif. 2015;48:259–269. doi: 10.1111/cpr.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspuria PJ, Lunt SY, Varemo L, Vergnes L, Gozo M, Beach JA, Salumbides B, Reue K, Wiedemeyer WR, Nielsen J, et al. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab. 2014;2:21. doi: 10.1186/2049-3002-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta. 2011;1807:1432–1443. doi: 10.1016/j.bbabio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005;2(Suppl 1):S4–11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- Briere JJ, Favier J, Benit P, El Ghouzzi V, Lorenzato A, Rabier D, Di Renzo MF, Gimenez-Roqueplo AP, Rustin P. Mitochondrial succinate is instrumental for HIF1alpha nuclear translocation in SDHA-mutant fibroblasts under normoxic conditions. Hum Mol Genet. 2005;14:3263–3269. doi: 10.1093/hmg/ddi359. [DOI] [PubMed] [Google Scholar]
- Burgess RJ, Agathocleous M, Morrison SJ. Metabolic regulation of stem cell function. J Intern Med. 2014;276:12–24. doi: 10.1111/joim.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012;2:881–898. doi: 10.1158/2159-8290.CD-12-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518:413–416. doi: 10.1038/nature13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies L, Morris LG, Haymart M, Chen AY, Goldenberg D, Morris J, Ogilvie JB, Terris DJ, Netterville J, Wong RJ, et al. American Association of Clinical Endocrinologists and American College of Endocrinology Disease State Clinical Review: The Increasing Incidence of Thyroid Cancer. Endocr Pract. 2015;21:686–696. doi: 10.4158/EP14466.DSCR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denkert C, Budczies J, Weichert W, Wohlgemuth G, Scholz M, Kind T, Niesporek S, Noske A, Buckendahl A, Dietel M, et al. Metabolite profiling of human colon carcinoma--deregulation of TCA cycle and amino acid turnover. Mol Cancer. 2008;7:72. doi: 10.1186/1476-4598-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edalat A, Schulte-Mecklenbeck P, Bauer C, Undank S, Krippeit-Drews P, Drews G, Dufer M. Mitochondrial succinate dehydrogenase is involved in stimulus-secretion coupling and endogenous ROS formation in murine beta cells. Diabetologia. 2015;58:1532–1541. doi: 10.1007/s00125-015-3577-9. [DOI] [PubMed] [Google Scholar]
- Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Hobert JA, Eng C. PTEN hamartoma tumor syndrome: an overview. Genet Med. 2009;11:687–694. doi: 10.1097/GIM.0b013e3181ac9aea. [DOI] [PubMed] [Google Scholar]
- Jardim-Messeder D, Caverzan A, Rauber R, de Souza Ferreira E, Margis-Pinheiro M, Galina A. Succinate dehydrogenase (mitochondrial complex II) is a source of reactive oxygen species in plants and regulates development and stress responses. New Phytol. 2015;208:776–789. doi: 10.1111/nph.13515. [DOI] [PubMed] [Google Scholar]
- Kitahara CM, Sosa JA. The changing incidence of thyroid cancer. Nat Rev Endocrinol. 2016;12:646–653. doi: 10.1038/nrendo.2016.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakabe T, Kawaguchi A, Kawaguchi R, Feigenbaum L, Kimura S. Thyrocyte-specific expression of Cre recombinase in transgenic mice. Genesis. 2004;39:212–216. doi: 10.1002/gene.20043. [DOI] [PubMed] [Google Scholar]
- Leenhardt L, Grosclaude P, Cherie-Challine L. Increased incidence of thyroid carcinoma in france: a true epidemic or thyroid nodule management effects? Report from the French Thyroid Cancer Committee. Thyroid. 2004;14:1056–1060. doi: 10.1089/thy.2004.14.1056. [DOI] [PubMed] [Google Scholar]
- Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- Li Q, Ye L, Zhang X, Wang M, Lin C, Huang S, Guo W, Lai Y, Du H, Li J, et al. FZD8, a target of p53, promotes bone metastasis in prostate cancer by activating canonical Wnt/beta-catenin signaling. Cancer Lett. 2017;402:166–176. doi: 10.1016/j.canlet.2017.05.029. [DOI] [PubMed] [Google Scholar]
- Lu C, Venneti S, Akalin A, Fang F, Ward PS, Dematteo RG, Intlekofer AM, Chen C, Ye J, Hameed M, et al. Induction of sarcomas by mutant IDH2. Genes Dev. 2013;27:1986–1998. doi: 10.1101/gad.226753.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, Watson DG, Gottlieb E. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol. 2007;27:3282–3289. doi: 10.1128/MCB.01927-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh DJ, Coulon V, Lunetta KL, Rocca-Serra P, Dahia PL, Zheng Z, Liaw D, Caron S, Duboue B, Lin AY, et al. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet. 1998;7:507–515. doi: 10.1093/hmg/7.3.507. [DOI] [PubMed] [Google Scholar]
- Millan-Ucles A, Diaz-Castro B, Garcia-Flores P, Baez A, Perez-Simon JA, Lopez-Barneo J, Piruat JI. A conditional mouse mutant in the tumor suppressor SdhD gene unveils a link between p21(WAF1/Cip1) induction and mitochondrial dysfunction. PLoS One. 2014;9:e85528. doi: 10.1371/journal.pone.0085528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison PJ, Atkinson AB. Genetic aspects of familial thyroid cancer. Oncologist. 2009;14:571–577. doi: 10.1634/theoncologist.2009-0046. [DOI] [PubMed] [Google Scholar]
- Nagayama Y, Shimamura M, Mitsutake N. Cancer Stem Cells in the Thyroid. Front Endocrinol (Lausanne) 2016;7:20. doi: 10.3389/fendo.2016.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes. Oncogene. 2004;23:6445–6470. doi: 10.1038/sj.onc.1207714. [DOI] [PubMed] [Google Scholar]
- Ni Y, Seballos S, Ganapathi S, Gurin D, Fletcher B, Ngeow J, Nagy R, Kloos RT, Ringel MD, LaFramboise T, et al. Germline and somatic SDHx alterations in apparently sporadic differentiated thyroid cancer. Endocr Relat Cancer. 2015;22:121–130. doi: 10.1530/ERC-14-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Y, Zbuk KM, Sadler T, Patocs A, Lobo G, Edelman E, Platzer P, Orloff MS, Waite KA, Eng C. Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am J Hum Genet. 2008;83:261–268. doi: 10.1016/j.ajhg.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nose V. Familial thyroid cancer: a review. Mod Pathol. 2011;24(Suppl 2):S19–33. doi: 10.1038/modpathol.2010.147. [DOI] [PubMed] [Google Scholar]
- Pardal R, Molofsky AV, He S, Morrison SJ. Stem cell self-renewal and cancer cell proliferation are regulated by common networks that balance the activation of proto-oncogenes and tumor suppressors. Cold Spring Harb Symp Quant Biol. 2005;70:177–185. doi: 10.1101/sqb.2005.70.057. [DOI] [PubMed] [Google Scholar]
- Pringle DR, Vasko VV, Yu L, Manchanda PK, Lee AA, Zhang X, Kirschner JM, Parlow AF, Saji M, Jarjoura D, et al. Follicular thyroid cancers demonstrate dual activation of PKA and mTOR as modeled by thyroid-specific deletion of Prkar1a and Pten in mice. J Clin Endocrinol Metab. 2014;99:E804–812. doi: 10.1210/jc.2013-3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pringle DR, Yin Z, Lee AA, Manchanda PK, Yu L, Parlow AF, Jarjoura D, La Perle KM, Kirschner LS. Thyroid-specific ablation of the Carney complex gene, PRKAR1A, results in hyperthyroidism and follicular thyroid cancer. Endocr Relat Cancer. 2012;19:435–446. doi: 10.1530/ERC-11-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Torres M, Allan AL. Aldehyde dehydrogenase as a marker and functional mediator of metastasis in solid tumors. Clin Exp Metastasis. 2016;33:97–113. doi: 10.1007/s10585-015-9755-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K, Hiltunen M, Kauppinen A. Krebs cycle dysfunction shapes epigenetic landscape of chromatin: novel insights into mitochondrial regulation of aging process. Cell Signal. 2014;26:1598–1603. doi: 10.1016/j.cellsig.2014.03.030. [DOI] [PubMed] [Google Scholar]
- Sciacovelli M, Frezza C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic Biol Med. 2016;100:175–181. doi: 10.1016/j.freeradbiomed.2016.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szarek E, Ball ER, Imperiale A, Tsokos M, Faucz FR, Giubellino A, Moussallieh FM, Namer IJ, Abu-Asab MS, Pacak K, et al. Carney triad, SDH-deficient tumors, and Sdhb+/− mice share abnormal mitochondria. Endocr Relat Cancer. 2015;22:345–352. doi: 10.1530/ERC-15-0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomsic J, He H, Akagi K, Liyanarachchi S, Pan Q, Bertani B, Nagy R, Symer DE, Blencowe BJ, de la Chapelle A. A germline mutation in SRRM2, a splicing factor gene, is implicated in papillary thyroid carcinoma predisposition. Sci Rep. 2015;5:10566. doi: 10.1038/srep10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson SR, Eble JN, Amin MB, Gupta NS, Smith SC, Sholl LM, Montironi R, Hirsch MS, Hornick JL. Succinate dehydrogenase-deficient renal cell carcinoma: detailed characterization of 11 tumors defining a unique subtype of renal cell carcinoma. Mod Pathol. 2015;28:80–94. doi: 10.1038/modpathol.2014.86. [DOI] [PubMed] [Google Scholar]
- Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeager N, Klein-Szanto A, Kimura S, Di Cristofano A. Pten loss in the mouse thyroid causes goiter and follicular adenomas: insights into thyroid function and Cowden disease pathogenesis. Cancer Res. 2007;67:959–966. doi: 10.1158/0008-5472.CAN-06-3524. [DOI] [PubMed] [Google Scholar]
- Yu W, He X, Ni Y, Ngeow J, Eng C. Cowden syndrome-associated germline SDHD variants alter PTEN nuclear translocation through SRC-induced PTEN oxidation. Hum Mol Genet. 2015;24:142–153. doi: 10.1093/hmg/ddu425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Metabolite levels in SDHD depleted cells. A. Mass spectrometry analysis of TCA cycle metabolites in SDHD depleted FTC133 cells. B. Mass spectrometry analysis of TCA cycle metabolites in SDHD depleted NthyOri 3.1 cells. C. Relative levels of ATP in FTC133 cells as analyzed by mass spectrometry. D. Relative levels of ATP in NthyOri 3.1 cells as analyzed by mass spectrometry. Graphs represent mean± SD of four biological replicates. Statistical analyses were performed by two-tailed student’s t-test. (*P value ≤ 0.05, **P value ≤ 0.01, ns= non significant).
SDHD depletion does not induce ROS in vitro. A. ROS levels in FTC133 cells as analyzed by flow cytometry. B. ROS levels in NthyOri 3.1 cells as analyzed by flow cytometry. C. Graph represents mean of percent FITC positive cells calculated from three independent experiments gated at 5% of untreated Sh-control cells. NAC (1mM) treatment was used as a negative control and H2O2 (1mM) treatment was used as a positive control. Statistical analyses were performed by two-tailed student’s t-test. (P value = non significant).
SDHD deletion activates mTOR pathway in vivo. A. p-mTOR staining by immunohistochemistry in formalin fixed thyroid sections of WT, Sdhd-TpoKO, Pten-TpoKO and SP-TpoKO mice (200x magnification). B. Western blot of mTOR in SDHD-KD cell lines.